

ABSTRACT
Reversed-phase liquid chromatography (RPLC) separations of proteins using optical detection generally use trifluoroacetic acid (TFA) because it is a strong, hydrophobic acid and a very effective ion-pairing agent for minimizing chromatographic secondary interactions. Conversely and in order to avoid ion suppression, analyses entailing mass spectrometry (MS) detection is often performed with a weaker ion-pairing modifier, like formic acid (FA), but resolution quality may be reduced. To gain both the chromatographic advantages of TFA and the enhanced MS sensitivity of FA, we explored the use of an alternative acid, difluoroacetic acid (DFA). This acid modifier is less acidic and less hydrophobic than TFA and is believed to advantageously affect the surface tension of electrospray droplets. Thus, it is possible to increase MS sensitivity threefold by replacing TFA with DFA. Moreover, we have observed DFA ion pairing to concomitantly produce higher chromatographic resolution than FA and even TFA. For this reason, we prepared and used MS-quality DFA in place of FA and TFA in separations involving IdeS digested, reduced NIST mAb and a proprietary antibody-drug conjugate (ADC), aiming to increase sensitivity, resolution and protein recovery. The resulting method using DFA was qualified and applied to two other ADCs and gave heightened sensitivity, resolution and protein recovery versus analyses using TFA. This new method, based on a purified, trace metal free DFA, can potentially become a state-of-the-art liquid chromatography-MS technique for the deep characterization of ADCs.
KEYWORDS: Difluoroacetic acid, DFA, formic acid, FA, trifluoroacetic acid, TFA, antibody-drug conjugate, ADC, IdeS digestion, monoclonal antibody, mAb, NIST mAb, reversed-phase chromatography, subunit profiling, LC-MS, peak capacity, protein recovery, MS sensitivity, disulfide isoforms, drug-to-antibody ratio, DAR, salt adducts, metal adducts, sodium, potassium
Introduction
Rapid advances in the biopharmaceutical industry have led to a growing demand for novel technologies to support the characterization of protein therapeutics, such as monoclonal antibodies (mAbs). Development of these characterization strategies are warranted given that the variants of a mAb therapeutic can affect its efficacy and safety.1 In fact, numerous types of variants and their associated post-translational modifications are risk assessed and defined as critical quality attributes (CQAs).2,3 While mAbs remain a prominent modality in their own right, they are also used as scaffolds for drug conjugation. These antibody-drug conjugates (ADCs) are finding applicability in the targeted treatment of cancer, but exhibit an even higher degree of complexity due to the heterogeneous results of linking cytotoxins onto an antibody.4 With an increased focus on the development of complex protein-based molecules, the biopharmaceutical industry has an ever increasing demand for sensitive analytical techniques.
Reversed-phase liquid chromatography (RPLC) is a technique routinely used to characterize biopharmaceuticals, such as mAbs and ADCs. Unlike many other separation mechanisms, it yields high resolution using volatile, mass spectrometry (MS)-compatible mobile phases and can be implemented to gain information at different molecular levels, from intact protein to subunits to peptides.3,5,6 This proves especially useful for ADC characterization, as it is imperative to monitor and report CQAs related to the cytotoxic payloads.4,7 For instance, the drug-to-antibody ratio (DAR), or the average number of drugs conjugated to the antibody, must be known since it can affect the potency and toxicity of the ADC.4,7,8 Other CQAs, such as drug load distribution and residual drug concentration, are also important.4,7 The versatility of protein RPLC, especially when coupled to MS for accurate mass analysis, allows the characterization of these CQAs. However, as the biotherapeutic industry matures even further, protein RPLC must also improve to support the need for higher resolution, enhanced sensitivity and faster throughput characterization.
While increases in resolution and speed can be conferred by new column technologies, increases in MS sensitivity can often be more challenging to achieve. Typically, protein RPLC-MS separations are performed with acidic mobile phase modifiers. Being a strong ion-pairing agent capable of mitigating secondary interactions, trifluoroacetic acid (TFA) is favored for optimizing chromatographic resolution. However, formic acid (FA) is preferred over TFA for MS analyses because it tends to give less ion suppression and adduct formation. Because each acid modifier has both benefits and drawbacks, many researchers have tried to combine them at varying ratios in an attempt to balance separation quality and MS sensitivity.9,10 Alternative acids or other mobile phase additives have also been investigated over the years. There have also been proposals to supplement TFA with additional reagents to increase signal quality, but these reagents are generally not LC-UV friendly.11 Ultimately, the attempts to make use of unconventional reagents demonstrate the need for a more optimal ion-pairing agent for RPLC.
We have investigated the use of difluoroacetic acid (DFA) as an acid modifier for RPLC-MS-based characterization of protein therapeutics. Previous studies proposed the use of either monofluoroacetic acid or DFA for use in LC-MS.12,13 Monohalogenated acids are extremely toxic14 and should not be applied to everyday use. DFA, on the other hand, could be more broadly implemented. With properties intermediate to both those of TFA and FA, it is reasonable to assume that DFA can yield LC-MS separations with only some compromise to TFA resolving power and FA sensitivity. In addition, it is also only slightly more hazardous than TFA, with a toxicity level similar to that of hexafluoro-2-propanol (HFIP), which is often used in LC separations.14
Despite its appeal, the use of DFA has not yet become routine in protein LC-MS. Current sources of DFA have been found to contain high sodium and potassium concentrations. These trace salt contaminants do not adversely affect separations, but they do disrupt the interpretability of mass spectra. Thus, for our study, we purified DFA before using it to develop an LC-MS technique with an unexpected balance of chromatographic resolution and MS sensitivity. Most interestingly, the results presented herein show that not only does DFA afford higher MS sensitivity than TFA, but that it can also provide better chromatographic resolution.
As described here, we used these gains in analytical capabilities to develop a new LC-MS method suitable for subunit-level characterization of mAb-based therapeutics, including a highly hydrophobic cysteine-linked ADC. Unlike previous methods, this DFA-based RPLC-MS method showed little to no on-column sample degradation, complete analyte recovery, noteworthy proteoform resolution and threefold higher MS sensitivity, which in sum made it possible to detect and monitor trace levels of product-related impurities with higher fidelity. Finally, we performed method qualification experiments, including applying this method to the characterization of additional ADCs, to demonstrate the robustness of this new method.
Results
Purification of DFA
In order to compare separations using DFA-modified mobile phases against those using FA- or TFA-modified mobile phases, we distilled reagent-grade DFA to a purity on par with LC-MS quality FA and TFA. Figure 1(a) indicates that, by means of this distillation, the sodium and potassium content of the purified DFA afforded spectra with three to fourfold less intense adduct signals when used for the LC-MS separation of IdeS digested, reduced NIST mAb. To estimate the limit of detection for sodiated and potassiated adducts, a subsequent study was performed where known salt concentrations in the mobile phase were correlated to the percentage of adducts detected from the deconvoluted MS spectra of the light chain. From this study, a concentration of about 150 ppb was estimated as the limit of detection for sodium, while there was a nearly negligible impact when increasing the concentration of potassium. Since DFA consisted of 0.1% of the mobile phase in this experiment, this suggested that the sodium concentration of DFA can be quite high (ppm levels) before sodiated adducts become problematic.
Figure 1.
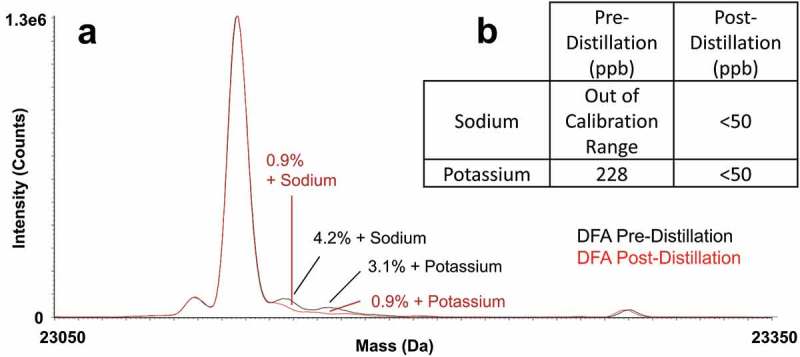
(a) The influence of sodium and potassium content on mass spectral quality as depicted by an overlay of the deconvoluted mass spectra of the NIST mAb light chain subunit obtained using as-received DFA and distilled DFA and (b) ICP-MS quantitation for as-received (reagent-grade) versus distilled DFA. Separations were performed with a high coverage phenyl-bonded superficially porous silica 450 Å, 2.7 μm, 2.1 × 50 mm column using a flow rate of 0.2 mL/min, column temperature of 80°C, and 0.25 μg mass loads.
To quantitatively measure the levels of sodium and potassium, inductively coupled plasma mass spectrometry (ICP-MS) was performed. ICP-MS confirmed the levels of both sodium and potassium in distilled DFA to be below 50 ppb, but was unable to provide accurate quantitation for the starting, reagent-grade material because the results were out of the calibration range (Figure 1(b)). As certain protein species may be more prone to sodium adduct formation (due to differences in isoelectric points, acidic sites, hydrophobicity and molecular weight), we compared pre-distillation DFA and post-distillation DFA in a peptide mapping LC-MS analysis (Supplementary Figure S1). In this experiment, the level of sodiated adducts was found to be much higher (at 21% for light chain tryptic peptide number 37) in comparison to the subunit analysis using pre-distillation DFA. Use of distilled DFA drastically reduced the amount of sodiated adducts to less than 0.5%. More information on these experiments are provided in the Supplementary Materials.
Resolution and sensitivity comparison for acidic modifiers
Next, we set out to investigate the utility of various mobile phase systems by exploring LC-UV-MS separations of reduced, IdeS-digested NIST mAb (Reference Material 8671) on 2.1 mm ID columns using 0.1% (v/v) modified water and acetonitrile mobile phases containing FA, DFA and TFA. Protein RPLC separations are dependent on numerous factors, including column temperature and flow rate; our tests were performed at a high temperature (80 °C) and a low flow rate (0.2 mL/min). The UV chromatograms resulting from these three separations are displayed in Figure 2(a). The peak shapes of the three subunits in the sample (Fc/2, light chain and Fd′) were seen to be broad and tailing in the FA chromatogram. DFA and TFA chromatograms showed improved separation quality in terms of peak capacity, peak width at 50%, return to baseline and indications of being able to resolve the lysine variant of the Fc/2 subunit. Interestingly, when comparing these analyses, while both acidic modifiers produced high-resolution separations, DFA that gave the highest peak capacities, not the historically preferred TFA ion-pairing reagent.
Figure 2.

(a) UV chromatograms of NIST mAb subunits obtained using 0.1% FA, DFA, or TFA-modified mobile phases, and (b) total ion current chromatograms (TICs) of NIST mAb subunits obtained using 0.1% FA, DFA, or TFA-modified mobile phases. Separations were performed with a high coverage phenyl-bonded superficially porous silica 450 Å, 2.7 μm, 2.1 × 50 mm column using a flow rate of 0.2 mL/min, column temperature of 80°C, and 0.25 μg mass loads.
Mass spectrometric analysis was also assessed as performed with electrospray ionization and high sensitivity quadrupole time-of-flight instrumentation. Total ion current chromatograms (TICs) from each separation are shown in Figure 2(b) along with the observed signal intensities. FA-modified mobile phases provided the highest intensities, followed by mobile phases containing DFA. Because the charge state distribution from DFA was in fact the same as FA, the ionization efficiencies from DFA and FA for this subunit separation were very similar. Separations using TFA gave the lowest sensitivity of the three acids, with intensities that decreased about 5–6 fold when compared to FA and two to threefold versus DFA. This is consistent with TFA being the strongest ion-pairing reagent, and thus the reagent most likely to cause pronounced ion suppression.15,16 FA, the weakest acid, produced the most intense MS signal. However, the usability of ESI-MS data is still reliant on the quality of the chromatographic separation. In this respect, DFA can be strongly considered as an ion-pairing alternative because it gives equivalent or better separation performance versus TFA with a two to threefold increase in MS sensitivity.
Characterization of an ADC
The advantages of a DFA-based LC-MS method can be seen in its application to the characterization of an ADC. RPLC is useful for ADC separations due to its ability to separate DAR species based on their hydrophobicity. To access this information for a middle-up/down analysis, limited digestion and reduction can be used to break the ADC down such that the extent of payload conjugation per subunit can be quickly analyzed via UV and MS.17 For this work, IdeS was used for its quick kinetics and high fidelity in producing F(ab′)2 and Fc/2 fragments that can be further reduced to Fc/2, light chain and Fd′ subunits.3,5
For this study, we chose to investigate methods for the analysis of an ADC that has previously proven difficult to characterize due to a highly hydrophobic Fd′ domain. This hydrophobicity is only exacerbated when it is modified to have up to three cysteine-linked auristatin payloads. With the application of IdeS digestion followed by reduction, a sample was obtained that contained this and other payload-bearing forms of Fc/2, light chain and Fd′ subunits (Figure 3). Separations of this sample were performed using 0.1% FA-, 0.1% DFA- and 0.1% TFA-modified mobile phases. The UV chromatograms for these separations are shown in Supplementary Figure S2. Similar to the mAb subunit analyses, separations using DFA and TFA were much improved versus those obtained with FA. Both DFA- and TFA-modified mobile phases performed similarly in terms of resolution. Peak capacity values for 0.1% DFA and 0.1% TFA were determined to be 462 and 465, respectively. Using these highly detailed LC-MS data, 12 different subunit species could be quickly identified down to a relative abundance of 0.6%. The limits of quantitation were estimated to be between 0.1% and 0.2%. A comprehensive accounting of the identified peaks, their retention times, widths and deconvoluted masses are provided in Supplementary Table S1 based on the data from Figure 3(c).
Figure 3.
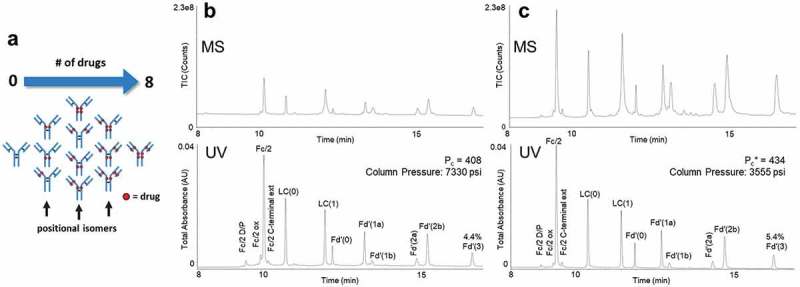
(a) Representation of the different possible drug load distributions of cysteine-conjugated ADCs.18 Subunits from a cysteine-linked auristatin-conjugated antibody as separated with (b) a C4-bonded organosilica 300 Å fully porous stationary phase, 0.6 mL/min flow rate, 80°C temperature, 0.1% TFA-modified mobile phases, and 90:10 acetonitrile/IPA eluent versus (c) a method consisting of a phenyl bonded 2.7 µm superficially porous 450 Å stationary phase, 0.6 mL/min flow rate, 70°C temperature, and 0.15% DFA-modified mobile phases.
Along with peak capacity, the recovery of analytes from each separation was critically examined. In particular, the relative peak area of the 3-payload modified Fd′ subunit, Fd′(3), was examined where a difference was observed for the use of DFA versus both FA and TFA. The relative peak area for this subunit was found to be 5.2% with DFA, but only 4.3% and 4.9% with FA and TFA, respectively. This suggests that separations performed in DFA might provide useful gains in recovery over FA and TFA.
MS performance seen in the analysis of NIST mAb subunits translated to the detection of ADC subunits. As shown in Supplementary Figure S3, total ion chromatograms and raw mass spectra of the unmodified light chain show that MS sensitivity increases in going from TFA to DFA to FA. Deconvoluted mass spectra of the light chain subunit also show that each acid, i.e., distilled DFA and MS-grade FA and TFA, gave low intensities for sodium and potassium adducts (Supplementary Figure S4). All spectra appeared to be clear of other interference peaks, with a notable exception produced with TFA. With TFA-modified mobile phases, we observed a small +114 Da peak, which correlates to undesirable gas phase ion pairing. This phenomenon was not apparent with either FA or DFA, although it must be pointed out that the optimization of collisional activation plays a role in how much gas phase ion pair is observed. Water loss is also observed with all acids; however, this is a common phenomenon that is thought to be a consequence of heated desolvation.19
We also examined levels of on-column degradation that occurred under various conditions. Elevated temperatures and stronger acidic conditions improve protein recovery but may induce backbone hydrolysis.9,20,21 This on-column degradation is exacerbated when proteins adsorb too strongly onto the stationary phase. As can be seen in Supplementary Figure S2, on-column degradation of the ADC increases with increasing acid strength. The D/P cleavage of the Fc/2 is marginal in FA conditions and increases as the conditions change to DFA and TFA. In all, it was necessary to lower column temperature to afford more accurate, higher fidelity information. So long as DFA was increased to a concentration of 0.15%, a column temperature of 70°C could be used with no apparent decrease in protein recovery. With this change, on-column degradation was markedly reduced (by over 50%) versus the use of TFA mobile phase at an 80 °C temperature. Notably, an increase from 0.1% to 0.15% DFA did not result in any significant compromises to LC resolving power, MS sensitivity or spectra quality (Supplementary Figure S5). However, since protein recovery with 0.15% DFA and a column temperature of 70°C was marginally higher (by about 4%) versus using 0.1% DFA, the latter concentration was chosen going forward for any work on additional ADCs.
New method for ADC characterization
From the above results, we propose here a new method for mAb and ADC subunit analysis. Prior to the insights made herein, the subunit samples had been routinely characterized by separations with a sub-2 µm C4-bonded organosilica 300 Å fully porous stationary phase, a separation temperature of 80°C, 0.1% TFA mobile phases, and eluent composed of 90% acetonitrile and 10% isopropanol (IPA), the latter being needed to facilitate the recovery of hydrophobic proteins.21,22 A result typical of this method is provided in Figure 3(b).
Using this separation as a starting point, we explored stepwise changes to move toward newer techniques that simplified the method, accelerated turnaround, or improved sensitivity. By using a modern column technology, higher resolution and improved selectivity was gained along with a reduction in backpressure and ability to use faster chromatographic velocities.23,24 With this change, it was also possible to exclude IPA from the mobile phase without significantly affecting peak capacity or protein recovery. Along with the adoption of DFA, it was also possible to reduce the separation temperature.
In turn, a new method was established, and its capabilities are exemplified by the chromatograms displayed in Figure 3(c). It is notable that, while the sub 2 µm column required ultrahigh-pressure compatible instrumentation, the 2.7 µm-based approach could be transferred to other less specialized instrumentation because operational back pressures were lower. Furthermore, using the latter method, it was possible to optimize nearly all facets of the chromatographic separation and to facilitate more strenuous examples of deep-level characterization. Two examples of low abundance variants are discoverable within the shoulder peaks adjacent to the unmodified Fc/2 subunit. Mass spectra corresponding to these species are displayed in Figure 4. The DFA method produced higher signal-to-noise spectra, which could be used to more confidently confirm +16 Da (pre-peak) and +673 Da (post-peak) mass additions and the corresponding identifications of Fc domain oxidation and low-level C-terminal sequence extension.25
Figure 4.

MS peak intensities of oxidized and extended C-terminal Fc/2 subunits from a cysteine-linked auristatin conjugated as determined by the (a) method consisting of a C4-bonded organosilica 300 Å fully porous stationary phase, 0.6 mL/min flow rate, 80°C temperature, 0.1% TFA-modified mobile phases, and 90:10 acetonitrile/IPA eluent versus (b) the new method consisting of a phenyl bonded 2.7 µm superficially porous 450 Å stationary phase, 0.6 mL/min flow rate, 70°C temperature, and 0.15% DFA-modified mobile phases.
In another respect, this DFA-based method has also facilitated quick assessments of DAR, which must be monitored to ensure the potency of the molecule. While DAR values are frequently assessed by hydrophobic interaction chromatography (HIC) or intact mass analysis,4 subunit-level RPLC analysis can also be used and is helpful for corroborating other assay results.26,27 The assessment depends on the use of individual peak areas, a calculation of subunit-specific DAR values, and an extrapolation of DARs to one intact molecule DAR value (see Equations 2–4). Supplementary Table S2 lists representative peak areas observed from the DFA method for the unconjugated and conjugated subunits of the cysteine-linked auristatin ADC, which in turn can be used to infer an average DAR value of 4.2. This particular value is in close agreement to DAR values that have been estimated for this molecule using HIC and intact mass analysis.
Method qualification
To demonstrate the robustness of this technique, method qualification experiments were performed. According to standard practices for robustness testing, we assessed different batches of chromatographic media, column, and sources of DFA. Lifetime studies were also performed to 1000 injections, and method parameters were varied by ±5%. Table 1 shows the % relative standard deviation (RSD) changes in resolution, protein recovery, retention time and average DAR. RSD values of less than 5% were recorded with each parameter test. This is in line with the general allowance for biopharmaceuticals.28-30
Table 1.
Variation in method output (RSD, %) as a function of robustness testing for a technique comprised of evaluating changes in stationary phase batches, DFA batches, LC systems, columns, temperature, mass load, flow rate, DFA concentration, and lifetime for the separation observed in Figure 3(c).
| Variation in Method Output (RSD, %) | ||||||
|---|---|---|---|---|---|---|
| Peak Capacity | Percent Peak Area of Light Chain(0) | Percent Peak Area of Fd′(3) | Retention Time of Light Chain(0) | Retention Time of Fd′(3) | DAR | |
| Stationary Phase Batches (3) | 3.7 | 1.4 | 3.1 | 1.9 | 1.8 | 0.2 |
| DFA Batches (3) | 0.6 | 0.8 | 2.6 | 0.7 | 0.4 | 0.4 |
| LC Systems (3 Systems, 9 Columns) | 1.7 | 3.1 | 4.3 | 0.5 | 0.4 | 0.5 |
| Temperature (±5%) | 3.7 | 1 | 1.4 | 1.1 | 0.3 | 1.1 |
| Mass Load (±5%) | 0.9 | 0.4 | 1.9 | 0.2 | 0.2 | 0.2 |
| Flow Rate (±5%) | 0.7 | 0.2 | 0.8 | 1.1 | 0.8 | 0.1 |
| Percent DFA (±5%) | 4.7 | 2.7 | 4.6 | 2.0 | 0.9 | 0.9 |
| Lifetime Study (1) | 1.6 | 1.8 | 4.0 | 0.5 | 0.3 | 0.0 |
| Lifetime Study (2) | 0.3 | 0.2 | 1.2 | 0.7 | 0.8 | 0.4 |
We also qualified this method for reversed-phase subunit separations using additional cysteine-conjugated ADCs. As shown in Figure 5(b), the new method gives high-resolution separations of a commercially available cysteine linked, dansyl-cadaverine-SMCC conjugated ADC mimic, and the DAR can be calculated from the peak areas observed. In another instance, we performed this method on a second discontinued cysteine-linked ADC, proprietary to Pfizer and extremely hydrophobic, even more so than the first. For this ADC, the separation performed best when the gradient conditions were run at a higher column temperature (Figure 5(c)). This suggests that for some unique samples, certain method parameters may need to be adjusted to provide an optimal separation. The proposed new method can therefore provide a suitable starting point for further method development.
Figure 5.
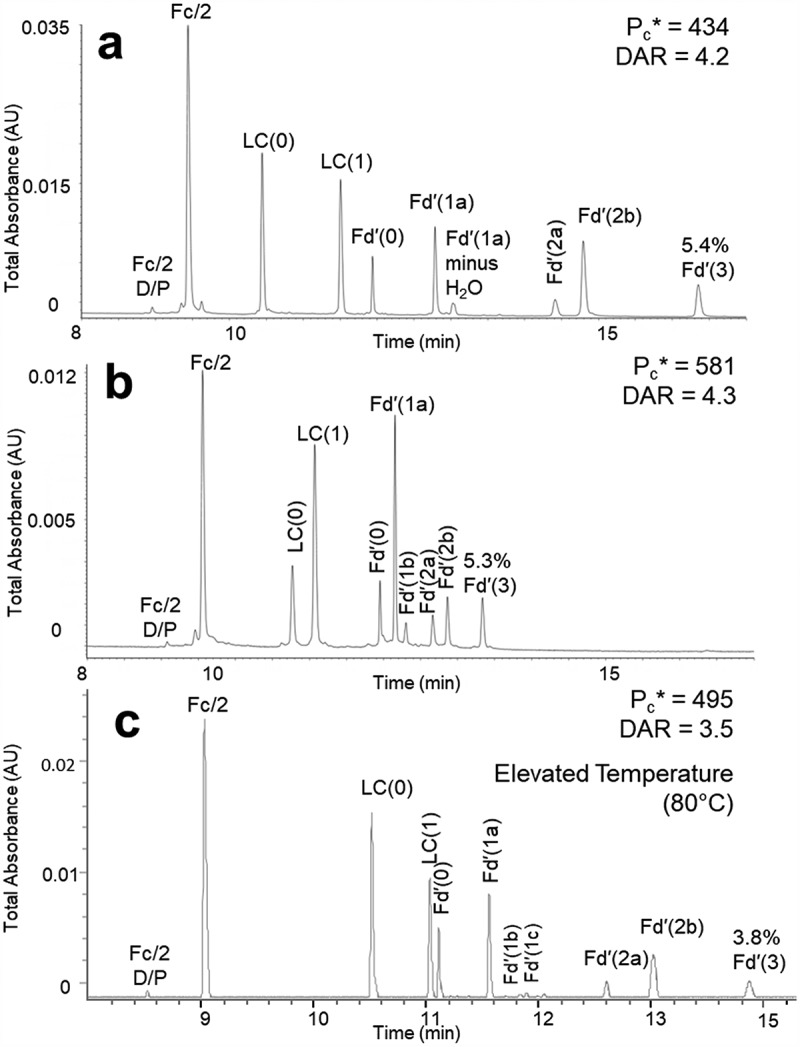
Qualification of the proposed method using 0.15% DFA-modified mobile phases with different cysteine-linked ADCs. (a) Subunits from a cysteine-linked auristatin conjugated antibody previously used in the proposed method. (b) Subunits from a commercially available cysteine linked, dansyl-cadaverine-SMCC conjugated ADC mimic. (c) Subunits from a second cysteine-linked ADC, separated using higher temperature. Separations were performed with a 2.1 × 150 mm column packed with a high coverage phenyl bonded 2.7 µm superficially porous 450 Å stationary phase using a flow rate of 0.6 mL/min, column temperature of 70°C or 80°C, and 1 μg mass loads.
The method was also applied to the analysis of the lysine-conjugated ADC trastuzumab emtansine (Kadcyla®). Due to the extreme heterogeneity of lysine-conjugated ADCs, characterization is difficult,31,32 especially at the subunit level. With this method, the heterogeneity of the molecule can indeed be readily observed (Supplementary Figure S6). While highly complicated, these LC-MS data might be a useful route to efficient comparative analyses.
Discussion
The increasing complexity of biopharmaceutical modalities requires improvements in analytical methodologies. RPLC is a powerful technique for the separation of proteins at all molecular levels, and it becomes substantially more powerful when coupled to MS. However, depending on the use of conventional acid modifiers, such as TFA and FA, protein RPLC often exhibits excellent chromatographic resolution but compromised MS sensitivity or, vice versa, i.e., excellent MS sensitivity but compromised separation quality.
Our work, as described here, portends a new approach to LC-MS analyses based on the use of highly purified DFA. This alternative ion-pairing modifier has now shown that it can achieve an optimization between chromatography and mass spectrometry that is unattainable when using TFA and FA. By distilling reagent-quality DFA, we could drastically reduce the sodium and potassium content to trace level quantities typical of LC-MS solvents. Higher resolution separations of NIST mAb subunits were achieved when using DFA over TFA, and DFA also provided 2–3 fold as much MS sensitivity with similar percentages of sodium and potassium adducts. A possible explanation for the increase in peak capacity could be optimization of chromatographic adsorption, where there is a benefit to a less sterically bulky ion pair or an advantage to having a slightly less hydrophobic ion pair. The former could be due to more complete ion-pairing coverage across the subunits and the latter could be an effect caused by a specific tuning of stationary phase retentivity.
By using DFA, we were also able to discover step changes in improving protein LC-MS capabilities, where a DFA-based method greatly improved subunit-level profiling of hydrophobic ADCs. We believed that this method has produced one of the first published examples of an RPLC chromatogram of subunit-digested trastuzumab emtansine. Moreover, in addition to providing benefits to chromatographic resolution and MS sensitivity, this robust method was found to greatly increase protein recovery without the need to use alcohol co-solvents for elution or excessively high column temperatures. We theorize this to be an effect resulting from DFA being sufficiently acidic so as to minimize ionic secondary interactions (unlike FA), but not as hydrophobic as TFA to force excessively strong adsorption.
We have also seen a benefit in applying this purified DFA to other applications, such as intact mass analysis and peptide mapping. Many of the benefits seen herein carry over to these other applications, where replacement of TFA with DFA yielded MS sensitivity gains with no to little loss in chromatographic resolution. No matter the nature of the application, it is, however, important to use purified DFA and mobile phase modifiers with low metal concentrations to avoid complications from the formation of gas-phase ion adducts.
In summary, we purified DFA so that it contained only trace levels of sodium and potassium and demonstrated its application for ADC subunit profiling. Our method shows robustness and has been successfully applied to characterize multiple ADCs. Additional studies are underway to evaluate the advantages of DFA for other biopharmaceutical applications, such as glycans, and for small molecule applications. Ultimately, we hope our demonstration that highly attractive protein MS data can be obtained via the use of stringently purified DFA will encourage broader use of this acid, and our method will be implemented in additional types of LC-MS work.
Materials and methods
Purification and qualification of LC-MS DFA
DFA was acquired from Oakwood Chemical and distilled with a DST-1000 acid purification system (Savillex). Reagent purity was confirmed through evaluations of deconvoluted light chain MS spectra resulting from separations of reduced, IdeS-digested NIST mAb (mAb Subunit Standard; Waters Corporation). Sodium and potassium were quantified via ICP-MS (Element 2 SF-ICP-MS, Thermo Fisher, Waltham, MA) using a 12-point linear calibration curve ranging from 0.05 to 100 parts per billion (ppb). Further data can be found in the Supplementary Materials.
IdeS digestion and reduction of cysteine-linked ADC
Reduced, IdeS (Immunoglobulin Degrading Enzyme) digested NIST mAb (Reference Material 8671) were obtained in the form of a ready-to-use standard (mAb Subunit Standard, Waters Corporation). Other reduced, IdeS-digested samples consisted of two discontinued cysteine-linked ADCs with auristatin payloads provided by Pfizer, Inc. A cysteine linked, dansyl-cadaverine-SMCC conjugated mAb (SigmaMAb Antibody Drug Conjugate Mimic, Millipore Sigma) and trastuzumab emtansine (Kadcyla®, Besse Medical) were reduced and digested via standard IdeS digestion and reduction protocols.
LC-UV-MS of NIST mAb and ADC subunits
Reduced, IdeS-digested NIST mAb and ADC subunits were analyzed by LC-UV-MS with an ACQUITY UPLC H-Class Bio chromatograph, an ACQUITY UPLC TUV detector, and a Xevo G2-XS QTOF mass spectrometer (Waters Corporation). mAb subunit separations were performed on a 2.7 µm, 2.1 × 50 mm high coverage phenyl-bonded superficially porous silica stationary phase (BioResolve RP mAb Polyphenyl column, Waters Corporation) using 0.1% or 0.15% FA, DFA, or TFA in LC-MS grade water (mobile phase A) and the same percent modifier in LC-MS grade acetonitrile (mobile phase B). NIST mAb samples were injected at a mass load of 0.25 µg and run at a temperature of 80°C, flow rate of 0.2 mL/min, and gradient from 15% to 55% B in 20 min. ADC subunit separations were performed on a 2.7 µm, 2.1 × 150 mm high coverage phenyl-bonded superficially porous silica stationary phase or a 1.7 µm, 2.1 × 150 mm C4-bonded fully porous organosilica stationary phase (ACQUITY UPLC BEH C4 column, Waters Corporation) using 0.1% or 0.15% FA, DFA, or TFA in LC-MS grade water (mobile phase A). For this column, the same percent modifier in LC-MS grade acetonitrile was used for mobile phase B, and for the C4-bonded fully porous organosilica column, 0.1% TFA in 90:10 LC-MS grade acetonitrile:IPA for mobile phase B was employed. ADC samples were injected at a mass load of 1 µg and run at a temperature of 80°C or 70°C, flow rate of 0.6 mL/min and gradient from 15% to 55% B in 20 min.
Analyses were performed with UV detection at 280 nm using MassLynx 4.1 and UNIFI 1.8 for data analysis. MS detection settings were optimized with: a capillary voltage of 3.0 kV, sampling cone and source offset at 80, a source temperature of 100°C, a desolvation temperature of 450°C, a cone gas flow at 0 L/h, desolvation gas flow set at 100 L/h, and collision energy set at 10 eV. Mass spectra were processed via MaxEnt1 deconvolution with a resolution of 20000 over a range of 850 − 1450 m/z at a rate of 10 Hz. Duplicate injections were analyzed.
Method qualification and robustness
Method qualification and robustness testing were performed at Waters Corporation in Milford, MA, and Pfizer, Inc. in St. Louis, MO. LC-UV characterization of the ADC was performed on three different chromatographic systems consisting of an ACQUITY UPLC H-Class Bio or H-Class chromatograph and an ACQUITY UPLC TUV detector or ACQUITY UPLC PDA detector. Each system was tested with three 2.1 × 150 mm high coverage phenyl-bonded superficially porous silica columns from the same batch of chromatographic material. Three different batches of chromatographic material and three separate sources of DFA were also evaluated. The method was qualified using two additional cysteine-conjugated ADCs (a commercially available mimic acquired from Sigma Aldrich and a discontinued proprietary ADC from Pfizer, Inc.) alongside the lysine-conjugated trastuzumab emtansine (Kadcyla®).
Robustness of the method conditions was investigated by varying the separation temperature, mass load, flow rate and concentration of DFA by ± 5% based on the proposed LC-UV-MS method for the separation of ADC subunits consisting of the following: the use of a 2.7 µm, 2.1 × 150 mm high coverage phenyl-bonded superficially porous silica column run with 0.15% DFA in LC-MS grade water (mobile phase A) and in LC-MS grade acetonitrile (mobile phase B) at a temperature of 70°C, flow rate of 0.6 mL/min, mass load of 1 µg, and gradient from 15% to 55% B. A column lifetime study was performed by running 1000 injections of the above method on two separate columns.
Equations
Gradient peak capacities (Pc) for the NIST mAb subunit and ADC subunit separations were calculated from Equation 1:
where Δt is the gradient time and W50%, avg is the average peak width at half height for all subunits.
The average DAR was calculated by using Equations 2 and 3 for both light chain and Fd′ conjugated subunits, with A representing the peak area of the subunit of interest and n representing the number of payloads:
In this equation, each payload modified subunit, for example, nA(Light Chain)n, is assessed in relation to the total area of the subunit as a whole – in this case, Ʃ A(Light Chain). The sum of these values results in the average DAR based on the light chain subunit or the Fd′ subunit. These DAR values can then be combined for an approximation of the total average DAR. Note that the Fc/2 subunits need not be included, as they do not contain payload modifications for the cysteine-linked ADC. Thus, by combining the above results, Equation 4 was used to estimate the total average DAR of the intact ADC. For this equation, the sum of DAR (Light Chain) and DAR (Fd′) was doubled as digestion and reduction results in two sets of light chain and Fd′ per IgG/ADC molecule:
Abbreviations
| ADC | Antibody-drug conjugate |
| CQA | Critical quality attribute |
| DAR | Drug-to-antibody ratio |
| DFA | Difluoroacetic acid |
| FA | Formic Acid |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| LC | Liquid chromatography |
| LC-MS | Liquid chromatography-mass spectrometry |
| mAb | Monoclonal antibody |
| MS | Mass spectrometry |
| ppb | Parts per billion |
| ppm | Parts per million |
| RPLC | Reversed-phase liquid chromatography |
| TIC | Total ion current |
| TFA | Trifluoroacetic acid |
| UV | Ultraviolet |
Acknowledgments
We would like to thank Olga Friese and Jason Rouse of Pfizer for their manuscript review. We would also like to acknowledge Nilini Ranbudage, Ximo Zhang, Henry Shion and Robert Birdsall for their initial work in understanding the benefits and drawbacks of difluoroacetic acid for reversed-phase separations of mAbs, as well as Robert’s permission to use Figure 3(a). We are also appreciative to Dan Walsh and Jim Cook for their contribution to the particle synthesis. Finally, we thank Monica Redente of ERA, part of Waters Corporation, for her expertise and provision of ICP-MS data.
Disclosure of Potential Conflicts of Interest
The work in this article was financially supported by Waters Corporation, and authors JMN, SR and MAL are currently employed by Waters Corporation. JS is employed by Pfizer, Inc. Waters equipment and ADCs developed by Pfizer were used for this research.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Berkowitz SA, Engen JR, Mazzeo JR, Jones GB.. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11:527–40. doi: 10.1038/nrd3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schiel JE, Mire-Sluis A, Davis DL . Monoclonal Antibody Therapeutics: The Need for Biopharmaceutical Reference Materials. In: Shiel JE, Davis DL, Borisov OV, editors. State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 1. Monoclonal Antibody Therapeutics: Structure, Function, and Regulatory Space. Washington (DC): The American Chemical Society; 2014. p. 1–34. [Google Scholar]
- 3.Zhang B, Jeong J, Burgess B, Jazayri M, Tang Y, Taylor Zhang Y. Development of a rapid RP-UHPLC-MS method for analysis of modifications in therapeutic monoclonal antibodies. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1032:172–81. doi: 10.1016/j.jchromb.2016.05.017. [DOI] [PubMed] [Google Scholar]
- 4.Wagh A, Song H, Zeng M, Tao L, Das TK. Challenges and new frontiers in analytical characterization of antibody-drug conjugates. mAbs. 2018;10:222–43. doi: 10.1080/19420862.2017.1412025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.An Y, Zhang Y, Mueller HM, Shameem M, Chen X. A new tool for monoclonal antibody analysis: application of IdeS proteolysis in IgG domain-specific characterization. mAbs. 2014;6:879–93. doi: 10.4161/mabs.28762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bobaly B, Sipko E, Fekete J. Challenges in liquid chromatographic characterization of proteins. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1032:3–22. doi: 10.1016/j.jchromb.2016.04.037. [DOI] [PubMed] [Google Scholar]
- 7.Friese OV, Smith JN, Brown PW, Rouse JC. Practical approaches for overcoming challenges in heightened characterization of antibody-drug conjugates with new methodologies and ultrahigh-resolution mass spectrometry. mAbs. 2018;10:335–45. doi: 10.1080/19420862.2018.1433973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sochaj AM, Swiderska KW, Otlewski J. Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol Adv. 2015;33:775–84. doi: 10.1016/j.biotechadv.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Bobaly B, D’Atri V, Lauber M, Beck A, Guillarme D, Fekete S. Characterizing various monoclonal antibodies with milder reversed phase chromatography conditions. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1096:1–10. doi: 10.1016/j.jchromb.2018.07.039. [DOI] [PubMed] [Google Scholar]
- 10.Fachi MM, Leonart LP, Cerqueira LB, Pontes FLD, de Campos ML, Pontarolo R. A systematic and critical review on bioanalytical method validation using the example of simultaneous quantitation of antidiabetic agents in blood. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1055–1056:61–71. doi: 10.1016/j.jchromb.2017.04.024. [DOI] [PubMed] [Google Scholar]
- 11.Iavarone AT, Jurchen JC, Williams ER. Supercharged protein and peptide ions formed by electrospray ionization. Anal Chem. 2001;73:1455–60. doi: 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monroe ME. Development of instrumentation and applications for microcolumn liquid chromatography coupled to time-of-flight mass spectrometry. Chapel Hill (NC): The University of North Carolina at Chapel Hill; 2002. [Google Scholar]
- 13.Yamamoto E, Ishihama Y, Asakawa N. Application of partially fluorinated carboxylic acids as ion-pairing reagents in LC/ESI-MS. Talanta. 2014;127:219–24. doi: 10.1016/j.talanta.2014.03.060. [DOI] [PubMed] [Google Scholar]
- 14.Elliott J. Fluorinated acetic acids. Encyclopedia of chemical technology. Hoboken (NJ): John Wiley & Sons; 2001. [Google Scholar]
- 15.Annesley TM. Ion suppression in mass spectrometry. Clin Chem. 2003;49:1041–44. doi: 10.1373/49.7.1041. [DOI] [PubMed] [Google Scholar]
- 16.Banerjee S, Mazumdar S. Electrospray ionization mass spectrometry: a technique to access the information beyond the molecular weight of the analyte. Int J Anal Chem. 2012;2012:282574. doi: 10.1155/2012/282574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma A, Thavathiru E, Benbrook DM, Woo S. Bioanalytical method development and validation of HPLCUV assay for the quantification of SHetA2 in mouse and human plasma: application to pharmacokinetics study. J Pharm Technol Drug Res. 2017:6. doi: 10.7243/2050-120X-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birdsall RE, Shion H, Kotch FW, Xu A, Porter TJ, Chen W. A rapid on-line method for mass spectrometric confirmation of a cysteine-conjugated antibody-drug-conjugate structure using multidimensional chromatography. mAbs. 2015;7:1036–44. doi: 10.1080/19420862.2015.1083665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheung MS, Garcia AE, Onuchic JN. Protein folding mediated by solvation: water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc Natl Acad Sci USA. 2002;99:685–90. doi: 10.1073/pnas.022387699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cohen KA, Schellenberg K, Benedek K, Karger BL, Grego B, Hearn MT. Mobile-phase and temperature effects in the reversed phase chromatographic separation of proteins. Anal Biochem. 1984;140:223–35. doi: 10.1016/0003-2697(84)90158-1. [DOI] [PubMed] [Google Scholar]
- 21.Fekete S, Rudaz S, Veuthey JL, Guillarme D. Impact of mobile phase temperature on recovery and stability of monoclonal antibodies using recent reversed-phase stationary phases. J Sep Sci. 2012;35:3113–23. doi: 10.1002/jssc.201200297. [DOI] [PubMed] [Google Scholar]
- 22.Dillon TM, Bondarenko PV, Rehder DS, Pipes GD, Kleemann GR, Ricci MS. Optimization of a reversed-phase high-performance liquid chromatography/mass spectrometry method for characterizing recombinant antibody heterogeneity and stability. J Chromatogr A. 2006;1120:112–20. doi: 10.1016/j.chroma.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Bobaly B, Lauber M, Beck A, Guillarme D, Fekete S. Utility of a high coverage phenyl-bonding and wide-pore superficially porous particle for the analysis of monoclonal antibodies and related products. J Chromatogr A. 2018;1549:63–76. doi: 10.1016/j.chroma.2018.03.043. [DOI] [PubMed] [Google Scholar]
- 24.Lippens JL, Egea PF, Spahr C, Vaish A, Keener JE, Marty MT, Loo JA, Campuzano IDG. Rapid LC-MS method for accurate molecular weight determination of membrane and hydrophobic proteins. Anal Chem. 2018;90:13616–23. doi: 10.1021/acs.analchem.8b03843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spahr CS, Daris ME, Graham KC, Soriano BD, Stevens JL, Shi SD. Discovery, characterization, and remediation of a C-terminal Fc-extension in proteins expressed in CHO cells. mAbs. 2018;10:1291–300. doi: 10.1080/19420862.2018.1511197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dodda S, Makula A, Polagani SR, Kandhagatla RN. Development and validation of bioanalytical liquid chromatography-tandem mass spectrometry method for the estimation of pentoxifylline in human plasma: application for a comparative pharmacokinetic study. Eur J Mass Spectrom. 2018:1469066718817929. doi: 10.1177/1469066718817929. [DOI] [PubMed] [Google Scholar]
- 27.Arnold ME, Booth B, King L, Ray C. Workshop report: Crystal city VI-bioanalytical method validation for biomarkers. Aaps J. 2016;18:1366–72. doi: 10.1208/s12248-016-9946-6. [DOI] [PubMed] [Google Scholar]
- 28.Smith G. European medicines agency guideline on bioanalytical method validation: what more is there to say? Bioanalysis. 2012;4:865–68. doi: 10.4155/bio.12.44. [DOI] [PubMed] [Google Scholar]
- 29.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CMV). Bioanalytical Method Validation: Guidance for Industry. Rockville (MD): Food and Drug Administration; 2018 May 24 [cited 2018 Aug 11]. https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf [Google Scholar]
- 30.Kadian N, Raju KS, Rashid M, Malik MY, Taneja I, Wahajuddin M. Comparative assessment of bioanalytical method validation guidelines for pharmaceutical industry. J Pharm Biomed Anal. 2016;126:83–97. doi: 10.1016/j.jpba.2016.03.052. [DOI] [PubMed] [Google Scholar]
- 31.Bobaly B, Fleury-Souverain S, Beck A, Veuthey JL, Guillarme D, Fekete S. Current possibilities of liquid chromatography for the characterization of antibody-drug conjugates. J Pharm Biomed Anal. 2018;147:493–505. doi: 10.1016/j.jpba.2017.06.022. [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Wang L, Shion H, Yu C, Yu YQ, Zhu L, Li M, Chen W, Gao K. In-depth structural characterization of Kadcyla(R) (ado-trastuzumab emtansine) and its biosimilar candidate. mAbs. 2016;8:1210–23. doi: 10.1080/19420862.2016.1204502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.