

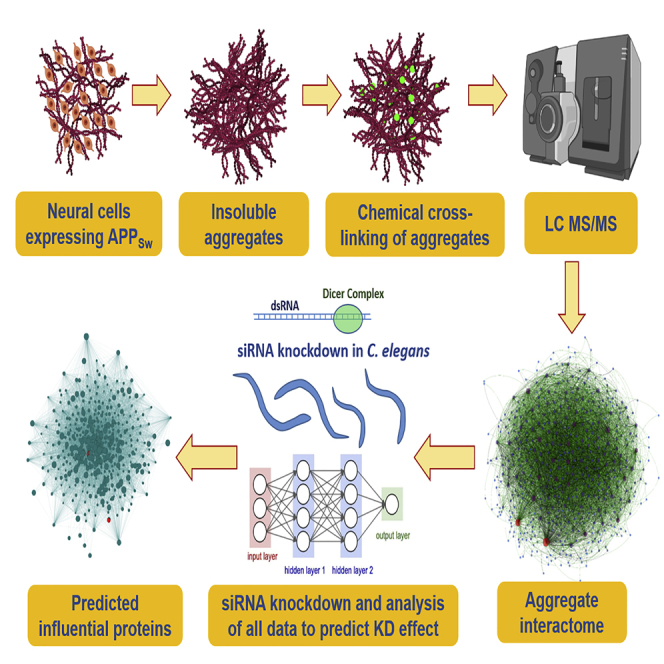
Summary
Diagnosis of neurodegenerative diseases hinges on “seed” proteins detected in disease-specific aggregates. These inclusions contain diverse constituents, adhering through aberrant interactions that our prior data indicate are nonrandom. To define preferential protein-protein contacts mediating aggregate coalescence, we created click-chemistry reagents that cross-link neighboring proteins within human, APPSw-driven, neuroblastoma-cell aggregates. These reagents incorporate a biotinyl group to efficiently recover linked tryptic-peptide pairs. Mass-spectroscopy outputs were screened for all possible peptide pairs in the aggregate proteome. These empirical linkages, ranked by abundance, implicate a protein-adherence network termed the “aggregate contactome.” Critical hubs and hub-hub interactions were assessed by RNAi-mediated rescue of chemotaxis in aging nematodes, and aggregation-driving properties were inferred by multivariate regression and neural-network approaches. Aspirin, while disrupting aggregation, greatly simplified the aggregate contactome. This approach, and the dynamic model of aggregate accrual it implies, reveals the architecture of insoluble-aggregate networks and may reveal targets susceptible to interventions to ameliorate protein-aggregation diseases.
Subject Areas: Neuroscience, Molecular Neuroscience, Neural Networks, Proteomics
Graphical Abstract
Highlights
-
•
Cross-link data support a preferred hierarchy of protein accrual into aggregates
-
•
Contact networks can predict proteins that contribute functionally to aggregation
-
•
RNAi knockdowns of key hubs and hub connectors imply functional roles in accrual
-
•
Aspirin opposes protein aggregation by reducing contactome interactions >5-fold
Neuroscience; Molecular Neuroscience; Neural Networks; Proteomics
Introduction
The presence of specific intra- and extra-cellular aggregates is the hallmark pathology for diverse neurodegenerative diseases, including Alzheimer disease (AD) (Davis et al., 2018). Such aggregates include a wide variety of proteins in addition to the “seed” proteins, i.e., Aβ42 and tau in AD, which are believed to initiate the formation of the diagnostic aggregates (Ayyadevara et al., 2016b). Many of these non-seed constituents, including 14-3-3 proteins, chaperones, phosphorylated TDP-43, and neurofilament and motor proteins, are common to a wide variety of pathological aggregates (Ayyadevara et al., 2015, Ayyadevara et al., 2016b, David et al., 2010). In previous studies, using C. elegans models of neurodegeneration-associated aggregation characteristic of AD and Huntington disease, we identified a wide variety of proteins in aggregates isolated by immuno-pulldown with antibodies to seed proteins. Importantly, many non-seed proteins have key functional roles that would be disrupted by their sequestration into aggregates (Ayyadevara et al., 2015, Ayyadevara et al., 2016b, Ayyadevara et al., 2016c, Balasubramaniam et al., 2018). In addition, post-translational modifications have been shown to result in structural changes that impact both protein function and aggregation by exposing buried/hydrophobic surfaces to other misfolded proteins (Fink, 1998, Huang et al., 2014, Karve and Cheema, 2011). As an example, we have documented interactions between aggregate proteins that were specific to their phosphorylated forms, e.g., 14-3-3 interactions with hyper-phosphorylated tau (hP-tau), observed only in aggregates from Alzheimer hippocampus (Ayyadevara et al., 2016b).
In the present study, we developed a cross-linking approach to define pathology-associated protein interactions involved in aggregation and to learn how post-synthetic modifications alter those interactions. We developed a two-stage cross-linking protocol that uses click-chemistry to add a high-affinity epitope for enrichment of linked peptide pairs before mass spectrometry. As a proof-of-principle study, we applied this to a cell-culture model of amyloid deposition: human neuroblastoma cells that express an AD-predisposing mutant of APP (SY5Y-APPSw). To identify cross-linked peptides and their phosphorylation state, liquid chromatography-tandem mass spectrometry (LC-MS/MS) data were analyzed by an algorithm targeting peptides previously identified in the aggregate proteome. This strategy allowed us to define the most abundant protein-protein interfaces within aggregates and thus to construct a nonfunctional interactome, herein termed the “aggregate contactome.” Using aspirin as a known positive drug documented to reduce aggregation (Ayyadevara et al., 2013), we then showed that this contactome shrinks after aspirin treatment, due in large part to disappearance of the majority of intra-aggregate interactions. We thus demonstrate that drug candidates and small-molecule libraries can be screened for disruption of key interfaces predicted to mediate the assembly of disease-specific aggregates.
Results
Analysis of Aggregate-Specific Contactomes
To characterize protein:protein contacts within amyloid aggregates, one of the principal diagnostic features of AD, we studied a well-defined model for amyloid accrual, the SY5Y-APPSw neuroblastoma cell line. These human cells express the “Swedish” dual mutation of Amyloid Precursor Protein (APPSw) observed in familial AD, and unlike their nontransgenic parental cell line SY5Y, they accumulate pericellular amyloid-like aggregates rich in β-pleated sheets (Figure 1A). Although SY5Y cells form a small amount of APP-containing aggregate, these foci are roughly a third as abundant as in SY5Y-APPSw cells (Figure 1B), based on fluorescence after thioflavin T staining.
Figure 1.

Analysis of Aggregate-Specific Interactomes
(A) Amyloid-like aggregates were stained with thioflavin T, in SY5Y neuroblastoma cells (top panel) and SY5Y-APPSw cells (bottom panels). Scale bar indicates 10 μm.
(B) Fluorescence intensity was quantified for cells stained with thioflavin-T; significance was determined by one-tailed t tests. Data are shown as mean ± S.E.M.
(C) Graphical view of the insoluble-aggregate interactome of SY5Y-APPSw cells. Node (protein) color is based on degree, the number of interacting partners (see key); the edge (interaction) color indicates whether the observed protein-protein contact is peculiar to SY5Y-APPSw cells (green) or is also present in untransformed (“WT”) SY5Y cells (black).
(D) Venn diagram indicating number of interactions that are specific to SY5Y-APPSw cells, specific to SY5Y, or present in both.
(E) Average spectral hits for observed peptide pairs (total per protein) quantified from SY5Y(WT) and SY5Y-APPSw cross-linking proteomics. “WT + APP” indicates hits for proteins shared by SY5Y(WT) and SY5Y-APPSw (see D).
(F) Proteins (nodes) from the aggregate interactome were categorized based on the number of interacting partners (degree), ranging from hub connectors that connect two to nine hub proteins, to mega-hubs (≥100 partners).
To define the aggregate contactome specific to AD-like amyloidosis, sarcosyl-insoluble aggregates were isolated using established protocols (Ayyadevara et al., 2015, Ayyadevara et al., 2016b, Ayyadevara et al., 2016c) from SY5Y and SY5Y-APPSw cells. In brief, aggregates were isolated by differential centrifugation before and following addition of 1% sarcosyl, incubated 0.5 h at 22oC with cross-linking reagents, and digested 14 h at 37°C (essentially to completion) with trypsin. A biotin moiety was coupled to the cross-linker by click chemistry (Chowdhury et al., 2009), enabling capture of linked peptide pairs on streptavidin-coated magnetic beads. Eluted peptide pairs were analyzed by high-resolution mass spectrometry, and the raw data files matched to m/z peaks predicted by pairwise addition of masses ([tryptic peptide 1] + linker + [tryptic peptide 2]) within the set of all proteins identified in the same aggregates without cross-linking. Because the latter process is computationally intensive for sets of >500 proteins, we created a multithreading version of Xlink-Identifier (Du et al., 2011) for high-throughput implementation to quantify linked peptide pairs from aggregates (see Supplemental Information, Transparent Methods, for details).
Peptide pairs confirmed as highly abundant in aggregates indicate specific proximal peptides at or near the interfaces of protein:protein interactions. These intra-aggregate cross-links neither exclude nor imply functional interactions between the soluble forms of the same proteins but rather suggest that such adherent surfaces may mediate aggregate accretion. In constructing aggregate contactomes (using GePhi graph-modeling software [Jacomy et al., 2014]), we included only those interactions with ≥10 spectral hits in at least two of three independent cross-linking experiments, thus eliminating protein-protein interfaces that are infrequent or irreproducible. With these constraints, we identified 535 proteins (nodes), comprising 471 unmodified (unphosphorylated) forms and 64 phosphorylated species. Of the 535 nodes, 29 were excluded as non-hub proteins (possessing only one cross-link partner), leaving 506 hubs involved in a total of 7,100 interactions. In Figure 1C, interactions are shown as edges (lines joining hubs), and each hub's color reflects the total number of its interactions to other proteins (see inset keys, Figure 1C).
Comparing hub interaction numbers (first-order connectivity) for aggregates from SY5Y versus SY5Y-APPSw cells, 5,994 of 7,100 interactions (84.4%) were specific to SY5Y-APPSw, whereas 5,643 of 6,749 (83.6%) were seen only in parental SY5Y cells (Figure 1D). These specific interactions thus depend positively or negatively, respectively, on the presence of mutated APP (APPSw). Although the total number of interactions is quite similar in SY5Y-APPSw and SY5Y cells, indicating a similar complexity of aggregate structure, the average spectral-hit count per linked protein pair unique to SY5Y-APPSw cells was twice the average for shared or SY5Y-unique pairs (Figure 1E). This abundance ratio is a bit smaller than the ratio estimated from thioflavin T staining (2.7-fold greater intensity for SY5Y-APPSw; Figures 1A and 1B). Thus, the transgenic expression of APPSw increases the total amount of aggregated protein, but this occurs at the expense of some interactions that had been observed in SY5Y cells lacking APPSw, presumably because key components of those aggregates have been diverted to APPSw-specific aggregates.
The aggregate contactome reveals a complex architecture comprising 506 nodes that vary in degree (number of interacting partners). For each protein node, characteristic descriptors were compiled, including previously reported graph modeling and statistical parameters (Jacomy et al., 2014, Demšar et al., 2013; see Methods for details). As an aid to visualization of candidates and their relative impacts on aggregation, hub proteins were partitioned into subcategories of mega, major, midi, and mini hubs based on the abundance of interacting nodes (defined in Figure 1F). Interestingly, the majority of mega-hub proteins (≥100 partners) are either structural or matrix proteins. Roughly half of all aggregate proteins fall into the midi-hub category (10–49 partners), whereas the hub categories just larger and smaller than that comprise 18.5% and 5%, respectively.
We also identified proteins with low levels of direct interaction (<10 partners), connecting higher-degree hubs (≥10 partners) that are not directly associated with one another. For each node in the contactome, we calculated the local, undirected clustering coefficient (CC) as the fraction of triplets (potential node triangles) directly connected to that node, which form closed triangles of three interconnected nodes (Latapy et al., 2008) (see Methods). We also calculated eigenvector centrality (EC, a measure of node connectivity to high-degree hubs, defined in Methods). Nodes with CC ≤ 0.5 and a ratio of EC/CC > k (see Methods) were considered to be influential “hub connectors,” which couple to two or more hub proteins that are not directly connected. Hub connectors comprise 134 proteins, or 26.5% of all nodes (Figure 1F).
Cross-link Contactomes Implicate Interactions between APP and Other Proteins
To assess the biological relevance of the cross-link-based contactome, we focused on the interactions of two key seed proteins that are widely accepted as sources of AD aggregate-initiating moieties: APP and tau. AD brain contains both extracellular amyloid plaque (enriched in Aβ42, a cleavage product of APP) and intra-neuronal neurofibrillary tangles (containing hP-tau fragments as paired helical filaments, or PHF). These two aggregate types appear at distinct sites in AD tissue; it remains controversial whether either or both drive disease progression and whether there is cross talk between them.
We recently reported evidence of tau:Aβ42 interactions within aggregates. Proximity-ligation amplification showed intracellular Aβ42 and tau situated within 40 nm of one another, supporting proteomic analyses that identified tau in aggregates purified by Aβ42 pulldown and vice versa (Ayyadevara et al., 2016b). We are now able to assess whether cross-linking profiles indicate direct or indirect interaction between tau and APP/Aβ42. Our analysis of APP interactions (enlarged box in Figure 2A) identified unexpected partners, including SRRM2 and SRSF6_P (pre-mRNA splicing factors), SRRT (miRNA processing component that binds capped primary transcripts), TOP1 (DNA topoisomerase 1), Ki-67 (a DNA-binding marker of cell proliferation), and LAMB2 (laminin B2, which tethers nuclei within the cytoplasm). Ki-67 and laminins were previously implicated in AD as they directly affect aggregation (Gauczynski et al., 2001, Jovanovic et al., 2014, Nagy et al., 1997, Smith and Lippa, 1995). Remarkably, the three largest (highest-degree) hubs among the eight proteins directly cross-linked to APP (SRRM2, SRSF6, and Ki-67) also have first-degree interactions with tau (Figure 2E) or phospho-tau (Figure 2F), implying that they often coalesce in the same aggregates.
Figure 2.

The Aggregate Interactome Identifies Indirect Interactions of APPSw and Tau
(A) Identified interacting partners of APP (E2 domain) in insoluble SY5Y-APPSw aggregates.
(B) Domain architecture of APP protein, as inferred from the Protein DataBank (PDB). ECM, extracellular matrix; 1mwp, 3nyl, 1iyt: APP domain IDs in PDB.
(C) Protein-protein docking of SRRM2 and the APP-E2 domain corroborates observed cross-linking data (yellow highlighted area).
(D) Protein-protein docking of TOP1 with APP-E2 corroborates observed cross-linking data (yellow highlighted oval indicates concordant evidence of interaction).
(E) Tau-interacting proteins identified from SY5Y-APPSw insoluble aggregates. Proteins in dashed boxes also interact with the APP-E2 domain.
(F) Interactions of phosphorylated tau peptides, characteristic of hP-TAU, identified in insoluble aggregates from SY5Y-APPSw. SRSF6_P (dashed box) also interacts with APP-E2.
APP fragments implicated or enriched in SY5Y-APPSw aggregates (with ≥10 spectral hits and corroborated in at least two independent cross-linking experiments) were mapped back to the intact protein, to define specific sites of all observed APP peptide interactions with other proteins. Surprisingly, all eight APP intra-aggregate interactions involve peptides in the central E2 domain (Lee et al., 2011) (Figure 2B). When we relaxed the threshold to ≥3 hits in at least two of three cross-linking experiments, we observed 191 interactions involving peptides within the E2 domain of APPSw but only six (3.1%) that involve tryptic fragments of the Aβ42 peptide. A known limitation of LC-MS/MS proteomics is that not all potential tryptic peptides are equally capable of contributing to identification of a protein; for example, very small or large peptides may never meet the Mascot threshold for high-confidence, unambiguous identification. Because two of the three possible Aβ42 peptides were detected abundantly, although in only a few interactions, the observed underrepresentation of Aβ42 is unlikely to be artifactual. We note that the two mutations in APPSw (K595N/M596L) lie adjacent to the β-secretase cleavage site, altering the Aβ42/Aβ40 ratio and greatly increasing the synthesis of APP itself (Pahrudin Arrozi et al., 2017). They could also have allosteric effects on APP structure elsewhere, similar to distal effects we reported for mutations in the C-terminal region of profilin (Kiaei et al., 2018). Because the E2 domain of APP contains a histidine tetrad with the potential to bind copper or zinc (Dahms et al., 2012), even moderate disturbance of E2 by K595N/M596L could profoundly influence APP's structure and potential for misfolding.
To visually compare observed and predicted interactions within the E2 domain of APPSw, we used HEX protein-protein docking software to model three of the eight observed molecular interactions in silico. Such in silico simulation results agree well with cross-linking data, providing support for the use of both methods to predict interactions. For example, the ovals in Figures 2C and 2D highlight identified peptide cross-link sites that coincide with computationally predicted protein-protein interfaces between APPSw-E2 and SRRM2 (Figure 2C) or TOP1 (Figure 2D). Similar agreement between peptide cross-linking results and protein docking predictions was also seen for RPS5 (40S ribosomal protein S5; data not shown).
Contactomes Identify Distinct Interacting Partners Specific to Either Unmodified Tau or Hyper-Phosphorylated Tau
Cross-link-based contactomes for tau fragments resolve them into two distinct mini-hubs with differing partners, one for unphosphorylated tau peptides that predominate in normal brain tissue (Figure 2E) and another for phosphorylated peptides (tau-P) typical of hyper-phosphorylated tau (hP-tau; Figure 2F) observed in AD (Ayyadevara et al., 2016b, Simic et al., 2016). Both unmodified tau and hP-tau interactions were found in wild-type SY5Y cells but were far more abundant in SY5Y-APPSw cells. Remarkably, of 7,255 spectral hits for tau interactions in aggregates (i.e., the sum of spectral hits for two independent isolations of linked-peptide pairs containing a tau or tau-P peptide), none were derived from the PHF region (tau residues 244–378), which is thought to initiate neurofibrillary tangle formation (Fitzpatrick et al., 2017). Our observation that both unmodified and phosphorylated Ki-67 peptides interact directly with unmodified tau fragments (Figure 2E) may help to explain the previously reported critical role of Ki-67 in tau hyperphosphorylation, which in turn leads to the generation of paired helical filaments (Smith and Lippa, 1995).
Protein Phosphorylation in the Aggregate Contactome
Hyperphosphorylation refers to the post-translational addition of multiple phosphate groups. It is typical of several diagnostic seed proteins, including tau, TDP-43, and α-synuclein (Fujiwara et al., 2002, Hasegawa et al., 2008), and is believed to alter protein structure to the detriment of function and stability, while promoting aggregation (Beyer and Ariza, 2013, McFarland et al., 2008, Ren et al., 2007, Rodriguez-Martin et al., 2013). Our cross-linking data distinguish between interactions involving unphosphorylated and phosphorylated peptides within aggregates and thus partition aggregation-associated hub proteins into three categories: 22% of hubs featured peptides that are always phosphorylated (red hubs in Figure 3A), 64% were identified entirely by unmodified peptides (blue hubs), and 14% consisted of mixtures of phosphorylated and unmodified peptides (purple hubs) (Figures 3A and 3B). Among 134 hub connectors, however (green circles in Figure 3A), 19% were exclusively identified through phosphopeptides, 79% entirely via unmodified peptides, and only 2% with both phosphorylated and unmodified peptides (Figure 3B). Scarcity of “ambivalent” hub connectors suggests that they are induced to aggregate by the hubs they connect, rather than through their own misfolding. The observation that 22% of peptides from aggregate-contactome hubs are always phosphorylated, and 36% are at least partially phosphorylated, is consistent with a previous report that phosphopeptides are highly enriched in aggregates (Ayyadevara et al., 2016b) and further implies that sites of abnormal phosphorylation are especially vulnerable to pro-aggregative interactions.
Figure 3.

Protein Phosphorylation in the Aggregate Interactome
(A) Aggregate protein-phosphorylation details inferred from SY5Y-APPSw proteomics. Three categories of aggregate proteins were observed: those identified via peptides that are always phosphorylated (red), those with peptides having both phosphorylated and unmodified forms (purple), and those whose peptides are always unmodified (blue). Hub connectors are shown in green.
(B) Venn diagram depicting the composition of aggregate proteins, based on peptide phosphorylation as in (A).
(C) Cross-linked contacts of phosphorylated CAND1 (arrow, cullin-associated and neddylation-dissociated protein 1), showing contactome linkages to other hub proteins, including DYHC1 and mega-hub SRRM2. Of 19 connected hubs, 12 are phosphorylation-state-specific (red or blue).
(D) Cross-linked contacts of phosphorylated DYHC1 (arrow, dynein heavy chain 1), showing that its contactome linkages with other hub proteins are mostly phosphorylation-specific (12 red/always, 17 blue/never, 11 purple/mixed).
(E) Cross-linked contacts of phosphorylated DYN2 (arrow, dynamin 2), showing its contactome linkages with 7 always-phosphorylated/red hub proteins, 4 never-phosphorylated/blue hubs and 2 mixed/purple hubs.
(F) Cross-linked contacts of phosphorylated SYNE2 (arrow, spectrin repeat containing nuclear envelope protein 2) showing complex, mega-hub interactions with other hubs and mega-hubs.
A substantial majority of the aberrantly phosphorylated proteins observed here had been implicated previously in neurodegenerative diseases. For example, CAND1, a component of E3 ubiquitin-ligase complexes, appears only in phosphorylated form in the APPSw aggregate contactome, allowing us to identify phosphospecific interacting partners (Figure 3C). CAND1 was previously shown to be a PIP3-binding protein that promotes aggregation; its suppression protects several C. elegans neurodegeneration models against aggregation and neurotoxicity (Ayyadevara et al., 2016a). Similarly, Dynein Heavy Chain 1 (DYHC1), Dynamin 2 (DYN2), and Nesprin 2 (SYNE2), all previously implicated in neurodegenerative diseases, appear in the aggregate contactome exclusively in phosphorylated forms (Figures 3D–3F).
Contactome Prediction of Critical Hubs for Aggregation Is Supported by Knockdown Effects
We previously showed that many proteins specifically enriched in hippocampal aggregates from patients with AD, relative to age-matched controls, contribute functionally to aggregate growth and progression of pathology in nematode models (Ayyadevara et al., 2016b). For this purpose, we modified two C. elegans models of AD-like amyloidosis (kindly provided by C. Link, University of Colorado, Boulder), replacing acute induction of human Aβ42 in late development, with leaky (uninduced) expression, so as to better mimic age-progressive amyloid accrual. Strain CL4176, through low-level muscle expression of Aβ42, accrues amyloid with age and becomes paralyzed, whereas neuronal Aβ42 expression in strain CL2355 progressively disrupts chemotaxis (Ayyadevara et al., 2015, Ayyadevara et al., 2016b). Of 21 worm orthologs of AD-enriched aggregate proteins previously targeted by RNAi, 11 were also identified in the SY5Y-APPSw contactome as key hubs (seven proteins) or hub connectors (four proteins). To this group, we added 25 proteins implicated only by connectivity in the SY5Y-APPSw aggregate connectome (Figure 4A), for a total of 28 hubs and 8 hub connectors assessed for RNAi rescue of chemotaxis decline. These outputs were used to train a neural network to predict which hub-protein knockdowns would be most effective to reduce aggregation.
Figure 4.

Hub-Protein Knockdown Results Are Consistent with Network-Based Predictions of Hub Centrality and Efficacy
(A) Chemotaxis levels of uninduced C. elegans adults (strain CL2355, with pan-neuronal leaky expression of human Aβ42), declined to an average chemotaxis index (C.I.) of 0.27 at 5 days of adult age (FV bars). C.I. levels (shown as mean ± S.E.M. for triplicate experiments) are higher for RNAi-treated worms, indicating up to 54% rescue relative to day 1 adult worms (C.I. ≈ 0.9). Knockdown targets were nematode orthologs or homologs of randomly selected human proteins identified in each indicated hub category, from the cross-link-defined contactome of SY5Y-APPSw neuroblastoma cells. Numbers over bars indicate the unadjusted significance (p values) of hub RNAi knockdowns differing from FV controls, based on heteroscedastic two-tailed paired t tests, considering the C.I. from each of three independent experiments as a single data point. **Unadjusted chi-squared (χ2) significance of p ≤ 0.001, combined from three independent experiments, i.e., the product of three χ2 p values comparing treated worms to their simultaneous controls.
(B) Network diagram of the SY5Y-APPSw contactome, displaying for each hub its degree (number of interactions; see key for node sizes) and the neural network prediction of knockdown efficacy (rescue of chemotaxis as determined in [A]; see key for node colors).
(C) Network diagram of control data, a scale-free network generated with the same node sizes as in (B). In a scale-free network, the degree distribution follows a power law; real-world networks, including protein-protein interaction networks, are widely considered to be scale-free. The distributions of node sizes, edges, and knockdown efficacies are here far more uniform than in the interactome based on empirically observed interactions (B).
Of 36 genes tested, 27 (75%) significantly rescued worms from the chemotaxis decline seen in mock-treated aging controls. Significant rescue was observed after knockdown of genes that encode ankyrin2 (unc-44), a neurofilament chain (mel-28), clathrin heavy chain (chc-1), a splicing factor (SRSF6/rsp-2), two DNA helicases (chd-1; ddx-52), subunits of translation initiation factor EIF3A (egl-45) and a DNA replication factor (rfc-1), a zinc-finger tumor suppressor (ZN292/lin-29), and three heat-shock proteins. Based on chi-squared tests of ratio shifts in individual experiments, significant protection was conferred by RNAi knockdowns targeting 19 of 28 hubs (68%), but 8 of 8 hub connectors (100%). If each proportion is treated as a single point, reducing the comparison to three experimental groups versus three control groups, t tests still indicate that 10 target knockdowns (28%) significantly rescued chemotaxis. To assess whether this high frequency of rescue is peculiar to contactome hubs and hub connectors, 20 RNAi constructs were chosen at random from the Ahringer RNAi library (Kamath and Ahringer, 2003) and knocked down individually in worm groups (Figure S1A). None of these random-RNAi-treated groups differed significantly from simultaneous, mock-treated controls, consistent with our previous experience in which random RNAi targets were only rarely effective in reducing aggregation. This contrast between knockdown of random versus contactome-implicated genes implies that genes encoding hubs and hub connectors are greatly enriched for protective knockdown targets. The protective fractions of hubs (19/28) and hub connectors (8/8) differed from random targets (0/20) with Fisher Exact test p < 3 × 10−6 and <4 × 10−7, respectively. Taken together, these data support our hypothesis that the aggregate contactome can identify “non-seed” hub proteins that play key functional roles in aggregate progression and associated neurotoxicity.
Predicting Influential Proteins Based on Contactome Properties of Proteins
Several sequential strategies were combined to predict the functional importance of hubs and hub connectors, as indicated by the chemotactic index (C.I., expressed as fold-change from controls). We first identified 12 primary input variables (predictors) that affect C.I.; these include contactome parameters such as degree (number of direct interacting partners), clustering coefficient (fraction of potential triangles containing a given node as a vertex that are complete), eigenvector centrality (a measure of direct node connections to high-degree nodes); relevant protein descriptors such as molecular weight; and a variety of derived variables including interactions among the primary parameters.
To minimize redundancy (multicollinearity) among input variables, we first used principal-component analysis (PCA), including all 12 input descriptors for each node. PCA indicates that three principal components account for >90% of total C.I. variance (Figure S1B). Considering the variables that underlie each component, the number of triangles (X3) alone contributed >90% of the variance explained by principal component 1 (i.e., the predictive value of PC1), clustering coefficient (X4) and molecular weight (X10) together account for >85% of PC2's contribution, and X10 was the predominant contributor to PC3 (45% of its explained variance) (Figure S1C). We next performed stepwise, forward/reverse multivariate linear regression (F/R-MLR), using the PCA-reduced set of variables to identify linear models that provide maximal sensitivity and specificity in predicting the output phenotype. We obtained the most reliable predictions with a model using five inputs: X1/degree, X4/clustering_coefficient, X10/molecular_weight, X1:X4 interactions, and X4:X10 interactions. Virtually identical F/R-MLR results were obtained if X3/triangle_count was substituted for X1/degree; both models gave a Root-Mean-Square Error (RMSE) of 0.25. Although hub degree (number of direct partners, X1) is considered an important variable for the analysis of any network (Lawyer, 2015), it did not appear in PCA as a major determinant of C.I. This may simply reflect the high correlation between degree (X1) and number of triangles (X3) as illustrated in panel D of Figure S1, since PCA is sensitive to the entry order of redundant input variables. In view of their operational equivalence, the X1-substituted parameter set was carried forward for nonlinear, neural-network optimization.
This sparse set of five inputs was employed to train and test a multi-layer perceptron neural-network algorithm with backpropagation, for the ability to predict knockdown efficacy for each network node. The Orange neural network (Demšar et al., 2013) conducted 50 cycles, in each of which the sample set was randomly partitioned into 70% for training and 30% for testing (see Methods). All hub and hub-connector proteins in the network were thus ranked by the extent of protection predicted for their knockdowns, as listed in Table S1. The contactome plot (Figure 4B), while displaying wide variation in node size, illustrates remarkable clustering or “centrality” of those hubs with the greatest predicted knockdown efficacy. In marked contrast, a control plot generated with the same number of nodes in scale-free mode shows a random distribution of nodes, edges, and degree (interactions/node). Predicted knockdown efficacy (C.I. fold change) is also distributed randomly across the network (Figure 4C), implying that the empirical network derived from cross-linking data (Figure 4B) reflects a distinctly non-random aggregation process. Training and testing the neural network with all 12 initial descriptors (i.e., without dimensional reduction by PCA and MLR modeling) did not improve prediction.
Knockdown of Hub Connectors Is Especially Protective against Chemotaxis Decline
Our initial RNAi screening identified a category of proteins we termed “hub connectors”—hubs of relatively low degree (few direct partners) but high connectivity (many indirect partners, which might otherwise remain unconnected). A formal topological test for this property asks whether a node connects two large hubs without forming a triangle. Hub connectors can thus be defined by two criteria: Clustering Coefficient (CC) < 0.5 and a ratio of CC to Eigenvector Centrality (EC) < 100. On this basis, 135 proteins in the SY5Y-APPSw aggregate contactome qualify as hub connectors (Figure 1F). Six examples are illustrated as subpanels of Figure 5A. The 40S ribosomal protein S5 (RPS5 or RS5) is a hub connector linking PPIG (mega-hub) with ATRX (major hub), which do not interact directly. Similarly, hub connector COPG1 (coatomer subunit γ-1) connects midi-hub PHF6 to mega-hubs TITIN and PPIG; and DRG1 (Developmentally Regulated GTP-binding protein 1) links four hub proteins that are otherwise not connected (i.e., the DRG1 triangle count = 0). Actinin 1 and SERF2 are examples of hub connectors that possess triangles but also connect otherwise unconnected hubs (degree > triangles). We reported previously that knockdown of SERF2 in SY5Y-APPSw cells, or of its ortholog CRAM-1 in C. elegans, confers significant protection against aggregation and associated cytotoxicity (Ayyadevara et al., 2015, Balasubramaniam et al., 2018).
Figure 5.

Hub-Connector Knockdowns Rescue Age-Dependent Chemotaxis Decline
(A) SY5Y-APPSw interactome showing the distribution of hub connectors (green dots) across the network. Insets show the local contactomes of six hub proteins: SERF2 (small EDRK-rich factor 2 [Balasubramaniam et al., 2018]); PP1A (α catalytic subunit of protein phosphatase 1, PP1, a serine/threonine protein phosphatase involved in cardiac function, learning, and memory); DRG1 (developmentally regulated GTP-binding protein 1, expressed in neural precursor cells); ACTN1 (actinin α1, a cytoskeletal protein related to spectrins and dystrophins); RPS5 (ribosomal protein S5); and COPG1 (coatomer complex subunit γ1, required for budding from Golgi membranes and for retrograde Golgi-to-ER transport of dilysine-tagged proteins).
(B) Rescue of age-dependent chemotaxis loss, by knockdown of hub connectors in C. elegans strain CL2355 (uninduced, pan-neuronal Aβ42 expression). Results are shown for three independent experiments (different-colored bars). *p < 0.04 by one-tailed paired t test, comparing each knockdown to its corresponding, simultaneous FV control.
(C) Computational model of ATRX interacting weakly but directly with PPIG (ΔEinteraction = −73.2 kcal/mol).
(D) Computational model of ATRX interacting strongly with PPIG via the hub connector RPS5 (ΔEinteraction = −120.9 kcal/mol).
To evaluate their functional importance, five connectome-identified hub connectors were individually suppressed by RNAi in C. elegans strain CL2355. Knockdown of each tested hub connector (RPS5, COPG1, DRG1, ACTN1, and SERF2) conferred significant protection against age-associated aggregation and chemotactic decline in each of three independent experiments (Figure 5B). Hub-connector predictions based on contactome topology are also supported by protein-docking simulations. For example, direct interaction between PPIG and ATRIX is unstable (Figure 5C) but is effectively stabilized when RPS5 serves as a connecting bridge (Figure 5D). Such bridging interactions, which have been largely ignored until now, appear in our analysis to be critical for the growth of pathological aggregates. Moreover, neural-network prediction has revealed many other hub connectors such as HEAT1 and EEF2, which are implicated by our data as key mediators of aggregation (Table S1). Targeting such hub connectors to disrupt their interactions may isolate large-hub clusters and thereby alleviate the aggregate burden.
Aspirin Treatment Reduces Aggregate Complexity
Aspirin (acetylsalicylic acid, or ASA) is an anti-inflammatory acetylating agent that exhibits a wide variety of biological activities (Ayyadevara et al., 2013, Ayyadevara et al., 2017, Khaidakov et al., 2010, Stark et al., 2007). In large prospective trials, aspirin reduced or delayed many age-dependent disorders, including neurodegenerative diseases, cardiovascular diseases, and diverse cancers (Jacobs et al., 2012). Its efficacy may be mediated by protection from age-dependent protein aggregation, as we previously demonstrated in several human-cell-culture and C. elegans models of neurodegenerative aggregation (Ayyadevara et al., 2017). For example, amyloid deposits in SY5Y-APPSw neuroblastoma cells, detected and quantified by fluorescence upon staining with thioflavin T, declined by 27% (p < 0.02) after cells were treated with ASA (Figures 6A and 6B), consistent with our earlier report (Ayyadevara et al., 2017). In view of these and other results supporting ASA antagonism to aggregation, we constructed the aggregate contactome for aspirin-treated SY5Y-APPSw cells (Figure 6C). As is evident from a comparison between Figures 1C and 6C, the aggregate contactome became far sparser and less complex after ASA exposure. The number of protein interactions dropped >2-fold in aspirin-treated cells (Figure 6D), comparable with the >2-fold protection from chemotaxis decline seen in aspirin-treated worms (strain CL2355, with neuronal expression of human Aβ42; Figure 6E). Aspirin elicited a substantial decline in degree and connectivity of most hub proteins in the SY5Y-APPSw contactome, including mega-hubs, consistent with aspirin disrupting aggregation via multiple targets.
Figure 6.

Aspirin Treatment Reduces Aggregate Complexity
(A) Thioflavin-T staining of amyloid in SY5Y-APPSw cells (images on left), ± simultaneous exposure to 0.5 mM aspirin. DAPI staining of nuclei (images on right) demonstrates similar cell density. Scale bar indicates 10 μm.
(B) Normalized quantitations of amyloid-like aggregation per cell in SY5Y-APPSw cells, with or without aspirin treatment. Data are shown as mean ± S.E.M. *p < 0.02 by one-tailed t test.
(C) The insoluble-aggregate interactome of SY5Y-APPSw cells exposed to 0.5 mM aspirin for 48 h shows substantially reduced hub degree and complexity relative to untreated cells (compare with Figure 1C). See also Figure S2.
(D) Number of aggregate-network interactions is reduced by half in SY5Y-APPSw cells exposed to 0.5 mM aspirin, relative to untreated control cells.
(E) Aspirin (1 mM) protects SY5Y-APPSw cells against chemotaxis decline in C. elegans strain CL2355 (pan-neuronal Aβ42 expression) relative to vehicle-only controls. ***p < 2.5 × 10−5, significance by chi-squared (χ2) test.
(F) Interaction energies (ΔGbinding) predicted by computational docking of aspirin with candidate proteins (blue bars, network-implicated hub proteins) versus previously reported ASA-binding targets (tan bars, five positive controls to confirm correct docking parameters).
(G–J) Aspirin docking poses of proteins predicted to be direct ASA-binding targets (Ayyadevara et al., 2017): (G) HSP90A (heat shock protein 90α); (H) PP1A (see Figure 5A legend); (I) TUBB4A (tubulin beta chain 4A, a major constituent of microtubules); and (J) LMNA (lamin 4 A/C, a constituent of nuclear lamina; mutations are implicated in several muscular and cardiac dystrophies and in Hutchinson-Gilford progeria).
Aspirin is a potent acetyl donor, which suggests a plausible mechanism for protection from Aβ aggregation, since we previously showed that Aβ42 cannot form globular aggregates when acetylated (Ayyadevara et al., 2017). More generally, aggregation is reduced by interventions that favor protein acetylation and promoted by drugs that disrupt acetylation (Ayyadevara et al., 2017). In the SY5Y-APPSw aggregate contactome, 84 protein hubs (with two or more interacting partners) were no longer interconnected after aspirin treatment. Among the proteins that aspirin treatment rendered noninteractive, a substantial number had been previously implicated in neurodegenerative diseases, e.g., ROA3, SERF2, COF2, TBB4A, TBB4B, phosphorylated forms of HSP90α, LMNA, RS11, and protein phosphatase PP1A (a “hub-connector”) (Blair et al., 2014, Jovanovic et al., 2014, Martin et al., 2014, Salama et al., 2018).
In recent studies, aspirin-binding regions were identified in a number of ASA-target proteins, including phospholipases (Dai et al., 2016, Singh et al., 2005). This suggested that aspirin might also interact directly with some of the proteins that vanished from the aggregate contactome of aspirin-treated cells. We selected five protein-aspirin complexes with structures resolved experimentally and documented in Protein DataBank (PDB; www.rcsb.org) as positive examples to validate parameters for computational docking and modeled 19 protein structures. Under the same standard conditions that replicated known aspirin binding to all five positive-control proteins, protein-ligand docking predicted direct, stable aspirin binding to 9 of the 19 tested proteins removed from the contactome by ASA: FUBP3, HSP90A, PP1A, RBN4B, TBB4A, GBB2, HNRPD, LMNA, and NNTM (Figures 6F–6J). The predicted stability of aspirin binding was similar for these nine proteins (blue bars in Figure 6F) and the five positive controls (tan bars), such that the ΔGbinding ranges are almost identical. It is noteworthy that HSP90A phosphorylation, which has been implicated in AD (Blair et al., 2014), is predicted to be blocked by ASA (Figure 6G). Likewise, phosphorylated forms of LMNA are also missing from the aspirin-treated contactome, suggesting that aspirin binding may inhibit phosphorylation and thus block LMNA entry into the contactome. These data highlight the importance of post-translational modification—in particular phosphorylation and acetylation—in aggregate accrual, and suggest that aspirin and other therapeutic drugs may prevent or slow aggregate accumulation and its associated neurotoxicity.
We repeated the above-mentioned contactome analyses after restricting consideration to only those aggregate-protein interactions shared by SY5Y-APPSw and SY5Y cells (Figure S2). Results were essentially unchanged, with aspirin treatment reducing the number and connectivity (degree) of hubs by > 5-fold for this subset of confirmed interactions.
Discussion
We have developed tools, and an integrated strategy, that enabled us to define the molecular interfaces formed in the accrual of insoluble protein aggregates. This approach comprised the design and synthesis of click reagents for efficient cross-linking and recovery of proximal peptides within aggregates, analysis of cross-linked peptide pairs by LC-MS/MS, and the development and application of improved bioinformatics tools to identify cross-linked peptides. The resulting data were used to construct nonfunctional interactomes (“contactomes”) that define the internal architecture of aggregates from SY5Y-APPSw, human neuronal cells expressing AD-prone mutations of Amyloid Precursor Protein.
In previous studies, we identified and quantified constituent proteins that are enriched in aggregates from AD-affected human hippocampus, relative to controls, and demonstrated functional roles for many of these proteins by RNAi knockdown of orthologous genes in C. elegans models of AD and other neurodegenerative diseases (Ayyadevara et al., 2016b). Significant protection against aggregation was observed upon knockdown of many aggregate-resident proteins, extending well beyond the known seed proteins thought to initiate aggregation (Aβ42 and tau for AD, α-synuclein for Parkinson disease, and huntingtin protein with expanded polyglutamine tracts for Huntington Disease) (Ayyadevara et al., 2015, Ayyadevara et al., 2016b). These results implied that many additional proteins play functional roles in aggregate initiation and/or progression. It remained uncertain, however, what those roles are and how diverse aggregate proteins coalesce into insoluble complexes.
Not all protein pairs can interact to form a stable interface, but a remarkably large fraction of those that do contribute causally to aggregate accrual (i.e., their knockdowns relieve aggregation), leading us to postulate that there is a preferred order of accretions. To elucidate this process, we analyzed cross-linked peptides to define the most abundant protein-protein interfaces in aggregates and thus to construct aggregate-interaction networks. The resulting contactomes reflect only peptide proximity at protein-protein interfaces or adhesion sites rather than functional interactions. Because the contactome is based on semi-quantitative data from cross-linking of aggregates that are likely to be heterogeneous within each cell and between cells, we set abundance thresholds to eliminate rarer interactions and also separated proteins into hub categories (mini-, midi-, major, and mega-hubs), bins that reflect the number of directly interacting partners for each hub. This analysis identified proteins that are predicted to strongly affect aggregate size and stability, owing to high abundance and diversity of either direct contacts (degree) or indirect partners (connectivity). Table S1 lists parameters that best predicted the extent of RNAi rescue from aggregation-mediated pathology. Neural-network predictions from these features correlated quite well (R = 0.77) with observed rescue efficacies.
Within the cross-link-based interactome, mega-hubs (nodes with ≥100 partners) include many proteins that bind DNA or RNA (enrichment p < 2E–20 and p < 3E–150, respectively). High cross-link frequencies were also observed for large structural proteins (e.g., TITIN, SRRM2, SRSF6), which because of their massive size yield large numbers of tryptic peptides with the potential to appear in cross-linked peptide pairs. These large macromolecules should not be discounted, however, since they are often components of intracellular structures such as microtubules and actin filaments, which may provide potent nexi of aggregate accrual upon partial denaturation.
In a model system with high propensity for amyloid aggregation (SY5Y-APPSw cells), but not in the nontransgenic parental cell line SY5Y, the aggregate contactome shows APP proximity to laminin B2 (Figure 2A). Integrins normally anchor cells to laminins of the extracellular matrix (ECM), and both protein families have been previously implicated in several neurological disorders (Sethi and Zaia, 2017). Kainic acid induces excitotoxicity, leading to laminin degradation, neuronal loss, and eventual degeneration of hippocampal neurons (Bonneh-Barkay and Wiley, 2009). Laminin B2, by interacting with APP, may alter its interactions to other proteins, dislodging cells from their normal ECM anchoring.
Hyperphosphorylated tau (hP-tau, with phosphorylated sites typical of AD) also shows many interactions specific to the SY5Y-APPSw transgenic cell line (Figure 2F), which differ strikingly from the connectome of tau carrying only normal phosphorylations within the same cells (Figure 2E). Curiously, the contactome derived from SY5Y-APPSw aggregates includes no peptides from the PHF region, widely believed to initiate tau seeding of neurofibrillary tangles (Fitzpatrick et al., 2017). It is noteworthy that all interactions of AD-like hP-tau, seen in SY5Y-APPSw cells but not in SY5Y, were lost upon exposure to aspirin (Figure 6).
Of the proteins identified in SY5Y-APPSw aggregates, 26% are disordered (e.g., 14-3-3, ROA3, SERF2), drawn from all hub categories, but chiefly midi-hubs, major hubs, and hub connectors. Being largely disordered or unstructured, these proteins can adopt multiple conformations and interact with multiple protein partners, making them difficult to target by structure-based drug design. Nevertheless, disordered proteins may adopt stable conformations when bound to other proteins (Ayyadevara et al., 2015, Balasubramaniam et al., 2018, Hu et al., 2014). Protein-protein interaction inhibitors (PPIIs), in particular those that target specific protein interfaces, have received considerable attention as potential therapeutic drugs for neurodegenerative and other diseases (Jnoff et al., 2014, Petta et al., 2016). Preferred interacting partners of disordered proteins, identified by aggregate cross-linking, may reveal stable or meta-stable conformations of these proteins within such complexes, which can be targeted by PPIIs.
We previously identified post-translationally modified (phosphorylated) proteins that are highly enriched in Alzheimer brain aggregates, using standard proteomic methods (Ayyadevara et al., 2016b). The present study corroborates many of those observations in a cell culture model of AD-like amyloid formation and demonstrates that key aggregate proteins adhere to different partners when phosphorylated. Key contactome hubs and hub connectors include proteins previously implicated in neurodegenerative diseases, such as HSP90 (vulnerable to phosphorylation and implicated in AD [Blair et al., 2014]). HSP90 was identified in insoluble AD aggregates, especially in the phosphorylated form, interacting with a variety of proteins (Ayyadevara et al., 2016b). Interestingly, aspirin treatment of SY5Y-APPSw cells removed HSP90 from the interactome. Small-molecule docking simulations indicate that aspirin could directly bind to HSP90, which is especially intriguing in view of prior reports that ATPase inhibition of HSP90 reduces tau aggregation (Blair et al., 2014).
Knockdowns in C. elegans, of orthologs or homologs of human proteins in each hub category from the aggregate contactome, demonstrated their functional roles in aggregate progression and maintenance. Network analyses of SY5Y-APPSw aggregates specify a variety of node descriptors, including degree and eigenvector centrality, for all nodes identified in the contactome. Because the dependent variable used for machine learning (fold protection against chemotaxis loss) was based on empirical data quantifying a behavioral consequence of amyloid deposition, and its relief by an intervention (RNAi-mediated knockdown), these neural-network predictions provide an unexplored approach to the future development of PPII interventions.
The cross-link-based aggregate contactome identifies peptide pairs within or adjacent to sites of dysfunctional protein-protein adhesion, creating intriguing opportunities for intervention. PPIIs targeting such interfaces might block key aggregation steps without affecting normal protein functions, whereas drugs targeting active sites or cofactor-binding sites are likely to inhibit the normal functions of their target proteins and also of structurally homologous “off-target” protein sites that bind the same or similar ligands.
Hub connectors constitute an interesting category in contactome analysis. Although hub proteins can augment aggregate networks in proportion to the abundance of hub-interacting partners, their coalescence into large, autophagy-resistant aggregates presumably requires linkage among hubs. Hub connectors can allow otherwise isolated hubs to interact indirectly with large assemblies of inter-connected hubs (Figure 5). Owing to the paucity of direct partners, hub connectors and their interfaces with large hubs may be valuable targets for drugs, which may have lower toxicity than molecules targeting mega-hubs (Agarwal et al., 2010), while at the same time achieving high efficacy owing to disruption of the assembly of large aggregates resistant to degradation via autophagy.
Only a small subset of protein pairs interact to form stable protein-protein interfaces, but a remarkably large fraction of those that do contribute causally to aggregate accrual (i.e., based on our data 77% of their knockdowns relieve aggregation). This surprising result led us to postulate that there is a preferred order of protein-protein interfaces/accretions, which can be interrupted at multiple steps. By identifying cross-linked peptides, we enumerated the most abundant protein-protein interfaces in an aggregate, enabling us to construct aggregate-interaction networks. The resulting protein-protein contactomes reflect only peptide proximity at protein-protein adhesion sites, rather than functional interactions. Because the contactome is based on semi-quantitative data from cross-linking of aggregates that are likely to be heterogeneous within each cell and between cells, we set stringent abundance and replication thresholds to eliminate rarer interactions. Our analysis identified proteins that are predicted to be strong determinants of aggregate size and stability, owing to their high abundance and diversity of either direct or indirect connections.
Numerous reports have identified anti-aggregation effects of aspirin (Ayyadevara et al., 2013, Ayyadevara et al., 2017, Moafian et al., 2016). Acetylation of proteins may, in a site-specific manner, alter protein structure. These altered structures may be either predisposed or resistant to aggregation, with resistance more likely if the acetylated residue would otherwise be a target for hyperphosphorylation (Ayyadevara et al., 2017). Aspirin treatment was protective in several C. elegans models of neurodegenerative aggregation (Ayyadevara et al., 2017), but little is known about its targets or mechanism of protection. A comparison of cross-link-based aggregate contactomes (Figures 1C versus 6C) demonstrates that aspirin exposure reduces interaction frequency and complexity. Moreover, 84 proteins were removed from the contactome by aspirin exposure, and molecular-dynamic modeling presented here supports the likelihood that they are aspirin targets. Of these 84 proteins, 26 were phosphorylated in untreated SY5Y-APPSw cells but not in aspirin-exposed cells. Aspirin could block phosphorylation in several ways: by interacting with kinase-targeted sites of these proteins (by either binding or acetylating them) or by similar interactions disrupting upstream kinases that are required to phosphorylate such proteins.
The approach described here defines the physical contactome of AD-like aggregates and thus the local affinities of proximal protein regions. Many aggregate components had never previously been implicated in neurodegeneration, and many interactions are specific to cells expressing the APPSw protein thought to foster seeding of AD-associated amyloid deposits. Identification of protein pairs that contribute disproportionately to aggregation may reveal attractive targets for interface-binding drugs to oppose protein aggregation. The strategies described here should also be quite generally applicable to the wide variety of neuropathies and other aging-associated diseases that feature progressive protein aggregation.
Limitations of the Study
Use of a neuronal cell culture predisposed to amyloid deposition provides a partial model of AD but is here intended as a well-controlled source of aggregates to validate our protocol. Cross-linking results from AD and control aggregates will be presented elsewhere.
Recovery of peptides from aggregates depends on the ability of trypsin to digest coalesced proteins. We reasoned that protease digestion should progressively degrade aggregates and tested this premise initially with Q40::YFP seeded by, and largely comprising, Q40::YFP (expressed in muscle of C. elegans strain AM141), isolated by YFP pulldown. Within 15–30 min of tryptic digestion under our standard conditions, YFP fluorescence declined to ∼12% of the control level, implying that as much as 12% of aggregates may be trypsin resistant. Similarly, sarcosyl-insoluble aggregates from AD hippocampus indicated ∼6% resistance to tryptic digestion, based on material failing to enter an electrophoretic gel. Such material will not be included in analyses conducted under the present protocol, although we note that Proteinase K digestion under denaturing conditions, 2% SDS at 65°C, leaves no residual aggregate protein (<0.1% undigested).
In addition, cross-linking of aggregate proteins depends on penetration of the cross-linking reagents, which may limit the representation of cross-linked peptides from aggregate interiors. We anticipated that cross-linking would quench YFP fluorescence in Q40::YFP aggregates from AM141, and indeed fluorescent emissions from mature aggregates were reduced to <1% of the control level within 30 min. We note that the click reagents we employed are much smaller than trypsin, which may account for their superior penetrance.
One additional caveat is that the bioinformatics procedure for identification of cross-linked peptide pairs (Xlink-Identifier; Du et al., 2011) is entirely dependent on the availability of a reference list of proteins and their post-translational modifications, identified in uncross-linked aggregates. Peptides from proteins not represented in this lookup table, including any isoforms not present or any PTMs not identified, cannot be detected by the software. It should also be noted that analysis requires high-resolution mass spectrometry outputs and may be quite computationally demanding for large reference lists.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by grants (Merit 2 I01 BX001655 and Senior Research Career Scientist Award) to R.J.S.R. from the U.S. Dept. of Veteran Affairs, to M.B. from the Inglewood Scholars Program, and by Program Project grant 2P01AG012411-17A1 (W.S.T. Griffin, P.I.) from the National Institute on Aging (NIA/NIH). The authors thank the Windgate Foundation and the Philip R. Jonsson Foundation for additional support, Samuel G. Macintosh for running proteomic analyses of cross-linked samples, and Ramani Alla for a variety of technical assistance including tryptic digestion of all cross-linked samples.
Author Contributions
R.J.S.R., S.A., and M.B. planned and designed the study; N.R.P. synthesized cross-linker and biotin-azide reagents with guidance from P.A.C.; A.G. performed three-dimensional modeling of proteins; S.K. performed PCA, MLR, and statistical modeling of the contactome; X.D. provided the primary scripts for Xlink-Identifier; M.B. performed cross-linking, network modeling, RNAi knockdown, machine learning/neural network algorithms, and other experiments mentioned in the article. R.J.S.R. and M.B. wrote the manuscript with contributions from S.T.G., S.A., and P.A.C.
Declaration of Interests
The authors declare no competing interests.
Published: October 25, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.09.026.
Contributor Information
Meenakshisundaram Balasubramaniam, Email: mbalasubramaniam@uams.edu.
Srinivas Ayyadevara, Email: ayyadevarasrinivas@uams.edu.
Robert J. Shmookler Reis, Email: rjsr@uams.edu.
Data and Code Availability
The link (database: accession number) for our raw data file reported in this paper, comprising all detected matches to predicted m/z values of linked peptide pairs, is Mendeley: http://dx.doi.org/10.17632/pw92vvj9rz.1#folder-d89b63a4-2d13-4eac-85af-cc082696412f
Supplemental Information
References
- Agarwal S., Deane C.M., Porter M.A., Jones N.S. Revisiting date and party hubs: novel approaches to role assignment in protein interaction networks. PLoS Comput. Biol. 2010;6:e1000817. doi: 10.1371/journal.pcbi.1000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara S., Bharill P., Dandapat A., Hu C., Khaidakov M., Mitra S., Shmookler Reis R.J., Mehta J.L. Aspirin inhibits oxidant stress, reduces age-associated functional declines, and extends lifespan of Caenorhabditis elegans. Antioxid. Redox Signal. 2013;18:481–490. doi: 10.1089/ars.2011.4151. [DOI] [PubMed] [Google Scholar]
- Ayyadevara S., Balasubramaniam M., Gao Y., Yu L.R., Alla R., Shmookler Reis R.J. Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome-blocking complexes. Aging Cell. 2015;14:35–48. doi: 10.1111/acel.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara S., Balasubramaniam M., Johnson J., Alla R., Mackintosh S.G., Shmookler Reis R.J. PIP3-binding proteins promote age-dependent protein aggregation and limit survival in C. elegans. Oncotarget. 2016;7:48870–48886. doi: 10.18632/oncotarget.10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara S., Balasubramaniam M., Parcon P.A., Barger S.W., Griffin W.S., Alla R., Tackett A.J., Mackintosh S.G., Petricoin E., Zhou W. Proteins that mediate protein aggregation and cytotoxicity distinguish Alzheimer's hippocampus from normal controls. Aging Cell. 2016;15:924–939. doi: 10.1111/acel.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara S., Mercanti F., Wang X., Mackintosh S.G., Tackett A.J., Prayaga S.V., Romeo F., Shmookler Reis R.J., Mehta J.L. Age- and hypertension-associated protein aggregates in mouse heart have similar proteomic profiles. Hypertension. 2016;67:1006–1013. doi: 10.1161/HYPERTENSIONAHA.115.06849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara S., Balasubramaniam M., Kakraba S., Alla R., Mehta J.L., Shmookler Reis R.J. Aspirin-mediated acetylation protects against multiple neurodegenerative pathologies by impeding protein aggregation. Antioxid. Redox Signal. 2017;27:1383–1396. doi: 10.1089/ars.2016.6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam M., Ayyadevara S., Shmookler Reis R.J. Structural insights into pro-aggregation effects of C. elegans CRAM-1 and its human ortholog SERF2. Sci. Rep. 2018;8:14891. doi: 10.1038/s41598-018-33143-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer K., Ariza A. alpha-Synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol. Neurobiol. 2013;47:509–524. doi: 10.1007/s12035-012-8330-5. [DOI] [PubMed] [Google Scholar]
- Blair L.J., Sabbagh J.J., Dickey C.A. Targeting Hsp90 and its co-chaperones to treat Alzheimer's disease. Expert Opin. Ther. Targets. 2014;18:1219–1232. doi: 10.1517/14728222.2014.943185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneh-Barkay D., Wiley C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009;19:573–585. doi: 10.1111/j.1750-3639.2008.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S.M., Du X., Tolic N., Wu S., Moore R.J., Mayer M.U., Smith R.D., Adkins J.N. Identification of cross-linked peptides after click-based enrichment using sequential collision-induced dissociation and electron transfer dissociation tandem mass spectrometry. Anal. Chem. 2009;81:5524–5532. doi: 10.1021/ac900853k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahms S.O., Konnig I., Roeser D., Guhrs K.H., Mayer M.C., Kaden D., Multhaup G., Than M.E. Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain. J. Mol. Biol. 2012;416:438–452. doi: 10.1016/j.jmb.2011.12.057. [DOI] [PubMed] [Google Scholar]
- Dai S.X., Li W.X., Li G.H., Huang J.F. Proteome-wide prediction of targets for aspirin: new insight into the molecular mechanism of aspirin. Peer J. 2016;4:e1791. doi: 10.7717/peerj.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David D.C., Ollikainen N., Trinidad J.C., Cary M.P., Burlingame A.L., Kenyon C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A.A., Leyns C.E.G., Holtzman D.M. Intercellular spread of protein aggregates in neurodegenerative disease. Annu. Rev. Cell Dev. Biol. 2018;34:545–568. doi: 10.1146/annurev-cellbio-100617-062636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demšar U., Harris P., Brunsdon C., Stewart Fotheringham A., McLoone S. Principal component analysis on spatial data: an overview. Ann. Assoc. Am. Geogr. 2013;103:106–128. [Google Scholar]
- Du X., Chowdhury S.M., Manes N.P., Wu S., Mayer M.U., Adkins J.N., Anderson G.A., Smith R.D. Xlink-Identifier: an automated data analysis platform for confident identifications of chemically cross-linked peptides using tandem mass spectrometry. J. Proteome Res. 2011;10:923–931. doi: 10.1021/pr100848a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink A.L. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold. Des. 1998;3:R9–R23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick A.W.P., Falcon B., He S., Murzin A.G., Murshudov G., Garringer H.J., Crowther R.A., Ghetti B., Goedert M., Scheres S.H.W. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature. 2017;547:185–190. doi: 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H., Hasegawa M., Dohmae N., Kawashima A., Masliah E., Goldberg M.S., Shen J., Takio K., Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gauczynski S., Peyrin J.M., Haik S., Leucht C., Hundt C., Rieger R., Krasemann S., Deslys J.P., Dormont D., Lasmezas C.I. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001;20:5863–5875. doi: 10.1093/emboj/20.21.5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M., Arai T., Nonaka T., Kametani F., Yoshida M., Hashizume Y., Beach T.G., Buratti E., Baralle F., Morita M. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G., Li H., Liu J.Y., Wang J. Insight into conformational change for 14-3-3sigma protein by molecular dynamics simulation. Int. J. Mol. Sci. 2014;15:2794–2810. doi: 10.3390/ijms15022794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q., Chang J., Cheung M.K., Nong W., Li L., Lee M.T., Kwan H.S. Human proteins with target sites of multiple post-translational modification types are more prone to be involved in disease. J. Proteome Res. 2014;13:2735–2748. doi: 10.1021/pr401019d. [DOI] [PubMed] [Google Scholar]
- Jacobs E.J., Newton C.C., Gapstur S.M., Thun M.J. Daily aspirin use and cancer mortality in a large US cohort. J. Natl. Cancer Inst. 2012;104:1208–1217. doi: 10.1093/jnci/djs318. [DOI] [PubMed] [Google Scholar]
- Jacomy M., Venturini T., Heymann S., Bastian M. ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS one. 2014;9:e98679. doi: 10.1371/journal.pone.0098679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jnoff E., Albrecht C., Barker J.J., Barker O., Beaumont E., Bromidge S., Brookfield F., Brooks M., Bubert C., Ceska T. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. Chem. Med. Chem. 2014;9:699–705. doi: 10.1002/cmdc.201300525. [DOI] [PubMed] [Google Scholar]
- Jovanovic K., Loos B., Da Costa Dias B., Penny C., Weiss S.F. High resolution imaging study of interactions between the 37 kDa/67 kDa laminin receptor and APP, beta-secretase and gamma-secretase in Alzheimer's disease. PLoS One. 2014;9:e100373. doi: 10.1371/journal.pone.0100373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath R.S., Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Karve T.M., Cheema A.K. Small changes, huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J. Amino Acids. 2011;2011:207691. doi: 10.4061/2011/207691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaidakov M., Szwedo J., Mitra S., Ayyadevara S., Dobretsov M., Lu J., Mehta J.L. Antiangiogenic and antimitotic effects of aspirin in hypoxia-reoxygenation modulation of the LOX-1-NADPH oxidase axis as a potential mechanism. J. Cardiovasc. Pharmacol. 2010;56:635–641. doi: 10.1097/FJC.0b013e3181f801e4. [DOI] [PubMed] [Google Scholar]
- Kiaei M., Balasubramaniam M., Govind Kumar V., Shmookler Reis R.J., Moradi M., Varughese K.I. ALS-causing mutations in profilin-1 alter its conformational dynamics: a computational approach to explain propensity for aggregation. Sci. Rep. 2018;8:13102. doi: 10.1038/s41598-018-31199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latapy M., Magnien C., del Vecchio N. Basic notions for the analysis of large two-mode networks. Social Networks. 2008;30:31–48. [Google Scholar]
- Lawyer G. Understanding the influence of all nodes in a network. Sci. Rep. 2015;5:8665. doi: 10.1038/srep08665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Xue Y., Hu J., Wang Y., Liu X., Demeler B., Ha Y. The E2 domains of APP and APLP1 share a conserved mode of dimerization. Biochemistry. 2011;50:5453–5464. doi: 10.1021/bi101846x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I., Kim J.W., Lee B.D., Kang H.C., Xu J.C., Jia H., Stankowski J., Kim M.S., Zhong J., Kumar M. Ribosomal protein S15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson's disease. Cell. 2014;157:472–485. doi: 10.1016/j.cell.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland M.A., Ellis C.E., Markey S.P., Nussbaum R.L. Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol. Cell. Proteomics. 2008;7:2123–2137. doi: 10.1074/mcp.M800116-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moafian Z., Khoshaman K., Oryan A., Kurganov B.I., Yousefi R. Protective effects of acetylation on the pathological reactions of the lens crystallins with homocysteine thiolactone. PLoS One. 2016;11:e0164139. doi: 10.1371/journal.pone.0164139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Z., Esiri M.M., Smith A.D. Expression of cell division markers in the hippocampus in Alzheimer's disease and other neurodegenerative conditions. Acta Neuropathol. 1997;93:294–300. doi: 10.1007/s004010050617. [DOI] [PubMed] [Google Scholar]
- Pahrudin Arrozi A., Shukri S.N.S., Wan Ngah W.Z., Mohd Yusof Y.A., Ahmad Damanhuri M.H., Makpol S. Evaluation of the expression of Amyloid Precursor Protein and the ratio of secreted Amyloid Beta 42 to Amyloid Beta 40 in SH-SY5Y cells stably transfected with wild-type, single-mutant and double-mutant forms of the APP gene for the study of Alzheimer's disease pathology. Appl. Biochem. Biotechnol. 2017;183:853–866. doi: 10.1007/s12010-017-2468-6. [DOI] [PubMed] [Google Scholar]
- Petta I., Lievens S., Libert C., Tavernier J., De Bosscher K. Modulation of protein-protein interactions for the development of novel therapeutics. Mol. Ther. 2016;24:707–718. doi: 10.1038/mt.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q.G., Liao X.M., Chen X.Q., Liu G.P., Wang J.Z. Effects of tau phosphorylation on proteasome activity. FEBS Lett. 2007;581:1521–1528. doi: 10.1016/j.febslet.2007.02.065. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Martin T., Cuchillo-Ibanez I., Noble W., Nyenya F., Anderton B.H., Hanger D.P. Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging. 2013;34:2146–2157. doi: 10.1016/j.neurobiolaging.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama M., Shalash A., Magdy A., Makar M., Roushdy T., Elbalkimy M., Elrassas H., Elkafrawy P., Mohamed W., Abou Donia M.B. Tubulin and Tau: possible targets for diagnosis of Parkinson's and Alzheimer's diseases. PLoS One. 2018;13:e0196436. doi: 10.1371/journal.pone.0196436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi M.K., Zaia J. Extracellular matrix proteomics in schizophrenia and Alzheimer's disease. Anal. Bioanal. Chem. 2017;409:379–394. doi: 10.1007/s00216-016-9900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic G., Babic Leko M., Wray S., Harrington C., Delalle I., Jovanov-Milosevic N., Bazadona D., Buee L., de Silva R., Di Giovanni G. Tau protein hyperphosphorylation and aggregation in Alzheimer's Disease and other tauopathies, and possible neuroprotective strategies. Biomolecules. 2016;6:6. doi: 10.3390/biom6010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.K., Ethayathulla A.S., Jabeen T., Sharma S., Kaur P., Singh T.P. Aspirin induces its anti-inflammatory effects through its specific binding to phospholipase A2: crystal structure of the complex formed between phospholipase A2 and aspirin at 1.9 angstroms resolution. J. Drug Target. 2005;13:113–119. doi: 10.1080/10611860400024078. [DOI] [PubMed] [Google Scholar]
- Smith T.W., Lippa C.F. Ki-67 immunoreactivity in Alzheimer's disease and other neurodegenerative disorders. J. Neuropathol. Exp. Neurol. 1995;54:297–303. doi: 10.1097/00005072-199505000-00002. [DOI] [PubMed] [Google Scholar]
- Stark L.A., Reid K., Sansom O.J., Din F.V., Guichard S., Mayer I., Jodrell D.I., Clarke A.R., Dunlop M.G. Aspirin activates the NF-kappaB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis. 2007;28:968–976. doi: 10.1093/carcin/bgl220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The link (database: accession number) for our raw data file reported in this paper, comprising all detected matches to predicted m/z values of linked peptide pairs, is Mendeley: http://dx.doi.org/10.17632/pw92vvj9rz.1#folder-d89b63a4-2d13-4eac-85af-cc082696412f