

Abstract
Background & Aims:
Obesity is a risk factor for pancreatic cancer. In mice, a high-fat diet (HFD) and expression of oncogenic KRAS lead to development of invasive pancreatic ductal adenocarcinoma (PDAC) by unknown mechanisms. We investigated how oncogenic KRAS regulates the expression of fibroblast growth factor 21 (FGF21), a metabolic regulator that prevents obesity, and the effects of recombinant human FGF21 (rhFGF21) on pancreatic tumorigenesis.
Methods:
We performed immunohistochemical analyses of FGF21 levels in human pancreatic tissue arrays, comprising 59 PDAC specimens and 45 non-tumor tissues. We also studied mice with tamoxifen-inducible expression of oncogenic KRAS in acinar cells (KrasG12D/+ mice) and fElasCreERT mice (controls). KrasG12D/+ mice were placed on a HFD or regular chow diet (control) and given injections of rhFGF21 or vehicle; pancreata were collected and analyzed by histology, immunoblots, quantitative PCR, and immunohistochemistry. We measured markers of inflammation in the pancreas, liver, and adipose tissue. Activity of RAS was measured based on the amount of bound GTP.
Results:
Pancreatic tissues of mice expressed high levels of FGF21 compared with liver. FGF21 and its receptor proteins were expressed by acinar cells. Acinar cells that expressed KrasG12D/+ had significantly lower expression of Fgf21 mRNA, compared with acinar cells from control mice, partly due to downregulation of PPARG expression—a transcription factor that activates Fgf21 transcription. Pancreata from KrasG12D/+ mice on a control diet and given injections of rhFGF21 had reduced pancreatic inflammation, infiltration by immune cells, and acinar-to-ductal metaplasia compared with mice given injections of vehicle. HFD-fed KrasG12D/+mice given injections of vehicle accumulated abdominal fat, developed extensive inflammation, pancreatic cysts, and high-grade pancreatic intraepithelial neoplasias (PanINs); half the mice developed PDAC with liver metastases. HFD-fed KrasG12D/+ mice given injections of rhFGF21 had reduced accumulation of abdominal fat and pancreatic triglycerides, fewer pancreatic cysts, reduced systemic and pancreatic markers of inflammation, fewer PanINs, and longer survival— only about 12% of mice developed PDACs and none of the mice had metastases. Pancreata from HFD-fed KrasG12D/+ mice given injections of rhFGF21 had lower levels of active RAS than from mice given vehicle.
Conclusions:
Normal acinar cells from mice and humans express high levels of FGF21. In mice, acinar expression of oncogenic KRAS significantly reduces FGF21 expression. When these mice are placed on a HFD, they develop extensive inflammation, pancreatic cysts, PanINs, and PDACs, which are reduced by injection of FGF21. FGF21 also reduces the GTP binding capacity of RAS. FGF21 might be used in prevention or treatment of pancreatic cancer.
Keywords: gene regulation, FGFR1, KLB, signaling
Graphical Abstract
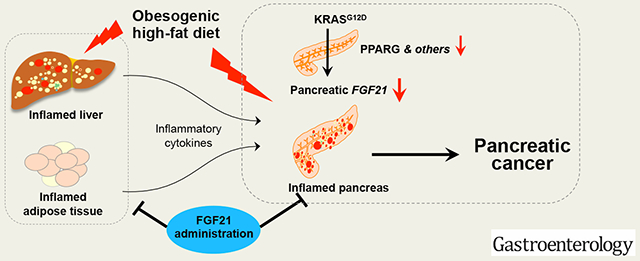
Lay Summary
A high-fat diet promotes development of pancreatic cancer. We found that acinar cells that express oncogenic KRAS reduce expression of fibroblast growth factor 21. In mice on a high-fat diet, injection of fibroblast growth factor 21 reduces pancreatic inflammation and tumor development.
Introduction
Pancreatic cancer is a fatal disease and is predicted to become the second leading cause of cancer-related deaths within a decade1. It is widely observed that mutations of KRAS (e.g., KRASG12D) are an essential initiating event for pancreatic cancer2. Previous studies have shown that mutant Kras, when expressed in pancreatic progenitor cells from the embryonic stage, is sufficient to drive PDAC in the Pdx-1Cre;KrasLSL-G12D/+ (KC) mice3. However, human PDAC likely originates from somatic mutations in KRAS in acinar cells during adulthood, suggesting that mice expressing mutant KRAS in acinar cells after birth may be more clinically relevant. Notably, mutant Kras expressed at an endogenous level in adult acinar cells does not, on its own, lead to PDAC4, 5, suggesting that an additional challenge is required.
Obesity is a modifiable risk factor for PDAC, and HFD is a major dietary component that drives obesity. Recent studies have shown that when fed a HFD, mice expressing an endogenous level of KrasG12D/+ in acinar cells developed marked inflammation, PanIN lesions, and invasive PDAC leading to lethality with high penetrance6. However, the mechanism underlying the cooperation between HFD and oncogenic KRAS remains a major knowledge gap.
The endocrine FGF21 has emerged as a novel metabolic regulator for the maintenance of lipid, glucose, and energy homeostasis7, 8. Studies have shown that the liver is one of the major sources of endocrine FGF21 under stress conditions9–11. By serving as an endocrine factor, FGF21 promotes the clearance of systemic lipids and glucose, while enhancing insulin sensitivity, fatty acid oxidation, and energy expenditure without apparent effects on cell proliferation and growth7, 12. Therefore, FGF21 has the pharmacological potential to treat obesity, diabetes, and meta-inflammation7, 12–14. The effects of endocrine FGF21 are mediated by binding to the transmembrane binary complex of fibroblast growth factors receptor 1 (FGFR1) and β-Klotho (KLB)15–17. In the pancreas, under wild-type KRAS conditions, FGF21 acts directly on acinar cells to protect the pancreas from pancreatitis and proteotoxic stress18, 19; however, the interaction between pancreatic FGF21 and oncogenic KRAS in pancreatic tumorigenesis has not been previously shown.
In the current study, by using mice with conditional knock-in of KrasLSL-G12D/+ under the control of a TM-inducible fElasCreERT driver as described6, 20, 21, we report that FGF21, its target receptor FGFR1, and its co-receptor KLB were abundantly expressed in normal acinar cells, suggesting that acinar cells are both a source and a target of FGF21. The expression of pancreatic FGF21 was dramatically downregulated, partly through a PPARG-dependent mechanism, upon an endogenous level of KrasG12D/+ expression. Remarkably, under chronic HFD challenge, administration of rhFGF21, which compensates for the loss of FGF21 in acinar cells, significantly suppressed both pancreatic and systemic inflammation, inhibited RAS activation, PanIN lesions, tumor incidence, and liver metastasis, as well as extended the survival of KrasG12D/+ mice in comparison to those without rhFGF21 treatment. Our results reveal that pancreatic FGF21 is downregulated by KRASG12D, which renders the pancreas vulnerable to obesogenic HFD challenge, leading to exacerbated inflammation and invasive PDAC, while pharmacological FGF21 counteracts the damaging effects of HFD and mitigates this vulnerability.
Materials and Methods
Mouse strains
CreERT-LoxP recombination approaches were used to control gene expression in pancreatic acinar cells. KrasLSL-G12D/+ mice, which possess the conditional knock-in of mutant KrasG12D, were obtained as described6. fElasCreERT mice, which express TM-regulated Cre recombinase under a full-length Elastase promoter specifically in acinar cells, were described previously22. KrasLSL-G12D/+ mice and fElasCreERT mice were cross-bred to generate fElasCreERT;KrasLSL-G12D/+ double-transgenic mice (called KrasG12D/+ after TM)6, 23. Alternatively, Ptf1aCreERT mice24 were crossed with KrasLSL-G12D/+ mice to generate Ptf1aCreERT;KrasLSL-G12D/+ mice (called Ptf1aCreERT;KrasG12D/+ after TM)25. Trp53LSL-R172H mice26 were crossed with fElasCreERT mice to generate fElasCreERT;Trp53LSL-R172H/+ (called Trp53R172H/+ after TM) mice. All animal experiments were reviewed and approved by Stony Brook University Institutional IACUC and the University of Texas, MD Anderson Cancer Center IACUC.
Additional experimental details are provided in the Supplementary Information.
Results
Pancreatic acinar cells are both a potential source and a target of FGF21
Previous studies have shown that HFD and KRASG12D cooperate to promote PDAC with high penetrance6, suggesting that acinar cells containing mutant KRAS are susceptible to chronic HFD challenge. FGF21 is an important regulator of lipid metabolism and elicits anti-obesity effects8, 12 while also protecting the pancreas from pancreatitis and metabolic stress18, 19. In order to understand the role of FGF21 in the pancreas and pancreatic tumorigenesis, we employed the established fElasCreERT and fElasCreERT;KrasLSL-G12D/+ mice6, 23. We first compared the pancreatic and hepatic Fgf21 expression in young adult fElasCreERT control mice one week post-TM. Pancreatic Fgf21 expression was significantly higher (>80 times) than that of hepatic tissue, which is known as the primary source of endocrine FGF21 under stress conditions (Figure 1A). This result concurs with a previous report showing that Fgf21 is highly expressed in the normal pancreas27. Immunohistochemistry (IHC) and Western blot further confirmed the presence of FGF21 in acinar cells (Figure 1B–C). Correspondingly, the expression of pancreatic FGF21 receptor, Fgfrl, was significantly higher (>10 times) than that of the liver (Figure 1D–E). The expression of FGF21 co-receptor, Klb, was comparable to that of the liver (Figure 1F), which is known to express a high level of Klb28, 29. Western blot (Figure 1G) and IHC (Figure 1H, open arrow) studies further confirmed the abundant presence of KLB in acinar cells. Comparison of Fgf21, Fgfr1, and Klb expression across the liver, pancreas, and adipose tissues in 7-month-old fElasCreERT mice after five months of TM induction revealed that pancreatic Fgf21 expression remained the highest (Figure 1I). Both the pancreas and adipose tissue expressed significantly higher levels of Fgfr1 than the liver (Figure 1J). Klb expression levels were comparable across all tissues (Figure 1K). Pancreatic Fgf21 expression was stable regardless of the age and the duration post TM treatment (Supplementary Figure 1A). All these data reveal that FGF21, FGFR1, and KLB are highly expressed in normal acinar cells, suggesting that pancreatic acinar cells are both a source and a target of FGF21.
Figure 1. Pancreatic acinar cells are both a source and a target of FGF21.
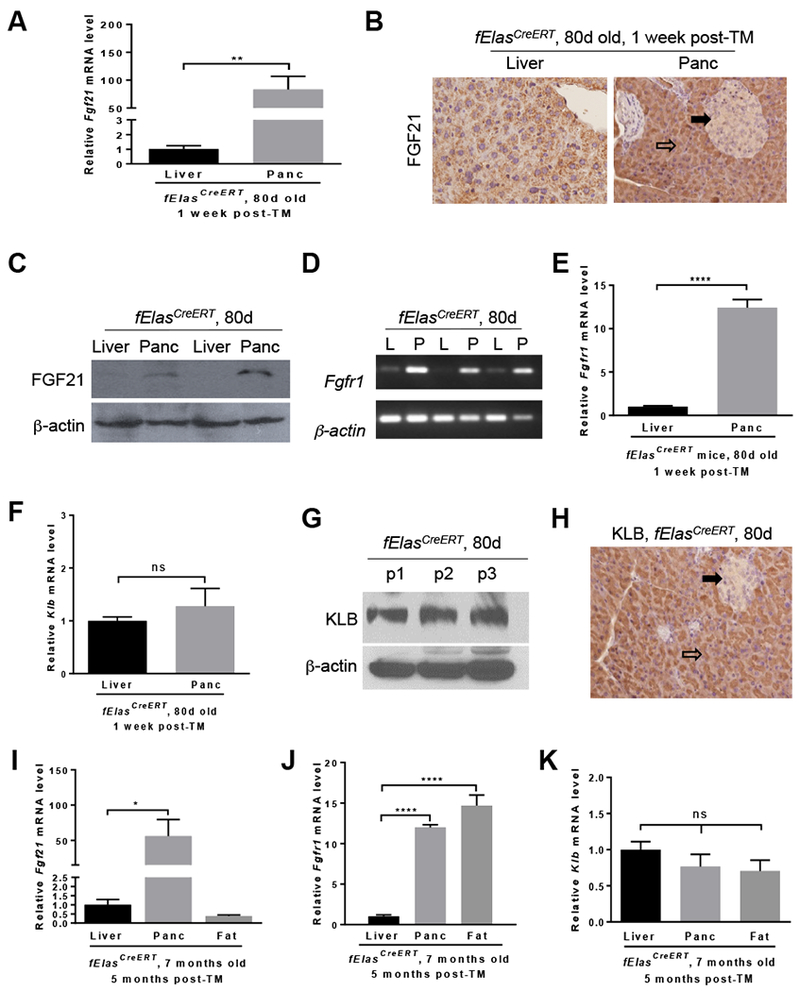
All mice in A-H were fed a ND, induced by TM at 70 days of age, and sacrificed one week post-TM. (A) qRT-PCR analysis of Fgf21 expression in the liver (n=11) and pancreas (Panc, n=11) of fElasCreERT mice. (B) IHC analysis of FGF21 on the liver and pancreas sections. 200x. (C) Western blot analysis for FGF21 levels. (D) Semi-quantitative RT-PCR analysis for Fgfrl in the liver (L) and pancreas (P). (E-F) qRT-PCR analysis for Fgfrl and Klb in the liver (n=9) and pancreas (n=9). (G) Western blot analysis for KLB in the pancreas (p). (H) IHC analysis of KLB in pancreatic sections. 200x. All mice in I-K were fed a ND, induced by TM at 70 days of age, and sacrificed at 7 months of age. Comparative qRT-PCR analysis for Fgf21 (I), Fgfr1 (J) and Klb (K) in the liver (n=7), pancreas (n=7), and adipose tissue (n=7) of fElasCreERT mice. Open arrow, acinar cells. Closed arrow, islet cells. Data are means ± SEM. *, p<0.05; **, p<0.01; ****, p<0.0001; ns, not significant.
FGF21 is a downstream target of oncogenic KRASG12D
To test if FGF21 plays a role in human pancreatic cancer, we performed IHC analysis of FGF21 in human pancreatic tissue arrays and found that the PDAC specimens (n=59) had substantially lower levels of FGF21 than normal human pancreatic tissues (n=45), which exhibited strong FGF21 positive staining in acinar cells (Figure 2A–B). FGF21 was also stained positively in normal ducts of both the normal human pancreas and PDAC specimens (Supplementary Figure 2A, inset). In addition, from the human GEO database (https://www.ncbi.nlm.nih.gov/geoprofiles/78912317), FGF21 expression in human PDAC tissues dropped significantly in comparison to the paired normal pancreata (n=39 pairs) (Figure 2C). Furthermore, FGF21 expression in human pancreatic cancer cell lines carrying KRAS mutations decreased significantly in comparison to that of normal human pancreatic ductal epithelial (HPDE) cells (Figure 2D), suggesting an inverse association between pancreatic FGF21 and oncogenic KRAS.
Figure 2. KRASG12D downregulates Fgf21 gene expression in acinar cells.
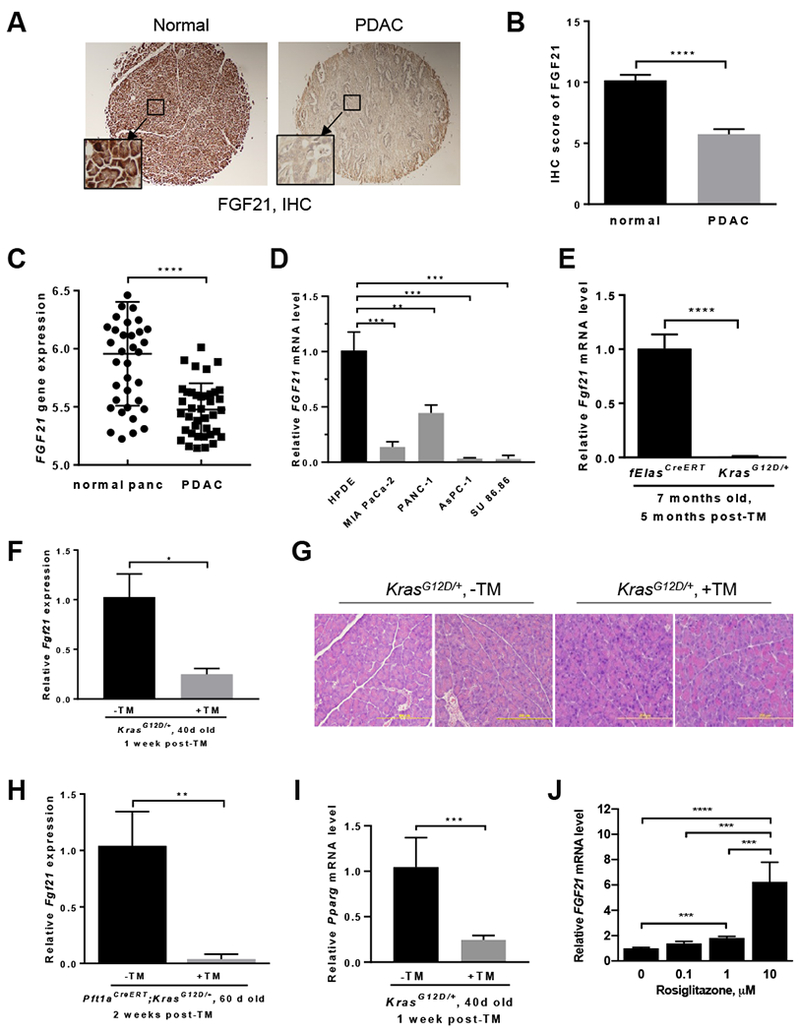
(A) IHC analysis for FGF21 levels in tissue arrays of human normal pancreas (n=45) and PDAC (n=59) (40x). Inset: 200x. (B) Quantitation of human FGF21 levels as described in 2A. (C) Human FGF21 expression in 39 pairs of normal and tumor tissues resected from pancreatic cancer patients as collected in the GEO database (https://www.ncbi.nlm.nih.gov/geo/tools/profileGraph.cgi?ID=GDS4103:221433_at). (D) qRT-PCR analysis of FGF21 in human pancreatic cancer cell lines carrying KRAS mutations. Human normal pancreatic ductal epithelial (HPDE) cells served as a control. All mice in E-I were fed a ND. (E) qRT-PCR analysis of pancreatic Fgf21 in 7-month-old KrasG12D/+ (n=11) or fElasCreERT mice (n=5) mice five months post-TM. (F) qRT-PCR analysis of pancreatic Fgf21 in 40-day-old KrasG12D/+ mice one week post-TM (n=7) or without TM (n=9). (G) Representative histology of the pancreata as in 2F. 200x. (H) qRT-PCR analysis of pancreatic Fgf21 of two-month-old Ptf1aCreERT;KrasG12D/+ mice two weeks post-TM (n=3) or without TM (n=3). (I) qRT-PCR analysis of pancreatic Pparg in 40-day-old KrasG12D/+ mice one week post-TM (n=7) or without TM (n=9). (J) qRT-PCR analysis of FGF21 in Panc-1 cells with Rosiglitazone treatment. Data are mean ± SD. *, p<0.05; **, p<0.01; ***, p<0.001; **** p<0.0001.
Given that PDAC likely originates from acinar cells with somatic mutations in KRAS occurring after birth, we next analyzed the potential changes of FGF21 in the fElasCreERT;KrasLSL-G12D/+ mice that express an endogenous level of KrasG12D/+ in nearly 100% acinar cells upon TM induction6. A striking decrease of >100 fold in Fgf21 expression was observed in mice five months post-TM relative to the age-matched fElasCreERT mice (Figure 2E). IHC analysis confirmed the substantially lower levels of FGF21 in the pancreatic tissues of KrasG12D/+ mice compared to that of fElasCreERT mice (Supplementary Figure 2B). To determine whether the drastic reduction of pancreatic Fgf21 is an early event related to KrasG12D/+ expression, we first compared Fgf21 expression in the 40-day-old fElasCreERT control mice one week post-TM to that without TM. No significant differences were observed between the two groups, suggesting that neither TM nor Cre recombinase affected Fgf21 expression (Supplementary Figure 2C). We then compared Fgf21 expression one week post-TM induction of KrasG12D/+ to that of the age-matched (40 days old) mice without TM and found that Fgf21 expression was significantly reduced upon KrasG12D/+ induction (Figure 2F), suggesting that the downregulation of Fgf21 is specific to KrasG12D/+. Notably, this occurred when no obvious alterations of pancreatic histology were observed (Figure 2G).
To further confirm our observation that KRASG12D specifically downregulates Fgf21 expression, we employed another mouse strain, the Ptf1aCreERT;KrasLSL-G12D/+ mice25. qRT-PCR data showed that Fgf21 expression was significantly reduced in these mice two weeks post-TM in comparison to the age-matched (two months of age) mice without TM induction (Figure 2H). No changes to the pancreatic histology were observed in the TM-induction group (Supplementary Figure 2D). Consistent with a role in tumorigenesis, the p53 mutations occur in 50–75% of human PDAC following an initiating mutation in KRAS. To determine if mutant p53 could also inhibit pancreatic Fgf21, we employed the fElasCreERT;Trp53LSL-R172H/+ mouse strain. Five months post-TM, no significant differences in pancreatic Fgf21 expression were observed between the age-matched Trp53R172H/+ and fElasCreERT mice (Supplementary Figure 2E), indicating that KrasG12D/+, but not Trp53R172H/+, specifically induces the marked downregulation of Fgf21. Furthermore, we compared Fgfr1 and Klb expression in the age-matched fElasCreERT and KrasG12D/+ mice one week post-TM. No significant changes for both genes were noticed (Supplementary Figure 2F–G).
Oncogenic KRAS reduces pancreatic Fgf21 expression partially through downregulating Pparg
To understand how oncogenic KRAS may potentially intervene to suppress FGF21 expression, we analyzed the expression of the transcription factors that are known to possess consensus binding sites on the Fgf21 promoter region and responsible for Fgf21 expression9, 10, 30–32. Among them, peroxisome proliferator-activated receptor gamma (Pparg, which encodes PPAR-gamma or PPARG), peroxisome proliferator-activated receptor alpha (Ppara), and retinoic acid receptor-related orphan receptor (Rora) were significantly downregulated (Figure 2I, Supplementary Figure 2H–I) in the same pancreatic tissues where Fgf21 was reduced by KrasG12D. However, other known Fgf21-regulating factors, such as Rarb and ChREBP (Supplementary Figure 2J–K), as well as SREBP1 and Lxrb (data not shown), were not changed. Treatment of mutant KRAS-bearing Panc-1 cells that have a weak FGF21 expression with a specific PPARG agonist, Rosiglitazone, resulted in a dose-dependent increase in FGF21 expression (Figure 2J), suggesting that PPARG is a potential mediator of KRASG12D-downregulated expression of pancreatic FGF21.
Replenishment of FGF21 inhibits KRASG12D-mediated pancreatic inflammation and PanIN lesions under normal dietary conditions
Studies have demonstrated that under wild-type KRAS conditions, FGF21 signaling was induced in acinar cells upon experimental induction of pancreatitis and that genetic deletion of Fgf21 exacerbated pancreatic injury19. FGF21 acts directly on acinar cells to promote exocrine secretion without increasing protein synthesis, thereby mitigating proteotoxic stress18, suggesting that acinar cell FGF21 alleviates metabolic stress and associated pathogenic effects in the presence of wild-type KRAS. By contrast, our above data reveal that oncogenic KRAS downregulates acinar cell FGF21.
To determine if FGF21 replenishment plays a role in restricting KrasG12D/+-mediated pancreatic inflammation and fibrosis under the normal diet (ND) conditions, we administered rhFGF21 to KrasG12D/+ mice for 10 weeks to compensate for the loss of acinar cell FGF21 (Figure 3A). rhFGF21 treatment significantly inhibited pancreatic inflammation and immune cell infiltration as measured by IHC staining of inducible cyclooxygenase-2 (COX-2) and F4/80, respectively, compared to those without rhFGF21 treatment (n=8) (Figure 3B, Supplementary Figure 3A–B). rhFGF21 also significantly reduced pancreatic fibrosis as measured by Sirius Red staining of collagen fibrils (Figure 3B, Supplementary Figure 3C). These data suggest that rhFGF21 supplementation effectively inhibits pancreatic inflammation and fibrosis induced by KRASG12D under the ND conditions.
Figure 3. Replenishment of FGF21 suppresses KRASG12D-induced pancreatic inflammation and PanIN lesions under ND conditions.
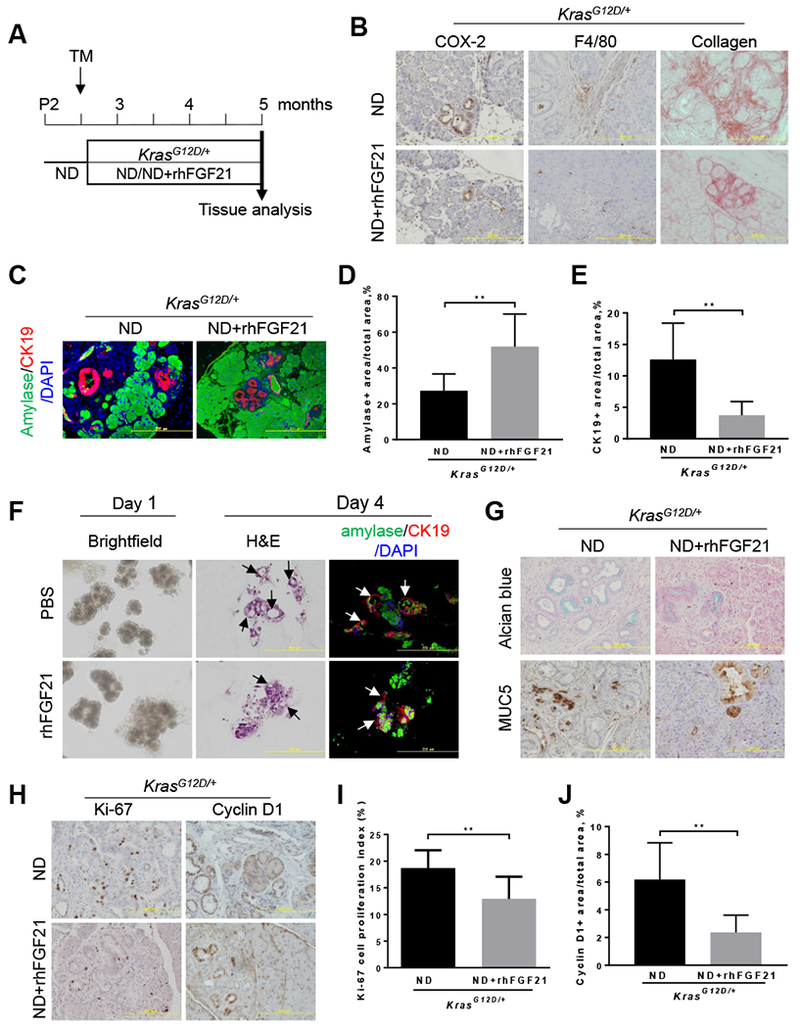
(A) Experimental scheme for B-E and G-J: 70-day-old ND-fed KrasG12D/+;fElasCreERT mice were treated with TM at 70 days of age to induce KrasG12D/+ expression in acinar cells, and then treated with rhFGF21 (0.5 mg/kg body weight/day) (n=10) or PBS (n=8) for ten weeks. (B) IHC staining of COX-2 and F4/80, and Sirius Red staining of collagen on pancreatic sections of KrasG12D/+ mice with indicated treatments. 200x. (C) Co-immunofluorescence (Co-IF) analysis of amylase and CK19 as indicated. 200x. (D-E) Quantitation of amylase+ areas or CK19+ areas on pancreatic sections (n=4) as described in 3C. (F) The explanted acinar cell 3D clusters isolated from 80-day-old KrasG12D/+ mice one week post-TM were treated daily with rhFGF21 or PBS and then analyzed by brightfield images on day 1, H&E staining on day 4, and Co-IF staining of amylase and CK19 on day 4. (G) Alcian blue staining of acidic mucins and IHC staining of MUC5. (H) IHC analyses of Ki-67 (ND, n=4; ND+rhFGF21, n=4) and Cyclin D1 (ND, n=5; ND+rhFGF21, n=3). (I-J) Quantitation of Ki-67 and Cyclin D1 levels in 3H. Results were expressed as mean ± SD. **, p<0.01.
To test whether rhFGF21 administration could inhibit KRASG12D-mediated acinar-to-ductal metaplasia (ADM), we evaluated acinar and duct cell markers in the ND-fed KrasG12D/+ mice upon rhFGF21 treatment. Co-immunofluorescence (Co-IF) analyses of amylase and CK19, which mark acinar and duct cells, respectively, revealed that rhFGF21 treatment significantly reduced CK19+ staining and preserved amylase+ acinar cells compared to those without rhFGF21 treatment (Figure 3C–E). These data suggest that rhFGF21 suppresses the ADM reprogramming induced by oncogenic KRAS under normal feeding conditions.
To determine whether rhFGF21 directly acts on acinar cells, we isolated acinar cell clusters from 80-day-old KrasG12D/+ mice one week post-TM and seeded them in collagen plates for 3-dimensional (3D) explant cultures with daily rhFGF21 treatment. On day one, treatment with rhFGF21 had little effects on the acinar cell clusters; however, by day four, rhFGF21 significantly inhibited ductal cell formation compared to PBS treatment group (Figure 3F, Supplementary Figure 3D), suggesting that rhFGF21 directly acts on acinar cells to inhibit KRASG12D-mediated ADM reprogramming.
As ADM is regarded as the precursor to the development of PanIN lesions, we evaluated the effects of FGF21 administration on KRASG12D-mediated PanIN lesions using Alcian blue staining for acidic mucins and IHC staining for Mucin 5AC (MUC5). rhFGF21 injections caused a noticeable reduction in both acidic mucins and MUC5 compared to those without FGF21 treatment (Figure 3G, Supplementary Figure 3E–F). Using IHC analysis, we observed that rhFGF21 suppressed ductal cell proliferation as measured by the decreased levels of Ki-67 and cyclin D1 (Figure 3H–J). Altogether, our data indicate that the exogenous supplementation of FGF21 to KrasG12D/+ mice that have a dramatic reduction of FGF21 in acinar cells results in significant beneficial effects on the inhibition of pancreatic inflammation, ADM, and PanIN lesions under the ND conditions.
Pharmacological administration of FGF21 inhibits HFD and KRASG12D induced PanIN lesions and PDAC
Inspired by the above data, we proceeded to determine if FGF21 plays a suppressive role in HFD and KRAS mediated invasive PDAC by treating the KrasG12D/+ mice concurrently with HFD and rhFGF21 for 10 weeks (Figure 4A). The HFD-fed KrasG12D/+ mice accumulated abdominal fat and developed multiple pancreatic cysts (Figure 4B). In contrast, treatment with rhFGF21 alleviated abdominal fat accumulation and pancreatic cysts (Figure 4B). Consistently, rhFGF21 administration reduced the accumulation of pancreatic triglycerides in comparison to those without rhFGF21 treatment (Supplementary Figure 4A). Histological analysis revealed that the HFD-fed KrasG12D/+ mice displayed high-grade PanIN lesions and PDAC with 50% penetrance (Figure 4C). In marked contrast, with rhFGF21 treatment, most of the mice (87.5%) exhibited well-preserved parenchyma and acinar cell content with only early stage PanIN lesions (Figure 4C–D). Only one out of eight mice treated with rhFGF21 developed PDAC.
Figure 4. Supplementation of FGF21 suppresses HFD and KRASG12D induced PanIN lesions and PDAC.
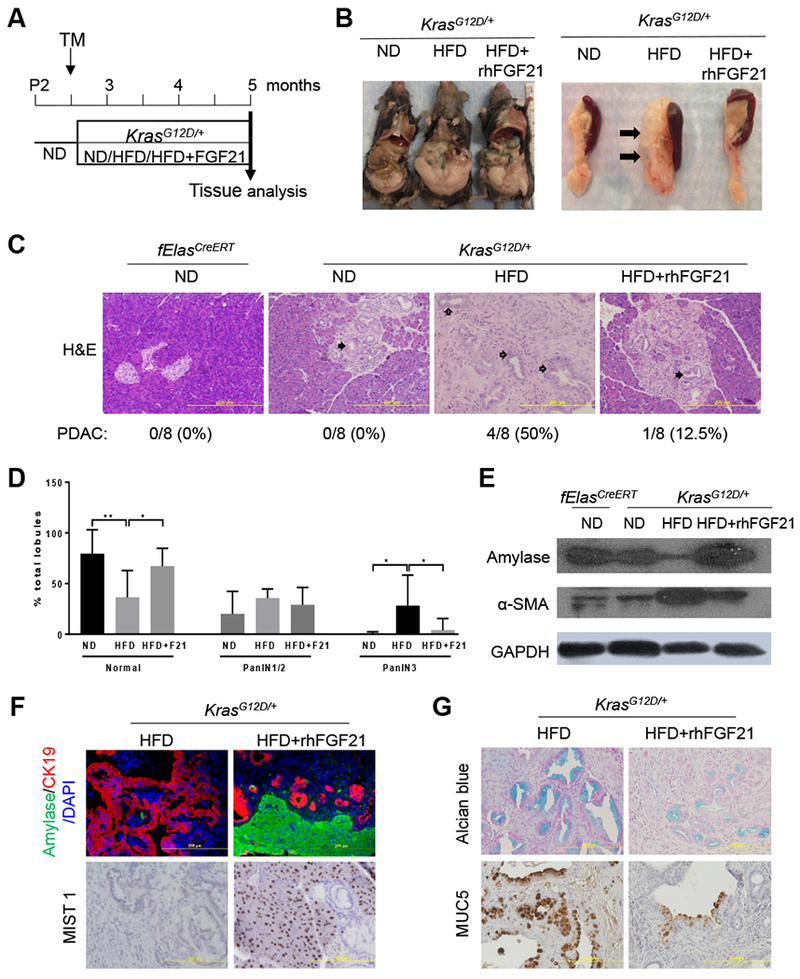
(A) Experimental scheme: 70-day-old ND-fed KrasG12D/+;fElasCreERT mice were treated with TM to induce KrasG12D/+ expression and randomly separated into ND, HFD, or HFD with rhFGF21 treatment groups (n=8 per group) for ten weeks. The age-matched ND-fed fElasCreERT mice (n=8) served as controls. (B) Gross images of KrasG12D/+ mice (left) and the pancreata (right) after the indicated treatments. Closed arrows, multiple pancreatic cysts. (C) Histology of representative pancreata. Closed arrows, PanIN lesions. Open arrows, cancerous ductal structure. 100x. (D) Average grades of PanIN1/2 and PanIN3 lesions as in 4C. F21, rhFGF21. (E) Western blot analysis of pancreatic amylase and α-SMA as indicated. (F) Co-IF analysis of pancreatic amylase and CK19 or IHC analysis of MIST1. 200x. (G) Alcian blue+ staining of acidic mucins and IHC staining of MUC5. 200x. Data are mean + SD. *, p<0.05; **, p<0.01.
Assessment of damage to pancreatic tissues revealed that chronic HFD consumption led to a significant decrease of amylase, which marks acinar cell content, while rhFGF21 treatment restored the lost amylase content (Figure 4E, Supplementary Figure 4B). Alpha-smooth muscle actin (α-SMA), which marks the levels of pancreatic stellate cell activation, was strongly elevated in the HFD-fed KrasG12D/+ mice. However, this elevation was significantly suppressed by chronic treatment with rhFGF21 (Figure 4E, Supplementary Figure 4C). The Co-IF analysis further revealed that rhFGF21 greatly reduced CK19+ staining and preserved acinar cells (amylase+) compared to the HFD-fed KrasG12D/+ mice without rhFGF21 treatment (Figure 4F, Supplementary Figure 4D–E). A similar pattern to that observed for amylase was found for MIST1, a transcription factor essential for maintaining an adult acinar cell phenotype (Figure 4F, Supplementary Figure 4F). These data suggest that rhFGF21 preserved acinar cells and inhibited stromal activation induced by HFD in KrasG12D/+ mice. Furthermore, we observed prominent positive stains of acidic mucins and MUC5 in the pancreata of the HFD-fed KrasG12D/+ mice; however, rhFGF21 treatment significantly inhibited these PanIN markers (Figure 4G, Supplementary Figure 4G–H). Our data suggest that the replenishment of FGF21 suppresses HFD and KRASG12D induced rapid PanIN lesions and PDAC.
To determine whether the significant downregulation of pancreatic FGF21 by KRASG12D can lead to a reduction of serum FGF21 levels, which may induce systemic metabolic alterations, we measured serum FGF21 levels pre-TM, one week and 10 weeks post-TM induction of KrasG12D/+. No significant differences in serum FGF21 levels were observed (Supplementary Figure 4I–J), indicating that alteration of pancreatic FGF21 by KRASG12D does not influence circulating FGF21 levels.
Pharmacological FGF21 inhibits HFD and KRASG12D induced pancreatic inflammation
Chronic HFD causes inflammation, which cooperates with oncogenic KRAS to drive PDAC4. To understand whether the tumor-suppressive effect of rhFGF21 is through alleviation of pancreatic inflammation, we analyzed the protein expression of COX-2, which was up-regulated in the HFD-fed KrasG12D/+ mice6. Indeed, these mice exhibited intense focal staining of COX-2 along the pancreatic ducts; however, rhFGF21 treatment significantly decreased COX-2+ staining to levels comparable to the ND-fed KrasG12D/+ mice (Figure 5A, Supplementary Figure 5A). Furthermore, macrophages, as detected by F4/80+ staining, were abundant in the pancreata of the HFD-fed KrasG12D/+ mice; however, rhFGF21 significantly inhibited HFD-induced macrophage infiltration (Figure 5A, Supplementary Figure 5B). In a similar manner, CD3, as a marker of peripheral T cell infiltration, was significantly elevated under chronic HFD insult, while rhFGF21 alleviated T cell infiltration (Figure 5A, Supplementary Figure 5C). Analysis of extracellular matrix collagen deposition that marks pancreatic fibrosis revealed that although the ND-fed fElasCreERT mice displayed a minimal collagen staining that was primarily localized around the blood vessels and the peri-lobular spaces, the HFD-fed KrasG12D/+ mice exhibited intense collagen deposition surrounding the neoplastic ductal structures, indicating a robust fibrotic response. In contrast, FGF21 supplementation significantly reduced collagen levels (Figures 5A, Supplementary Figure 5D).
Figure 5. Pharmacological FGF21 inhibits HFD and KRASG12D induced pancreatic inflammation and ductal cell proliferation.
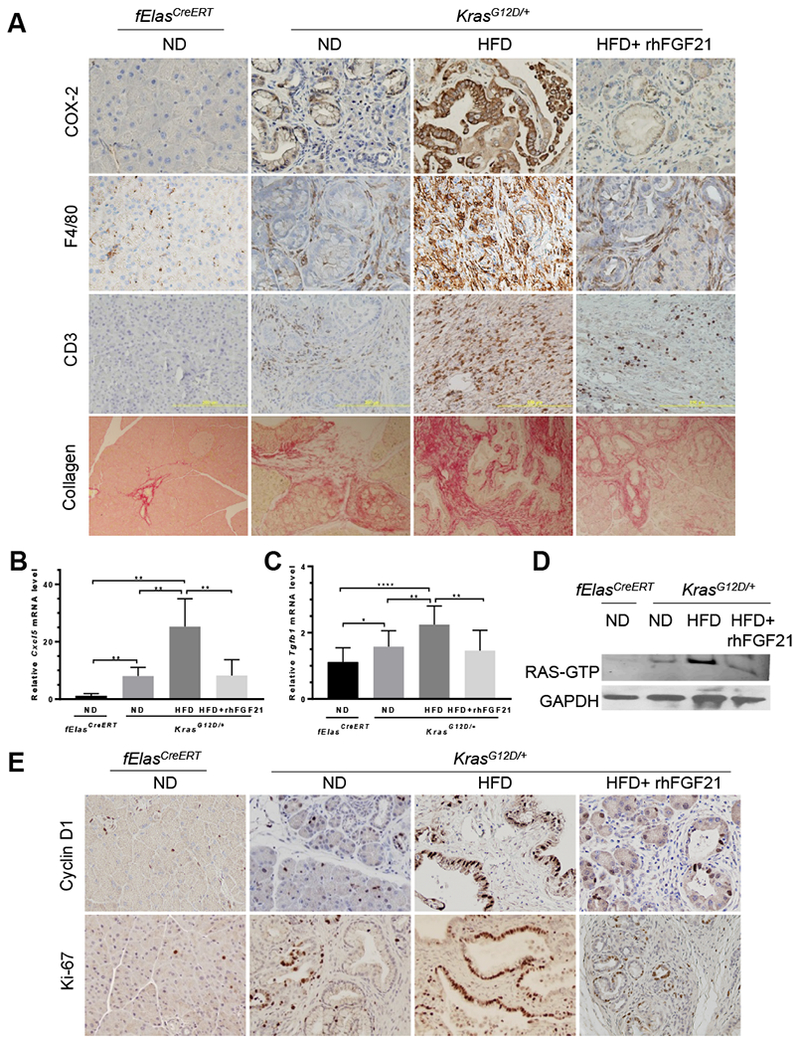
All the samples were described as in Figure 4A. (A) IHC staining of COX-2, F4/80, and CD3, as well as Sirius Red staining for collagen on pancreatic sections of KrasG12D/+ mice. 200x. (B-C) qRT-PCR analysis of pancreatic Cxc/5 and Tgfbl expression in KrasG12D/+ mice with ND (n=4), HFD (n=4) or HFD+rhFGF21 (n=5). The age-matched ND-fed fElasCreERT mice (n=5) served as a control. (D) Pancreatic RAS activity with the indicated treatments. (E) IHC staining of pancreatic cyclin D1 and Ki-67. 200x. Data are mean ± SD. *, p<0.05; **, p<0.01; ****, p<0.0001.
To better understand the suppressive role of FGF21 in pancreatic inflammation promoted by the cooperative interaction between HFD and KRASG12D, we analyzed the expression of an array of inflammation-associated cytokines/chemokines and receptors comparatively by qRT-PCR. Of note, the pancreata of the ND-fed KrasG12D/+ mice showed significant increases in the expression of Cxcl5, Tgfb1, Ccl2, Csf2, Tnf, and Tnfrsf11b in comparison to those of the ND-fed fElasCreERT mice, suggesting that pancreatic inflammation is mediated by oncogenic KRAS in this context (Figure 5B–C, Supplementary Figure 5E–H). HFD treatment further enhanced the expression of these genes (Figure 5B–C, Supplementary Figure 5E–H), indicating a significant inflammatory effect of obesogenic HFD on the pancreas. Interestingly, although pancreatic Ccl17 was not affected by KRASG12D under the ND conditions, it was markedly upregulated in response to the HFD challenge in the KrasG12D/+ mice (Supplementary Figure 5I). Importantly, rhFGF21 administration significantly suppressed the augmented expression of all these genes (Figure 5B–C, Supplementary Figure 5E–I). These data suggest a prominent anti-inflammatory role of FGF21 in the pancreas. On the other hand, the expression levels of the cytokines that have anti-inflammation or anti-neoplastic functions, including Ccl19, Ifng, Il21, Cd40l, Ccl21a, and Cxcl9, were significantly downregulated in the HFD-fed KrasG12D/+ mice, while administration of rhFGF21 effectively restored their expression (Supplementary Figure 5J–O).
We have previously shown that RAS activity is an important determinant in pancreatic tumorigenesis, and that inflammation enhances RAS activity21, 33. As pharmacological rhFGF21 significantly suppressed HFD-promoted pancreatic inflammation, we tested if this would lead to a reduction of RAS activity in the pancreata of the HFD-fed KrasG12D/+ mice. Indeed, we observed that the HFD-enhanced RAS hyperactivity was significantly inhibited by rhFGF21 (Figure 5D, Supplementary Figure 5P). These data indicate that the beneficial effects of FGF21 in protecting the pancreas from neoplastic progression are attributable to the inhibition of the accentuated inflammation and RAS hyperactivation induced by HFD.
Correspondingly, rhFGF21 treatment significantly reduced cyclin D1 and Ki-67 levels in the pancreatic ductal cells of the HFD-fed KrasG12D/+ mice compared to those without rhFGF21 treatment (Figure 5E, Supplementary Figure 5Q–R). These data indicate that the supplementation of rhFGF21 to compensate for the loss of pancreatic FGF21 inhibits pancreatic tumor cell proliferation induced by the synergistic cooperation between KRASG12D and HFD.
Pharmacological FGF21 inhibits HFD-induced liver and adipose tissue inflammation
As chronic HFD imposes adverse metabolic effects on adipose tissue and the liver, which are among the direct or indirect targets of endocrine FGF219, 10, 16, we therefore tested the anti-inflammatory effects of rhFGF21 on both tissues. The ND-fed KrasG12D/+ mice showed no significant differences in the inflammation markers compared to the ND-fed fElasCreERT mice in either the liver or the adipose tissue (Figure 6A–I, Supplementary Figure 6A–B), suggesting that the liver and adipose tissue do not impose significant inflammatory stress on the pancreas under the ND conditions, and therefore, the observed pancreatic inflammation and associated pathologies in Figure 3 are attributable to KRASG12D in this context. Thus, FGF21 plays a direct role in restricting KRASG12D-mediated pancreatic inflammation and associated pathogenesis.
Figure 6. Pharmacological FGF21 inhibits HFD-promoted liver and adipose tissue inflammation.
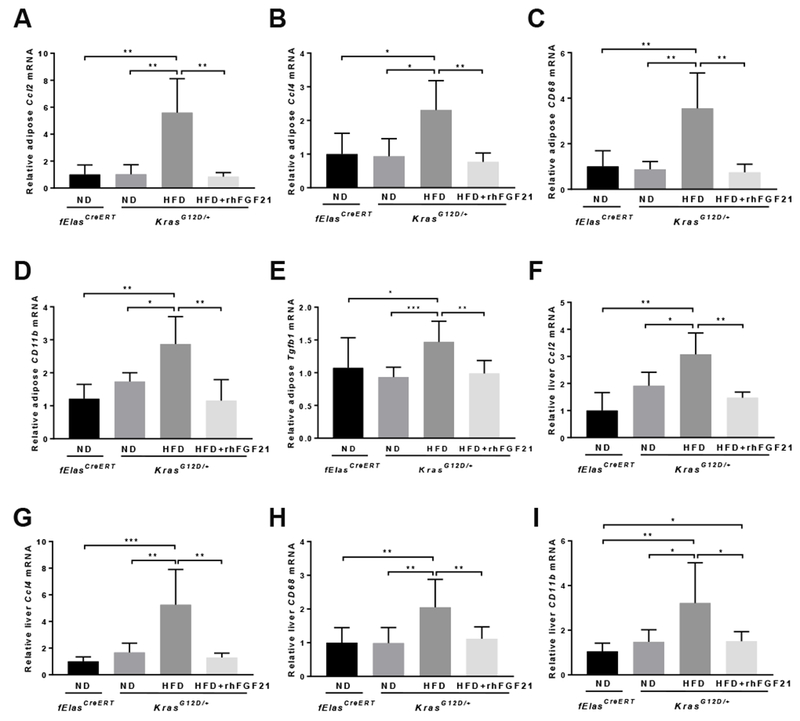
All mice were induced by TM at 70 days of age and then treated as indicated for 10 weeks prior to analyses. qRT-PCR analysis of Ccl2, Ccl4, CD68, CD11b, and Tgfbl in adipose tissues (A-E) and Ccl2, Ccl4, CD68, and CD11b in the liver (F-I) of KrasG12D/+ mice with ND (n=5), HFD (n=6), or HFD+rhFGF21 (n=5). The ND-fed fElasCreERT mice (n=5) were controls. Data are mean ± SD. *, p<0.05; **, p<0.01; ***, p<0.001.
Furthermore, chronic HFD significantly elevated the expression of Ccl2, Ccl4, CD68, and CD11b in both the liver and adipose tissues, as well as adipose Tgfbl, Tnf, and Ccl11, while rhFGF21 effectively suppressed these inflammation markers (Figure 6A–I, Supplementary Figure 6A–B). As a metabolic marker of adipose tissue response, adipose Ucp1 underwent a striking increase of >40 fold with rhFGF21 treatment (Supplementary Figure 6C). Furthermore, the anti-inflammatory and anti-fibrotic adipokine, Adipoq, was also significantly elevated upon rhFGF21 treatment (Supplementary Figure 6D). These data suggest that rhFGF21 inhibits the HFD-induced systemic inflammation that otherwise may impinge on the pancreas through secretory cytokines or adipokines.
Pharmacological FGF21 inhibits HFD and KRASG12D induced liver metastasis and prolongs survival
Next, we determined whether pharmacological administration of rhFGF21 would prolong the overall survival and inhibit liver metastasis in the HFD-fed KrasG12D/+ mice. We first observed that mice fed a HFD had a substantial increase in body weight; however, this was not evident under rhFGF21 treatment despite a continued HFD consumption (Figure 7A). Furthermore, the survival rate of the HFD-fed KrasG12D/+ mice significantly decreased compared to the age-matched ND-fed KrasG12D/+ mice, with median survival rates of 229 days and 352 days, respectively. Treatment with rhFGF21 prolonged the median survival of the HFD-fed KrasG12D/+ to 323 days (Figure 7B). KrasG12D/+ mice fed a ND and concurrently treated with rhFGF21 showed a better survival rate than the ND-fed KrasG12D/+ mice (Figure 7B), which do not spontaneously develop obesity. Regardless of the nature of the diets and the survival rates, the aged KrasG12D/+ mice eventually developed PDAC (Figure 7C). The cancerous cells formed ductal structures (closed arrow), showing severe architectural and cytological atypia with frequent mitotic figures and abundant interstitial fibrosis (Figure 7C).
Figure 7. Pharmacological FGF21 prevents liver metastasis and prolongs KRASG12D mice survival.
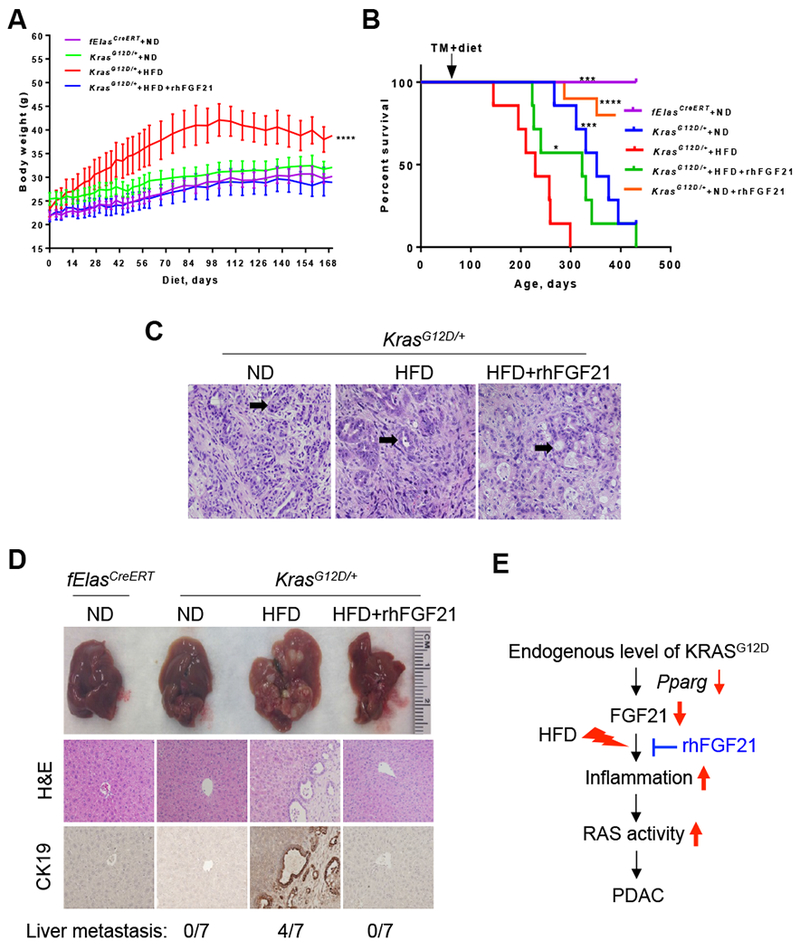
70-day-old mice were treated with TM to induce fElasCreERT or KrasG12D/+ expression, and then fed a ND or a HFD with or without rhFGF21 until they aged. The ND-fed fElasCreERT mice (n=7) were controls. (A) Body weight of the KrasG12D/+ mice with ND (n=7), HFD (n=7), or HFD+rhFGF21 treatment (n=7) until the HFD group aged. (B) Survival of the KrasG12D/+ mice with ND (n=7), ND+rhFGF21 (n=10), HFD (n=7), or HFD+rhFGF21 treatment (n=7). The HFD-fed KrasG12D/+ mice served as a control for statistical comparison. (C) Histological changes in pancreata of KrasG12D/+ mice with the indicated treatments upon aging. 200x. (D) Liver metastasis. Upper panel, gross images of the livers. Middle panel, H&E stained sections. Lower panel, IHC analysis of CK19. (E) Schematic summary of the role of pancreatic FGF21 in the HFD and oncogenic KRAS mediated pancreatic tumorigenesis. 200x. Data are mean ± SD. *, p<0.05; ***, p<0.001; ****, p<0.0001.
Over 50% of the HFD-fed KrasG12D/+ mice progressed to liver metastasis. Multiple metastatic nodules with distinct borders were mostly found located on the liver surface in the HFD treatment group (Figure 7D, upper panel). Histological analyses revealed prominent metastatic foci on liver sections of the HFD treatment mice (Figure 7D, middle panel), and CK19 positive staining further confirmed the liver metastases in the HFD-fed KrasG12D/+ mice. In contrast, the HFD-fed KrasG12D/+ mice concurrently treated with rhFGF21 failed to develop liver metastases (Figure 7D, lower panel, Supplementary Figure 7A).
In summary, our studies show that KRASG12D, partly through downregulating Pparg, reduces Fgf21, which in turn creates a vulnerability to obesogenic HFD challenge, leading to extensive inflammation, RAS hyperactivation, PanIN lesions, and invasive PDAC. Pharmacological supplementation of FGF21 mitigates both pancreatic and systemic inflammation, resulting in the suppression of RAS hyperactivity and PDAC development (Figure 7E).
Discussion
Obesity dramatically increases the risk of pancreatic cancer, which remains one of the most aggressive malignancies34, and the chronic consumption of a HFD is known as a primary dietary factor in promoting obesity. As the prevalence of obesity has been increasing at an alarming rate, obesity-associated pancreatic cancer poses an even greater threat. Thus, gaining a better understanding of HFD-promoted pancreatic cancer is urgent, as it will provide new insights into possible effective intervention strategies. Experimentally, HFD promotes oncogenic KRAS-mediated PDAC, indicating that mutations in KRAS render mice susceptible to obesogenic HFD insults leading to PDAC. However, the mechanism underlying such vulnerability is unclear. In this study, we made several novel observations concerning the roles of FGF21, metabolic regulator that prevents obesity, in the pancreas and oncogenic KRAS-mediated pancreatic tumorigenesis. First, we find that acinar cells express high levels of Fgf21, Fgfrl, and Klb under normal feeding conditions, suggesting that acinar cells are both a source and a target of FGF21. Second, we report for the first time that Fgf21 expression in acinar cells is highly sensitive to mutant KRASG12D, showing remarkable decrease upon the expression of KrasG12D/+ at an endogenous level when no overt alterations of pancreatic histology are noted. Third, we show that the significant downregulation of Fgf21 by mutant KRASG12D is partly mediated by the reduced PPARG. Fourth, we note that replenishment of the lost FGF21 directly inhibits pancreatic inflammation, transdifferentiation of acinar cells into ductal cells, and PanIN lesions under the ND conditions. Fifth, pharmacological FGF21 significantly mitigates inflammation in both the pancreas and systemic liver and adipose tissue under the HFD conditions. Sixth, FGF21 supplementation significantly inhibits HFD-promoted pancreatic RAS hyperactivity. Finally, pharmacological FGF21 significantly ameliorates pancreatic pathogenic responses, leading to the significant delay of PDAC development and extension of survival under obesogenic HFD challenge.
We have provided evidence to support that KRASG12D downregulates pancreatic FGF21 expression. First, we show that PADC patient tissues and mutant KRAS-bearing pancreatic cancer cells exhibit a significant reduction of FGF21 compared to their normal counterparts. Second, in several KrasG12D/+ mouse strains with different Cre drivers, we show that KRASG12D significantly inhibits Fgf21 expression. Third, we note that Pparg, Ppara, and Rora, which were shown to induce FGF21 expression9, 10, 30–32 are downregulated by KRASG12D expression. Finally, treatment of pancreatic cancer cells with PPARG agonist increased FGF21 expression in a dose-dependent manner, suggesting a role of PPARG in regulating pancreatic FGF21. It would be interesting to investigate further the functional roles of PPARG, PPARA, and RORA in regulating pancreatic FGF21 and pancreatic tumorigenesis.
Our results indicate a prominent role of FGF21 in directly protecting the pancreas from neoplastic progression induced by KRASG12D. First, we show that acinar cells express high levels of FGF21 and its receptor complex, indicating an autocrine/paracrine mode of FGF21 action in the pancreas, which aligns with previous findings showing that FGF21 directly targets acinar cells18, 19. Second, the pancreata of the ND-fed KrasG12D/+ mice, which do not develop obesity and systemic inflammation, were effectively protected from KRASG12D-driven inflammation and neoplastic progression by rhFGF21, indicating a direct protective effect of FGF21 on the pancreas with minimal systemic influence. Third, we show that rhFGF21 directly inhibits ADM in the in vitro 3D explant cultures of acinar clusters, which is not subject to systemic metabolic influence.
Pancreatic function and systemic metabolism are inextricably linked. In the KrasG12D/+ mice, chronic consumption of HFD leads to obesity and invasive PDAC, while rhFGF21 ameliorates these detrimental systemic and local pancreatic effects of HFD. We show that HFD induces inflammation in both the pancreas and systemic liver and adipose tissue, while these adverse effects are effectively inhibited by rhFGF21, emphasizing the broad anti-inflammatory role of FGF21. Decreased inflammation was accompanied by the suppression of inflammation-associated fibrosis, PanIN lesions, and PDAC development. Importantly, we observed that the HFD-enhanced pancreatic RAS activity was also significantly reduced by rhFGF21. Furthermore, previous studies have shown that inhibition of local pancreatic inflammation through ablation of COX-2 in acinar cells markedly suppressed HFD and KRASG12D mediated pancreatic neoplastic progression6, supporting that the inhibition of local pancreatic inflammation is critical to the suppression of pancreatic tumorigenesis. Collectively, our data suggest that in addition to the noticeable systemic anti-inflammatory effects, the anti-inflammatory effects of rhFGF21 on the pancreas are essential to the suppression of HFD-promoted inflammatory damage and RAS hyperactivation, leading to the protection of the pancreas from neoplastic progression.
Previous studies focused on the pathophysiological roles of FGF21 in the liver and pancreas in the setting of wild-type KRAS18, 19, 35, which showed that FGF21 exhibits a marked elevation in response to stress-inducing conditions, including the ingestion of a HFD11. Unlike these previous studies, however, our study shows that pancreatic FGF21 is negatively regulated by oncogenic KRAS. By abrogating FGF21, oncogenic KRAS allows the malevolent metabolic effect of chronic HFD consumption to take its maximal toll on the pancreas. In light of this fact, the loss of pancreatic FGF21 specifically by oncogenic KRAS is significant, as this breaks a stress-defensive barrier in the path towards PDAC development.
We observed multiple small pancreatic cysts in the HFD-fed KrasG12D/+ mice; however, rhFGF21 treatment effectively inhibited this phenomenon, suggesting that inflammation is likely the cause of pancreatic cysts. In patients, mucinous cystic lesions are precursors to invasive pancreatic cancer. Therefore, it is possible that FGF21 holds the potential to reverse mucinous pancreatic cysts and reduce the risk of pancreatic cancer in such patients.
In summary, our findings reveal a previously unknown critical vulnerability conferred by oncogenic KRAS, highlight pancreatic FGF21 as an important downstream target of oncogenic KRAS, and identify pharmacological FGF21 as a putative intervention strategy to hinder pancreatic inflammation and cancer development in the context of obesity.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT:
A high-fat diet (HFD) promotes obesity, which increases the risk of pancreatic cancer. In mice, a HFD and expression of oncogenic KRAS lead to the development of pancreatic cancer. We investigated how the expression of oncogenic KRAS regulates fibroblast growth factor 21 (FGF21), which prevents obesity, and the effects of FGF21 administration on pancreatic tumorigenesis.
NEW FINDINGS:
Normal acinar cells from mice and humans express high levels of FGF21. In mice, acinar expression of oncogenic KRAS significantly reduces FGF21 expression. When these mice are placed on a HFD, they develop extensive inflammation, pancreatic cysts, pancreatic intraepithelial neoplasias, and pancreatic adenocarcinomas—all were reduced by injection of FGF21. FGF21 reduces HFD-promoted RAS activity.
LIMITATIONS:
These studies were performed in mice—more studies of humans are needed.
IMPACT:
FGF21 is downregulated by oncogenic KRAS. FGF21 might be given to patients for the prevention or treatment of pancreatic cancer.
Acknowledgements
We would like to express our gratitude to Dr. David A. Tuveson (Cold Spring Harbor Laboratory, NY) and Dr. Anirban Maitra (MD Anderson Cancer Center, TX) for providing insight and expertise that greatly improved the manuscript.
Funding: This work was supported by grants from the Start-up Funds from the Department of Medicine at Stony Brook University (W.L.), Sheikh Ahmed Bin Zayed Al Nahyan Center for Pancreatic Cancer Research from the University of Texas MD Anderson Cancer Center (W.L.), Pilot Project Grant at Stony Brook University (W.L.), National Institutes of Health DK052067 (C.D.L.), the Lockton Foundation Endowment (C.D.L.), China Scholarship Council #201403170269 (M.L.), #201603170128 (D.W.), #201703170106 (Y.B.), #201506580014 (J.J.), and National Institutes of Health 5P20CA192994-02 (E.L.).
Abbreviations:
- ADM
acinar-to-ductal metaplasia
- CCL17
C-C chemokine ligand 17
- CCL21a
C-C chemokine ligand 21A
- CD3
cluster of differentiation 3
- CD11b
macrophage cluster of differentiation 11
- CD40L
Cluster of differentiation 40 ligand
- CD68
cluster of differentiation 68
- CK19
cytokeratin-19
- COX2
cyclooxygenase-2
- CXCL5
C-X-C motif chemokine ligand 5
- CXCL9
C-X-C motif chemokine ligand 9
- F4/80
EGF-like module-containing mucin-like hormone receptor-like 1
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- CSF2 (GM-CSF)
granulocyte-macrophage colony-stimulating factor
- HFD
high-fat diet
- HPDE
human pancreatic ductal epithelial cells
- IFNG
interferon gamma
- IL21
interleukin 21
- Ki-67
proliferation-related Ki-67 antigen
- KLB
Klotho beta
- KRAS
Kirsten rat sarcoma-2 viral (v-Kiras2) oncogene homolog
- CCL2 (MCP1)
Monocyte/macrophage chemoattractant protein 1
- CCL4 (MIP1b)
macrophage inflammatory protein 1 beta
- MIST1
muscle, intestine and stomach expression 1
- MUC5
mucin 5 subtypes A and C
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
- PPARA
peroxisome proliferator-activated receptor alpha
- PPARG
peroxisome proliferator-activated receptor gamma
- RORA
retinoid-related orphan receptor alpha
- TGFB1
transforming growth factor beta
- TNF
tumor necrosis factor alpha
- TNFRSF11b
TNF receptor superfamily member 11b
- UCP1
uncoupling protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors declare no potential conflicts of interest.
REFERENCE:
Author names in bold designate shared co-first authorship.
- 1.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913–21. [DOI] [PubMed] [Google Scholar]
- 2.Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122:639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50. [DOI] [PubMed] [Google Scholar]
- 4.Guerra C, Collado M, Navas C, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011;19:728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007;11:291–302. [DOI] [PubMed] [Google Scholar]
- 6.Philip B, Roland CL, Daniluk J, et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2013;145:1449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foltz IN, Hu S, King C, et al. Treating diabetes and obesity with an FGF21-mimetic antibody activating the betaKlotho/FGFR1c receptor complex. Sci Transl Med 2012;4:162ra153. [DOI] [PubMed] [Google Scholar]
- 8.Kharitonenkov A, Shiyanova TL, Koester A, et al. FGF-21 as a novel metabolic regulator. J Clin Invest 2005;115:1627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badman MK, Pissios P, Kennedy AR, et al. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 2007;5:426–37. [DOI] [PubMed] [Google Scholar]
- 10.Inagaki T, Dutchak P, Zhao G, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab 2007;5:415–25. [DOI] [PubMed] [Google Scholar]
- 11.Yang C, Lu W, Lin T, et al. Activation of Liver FGF21 in hepatocarcinogenesis and during hepatic stress. BMC Gastroenterol 2013;13:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaich G, Chien JY, Fu H, et al. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab 2013;18:333–40. [DOI] [PubMed] [Google Scholar]
- 13.Talukdar S, Zhou Y, Li D, et al. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab 2016;23:427–40. [DOI] [PubMed] [Google Scholar]
- 14.Wu AL, Kolumam G, Stawicki S, et al. Amelioration of type 2 diabetes by antibody-mediated activation of fibroblast growth factor receptor 1. Sci Transl Med 2011. ;3:113ra126. [DOI] [PubMed] [Google Scholar]
- 15.Ogawa Y, Kurosu H, Yamamoto M, et al. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc Natl Acad Sci U S A 2007;104:7432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams AC, Yang C, Coskun T, et al. The breadth of FGF21’s metabolic actions are governed by FGFR1 in adipose tissue. Mol Metab 2012;2:31–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang C, Jin C, Li X, et al. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PLoS One 2012;7:e33870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coate KC, Hernandez G, Thorne CA, et al. FGF21 Is an Exocrine Pancreas Secretagogue. Cell Metab 2017;25:472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson CL, Weston JY, Chadi SA, et al. Fibroblast Growth Factor 21 Reduces the Severity of Cerulein-Induced Pancreatitis in Mice. Gastroenterology 2009;137:1795–804 [DOI] [PubMed] [Google Scholar]
- 20.Huang H, Daniluk J, Liu Y, et al. Oncogenic K-Ras requires activation for enhanced activity. Oncogene 2014;33:532–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daniluk J, Liu Y, Deng D, et al. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest 2012;122:1519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji B, Song J, Tsou L, et al. Robust acinar cell transgene expression of CreErT via BAC recombineering. Genesis 2008;46:390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D, Bi Y, Hu L, Luo Y, et al. Obesogenic high-fat diet heightens aerobic glycolysis through hyperactivation of oncogenic KRAS. Cell Commun Signal 2019;17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan FC, Bankaitis ED, Boyer D, et al. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 2013;140:751–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He P, Yang JW, Yang VW, et al. Kruppel-like Factor 5, Increased in Pancreatic Ductal Adenocarcinoma, Promotes Proliferation, Acinar-to-Ductal Metaplasia, Pancreatic Intraepithelial Neoplasia, and Tumor Growth in Mice. Gastroenterology 2018;154:1494–1508 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–83. [DOI] [PubMed] [Google Scholar]
- 27.Adams AC, Coskun T, Cheng CC, et al. Fibroblast growth factor 21 is not required for the antidiabetic actions of the thiazoladinediones. Mol Metab 2013;2:205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fon Tacer K, Bookout AL, Ding X, et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol 2010;24:2050–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurosu H, Choi M, Ogawa Y, et al. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem 2007;282:26687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muise ES, Azzolina B, Kuo DW, et al. Adipose fibroblast growth factor 21 is up-regulated by peroxisome proliferator-activated receptor gamma and altered metabolic states. Mol Pharmacol 2008;74:403–12. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Qiang L, Farmer SR. Identification of a domain within peroxisome proliferator-activated receptor gamma regulating expression of a group of genes containing fibroblast growth factor 21 that are selectively repressed by SIRT1 in adipocytes. Mol Cell Biol 2008;28:188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Solt LA, Burris TP. Regulation of FGF21 expression and secretion by retinoic acid receptor-related orphan receptor alpha. J Biol Chem 2010;285:15668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji B, Tsou L, Wang H, et al. Ras activity levels control the development of pancreatic diseases. Gastroenterology 2009;137:1072–82, 1082, e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Incio J, Liu H, Suboj P, et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov 2016;6:852–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singhal G, Kumar G, Chan S, et al. Deficiency of fibroblast growth factor 21 (FGF21) promotes hepatocellular carcinoma (HCC) in mice on a long term obesogenic diet. Mol Metab 2018;13:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.