

Abstract
Introduction
Nucleic acids have gained recognition as promising immunomodulatory therapeutics. However, their potential is limited by several drug delivery barriers, and there is a need for technologies that enhance intracellular delivery of nucleic acid drugs. Furthermore, controlled and sustained release is a significant concern, as the kinetics and localization of immunomodulators can influence resultant immune responses. Here, we describe the design and initial evaluation of poly(lactic-co-glycolic) acid (PLGA) microparticle (MP) depots for enhanced retention and sustained release of endosomolytic nanoparticles that enable the cytosolic delivery of nucleic acids.
Methods
Endosomolytic p[DMAEMA]10kD-bl-[PAA0.3-co-DMAEMA0.3-co-BMA0.4]25kD diblock copolymers were synthesized by reversible addition-fragmentation chain transfer polymerization. Polymers were electrostatically complexed with nucleic acids and resultant nanoparticles (NPs) were encapsulated in PLGA MPs. To modulate release kinetics, ammonium bicarbonate was added as a porogen. Release profiles were quantified in vitro and in vivovia quantification of fluorescently-labeled nucleic acid. Bioactivity of released NPs was assessed using small interfering RNA (siRNA) targeting luciferase as a representative nucleic acid cargo. MPs were incubated with luciferase-expressing 4T1 (4T1-LUC) breast cancer cells in vitro or administered intratumorally to 4T1-LUC breast tumors, and silencing via RNA interference was quantified via longitudinal luminescence imaging.
Results
Endosomolytic NPs complexed to siRNA were effectively loaded into PLGA MPs and release kinetics could be modulated in vitro and in vivovia control of MP porosity, with porous MPs exhibiting faster cargo release. In vitro, release of NPs from porous MP depots enabled sustained luciferase knockdown in 4T1 breast cancer cells over a five-day treatment period. Administered intratumorally, MPs prolonged the retention of nucleic acid within the injected tumor, resulting in enhanced and sustained silencing of luciferase relative to a single bolus administration of NPs at an equivalent dose.
Conclusion
This work highlights the potential of PLGA MP depots as a platform for local release of endosomolytic polymer NPs that enhance the cytosolic delivery of nucleic acid therapeutics.
Keywords: Nucleic acid therapeutics, Local delivery, Intratumoral, Immunotherapy, RNA interference, Endosomal escape, PLGA, Biomaterial, Drug delivery depot
Introduction
Nucleic acids have emerged as a promising class of immunotherapeutics with potential to treat numerous diseases, including infections, inflammation, autoimmunity, and cancer.20,33,42,43,57,79,85 This broad and versatile class of biomacromolecular drugs can be leveraged to both activate and suppress the immune system. Notably, short-interfering RNA (siRNA) can be utilized to selectively inhibit expression of specific immunoregulatory proteins through RNA interference (RNAi),26,57,68,76,79 allowing for precision tailoring of immune responses. Additionally, nucleic acids that chemically or structurally mimic pathogenic genetic material can be harnessed to activate the innate immune system by targeting various nucleic acid sensing pathways, which have evolved to detect viral or bacterial invasion.13,20,33,42,43,84,85 Nucleic acids have been widely explored as adjuvants to bolster responses to vaccines,70 and more recently as cancer immunotherapeutics that initiate inflammatory programs at tumor sites to stimulate antitumor immunity.1,62 Despite their immense promise as immunomodulators, the clinical advancement of nucleic acid therapeutics has been relatively modest due to a multitude of challenges that hinder drug efficacy and/or patient safety.11,63
Inefficient intracellular delivery is a significant barrier to efficacy that is shared across virtually all types of nucleic acid therapeutics.11,34,68,69,72,76 Nucleic acids do not passively diffuse across the plasma membrane, are cleared rapidly after administration, and are endocytosed with relatively low efficiency. Additionally, while several immunostimulatory nucleic acids (e.g. CpG DNA, poly(I:C)) act through receptors residing in endosomal membranes, a larger number must access cytosolic targets to exert their immunoregulatory effects. This includes more common classes of nucleic acid therapeutics that can be leveraged for immunotherapy, such as siRNA, miRNA, and mRNA, but also an emerging family of immunostimulatory agents that engage cytosolic pattern recognition receptors (PRRs), such as RIG-I, MDA-5, cGAS, and STING.1,24,31 This pervasive challenge has led to the widespread development of synthetic nucleic acid carriers that enhance cellular uptake and promote endosomal escape of associated cargo.4,36,66 Our group, and others, have recently utilized pH-responsive, endosomolytic polymer nanoparticles (NPs) to enhance the cytosolic delivery and activity of siRNA and immunostimulatory 5′- triphosphate RNA.19,24,31,46 These NPs are assembled using amphiphilic diblock copolymers that self-assemble into micelles with a cationic dimethylaminoethyl methacrylate (DMAEMA) corona for electrostatic complexation of nucleic acids, and a pH-responsive, endosomolytic core comprising DMAEMA, butyl methacrylate (BMA), and propylacrylic acid (PAA) (Fig. 1b).18,19 While highly efficient at cytosolic delivery, the cationic corona has restricted the use of such NPs to local delivery applications, including tissue regeneration, vaccine delivery, and intratumoral cancer immunotherapy.7,31,73,77
Figure 1.

PLGA microparticle depots for controlled release of endosomolytic nanoparticles. (a) PLGA MP depots mediate local nanoparticle release and subsequent intracellular delivery of nucleic acid to local cell populations. (b) Structure and composition of the endosomolytic diblock copolymers used for cytosolic nucleic acid delivery. (c) Representative scanning electron microscopy (SEM) images of nonporous microparticles (left) and porous microparticles (right). Scale: 3 µm.
While systemic administration of nucleic acid therapeutics is necessary for many applications, directed, local delivery circumvents critical systemic delivery barriers and ensures sufficiently high doses reach target tissues, while also reducing systemic side effects.52,66 Indeed, local delivery is commonly used, and often preferred, for many immunotherapeutics, the most salient example being vaccines, which are delivered intradermally or intramuscularly.53,83 Additionally, image-guided, direct injection into lymph nodes (intranodal), considered the “command centers” of an immune response, is used clinically for treatment of allergy.67 Finally, intratumoral injection of immunotherapeutics, including several different nucleic acids, has become increasingly prevalent in recent clinical trials among substantial preclinical evidence that local immunotherapy can generate systemic immunity capable of eliminating distal, untreated tumors (e.g. abscopal effect).48,71 However, for nearly all of these applications, multiple, repeated injections are necessary to stimulate desired immune responses and attendant therapeutic activitiy.9,39,80 This requirement for multiple injections can pose a significant practical challenge for both physicians and patients and, in some cases, may not be feasible. Additionally, the timing, dose, and localization of immunomodulators plays a critical role in determining the magnitude and phenotype of the resultant immune response.5,10,65,81,82 Yet, locally administered biomacromolecules, including nucleic acids, typically rapidly clear from the injection site, which not only limits local bioavailability but can also result in systemic distribution with an increased risk of toxicity.37,39,80 These challenges have motivated the development of delivery technologies for controlled and sustained release of nucleic acid immunotherapeutics.2,3,32,55,58,75 These drug delivery depots can be either injectable or implantable scaffolds or microparticles, and are typically composed of biodegradable materials that release cargo in a controlled and sustained manner.64 Depots can also be engineered to exhibit a wide variety of drug release profiles by altering their chemical and physical properties.14
The NP system used here has been previously used in sustained and controllable release scaffolds aimed toward wound healing applications.50,54,73 Here, we describe an intratumorally-injectable nanoparticle-in-microparticle strategy for controlled, localized delivery of cytosolically-active nucleic acid therapeutics. Poly(lactic-co-glycolic) (PLGA) microparticle (MP) depots were designed for sustained release of endosomolytic NPs that can mediate the cytosolic delivery of various nucleic acids, exemplified here by intratumoral delivery of siRNA. Through enhanced retention, controlled and sustained release, and prolonged functionality of encapsulated NPs, this approach offers a simple and potentially universally applicable strategy for achieving enhanced spatial and temporal control of nucleic acid delivery for applications in immunotherapy.
Materials and Methods
Materials
All chemicals were supplied by Sigma Aldrich unless otherwise specified.
Synthesis of Endosomolytic Polymers
The amphiphilic diblock copolymer, poly(dimethylaminoethyl methacrylate)-block-[(propylacrylic acid)0.3-co-(dimethylaminoethyl methacrylate)0.3-co-(butyl methacrylate)0.4] (p[DMAEMA]-bl-[PAA0.3-co-DMAEMA0.3-co-BMA0.4]; D-PDB) was synthesized via reversible addition-fragmentation chain transfer (RAFT) polymerizations following a protocol adapted from Convertine et al.19 Briefly, 4-cyano-4-(ethylsulfanylthiocarbonyl)sulfanylpentanoic acid (ECT; Boron Molecular) was used as a chain transfer agent (CTA), and 2,2′-azobis(4-methoxy-2,4-dimethyl valeronitrile) (V-70; Wako Chemicals) was used as an initiator for RAFT polymerization. Mass measurements were performed using an analytical mass balance (XSE205DU DualRange; Mettler Toledo). Gravity filtration was employed in columns packed with aluminum oxide to remove inhibitors from monomer solutions. For the polymerization of the first block, DMAEMA, CTA, and initiator were dissolved in dioxane at a molar ratio of 100:1:0.05 at 40 wt% monomer, purged with nitrogen gas for 30 min on ice, and reacted at 30 °C for 18 h. The resultant polymer was then purified by precipitation (6x) in cold pentane followed by dialysis (3.5 kDa MWCO) in deionized water. Poly(DMAEMA) was then frozen at − 80 °C and then lyophilized for 3 days to obtain a dry powder.
For the polymerization of the second block, poly(DMAEMA) was used as a macroCTA (mCTA) and was added to DMAEMA, PAA, and BMA (30:30:40 mol%). PAA was synthesized as previously described using diethyl propylmalonate as the precursor.25 Using N,N-dimethylacetamide (DMAC) as the reaction solvent, initiator was added to mCTA and monomers at a molar ratio of 450:1:0.4 representing total monomer, mCTA, and initiator, respectively at 40 wt% mCTA and monomer. The reaction vessel was purged with nitrogen gas for 30 min on ice followed by reaction for 24 h at 30 °C in an oil bath. The resultant polymer was then purified by precipitation (6×) in pentane:ether (80:20) followed by dialysis in acetone (4 exchanges) and subsequent dialysis in deionized water. D-PDB was then frozen at − 80 °C and then lyophilized for 3 days. All lyophilized polymer was stored at − 20 °C until used.
The experimental degree of polymerization, polymer composition, and theoretical molecular weight were obtained by 1H nuclear magnetic resonance (NMR) Spectroscopy (CDCl3 with TMS, Bruker AV 400). Experimental molecular weight and polydispersity were determined via gel permeation chromatography (GPC) using HPLC-grade dimethylformamide (DMF) containing 0.1% LiBr as a mobile phase with inline light scattering (Wyatt Technology) and refractive index (Agilent) detectors. The ASTRA V Software (Wyatt Technology) was used for all GPC calculations. Hydrodynamic size of the polymer micelles at physiological pH 7.4 was measured via digital light scattering (DLS) using a Zetasizer Nano ZS Instrument (Malvern, USA). D-PDB used herein had a 1st block molecular weight of 10.3 kDa, a 2nd block molecular weight of 31.0 kDa, and a polydispersity index (PDI) of 1.24. The 2nd block composition was determined to be 28:33:39 for PAA, DMAEMA, and BMA, respectively. Additionally, the hydrodynamic diameter of the D-PDB micelles was ~ 100 nm by an intensity particle size distribution.
Formulation of Polymer Nanoparticles and Nucleic Acid Complexes for In Vitro Experiments
Micellar nanoparticles (NPs) were formulated according to a protocol adapted from Wilson et al.77 Lyophilized D-PDB was dissolved in ethanol to 50 mg/mL and rapidly diluted in 100 mM phosphate buffer (pH 7.0) to a concentration of 10 mg/mL to induce self-assembly into micelles. Polymer micelles were subsequently diluted in phosphate buffer saline (PBS; pH 7.4, 155 mM NaCl, 1.05 mM KH2PO3, 4 mM Na2HPO4, Gibco) to a concentration to 1 mg/mL. The micelles were then added to nucleic acid solutions at concentrations corresponding to a charge ratio (i.e. N/P ratio: molar charge from the polymer’s tertiary amines relative to the molar charge of phosphate from the nucleic acid backbone) of 4:1. Note that the N:P ratio is based on the poly(DMAEMA) first block and assuming 50% protonation of DMAEMA groups at pH 7.4. D-PDB micelles and nucleic acid were incubated at room temperature for 20 min to ensure complete electrostatic complexation.
Formulation of Polymer Nanoparticles and Nucleic Acid Complexes for In Vivo Experiments
D-PDB micelles were formulated as described above, followed by sterile filtration (0.2 µm polyethersulfone sterile filter) and subsequent concentration to 30–60 mg/mL in PBS via centrifugal filtration (Amicon® Ultra 0.5 mL Centrifugal Filter Units; Ultracel®—3 K, Regenerated Cellulose 3000 NMWL, Millipore) following manufacturer’s instructions. The final concentrated solution was collected, and an aliquot was used to determine the resultant polymer concentration using UV–Vis spectroscopy (Synergy H1 Multi-Mode Microplate Reader, Biotek) based on an absorbance-wavelength of 310 nm corresponding to ECT. The solution was added to nucleic acids at concentrations corresponding to a charge ratio (i.e. N/P ratio) of 4:1 as described above.
Cell Culture
All cell handling procedures were performed in accordance with published technical data sheets. Murine mammary epithelial 4T1-LUC tumor cells stably co-express destabilized copepod green fluorescent protein (cop-GFP) and firefly luciferase were generated using psuedotyped lentiviral particles. Briefly, a transfection mixture consisting of pGreenFire1-CMV (System Biosciences, Cat. No. TR011PA-1), psPAX2 (Addgene Plasmid #12260), and pCMV-VSV-G (Addgene Plasmid #8454) in water at a quantity of 10, 10, and 1 µg, respectively, was added to a final volume of 558 µL in Opti-MEM media (Gibco, Cat. No. 31985062) in a polypropylene tube, followed by the addition of 42 μL FuGENE 6 (Promega, Cat. No. E2691). The tube was gently flicked to mix the plasmids before and after the addition of FuGENE 6. The transfection mixture was added dropwise to a T-75 tissue culture flask at approximately 50% confluency of HEK-293-T cells in 11 mL Dulbecco’s modified eagle medium (DMEM, Gibco) supplemented to 10% heat-inactivated fetal bovine serum (HI-FBS) without antibiotics. Cells were incubated at 37 °C for 18 h, and then the media on the HEK-293-T cells was exchanged for DMEM supplemented with 10% HI-FBS and 1% penicillin/streptomycin (P/S). At 24 and 48 h after this media change, the viral supernatant was removed, clarified by centrifugation (1000×g, 5 min, room temperature) and syringe filtered (0.45 μm, nylon). To transduce 4T1 cells, viral supernatant was mixed 1:1 with fresh DMEM supplemented with 10% HI-FBS without antibiotics and applied to cells for 24 h. Cells were selected with 5 µg/mL puromycin for 2 weeks then sorted into approximately equal populations of low, medium, and high expressing cop-GFP cells using fluorescence activated cell sorting of GFP (BD FACSARIA IIIu, BD Biosciences) in the Vanderbilt Flow Cytometry Shared Resource Facility. The high expressing cop-GFP 4T1-LUC cells were used for all luminescent experiments herein. 4T1 and 4T1-LUC cells were maintained in DMEM supplemented with 2 mM l-glutamine, 4.5 g/L glucose, 10% HI-FBS, and 1% P/S. Cells were kept in a humidified environment at 37 °C with 5% CO2. Puromycin was added to 4T1-LUC cells after every cell passage at a concentration of 1 µg/mL for the continual selection of cells.
Preparation of PLGA Microparticles Encapsulating Micellar Nanoparticles
PLGA MPs encapsulating pH-responsive NPs were formed using a water-in-oil-in-water (W1/O/W2) double emulsion synthesis method previously reported.15,27,47,51,56 A fluorescently labelled double-stranded DNA (5′-[6FAM]ATAGGCGTATTATACGCGATTAACG-3′, negative control sequence) was used as representative cargo to determine the ideal conditions for the loading of NPs into PLGA MPs. Briefly, 100 mg of poly(d,l-lactide-co-glycolide) (PLGA, Resomer® RG 503, 50:50, ester-terminated, MW 24,000–38,000 Da) was dissolved in 750 μL of dichloromethane (DCM) for 30 min under continuous shaking at room temperature. 200 μL of NP solution (i.e. polyplexes prepared with D-PBD and various amounts of nucleic acid strands ranging in concentration from 1.9 nmol to 11.4 nmol) was added dropwise to the PLGA solution at a primary aqueous phase (W1) to oil phase (O) volume ratio of 0.27. The primary emulsion (W1/O) was prepared by sonicating the two phases for 30 s at 40% amplitude on ice using a Sonic Dismembrator (Fisher Scientific™ Model 120). The secondary emulsion (W1/O/W2) was formulated by homogenizing the primary emulsion into 15 mL of 1% polyvinyl alcohol solution (PVA) for 30 s at 20,000 rpm on ice using a T18 digital ULTRA-TURRAX®, equipped with a S18N-10G dispersing tool (IKA). The double emulsion was then transferred to a round bottom flask and rotary evaporated for 1 h at 400 torr to allow complete evaporation of DCM. MPs were collected by centrifugation (10,000×g, 10 min, 4 °C) and washed 3 times with sterile water. PLGA MPs were then frozen at − 80 °C for 5 h and then lyophilized for 3 days. The effervescent salt, ammonium bicarbonate was employed as a porogen to create porous MPs. 20 wt% NH4HCO3 was incorporated into the W1 aqueous phase along with the NPs and then emulsified with the oil phase as described. All PLGA MPs were stored at − 20 °C until used.
The hydrodynamic diameter of the PLGA MPs was measured by laser-diffraction size analysis using a Mastersizer 2000 (Malvern, USA). Approximately, 10–20 mg of PLGA MPs were dissolved in deionized water and used for analysis. Measurements detected within the acceptable range, between 10 and 15% obscuration, were deemed to be reproducible data points. Surface morphology and porosity of the PLGA MPs were analyzed using a Zeiss Merlin scanning electron microscope (SEM; Carl Zeiss Microscopy, LLC, ZEISS Group, Thornwood, NY) equipped with a GEMINI II column. SEM samples were prepared by reconstituting PLGA MPs in deionized water at a concentration of 2 mg/mL and then placing 20 µL of the solution on a strip of carbon tape (Ted Pella Inc.) adhered onto an aluminum SEM stub (Ø12.7 mm, Ted Pella Inc). After drying overnight, samples were sputter coated with gold–palladium for 120 s and immediately imaged via SEM.
Evaluation of Loading and Encapsulation Efficiency
To determine the nucleic acid loading and encapsulation efficiency, nucleic acids were extracted from PLGA MPs. In brief, 7.5 mg of PLGA MPs were dissolved in 400 µL DCM and continuously mixed for 45 min at room temperature. 400 µL of TE buffer supplemented with 100 mM NaCl was added to this mixture and vortexed vigorously for 5 min. The suspension was then centrifuged (15,000×g, 10 min, 4 °C). The aqueous layer was collected into a fresh tube, and the extraction was performed again. The two extracted layers were combined, incubated with 1% sodium dodecyl sulfate (SDS) for 10 min at room temperature to disassemble the any electrostatically associated nucleic acids, and nucleic acid concentration was determined via fluorescence spectroscopy (excitation/emission wavelengths of 495/525 nm for 6FAM-DNA or 650/685 nm for Alexa Fluor® 647 (A647)-siRNA). Nucleic acid loading and encapsulation efficiencies were determined based on the ratio of encapsulated nucleic acid to PLGA MPs (µg/mg) and percentage relative to the theoretical maximum loading (%), respectively.
In Vitro Release of Nanoparticles from PLGA Microparticles
To investigate the in vitro release profiles of NPs from porous and nonporous MPs, 20 mg of PLGA MPs was suspended 1 mL sterile PBS (pH 7.4, 0.02% sodium azide) in microcentrifuge tubes and maintained at 37 °C with constant rotation. At pre-determined time intervals, tubes were centrifuged (15,000 rpm, 10 min, 4 °C), and 900 µL of supernatant was removed for analysis, replaced by the same volume of fresh buffer, and frozen and lyophilized for further analysis. Each lyophilized sample was reconstituted in 220 µL TE buffer supplemented with 100 mM NaCl, pipetted into a UV-Star® microplate (100 µL/well), and quantified by a fluorescence plate reader (Synergy H1 Hybrid Multi-Mode Reader, BioTek) as described above. All samples were run in technical duplicates.
In Vivo Controlled Release of Nanoparticles from PLGA Microparticles
Female BALB/c mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained at the animal facilities of Vanderbilt University under conventional conditions. The mice were anesthetized with isoflurane gas and maintained at 37 °C while their flanks or abdomens were depilated and sterilized for subcutaneous or intratumoral administration. NPs were prepared with A647-DNA (negative control sequence, IDT DNA) and loaded into PLGA MPs with or without porogen for subcutaneous and intratumoral in vivo release studies. 6–8 week old mice were anesthetized with isoflurane gas and given a single subcutaneous injection of porous MP (n = 5), nonporous MP (n = 5), or NP (n = 3). For the murine tumor studies, 106 4T1-LUC cells were inoculated (50 µL injection volume) into the inguinal mammary fatpads of 6–8 week old mice anesthetized with isoflurane gas. Tumor volume was calculated using the equation: Volume = (Length × Width2)/2. When tumor volumes reached 50–100 mm3, mice were anesthetized with isoflurane gas and administered a single intratumoral injection of porous MPs (n = 3), nonporous MPs (n = 3), or NPs (n = 3). All treatments were administered at a 10 µg dose of nucleic acid in a 100 µL injection volume. Using constant image capture settings on an IVIS Spectrum (PerkinElmer), mice were imaged at predetermined time intervals to quantify A647 fluorescence. Relative release of NPs was determined by measuring the total fluorescent efficiency (cm2) of A647 overtime and normalizing to the respective initial (day 0) values.
In Vitro Evaluation of Luciferase Knockdown
NPs were prepared with the siRNA oligos, siLUC (anti-luciferase sequence, 5′-CAAUUGCACUGAUAAUGAACUCCTC[3AlexF647N]-3′; IDT DNA) or siNC (negative control sequence, 5′-[5AlexF647N]AUACGCGUAUUAUACGCGAUUAACGAC-3′; IDT DNA) and encapsulated into porous MPs as described above. 4T1-LUC cells were seeded in five black 24-well plates (a separate plate for each day of imaging) with clear tissue culture treated bottoms (Sensoplate REF:662892; Greiner Bio-One) at 2000 cells per well (500 µL seeding volume). NPs were complexed with either siLUC (siLUC/NP) or siNC (siNC/NP) and embedded in porous MPs (siLUC/MP and siNC/MP). Cells were treated 24 h later with free NPs or porous MPs at a final concentration of 50 nM nucleic acid per well. The supernatant in the free NP-treated wells was removed from all plates at 24 h to mimic the NP clearance observed in vivo. Every 24 h over the course of 5 days, Pierce d-luciferin (ThermoFisher Scientific) was administered to all the wells within the plate for the corresponding day to a final d-luciferin concentration of 0.15 mg/mL. 5 min after the addition of d-luciferin, plates were imaged for bioluminescent signal using an IVIS Lumina III (PerkinElmer). Images were captured at 24, 48, 72, 96, 120 h post-treatment, and luciferase knockdown was quantified for each day based on the percent decrease in bioluminescent signal (i.e. Total Flux, photons/second) relative to each respective negative control siRNA.
In Vivo Evaluation of Luciferase Knockdown
Female BALB/c mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained at the animal facilities of Vanderbilt University under conventional conditions. Orthotopic 4T1-LUC tumors were generated as described above. siLUC/NPs and siNC/NPs were prepared as described above and loaded into porous MPs (siLUC/MP and siNC/MP). When tumor volumes reached 50–100 mm3, mice were anesthetized with isoflurane gas and administered a single intratumoral injection of free NPs or porous MPs (n = 10 for all treatment groups). All treatments were administered at a 10 µg oligonucleotide dose (0.5 mg/kg) in a 100 µL injection volume. Using constant image capture settings on an IVIS Lumina III (PerkinElmer), mice were analyzed at predetermined time intervals for fluorescence and bioluminescence. Bioluminescence within the mice was measured 10 min after dorsal subcutaneous injection of 300 µL Pierce d-luciferin (15 mg/mL). After 14 days mice were euthanized and tumor samples were isolated postmortem for histological analysis.
Statistical Analysis
All data analysis was performed on Graphpad Prism (Version 7.0c). One-way analysis of variance (ANOVA) coupled with Tukey’s post-test was used to compare statistical significance among multiple groups (> 2). Differences between two groups were analyzed by unpaired t tests. In vivo experiments were performed with at least three biological replicates, with ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05 being considered statistically significant.
Results and Discussion
Design and In Vitro Characterization of PLGA Microparticle Depots
To generate depots for controlled release of cytosolically-active nucleic acids, we encapsulated endosomolytic polymer NPs complexed with nucleic acid (either double-stranded DNA or siRNA) within MPs of PLGA, a biocompatible, hydrolytically-degradable, and commonly used biomaterial for local and sustained therapeutic drug delivery.17,21–23,29,35,40,41,44,45,74 PLGA MPs were synthesized using DCM as a volatile organic solvent and PVA as a surfactant in a W1/O/W2 double emulsion as previously described.15,27,47,56 Sonication and homogenization were employed after the primary and secondary emulsions, respectively. NPs were incorporated into the W1 aqueous phase, resulting in a drug loading of approximately 1.8 ± 0.05 µg nucleic acid per mg PLGA and an encapsulation efficiency of about 75 ± 2%. To generate porous MPs with a faster release profile, the effervescent salt ammonium bicarbonate was added to the W1 aqueous phase. Following PLGA MP synthesis, SEM imaging was performed to characterize MP morphology, which confirmed that ammonium bicarbonate was an effective porogen for NP-loaded PLGA MPs (Fig. 1c). Laser diffraction size analysis was used to quantitatively characterize the particle size distribution (Fig. 2a). Nonporous MPs and porous MPs had an average diameter of 21.21 µm and 28.33 µm, respectively. An in vitro release assay was performed to characterize the release profiles of NPs from PLGA MP depots with varying porosity (Fig. 2b). As expected, the addition of pores and the associated increase in surface area within PLGA MPs resulted in faster release of the NP cargo, likely reflecting the shorter diffusion distance for release. While cationic excipients, such as polyethyleneimine or polyamines, have been incorporated into PLGA to increase nucleic acid loading and intracellular delivery,6,59,61,78 this represents the first demonstration of a PLGA MP depot used for sustained release of endosomolytic nanoparticles that enhance cytosolic nucleic acid delivery.
Figure 2.
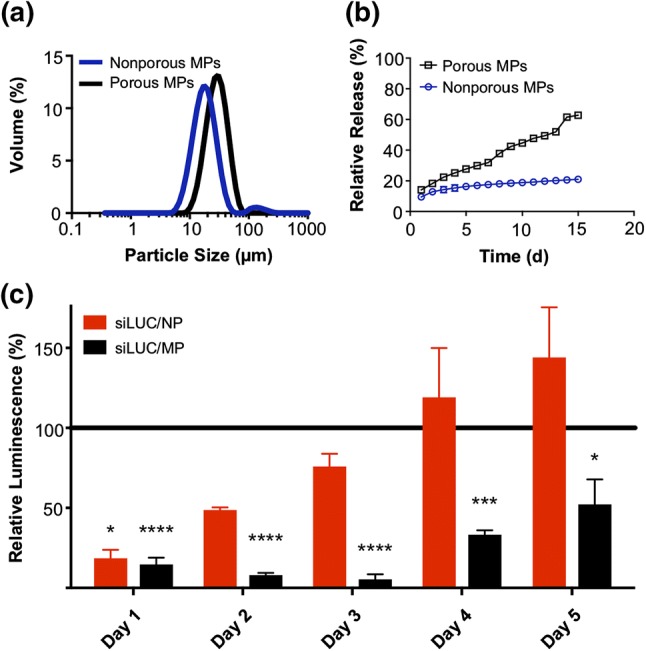
In vitro characterization of PLGA microparticle depots. (a) Particle size distribution of nonporous and porous MPs determined by laser diffraction particle sizing. (b) In vitro release profiles of NPs from porous and nonporous MP depots over a 15 day period. (c) Longitudinal analysis of luciferase silencing in 4T1-LUC breast cancer cells treated with a single administration of either free NPs or porous MPs. The NP treatments were removed after 24 h, while MPs were left in coculture with the cells throughout the experiment to mimic biological residence. Luminescent signal for each treatment group was normalized to that of an analogous treatment containing scrambled negative control RNA substituted for luciferase siRNA.
In Vitro RNAi Luciferase Silencing
An in vitro RNAi protein knockdown assay was performed to demonstrate that PLGA MP depots could sustain the release and biological activity of nucleic acid-loaded NPs (Fig. 2c). As a model nucleic acid cargo, siRNA specific for luciferase (siLUC) or a scrambled negative control siRNA (siNC) were complexed with D-PDB micelles (siRNA/NP) and loaded into porous MPs (siRNA/MP). 4T1-LUC breast cancer cells, engineered to constitutively express luciferase, were treated with free NP or porous MPs each complexed to either siLUC or siNC at 50 nM siRNA per well. Free NPs were removed after 24 h to approximate a transient residence time at an injected site, whereas cells were incubated with MP depots for an additional 4 days. Bioluminescence imaging was used to quantify luciferase expression each day, following an administration of d-luciferin. While comparable silencing was observed between free siLUC/NP and siLUC/MP after 1 day (~ 75% knockdown), continuous incubation with depots resulted in significantly greater knockdown on days 2–5. The luciferase expression of the cells treated for 24 h with siLUC/NP returned to near baseline intensity within 3 days. Due to the short doubling time of 4T1 cells, cultures approached confluence within 5 days, which therefore precluded evaluation of knockdown at later timepoints. Nonetheless, these data demonstrate the capacity of PLGA MP depots to sustain the release and silencing activity of encapsulated siRNA/NP complexes.
Cytotoxicity is a well-established challenge of all polycationic nucleic acid delivery platforms that can indeed limit their utility in local delivery applications. However, this may be advantageous or detrimental depending on the intended application of the system; for example, in an intratumoral setting, some toxicity can galvanize cancer cell antigen release and may therefore be beneficial toward priming an anti-cancer immune response. Notably, we observed similar expression of bioluminescence in both the NP and MP negative control groups, suggesting that there is no difference in cell viability between the various treatments and that the PLGA used to entrap the NPs does not contribute to cellular toxicity, which is consistent with its high cytocompatibility.
In Vivo Nanoparticle Release from PLGA Microparticle Depots
To monitor NP release and retention in vivo, NPs were electrostatically complexed with a fluorescently-labeled double-stranded DNA (dsDNA/NP) and then loaded into PLGA MP depots (dsDNA/MP). Fluorescent dsDNA was used as representative cargo as it is a cost-effective analog to other nucleic acid sequences of similar length such as fluorescent siRNA. Free dsDNA/NP, nonporous dsDNA/MP, and porous dsDNA/MP were administered subcutaneously (s.c.) at a dose of 10 µg DNA (0.5 mg/kg) into BALB/c mice, and fluorescence was monitored with an in vivo imaging system (IVIS) to track the retention of dsDNA at the injection site (Figs. 3a and 3c). Free dsDNA/NP rapidly cleared the injection site, with > 50% clearance within 24 h and undetectable levels present by 5 days. By contrast, both MP depots enhanced retention and sustained release of dsDNA/NP, with porous depots demonstrating faster release than analogous nonporous depots, particularly within the first week of administration. Gradual release from both depots was observed over the following month with significant fluorescence still evident at day 56.
Figure 3.

In vivo retention and release of nanoparticles from PLGA microparticles. In vivo analysis of injection site localization of free NPs, nonporous MP depots, and porous MP depots in BALB/c mice. (a) Relative fluorescence of Alexa Fluor® 647(A647)-labelled dsDNA cargo injected subcutaneously and monitored over 56 days. (b) Relative fluorescence of A647-labelled dsDNA cargo, releasing from an intratumoral injection site over 14 days. The fluorescent efficiency of each mouse was captured by IVIS imaging and was normalized to the respective initial (day 0) fluorescence. (c) Representative IVIS images of mice bearing subcutaneously administered particles containing fluorescent dsDNA (Red). (d) Representative IVIS images of the mice treated intratumorally with particles containing fluorescent dsDNA (Red).
We also evaluated NP retention in the context of intratumoral (i.t.) delivery, which is increasing in use both preclinically and clinically as an administration route for cancer immunotherapeutics, including several nucleic acid drugs.8,12,28 Here, we administered free dsDNA/NP, nonporous dsDNA/MP, and porous dsDNA/MP into 50 mm3 4T1 tumors growing in the inguinal mammary fat pad, fluorescence was monitored within the tumor over time with intravital fluorescence imaging via IVIS. Similar to the release profiles observed with s.c. administration, MP depots enabled sustained release of dsDNA/NP over a 2-week period, the longest possible time-frame based on the endpoint tumor volume (~ 1500 mm3). Again, the porous MP depots exhibited faster release with ~ 75% of cargo cleared within 2 weeks, whereas minimal release from nonporous MPs was observed (Figs. 3a and 3b). Notably, despite their cationic surface charge, free dsDNA/NP drained quickly with > 60% of nucleic acid cleared from the tumor site within 24 h. This rapid clearance may in part explain the need for multiple injections when using these or similar NPs for localized intratumoral delivery of siRNA or 5′ppp-RNA ligands of RIG-I.24,31 Moreover, these data add to a large body of evidence indicating that the fate of most intratumorally administered nanoparticles and/or macromolecular therapeutics is a short and often suboptimal intratumoral half-life followed by ultimate systemic clearance. This also further motivates the design of implantable or injectable depots for intratumoral administration16,60 or the incorporation of ligand to tether agents to local cells and/or extracellular matrix.30,38
In Vivo RNAi Luciferase Silencing
Based on their capacity to release ~ 50% of NP cargo into tumors within 1 week, we evaluated the ability of porous MP depots to sustain activity of a nucleic acid therapeutic, here, siRNA targeting luciferase. Inspired by several ongoing clinical trials exploring intratumoral immunotherapy,8,12,28,48 intratumoral injections were employed for protein knockdown studies to demonstrate the utility of PLGA MP depots in a cancer setting. While subcutaneous injections are undoubtedly easier for physicians to perform, recent advances in surgical intervention have made intratumoral injections more practical, as almost every site in the human body can be biopsied and therefore injected.49 Thus, both administration routes explored within the retention studies have potential for clinical translation. To evaluate luciferase knockdown, mice with 4T1-LUC tumors growing in the inguinal mammary fat pad were intratumorally administered a single 10 µg siRNA dose (0.5 mg/kg) of siLUC/NP either free or loaded into depots (siLUC/MP); siNC/NP and siNC/MP were used as negative controls. IVIS imaging of both luminescence and fluorescence demonstrated a qualitatively high degree of co-localization between siLUC/NP and tumor cells (Fig. 4b), and MPs could also be identified within cyrosections of resected tumors (Fig. 4a). Using longitudinal IVIS imaging, we also quantified bioluminescence to determine the degree of luciferase knockdown from the anti-luciferase siRNA cargo 1–4 days post-intratumoral injection (Fig. 4c). We found that porous MP depots loaded with siLUC/NP resulted in ~ 50% reduction in luminescent signal relative to analogous depots loaded with siNC/NP control complexes. By contrast, at a 10 µg siRNA dose, no luciferase knockdown was observed using free siLUC/NP, potentially reflecting the relatively short half-life of NPs within the tumor after local administration. Collectively, these data demonstrate that increasing intratumoral residence time of nucleic acid therapeutics via sustained release can enhance and prolong biological activity.
Figure 4.

In vivo activity of PLGA microparticle depots for siRNA delivery. In vivo activity of free NP and porous MPs delivering Alexa Fluor® 647 siRNA cargo was investigated in an orthotopic 4T1-LUC breast cancer model. (a) Fluorescent (top) and overlaid fluorescent and bright field (bottom) images of cyrosections of tumor tissue following intratumoral injection of porous MPs. Scale: 75 µm. (b) Representative IVIS images of mice bearing luciferase-expressing 4T1-LUC cells (Blue), treated intratumorally with fluorescent RNA (Red). (c) Longitudinal analysis of luciferase silencing in a 4T1-LUC breast cancer tumor model treated with a single intratumoral injection of either free NPs or porous MPs. Luminescent signal for each treatment group was normalized to that of an analogous treatment containing scrambled negative control RNA substituted for luciferase siRNA.
In these studies, we utilized PLGA MPs as a depot for siRNA/NP owing to their favorable biocompatibility and tunable biodegradability. However, inefficient loading of hydrophilic cargo during the W1/O/W2 emulsion synthesis is a known limitation of PLGA depots, which we found to be the case as well for the loading of NPs (~ 1.8 µg oligonucleotide per mg PLGA). This necessitates delivery of a relatively high volume of MPs to obtain relevant doses of NPs in the context of RNAi, which may restrict the applications of this approach. While we achieved ~ 50% luciferase silencing over 3 days using a single dose of MPs, further enhancements may be achieved using doses higher than those employed herein, which were restrained by the volume of MPs that could be physically injected into 4T1 tumors. Therefore, for cancer therapy applications, PLGA MP depots for NP release may be better suited for localized delivery into tumor resection cavities that can be filled with a larger volume of MPs. To establish proof-of-concept, we used siRNA as a well-established model nucleic acid cargo throughout our investigations, but in principle this approach can be used for local and sustained delivery of any cytosolically-active nucleic acid, including immunostimulatory agonists such as 5′ppp-RNA RIG-I ligands31 or immunostimulatory DNA ligands of cGAS.1 However, exploration of these promising immunotherapeutics is at a stage of relative infancy, and therefore much remains to be elucidated regarding dose and treatment regimens that result in optimal efficacy. Nonetheless, PLGA MP depots for release of endosomolytic NPs offer a promising strategy for enhancing the cytosolic delivery of such nucleic acids and locally sustaining their bioavailability and immunostimulatory activity in vivo.
Conclusion
Localized delivery of cytosolically-active nucleic acids offers a promising approach for spatiotemporal modulation of immune responses with broad potential applicability in the treatment of many diseases. However, efficacy in this setting is limited by inefficient cytosolic delivery as well as rapid clearance from the administration site. To address these challenges, we developed a nano-in-microparticle delivery platform using PLGA MPs as a depot for the controlled release of endosomolytic NPs that promote cytosolic delivery of electrostatically complexed nucleic acid cargo. Using siRNA as a model therapeutic, we demonstrated that the rate of release of siRNA/NP complexes both in vitro and in vivo could be increased using ammonium bicarbonate as a porogen during the fabrication process. Importantly, we found that release of siRNA/NP complexes from PLGA MP depots resulted in sustained protein silencing in vitro as well as in an orthotopic murine breast cancer model via intratumoral administration. The observed 50% protein knockdown in breast cancer tumors may indeed be sufficient for delivery of an immunomodulatory agent where only a portion of cells within a tumor need to be stimulated in order to produce a more immunogenic tumor microenvironment. Collectively, these studies demonstrate that controlled release of endosomolytic nanoparticles from porous MP depots may offer an enabling strategy for controlled and localized delivery of nucleic acid therapeutics that target cytosolic immunoregulatory machinery. While this technology holds promise for local administration, improved performance could be achieved with a higher degree of drug loading and more tightly controlled kinetics of drug release that might enable sustained silencing and/or enhanced cytosolic delivery of siRNA or innate immune agonists. Additionally, co-administering chemotherapy or radiotherapy to ablate the majority of the tumor cells and allow for decreased tumor burden at the site of injection would likely synergize well with this platform.
Acknowledgments
We gratefully acknowledge Dr. Bob Weinberg and Dr. Didier Trono for gifts of plasmids via Addgene.org. We thank Dr. Steven Goodbred Jr. and his laboratory for use of the Mastersizer 2000 (Malvern, USA). We thank Kyle Becker for his assistance with the orthotopic tumor inoculations. We thank the core facilities of Vanderbilt, including the Vanderbilt Institute of Nanoscale Sciences and Engineering (VINSE) for the use of both the Zetasizer Nano ZS Instrument (Malvern, USA) and the Zeiss Merlin SEM (Carl Zeiss Microscopy, LLC, ZEISS Group, Thornwood, NY), the Vanderbilt Translational Pathology Shared Resource (supported in part by the NCI/NIH Cancer Center Support Grant 5P30 CA684850-19) for cryosectioning of tumor samples, and Vanderbilt University Medical Center Flow Cytometry Shared Resource (supported by the Vanderbilt Ingram Cancer Center P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404) for cell sorting. This research was supported by grants from Alex’s Lemonade Stand Foundation ‘A’ Award SID924 (JTW) and Pediatric Oncology Student Training (POST) Award cosponsored by Love Your Mellon (KMG), the American Cancer Society Institutional Research Grant IRG-58-009-56 (JTW), the Congressionally-Directed Medical Research Program W81XWH-161-0063 (JTW) and W81XWH-161-0063 (RSC), the National Institutes of Health R01CA224241 (CLD) and R01EB019409 (CLD), and the National Science Foundation Graduate Research Fellowship Program 0909667 and 1445197 (KVK).
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
All animal experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committee (IACUC), and all surgical and experimental procedures were performed in accordance with the regulations and guidelines of the Vanderbilt University IACUC. Female BALB/cJ mice (6–8 weeks old; The Jackson Laboratory, Bar Harbor, ME) were maintained at the animal facilities of Vanderbilt University under specific pathogen-free conditions. Tumor volume, total mass, and animal well-being were monitored every other day.
Abbreviations
- BMA
Butyl methacrylate
- DCM
Dichloromethane
- DMAEMA
Dimethylaminoethyl methacrylate
- D-PDB
Poly[DMAEMA]10kD-block-[PAA0.3-co-DMAEMA0.3-co-BMA0.4]25kD
- ECT
4-Cyano-4-(ethylsulfanylthiocarbonyl)sulfanylpentanoic acid
- MP
Microparticle
- NP
Nanoparticle
- PAA
Propylacrylic acid
- PLGA
Poly(lactic-co-glycolic) acid
- PVA
Polyvinyl alcohol
- SEM
Scanning electron microscopy
- V-70
2,2′-Azobis(4-methoxy-2,4-dimethyl valeronitrile)
Footnotes
John T. Wilson is an Assistant Professor of Chemical and Biomolecular Engineering and Biomedical Engineering at Vanderbilt University. He received his B.S. in Bioengineering from Oregon State University and his Ph.D. from the Georgia Institute of Technology in the laboratory of Professor Elliot L. Chaikof, where he was awarded a Whitaker Foundation Graduate Fellowship. He then joined the laboratory of Professor Patrick Stayton at the University of Washington with support of a Cancer Research Institute Postdoctoral Fellowship. He started his independent laboratory at Vanderbilt in January of 2014, where his group works at the interface of molecular engineering and immunology to innovate technologies to improve human health. His multidisciplinary research program is supported by productive and synergistic collaborations with oncologists, cancer biologists, immunologists, chemists, and other engineers. Since establishing his lab at Vanderbilt, he has been awarded the NSF CAREER Award, an ‘A’ Award from Alex’s Lemonade Stand Foundation, a Melanoma Research Alliance Young Investigator Award, an Innovative Research Grant from Stand Up To Cancer, and has been named an Emerging Investigator by Biomaterials Science.
This article is part of the 2019 CMBE Young Innovators special issue.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kyle Garland and Sema Sevimli contributed equally to this paper.
References
- 1.Ahn J, Xia T, Rabasa Capote A, Betancourt D, Barber GN. Extrinsic phagocyte-dependent STING signaling dictates the immunogenicity of dying cells. Cancer Cell. 2018;33(5):862–873e5. doi: 10.1016/j.ccell.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nat. Mater. 2009;8(2):151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali OA, Verbeke C, Johnson C, Sands RW, Lewin SA, White D, Doherty E, Dranoff G, Mooney DJ. Identification of immune factors regulating antitumor immunity using polymeric vaccines with multiple adjuvants. Cancer Res. 2014;74(6):1670–1681. doi: 10.1158/0008-5472.CAN-13-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aliabadi HM. Natural polymers in nucleic acid delivery. In: Narain R, editor. Polymers and Nanomaterials for Gene Therapy. Cambridge: Woodhead Publishing; 2016. pp. 55–80. [Google Scholar]
- 5.Aliru ML, Schoenhals JE, Venkatesulu BP, Anderson CC, Barsoumian HB, Younes AI, Ls KM, Soeung M, Aziz KE, Welsh JW, Krishnan S. Radiation therapy and immunotherapy: what is the optimal timing or sequencing? Immunotherapy. 2018;10(4):299–316. doi: 10.2217/imt-2017-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amar-Lewis E, Azagury A, Chintakunta R, Goldbart R, Traitel T, Prestwood J, Landesman-Milo D, Peer D, Kost J. Quaternized starch-based carrier for siRNA delivery: from cellular uptake to gene silencing. J. Control. Release. 2014;185:109–120. doi: 10.1016/j.jconrel.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 7.Arany S, Benoit DS, Dewhurst S, Ovitt CE. Nanoparticle-mediated gene silencing confers radioprotection to salivary glands in vivo. Mol. Ther. 2013;21(6):1182–1194. doi: 10.1038/mt.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aznar MA, Tinari N, Rullan AJ, Sanchez-Paulete AR, Rodriguez-Ruiz ME, Melero I. Intratumoral delivery of immunotherapy-act locally, think globally. J. Immunol. 2017;198(1):31–39. doi: 10.4049/jimmunol.1601145. [DOI] [PubMed] [Google Scholar]
- 9.Bartlett DW, Davis ME. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006;34(1):322–333. doi: 10.1093/nar/gkj439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beyranvand Nejad E, Welters MJ, Arens R, van der Burg SH. The importance of correctly timing cancer immunotherapy. Expert Opin. Biol. Ther. 2017;17(1):87–103. doi: 10.1080/14712598.2017.1256388. [DOI] [PubMed] [Google Scholar]
- 11.Bobbin ML, Rossi JJ. RNA Interference (RNAi)-Based Therapeutics: delivering on the Promise? Annu. Rev. Pharmacol. Toxicol. 2016;56:103–122. doi: 10.1146/annurev-pharmtox-010715-103633. [DOI] [PubMed] [Google Scholar]
- 12.Brody JD, Ai WZ, Czerwinski DK, Torchia JA, Levy M, Advani RH, Kim YH, Hoppe RT, Knox SJ, Shin LK, Wapnir I, Tibshirani RJ, Levy R. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J. Clin. Oncol. 2010;28(28):4324–4332. doi: 10.1200/JCO.2010.28.9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013;13(8):551–565. doi: 10.1038/nri3479. [DOI] [PubMed] [Google Scholar]
- 14.Brudno Y, Mooney DJ. On-demand drug delivery from local depots. J. Control. Release. 2015;219:8–17. doi: 10.1016/j.jconrel.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 15.Chang E, McClellan AJ, Farley WJ, Li DQ, Pflugfelder SC, De Paiva CS. Biodegradable PLGA-based drug delivery systems for modulating ocular surface disease under experimental murine dry eye. J. Clin. Exp. Ophthalmol. 2011;2(11):191. doi: 10.4172/2155-9570.1000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Q, Wang C, Zhang X, Chen G, Hu Q, Li H, Wang J, Wen D, Zhang Y, Lu Y, Yang G, Jiang C, Wang J, Dotti G, Gu Z. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol. 2019;14(1):89–97. doi: 10.1038/s41565-018-0319-4. [DOI] [PubMed] [Google Scholar]
- 17.Cohen H, Levy RJ, Gao J, Fishbein I, Kousaev V, Sosnowski S, Slomkowski S, Golomb G. Sustained delivery and expression of DNA encapsulated in polymeric nanoparticles. Gene Ther. 2000;7(22):1896–1905. doi: 10.1038/sj.gt.3301318. [DOI] [PubMed] [Google Scholar]
- 18.Convertine AJ, Benoit DS, Duvall CL, Hoffman AS, Stayton PS. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J. Control. Release. 2009;133(3):221–229. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Convertine AJ, Diab C, Prieve M, Paschal A, Hoffman AS, Johnson PH, Stayton PS. pH-Responsive polymeric micelle carriers for siRNA drugs. Biomacromolecules. 2010;11(11):2904–2910. doi: 10.1021/bm100652w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooper C, Mackie D. Hepatitis B surface antigen-1018 ISS adjuvant-containing vaccine: a review of HEPLISAV safety and efficacy. Expert Rev. Vaccines. 2011;10(4):417–427. doi: 10.1586/erv.10.162. [DOI] [PubMed] [Google Scholar]
- 21.Cun D, Foged C, Yang M, Frokjaer S, Nielsen HM. Preparation and characterization of poly(d,l-lactide-co-glycolide) nanoparticles for siRNA delivery. Int. J. Pharm. 2010;390(1):70–75. doi: 10.1016/j.ijpharm.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 22.Cun D, Jensen DK, Maltesen MJ, Bunker M, Whiteside P, Scurr D, Foged C, Nielsen HM. High loading efficiency and sustained release of siRNA encapsulated in PLGA nanoparticles: quality by design optimization and characterization. Eur. J. Pharm. Biopharm. 2011;77(1):26–35. doi: 10.1016/j.ejpb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J. Control. Release. 2012;161(2):505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 24.Elion DL, Jacobson ME, Hicks DJ, Rahman B, Sanchez V, Gonzales-Ericsson PI, Fedorova O, Pyle AM, Wilson JT, Cook RS. Therapeutically active RIG-I agonist induces immunogenic tumor cell killing in breast cancers. Cancer Res. 2018;78(21):6183–6195. doi: 10.1158/0008-5472.CAN-18-0730. [DOI] [PubMed] [Google Scholar]
- 25.Ferritto MS, Tirrell DA. Photoregulation of the binding of an azobenzene-modified poly(methacrylic acid) to phosphatidylcholine bilayer membranes. Biomaterials. 1990;11(9):645–651. doi: 10.1016/0142-9612(90)90022-i. [DOI] [PubMed] [Google Scholar]
- 26.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 27.Frauke Pistel K, Breitenbach A, Zange-Volland R, Kissel T. Brush-like branched biodegradable polyesters, part III. Protein release from microspheres of poly(vinyl alcohol)-graft-poly(d,l-lactic-co-glycolic acid) J. Control. Release. 2001;73(1):7–20. doi: 10.1016/s0168-3659(01)00231-0. [DOI] [PubMed] [Google Scholar]
- 28.Hammerich L, Binder A, Brody JD. In situ vaccination: cancer immunotherapy both personalized and off-the-shelf. Mol Oncol. 2015;9(10):1966–1981. doi: 10.1016/j.molonc.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han FY, Thurecht KJ, Whittaker AK, Smith MT. Bioerodable PLGA-based microparticles for producing sustained-release drug formulations and strategies for improving drug loading. Front Pharmacol. 2016;7:185. doi: 10.3389/fphar.2016.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishihara J, Fukunaga K, Ishihara A, Larsson HM, Potin L, Hosseinchi P, Galliverti G, Swartz MA, Hubbell JA. Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl. Med. 2017;9(415):eaan0401. doi: 10.1126/scitranslmed.aan0401. [DOI] [PubMed] [Google Scholar]
- 31.Jacobson ME, Wang-Bishop L, Becker KW, Wilson JT. Delivery of 5′-triphosphate RNA with endosomolytic nanoparticles potently activates RIG-I to improve cancer immunotherapy. Biomater. Sci. 2019;7(2):547–559. doi: 10.1039/c8bm01064a. [DOI] [PubMed] [Google Scholar]
- 32.Jewell CM, López SCB, Irvine DJ. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc. Natl. Acad. Sci. USA. 2011;108(38):15745–15750. doi: 10.1073/pnas.1105200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang W, Zhu FG, Bhagat L, Yu D, Tang JX, Kandimalla ER, La Monica N, Agrawal S. A toll-like receptor 7, 8, and 9 antagonist inhibits Th1 and Th17 responses and inflammasome activation in a model of IL-23-induced psoriasis. J. Invest. Dermatol. 2013;133(7):1777–1784. doi: 10.1038/jid.2013.57. [DOI] [PubMed] [Google Scholar]
- 34.Johannes L, Lucchino M. Current challenges in delivery and cytosolic translocation of therapeutic RNAs. Nucleic Acid Ther. 2018;28(3):178–193. doi: 10.1089/nat.2017.0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan A, Benboubetra M, Sayyed PZ, Ng KW, Fox S, Beck G, Benter IF, Akhtar S. Sustained polymeric delivery of gene silencing antisense ODNs, siRNA, DNAzymes and ribozymes: in vitro and in vivo studies. J. Drug Target. 2004;12(6):393–404. doi: 10.1080/10611860400003858. [DOI] [PubMed] [Google Scholar]
- 36.Krhac Levacic A, Morys S, Wagner E. Solid-phase supported design of carriers for therapeutic nucleic acid delivery. Biosci. Rep. 2017;37(5):BSR20160617. doi: 10.1042/BSR20160617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwong B, Gai SA, Elkhader J, Wittrup KD, Irvine DJ. Localized immunotherapy via liposome-anchored Anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Can. Res. 2013;73(5):1547–1558. doi: 10.1158/0008-5472.CAN-12-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwong B, Liu H, Irvine DJ. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials. 2011;32(22):5134–5147. doi: 10.1016/j.biomaterials.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langer R. Drug delivery and targeting. Nature. 1998;392(6679 Suppl):5–10. [PubMed] [Google Scholar]
- 40.Langer R, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428(6982):487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 41.Luby TM, Cole G, Baker L, Kornher JS, Ramstedt U, Hedley ML. Repeated immunization with plasmid DNA formulated in poly(lactide-co-glycolide) microparticles is well tolerated and stimulates durable T cell responses to the tumor-associated antigen cytochrome P450 1B1. Clin. Immunol. 2004;112(1):45–53. doi: 10.1016/j.clim.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 42.Lurescia S, Fioretti D, Rinaldi M. Targeting cytosolic nucleic acid-sensing pathways for cancer immunotherapies. Front. Immunol. 2018;9:711. doi: 10.3389/fimmu.2018.00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lurescia S, Fioretti D, Rinaldi M. Nucleic acid sensing machinery: targeting innate immune system for cancer therapy. Recent Pat. Anticancer Drug Discov. 2018;13(1):2–17. doi: 10.2174/1574892812666171030163804. [DOI] [PubMed] [Google Scholar]
- 44.Luten J, van Nostrum CF, De Smedt SC, Hennink WE. Biodegradable polymers as non-viral carriers for plasmid DNA delivery. J. Control. Release. 2008;126(2):97–110. doi: 10.1016/j.jconrel.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 45.Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 2011;3(3):1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malcolm DW, Freeberg MAT, Wang Y, Sims KR, Awad HA, Benoit DSW. Diblock copolymer hydrophobicity facilitates efficient gene silencing and cytocompatible nanoparticle-mediated siRNA delivery to musculoskeletal cell types. Biomacromolecules. 2017;18(11):3753–3765. doi: 10.1021/acs.biomac.7b01349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao S, Xu J, Cai C, Germershaus O, Schaper A, Kissel T. Effect of WOW process parameters on morphology and burst release of FITC-dextran loaded PLGA microspheres. Int. J. Pharm. 2007;334(1–2):137–148. doi: 10.1016/j.ijpharm.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 48.Marabelle A, Kohrt H, Caux C, Levy R. Intratumoral immunization: a new paradigm for cancer therapy. Clin. Cancer Res. 2014;20(7):1747–1756. doi: 10.1158/1078-0432.CCR-13-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marabelle A, Tselikas L, de Baere T, Houot R. Intratumoral immunotherapy: using the tumor as the remedy. Ann. Oncol. 2017;28(suppl 12):xii33–xii43. doi: 10.1093/annonc/mdx683. [DOI] [PubMed] [Google Scholar]
- 50.Martin JR, Nelson CE, Gupta MK, Yu F, Sarett SM, Hocking KM, Pollins AC, Nanney LB, Davidson JM, Guelcher SA, Duvall CL. Local delivery of PHD2 siRNA from ROS-degradable scaffolds to promote diabetic wound healing. Adv. Healthc. Mater. 2016;5(21):2751–2757. doi: 10.1002/adhm.201600820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGinity JW, O’Donnell PB. Preparation of microspheres by the solvent evaporation technique. Adv. Drug Deliv. Rev. 1997;28(1):25–42. doi: 10.1016/s0169-409x(97)00049-5. [DOI] [PubMed] [Google Scholar]
- 52.Milling L, Zhang Y, Irvine DJ. Delivering safer immunotherapies for cancer. Adv. Drug Deliv. Rev. 2017;114:79–101. doi: 10.1016/j.addr.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.National Center for Immunization and Respiratory Diseases General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm. Rep. 2011;60(2):1–64. [PubMed] [Google Scholar]
- 54.Nelson CE, Gupta MK, Adolph EJ, Shannon JM, Guelcher SA, Duvall CL. Sustained local delivery of siRNA from an injectable scaffold. Biomaterials. 2012;33(4):1154–1161. doi: 10.1016/j.biomaterials.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishikawa M, Mizuno Y, Mohri K, Matsuoka N, Rattanakiat S, Takahashi Y, Funabashi H, Luo D, Takakura Y. Biodegradable CpG DNA hydrogels for sustained delivery of doxorubicin and immunostimulatory signals in tumor-bearing mice. Biomaterials. 2011;32(2):488–494. doi: 10.1016/j.biomaterials.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 56.Ogawa Y, Yamamoto M, Okada H, Yashiki T, Shimamoto T. A new technique to efficiently entrap leuprolide acetate into microcapsules of polylactic acid or copoly(lactic/glycolic) acid. Chem. Pharm. Bull. (Tokyo) 1988;36(3):1095–1103. doi: 10.1248/cpb.36.1095. [DOI] [PubMed] [Google Scholar]
- 57.Ozcan G, Ozpolat B, Coleman RL, Sood AK, Lopez-Berestein G. Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 2015;87:108–119. doi: 10.1016/j.addr.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pannier AK, Shea LD. Controlled release systems for DNA delivery. Mol. Ther. 2004;10(1):19–26. doi: 10.1016/j.ymthe.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 59.Pantazis P, Dimas K, Wyche JH, Anant S, Houchen CW, Panyam J, Ramanujam RP. Preparation of siRNA-encapsulated PLGA nanoparticles for sustained release of siRNA and evaluation of encapsulation efficiency. Methods Mol. Biol. 2012;906:311–319. doi: 10.1007/978-1-61779-953-2_25. [DOI] [PubMed] [Google Scholar]
- 60.Park CG, Hartl CA, Schmid D, Carmona EM, Kim HJ, Goldberg MS. Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci. Transl. Med. 2018;10(433):eear1916. doi: 10.1126/scitranslmed.aar1916. [DOI] [PubMed] [Google Scholar]
- 61.Patil Y, Panyam J. Polymeric nanoparticles for siRNA delivery and gene silencing. Int. J. Pharm. 2009;367(1–2):195–203. doi: 10.1016/j.ijpharm.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J, Schmidt A, Anz D, Bscheider M, Schwerd T, Berking C, Bourquin C, Kalinke U, Kremmer E, Kato H, Akira S, Meyers R, Häcker G, Neuenhahn M, Busch D, Ruland J, Rothenfusser S, Prinz M, Hornung V, Endres S, Tüting T, Hartmann G. 5′-Triphosphate-siRNA: turning gene silencing and RIG-I activation against melanoma. Nat. Med. 2008;14(11):1256–1263. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 63.Radovic-Moreno AF, Chernyak N, Mader CC, Nallagatla S, Kang RS, Hao L, Walker DA, Halo TL, Merkel TJ, Rische CH, Anantatmula S, Burkhart M, Mirkin CA, Gryaznov SM. Immunomodulatory spherical nucleic acids. Proc. Natl. Acad. Sci. USA. 2015;112(13):3892–3897. doi: 10.1073/pnas.1502850112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rathbone MJ, Hadgraft J, Roberts MS. Modified-release drug delivery technology. London: Taylor & Francis; 2002. [Google Scholar]
- 65.Rothschilds AM, Wittrup KD. What, why, where, and when: bringing timing to immuno-oncology. Trends Immunol. 2019;40(1):12–21. doi: 10.1016/j.it.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 66.Sarett SM, Nelson CE, Duvall CL. Technologies for controlled, local delivery of siRNA. J. Control. Release. 2015;218:94–113. doi: 10.1016/j.jconrel.2015.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Senti G, Freiburghaus AU, Larenas-Linnemann D, Hoffmann HJ, Patterson AM, Klimek L, Di Bona D, Pfaar O, Ahlbeck L, Akdis M, Weinfeld D, Contreras-Verduzco FA, Pedroza-Melendez A, Skaarup SH, Lee SM, Cardell LO, Schmid JM, Westin U, Dollner R, Kundig TM. Intralymphatic immunotherapy: update and unmet needs. Int. Arch. Allergy Immunol. 2019;178(2):141–149. doi: 10.1159/000493647. [DOI] [PubMed] [Google Scholar]
- 68.Sioud M. RNA interference: mechanisms, technical challenges, and therapeutic opportunities. Methods Mol. Biol. 2015;1218:1–15. doi: 10.1007/978-1-4939-1538-5_1. [DOI] [PubMed] [Google Scholar]
- 69.Smith SA, Selby LI, Johnston APR, Such GK. The endosomal escape of nanoparticles: towards more efficient cellular delivery. Bioconjug. Chem. 2018;30(2):263–272. doi: 10.1021/acs.bioconjchem.8b00732. [DOI] [PubMed] [Google Scholar]
- 70.van den Boorn JG, Barchet W, Hartmann G. Nucleic acid adjuvants: toward an educated vaccine. Adv. Immunol. 2012;114:1–32. doi: 10.1016/B978-0-12-396548-6.00001-9. [DOI] [PubMed] [Google Scholar]
- 71.van den Boorn JG, Hartmann G. Turning tumors into vaccines: co-opting the innate immune system. Immunity. 2013;39(1):27–37. doi: 10.1016/j.immuni.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 72.Wang LL, Burdick JA. Engineered hydrogels for local and sustained delivery of RNA-interference therapies. Adv. Healthc. Mater. 2017;6(1):1601041. doi: 10.1002/adhm.201601041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, Malcolm DW, Benoit DSW. Controlled and sustained delivery of siRNA/NPs from hydrogels expedites bone fracture healing. Biomaterials. 2017;139:127–138. doi: 10.1016/j.biomaterials.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang D, Robinson DR, Kwon GS, Samuel J. Encapsulation of plasmid DNA in biodegradable poly(d,l-lactic-co-glycolic acid) microspheres as a novel approach for immunogene delivery. J. Control. Release. 1999;57(1):9–18. doi: 10.1016/s0168-3659(98)00099-6. [DOI] [PubMed] [Google Scholar]
- 75.Wang C, Sun W, Wright G, Wang AZ, Gu Z. Inflammation-triggered cancer immunotherapy by programmed delivery of CpG and anti-PD1 antibody. Adv. Mater. 2016;28(40):8912–8920. doi: 10.1002/adma.201506312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 2009;8(2):129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee C-C, Opara C, Convertine A, Stayton PS. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano. 2013;7(5):3912–3925. doi: 10.1021/nn305466z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woodrow KA, Cu Y, Booth CJ, Saucier-Sawyer JK, Wood MJ, Saltzman WM. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat. Mater. 2009;8(6):526–533. doi: 10.1038/nmat2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu SY, Lopez-Berestein G, Calin GA, Sood AK. RNAi therapies: drugging the undruggable. Sci. Transl. Med. 2014;6(240):240. doi: 10.1126/scitranslmed.3008362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu-Pong S, Rojanasakul Y. Biopharmaceutical Drug Design and Development. 2. Totowa: Humana Press; 2008. p. 375. [Google Scholar]
- 81.Yan J, Wang Z-Y, Yang H-Z, Liu H-Z, Mi S, Lv X-X, Fu X-M, Yan H-M, Zhang X-W, Zhan Q-M, Hu Z-W. Timing is critical for an effective anti-metastatic immunotherapy: the decisive role of IFNγ/STAT1-mediated activation of autophagy. PLoS ONE. 2011;6(9):e24705. doi: 10.1371/journal.pone.0024705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Young KH, Baird JR, Savage T, Cottam B, Friedman D, Bambina S, Messenheimer DJ, Fox B, Newell P, Bahjat KS, Gough MJ, Crittenden MR. Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. PLoS ONE. 2016;11(6):e0157164. doi: 10.1371/journal.pone.0157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang L, Wang W, Wang S. Effect of vaccine administration modality on immunogenicity and efficacy. Expert Rev. Vaccines. 2015;14(11):1509–1523. doi: 10.1586/14760584.2015.1081067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu FG, Jiang W, Bhagat L, Wang D, Yu D, Tang JX, Kandimalla ER, La Monica N, Agrawal S. A novel antagonist of Toll-like receptors 7, 8 and 9 suppresses lupus disease-associated parameters in NZBW/F1 mice. Autoimmunity. 2013;46(7):419–428. doi: 10.3109/08916934.2013.798651. [DOI] [PubMed] [Google Scholar]
- 85.Zhu X, Nishimura F, Sasaki K, Fujita M, Dusak JE, Eguchi J, Fellows-Mayle W, Storkus WJ, Walker PR, Salazar AM, Okada H. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J. Transl. Med. 2007;5:10. doi: 10.1186/1479-5876-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]