

Abstract
Background
Statins improve endothelial function, but their effects on arterial stiffness and aortic blood pressure in middle‐aged adults are uncertain.
Methods and Results
This was a prospective, randomized, double‐blind, placebo‐controlled trial of middle‐aged (40‐72 years old) adults who were randomly assigned to receive simvastatin 40 mg (n=44) or placebo (n=44) daily for 18 months to evaluate impact on dementia‐related biomarkers (primary end points) and measures of vascular health (secondary end points). This analysis focuses on the predetermined secondary end points of changes in central aortic blood pressure, aortic augmentation index, and brachial artery flow‐mediated dilation. Measurements were performed at baseline and after 6, 12, and 18 months. Multivariable models were used to identify predictors of these prespecified vascular end points. Study groups were similar at baseline; low‐density lipoprotein cholesterol declined in the statin group but not in the placebo group (P<0.01). There were no significant differences in changes in central blood pressure parameters or flow‐mediated dilation (all P>0.2). After 12 months, augmentation index decreased from baseline in the statin group compared with the placebo group (−2.3% [5.5%] versus 1.2% [5.7%], P=0.007), but by 18 months the response in both groups trend toward baseline (−1.1% [5.8%] versus 0.2% [4.8%], P=0.3). Low‐density lipoprotein cholesterol was not associated with changes in augmentation index at any time point.
Conclusions
Statin therapy led to a short‐term reduction in augmentation index after 12 months, but this effect did not persist after 18 months despite continued reduction in low‐density lipoprotein cholesterol levels. These findings suggest that statins may have a transient effect on aortic stiffness.
Clinical Trial Registration
URL: https://www.clinicaltrials.gov/. Unique identifier: NCT00939822.
Keywords: arterial stiffness, atherosclerosis, low‐density lipoprotein cholesterol, randomized controlled trial, statin therapy
Subject Categories: Clinical Studies, Inflammation, Lipids and Cholesterol, Vascular Biology
Clinical Perspective
What Is New?
Statin therapy significantly lowered low‐density lipoprotein cholesterol compared with placebo for the entire 18‐month study; augmentation index significantly decreased after 12 months in the treatment group, but the effect was transient and no longer significantly different from the placebo group at 18 months.
The transient effects of statin therapy on augmentation index are independent of low‐density lipoprotein cholesterol.
What Are the Clinical Implications?
We should continue to prescribe statin therapy for primary and secondary cardiovascular disease risk modification, which is unlikely to be mediated through long‐term improvements in augmentation index, endothelial function, or central aortic blood pressures.
Introduction
Arterial stiffness progresses with age and is associated with cardiovascular disease (CVD) events and stroke.1, 2, 3, 4 There is also a potential link between greater arterial stiffness and the progression to dementia due to Alzheimer disease (AD).5, 6, 7, 8 Statin therapy robustly reduces plasma cholesterol levels, including atherogenic low‐density lipoprotein (LDL) cholesterol and LDL particle concentrations,9 minimizing inflammation through a reduction of proinflammatory cytokines and reactive oxygen species,10 Changes in the lipid content and composition of elastin and collagen within the vessel wall also may have beneficial effects on vascular remodeling and reduced arterial stiffness.11, 12 Reduction in LDL concentration is directly proportional to a durable and long‐lasting reduction in CVD events,13, 14, 15 but the magnitude and timing of the lipid‐independent effects of statin therapy on the arterial wall that may result in changes in vessel properties are not fully understood. A major regulator of cholesterol metabolism is the cholesterol transporter ApoE (apolipoprotein E), and the APOE ε4 allele has been associated with both higher cholesterol levels and an increased risk of vascular disease. Similarly, APOE plays a role in arterial stiffness, primarily through expression of extracellular matrix genes in vascular smooth muscle cells, which help maintain arterial elasticity independently of cholesterol levels.16, 17 Trials examining the effects of statins on arterial stiffness have had inconsistent results.18, 19, 20, 21, 22, 23, 24, 25 To our knowledge there are no other randomized controlled trials with prolonged follow‐up evaluating the effects of statin therapy on central blood pressures and arterial function in healthy middle‐aged offspring of those with AD without significant CVD risk factors, including hypertension.
The study was designed to evaluate the impact of statin therapy on AD biomarkers in middle‐aged adults at increased risk for dementia due to their parental history of AD. Those findings are reported elsewhere. However a predefined secondary outcome of this study was to determine if 18 months of statin therapy improved augmentation index (Aix), an accepted measure of the enhancement of central aortic pressure caused by the reflected pulse wave in a stiffened artery, flow‐mediated dilation (FMD) a measure of endothelial function, and central aortic blood pressure compared with a placebo control in these middle‐aged adults with minimal CVD risk who were not on statin therapy at baseline.
Materials and Methods
Study Design and Population
Anonymized data and materials have been made publicly available through the US National Library of Medicine and can be accessed at ClinicalTrials.gov. Subjects in this prospective, double‐blind, randomized, placebo‐controlled trial were middle‐aged (40‐72 years old) children of individuals with Alzheimer disease (AD) who were recruited from the community through clinics, newsletters, educational talks, health fairs, and print‐media advertisement. The reported analysis was an exploratory component of a study designed to evaluate the effects of statin therapy on AD risk factors. Subjects were enrolled from November 2009 through April 2012. The key end points of the reported analysis were changes in central aortic blood pressure and AIx, estimated by tonometry, and brachial artery FMD. Data were collected, and measurements were performed, at baseline and after 6, 12, and 18 months. Fasting blood draws were collected for all participants at the prescreening study visit at least 2 weeks before baseline assessment and randomization and then at 6‐, 12‐, and 18‐month follow‐up time points. Laboratory values, including total cholesterol, high‐density lipoprotein cholesterol, LDL cholesterol, triglycerides, serum creatinine, serum glucose, and high‐sensitivity C‐reactive protein (hsCRP) were assessed at the University of Wisconsin Hospital and Clinics clinical chemistry laboratory.
This study was approved by the University of Wisconsin Health Sciences Institutional Review Board. It was conducted per the principles expressed in the Declaration of Helsinki. All subjects provided written informed consent. The trial was registered using the clinicaltrials.gov identifier NCT00939822. Data were collected from November 24, 2009 to September 17, 2013.
Inclusion and Exclusion Criteria
Inclusion criteria included a parental history of probable or definite AD in adults aged 40 to 72 years. Exclusion criteria included dementia or mild cognitive impairment on baseline screening; use or contraindications to use of statin medications; history of CVD that would require statin use; or active participation in another research study. Subjects also could not have a contraindication for magnetic resonance imaging or lumbar puncture because these were key primary outcomes for the study. Baseline cognitive evaluation was also performed, and individuals with a positive memory screen and those who performed greater than 1.5 SD below the age‐ and education‐predicted mean performance on the Wechsler Memory Scale also were excluded and referred for medical and neuropsychological evaluation.26
Randomization, Treatment Allocation, and Outcomes
Subjects were randomly assigned to a treatment group, which received simvastatin 40 mg daily, or a placebo group, which received a matching placebo daily for 18 months. Treatment randomization was conducted by the UW Pharmaceutical Research Center using a 1:1 design and was stratified based on sex and APOE4 allele status. Within each stratum a permuted blocked randomization scheme was used with block sizes of 4 alternating with 6. This was a double‐blind study in which participants, study personnel, and investigators were blinded to treatment group and lipid‐lowering effects throughout the course of the study. The primary outcomes of this trial will be reported elsewhere. The analyses presented here are predefined secondary investigations assessing the effects of statin therapy on changes in brachial artery FMD, central aortic blood pressure, and AIx. All subjects were analyzed in the group to which they were randomized in a typical intention‐to‐treat fashion.
Measurement of Endothelial Function and Arterial Stiffness
Endothelial function was measured by ultrasound brachial artery FMD. Subjects rested in a supine position in a temperature‐controlled room for 10 minutes before imaging was performed. A blood pressure cuff was placed on the widest part of the proximal right forearm. An 8‐MHz linear array vascular ultrasound transducer and an ultrasound system (Acuson Sequoia 512, Siemens Medical Solutions, Issaquah, WA) allowed the brachial artery to be located above the elbow and scanned longitudinally. After recording of B‐mode ultrasound images of the brachial artery and spectral Doppler velocities, the cuff was inflated to 250 mm Hg for 5 minutes to induce reactive hyperemia. Immediately after cuff deflation, spectral Doppler images were obtained to verify hyperemia. Brachial artery B‐mode images were reobtained 60 and 90 seconds after cuff release. Measures of microvascular function that have also been associated with cardiovascular events, including reactive hyperemia (RH), peak Doppler velocity, RH velocity‐time integral, and RH brachial flow, were also performed.23, 27 . Studies were recorded digitally; brachial artery diameters were measured in triplicate with a digital border‐tracing tool (Access Point Web 3.0, Freeland Systems, Westfield, IN). Studies were read blinded to study treatment group. Reproducibility of measurements from this laboratory using the same techniques is excellent, as reported recently.28, 29
Arterial stiffness was measured by arterial tonometry (AtCor SphygmoCor Px, AtCor Medical, Sydney, Australia). Tonometry recordings of the radial arterial pulses were taken when a reproducible signal with a clear upstroke was obtained. AIx and central aortic pressures were derived from radial tonometry using a validated, generalized transfer function and calibrated using oscillometric brachial artery blood pressures. AIx measurements were standardized to a heart rate of 75 bpm30, 31; however, the results of the unadjusted AIx data are shown in Table S1. Quality and stability of the tonometry signals were ensured by requiring an operator index greater than 85% for all analyzed tracings. This index is based on the weighted value of 5 quality‐control indices (average pulse height, pulse height variation, diastolic point variation, curve shape variation, and maximum dP/dT as the first derivative of the systolic upstroke). Serial radial artery tonometry was used preferentially over carotid artery tonometry for 3 main reasons: (1) the transfer function used to derive central aortic pressures has only been validated using radial tonometry36; (2) it is more accurate and reproducible over time; and (3) it is more comfortable for the participant.
Statistical Analysis
All statistical analyses were conducted using SAS software (Version 999.4; Cary, NC). Subject demographics and laboratory data are presented as means (SD). Baseline comparisons were performed using 2‐tailed Student t tests. Pearson correlations were used to determine cross‐sectional relationships between cholesterol values and the outcome variables at each time point. All associations were tested using unconditional 2‐level linear mixed‐effects regression models implemented in the SAS MIXED procedure. To determine if simvastatin treatment effected Alx at different study visits, an interaction term between treatment group and study visit was included in the models. Fixed effects included treatment group, study visit, centered age, sex, LDL, and hsCRP corresponding to each time point. Treatment group and study visit were treated as categorical variables. The placebo group and baseline visit served as comparison categories. Subject was included as a random effect. Random intercepts were included for within‐subject correlations due to repeated measures. Models performed unadjusted (model 1), adjusted for age and sex (model 2); age, sex, and LDL cholesterol (model 3); age, sex, and hsCRP (model 4); age, sex, LDL cholesterol, and hsCRP (model 5); age, sex, peripheral systolic blood pressure (model 6); age, sex, central aortic systolic blood pressure (model 7). Although treatment group was highly correlated with LDL, a low variance inflation factor does not suggest multicollinearity. Secondary analyses with general linear models were performed to determine if changes in LDL levels influenced Alx.
Results
At baseline both groups were similar in age, with the placebo group being mean (SD) 54.4 (7.8) years old and the simvastatin group being 56.0 (6.2) years old (P=0.31). Both groups were composed mostly of women with 32 (73%) in the placebo group and 31 (70%) in the simvastatin group (P=0.82). Both groups had normal blood pressure at baseline (placebo 123.7 [16.9]/72.7 [10.8] mm Hg versus simvastatin 125.1 [15.7]/72.3 [9.9] mm Hg; P>0.6). Only 1 (2%) individual in the placebo group and 3 (7%) people in the simvastatin group were taking antihypertensive medications (P=0.7), and there were no changes in blood pressure meds during the study period. The average total cholesterol was 206.6 (34.2) mg/dL with an LDL cholesterol of 122.8 (28.1) mg/dL in the placebo group and 207.2 (35.7) mg/dL and 122.3 (28.4) mg/dL, respectively, in the simvastatin group (P>0.9). The 2 groups both had low 10‐year atherosclerotic CVD risk estimates (placebo 3.3 [3.3]% versus statin 4.1 [3.8]%; P=0.35). At baseline, the 2 randomized study groups were also similar in the primary outcome measures including augmentation index, central blood pressure parameters, and FMD (all P>0.2, Table 1).
Table 1.
Descriptive Statistics at Baseline, 12, and 18 Months
| Mean (Standard Deviation) unless otherwise noted | Baseline | 12 Months | 18 Months | |||
|---|---|---|---|---|---|---|
| Placebo (n=44) | Simvastatin (n=44) | Placebo (n=44) | Simvastatin (n=44) | Placebo (n=41) | Simvastatin (n=42) | |
| Age, y | 54.4 (7.7) | 56.0 (6.2) | ‐ | ‐ | ‐ | ‐ |
| Female sex, n (%) | 32 (73%) | 31 (70%) | 32 (73%) | 31 (70%) | 31 (76%) | 29 (69%) |
| White, n (%) | 43 (98%) | 44 (100%) | 43 (98%) | 44 (100%) | 40 (97%) | 42 (100%) |
| Body mass index, kg/m2 | 27.6 (5.6) | 27.2 (5.6) | 28.0 (5.7) | 27.2 (5.7) | 28.7 (7.2) | 27.3 (5.7) |
| Heart rate, bpm | 59.6 (9.0) | 60.6 (9.2) | 58.9 (7.9) | 60.6 (10.2) | 59.6 (9.0) | 59.8 (9.5) |
| Total cholesterol, mg/dL | 206.6 (34.2) | 207.2 (35.7) | 202.1 (31.8)a | 146.1 (27.1)a | 196.8 (34.2)a | 153.2 (28.0)a |
| Triglycerides, mg/dL | 101.8 (40.1) | 112.4 (57.6) | 99.9 (42.8) | 91.9 (46.5) | 109.1 (80.5) | 88.0 (44.8) |
| High‐density lipoprotein cholesterol, mg/dL | 63.5 (17.2) | 62.5 (20.8) | 65.5 (18.3) | 64.8 (20.5) | 63.0 (21.4) | 66.4 (20.1) |
| Low‐density lipoprotein cholesterol, mg/dL | 122.8 (28.1) | 122.3 (28.4) | 116.5 (28.2)a | 62.9 (18.8)a | 114.2 (28.0)a | 69.2 (20.1)a |
| Systolic blood pressure, mm Hg | 123.7 (16.9) | 125.1 (15.7) | 124.7 (13.3) | 122.6 (14.5) | 122.2 (21.8) | 123.2 (15.0) |
| Diastolic blood pressure, mm Hg | 72.7 (10.8) | 72.3 (9.9) | 75.1 (11.3) | 71.0 (9.6) | 75.0 (11.8) | 72.1 (9.2) |
| High‐sensitivity C‐reactive protein, mg/L | 1.9 (2.2) | 1.8 (2.2) | 2.0 (2.2) | 1.8 (2.4) | 2.1 (2.3) | 1.4 (2.6) |
| Antihypertensive medication use, n (%) | 1 (2%) | 3 (7%) | 1 (2%) | 3 (7%) | 1 (2%) | 2 (5%) |
| Average brachial artery diameter, cm | 0.38 (0.06) | 0.38 (0.07) | 0.38 (0.06) | 0.39 (0.07) | 0.38 (0.06) | 0.39 (0.07) |
| Maximum absolute flow‐mediated dilation, cm | 0.017 (0.01) | 0.016 (0.01) | 0.018 (0.01) | 0.017 (0.01) | 0.017 (0.01) | 0.016 (0.01) |
| Maximum relative flow‐mediated dilation, % | 4.45 (2.54) | 4.35 (2.94) | 4.18 (2.72) | 4.63 (2.59) | 4.64 (2.56) | 4.24 (2.15) |
| Augmentation index, %b | 23.1 (11.4) | 23.4 (9.0) | 24.9 (10.9)a | 20.6 (9.4)a | 24.1 (11.3) | 21.8 (9.3) |
| Aortic systolic pressure, mm Hg | 118.3 (12.7) | 116.8 (14.2) | 118.2 (15.0) | 113.6 (10.3) | 117.9 (5.3) | 114.6 (10.6) |
| Aortic diastolic pressure, mm Hg | 74.3 (7.7) | 74.5 (10.2) | 74.4 (7.2) | 72.8 (7.2) | 74.6 (9.7) | 73.0 (7.4) |
P<0.01 between placebo and simvastatin at each time point.
At 18 months there were 40 patients with measures of Augmentation Index; of the 3 subjects missing AIx measurements, 2 were due to very low heart rates, and the third was in bigeminy at the time of the study.
During the treatment period, there was a significant reduction in LDL cholesterol that was seen at each time point in the statin group but not in the placebo group starting at 6 months (P <0.0001). At 18 months, LDL cholesterol had decreased by 43.4% (−51.9 [25.8] mg/dL; P<0.0001) in the statin group compared with only a 7% reduction (−8.8 [22.5] mg/dL P=0.14) in the placebo group (Figure 1). Changes in AIx in the statin group and placebo group at each time point are shown in Figure 2. At 12 months there was a significant difference in AIx in the statin group compared with the placebo group (−2.3% [5.5] versus 1.2% [5.7], P=0.007). At 18 months the Alx change from baseline increased slightly to −1.1% [5.8] in the treatment group and decreased slightly to 0.2% [4.8] in the placebo group. Thus, 18‐month Alx was not significantly different from baseline Alx in either the statin (t[39]=1.18, P=0.24) or the placebo group (t[39]=−0.29; P=0.77), and the treatment effect was no longer significant (P=0.3). Similar findings were observed in the mixed models, where we evaluated the effect of the interaction of treatment group×study visit on AIx in both an unadjusted model (12 months, β=−3.57 [1.21], P=0.004; 18 months, β=−1.44 [1.21], P=0.24) and age‐ and sex‐adjusted models (12 months, β=−3.52 [1.21], P=0.004; 18 months β=−1.42 [1.21], P=0.2) (Table 2). Secondary analysis evaluating the impact of LDL on Alx while adjusting for age and sex showed that changes in LDL cholesterol levels did not have a significant effect on Alx at any study visit. This suggests that the transient changes in AIx do not significantly vary with differences in LDL cholesterol levels over time. Scatter plots showing the effect of simvastatin treatment or LDL on AIx at each time point can be found in Figure S1.
Figure 1.
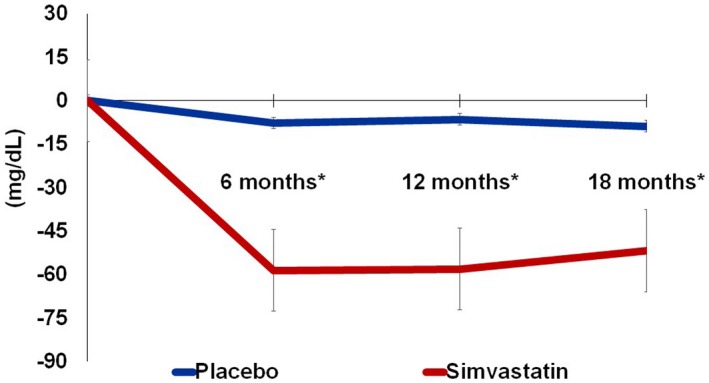
Absolute change in LDL cholesterol over 18 months. *P<0.001; Error bars represent standard error. LDL indicates low‐density lipoprotein.
Figure 2.
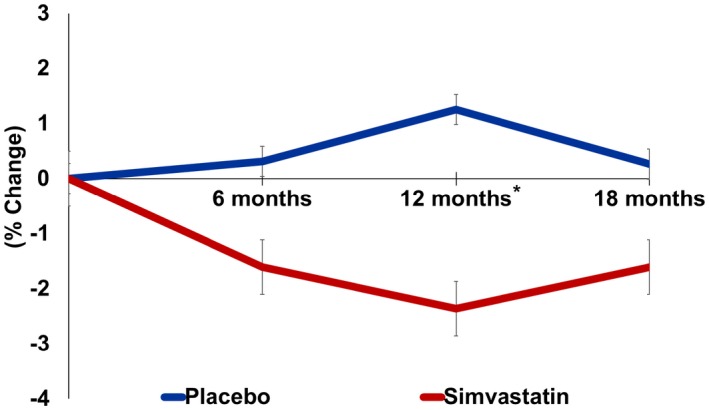
Percentage change in augmentation index over 18 months. *P<0.001; Error bars represent standard error.
Table 2.
Effects of Simvastatin Treatment on Central Augmentation Index Using Mixed Models
| Modela | 6 Months | 12 Months | 18 Months | ||||||
|---|---|---|---|---|---|---|---|---|---|
| β | SE | P Value | β | SE | P Value | β | SE | P Value | |
| Model 1: Treatment group×visit | −1.91 | 1.21 | 0.115 | −3.17 | 1.21 | 0.009 | −1.39 | 1.21 | 0.252 |
| Model 2: Treatment group×visit | −1.91 | 1.21 | 0.115 | −3.12 | 1.21 | 0.010 | −1.37 | 1.21 | 0.258 |
| Age | 0.60 | 0.12 | <0.0001 | 0.60 | 0.12 | <0.0001 | 0.60 | 0.12 | <0.0001 |
| Sex | 10.54 | 1.81 | <0.0001 | 10.54 | 1.82 | <0.0001 | 10.54 | 1.81 | <0.0001 |
| Model 3: Treatment group×visit | −1.68 | 1.46 | 0.251 | −2.89 | 1.47 | 0.051 | −1.18 | 1.40 | 0.399 |
| Age | 0.60 | 0.12 | <0.0001 | 0.60 | 0.12 | <0.0001 | 0.60 | 0.12 | <0.0001 |
| Sex | 10.52 | 1.81 | <0.0001 | 10.52 | 1.81 | <0.0001 | 10.52 | 1.81 | <0.0001 |
| LDL | 0.005 | 0.02 | 0.779 | 0.005 | 0.02 | 0.779 | 0.005 | 0.02 | 0.779 |
| Model 4: Treatment group×visit | −2.13 | 1.21 | 0.079 | −3.30 | 1.21 | 0.007 | −1.49 | 1.21 | 0.216 |
| Age | 0.59 | 0.12 | <0.0001 | 0.59 | 0.12 | <0.0001 | 0.59 | 0.12 | <0.0001 |
| Sex | 10.47 | 1.82 | <0.0001 | 10.47 | 1.82 | <0.0001 | 10.47 | 1.82 | <0.0001 |
| hsCRP | 0.11 | 0.06 | 0.088 | 0.11 | 0.06 | 0.088 | 0.11 | 0.06 | 0.088 |
| Model 5: Treatment group×visit | −1.83 | 1.46 | 0.209 | −2.99 | 1.46 | 0.042 | −1.24 | 1.39 | 0.371 |
| Age | 0.59 | 0.12 | <0.0001 | 0.59 | 0.12 | <0.0001 | 0.59 | 0.12 | <0.0001 |
| Sex | 10.44 | 1.82 | <0.0001 | 10.44 | 1.82 | <0.0001 | 10.44 | 1.82 | <0.0001 |
| LDL | 0.01 | 0.02 | 0.709 | 0.01 | 0.02 | 0.709 | 0.01 | 0.02 | 0.709 |
| hsCRP | 0.11 | 0.06 | 0.090 | 0.11 | 0.06 | 0.090 | 0.11 | 0.06 | 0.090 |
| Model 6: Treatment group×visit | −1.67 | 1.21 | 0.167 | −3.15 | 1.21 | 0.010 | −1.30 | 1.20 | 0.278 |
| Age | 0.58 | 0.12 | <0.0001 | 0.58 | 0.12 | <0.0001 | 0.58 | 0.12 | <0.0001 |
| Sex | 10.77 | 1.78 | <0.0001 | 10.77 | 1.78 | <0.0001 | 10.77 | 1.78 | <0.0001 |
| Peripheral systolic blood pressure | 0.05 | 0.02 | 0.021 | 0.05 | 0.02 | 0.021 | 0.05 | 0.02 | 0.021 |
| Model 7: Treatment group×visit | −1.66 | 1.18 | 0.160 | −2.70 | 1.18 | 0.024 | −1.19 | 1.18 | 0.316 |
| Age | 0.56 | 0.11 | <0.0001 | 0.56 | 0.11 | <0.0001 | 0.56 | 0.11 | <0.0001 |
| Sex | 11.38 | 1.72 | <0.0001 | 11.38 | 1.72 | <0.0001 | 11.38 | 1.72 | <0.0001 |
| Aortic systolic pressure | 0.13 | 0.03 | <0.0001 | 0.13 | 0.03 | <0.0001 | 0.13 | 0.03 | <0.0001 |
hsCRP indicates high‐sensitivity C‐reactive protein; LDL, low‐density lipoprotein; SE, standard error.
Mixed models adjusted and unadjusted.
No differences were seen between the 2 groups at any time point for any of the other outcome measures including brachial artery blood pressure, central aortic blood pressures, FMD, RH peak Doppler velocity, RH velocity‐time integral, or RH brachial flow with and without adjustment (all P>0.2). Additionally, hsCRP (0.10 [0.06]; P=0.09) was not associated with a change in AIx (0.10 [0.06]; P=0.3) Sensitivity analyses that included treatment with antihypertensive medication in the model did not have an impact on augmentation index over time (β=4.57, P=0.3).
Discussion
Statins decrease CVD events12 and are an established pillar for CVD prevention.12, 32, 33 There is a well‐defined relationship between atherosclerosis and arterial stiffness in which increasing atherosclerotic disease leads to increased stiffness over time. In our analyses of middle‐aged adults of offspring of those with AD but without CVD, the effects of statin therapy on augmentation indices are not related to their LDL cholesterol–lowering effects. Statin therapy also had no impact on vascular reactivity or inflammatory markers at any time point compared with those taking placebo.
The differences in AIx that emerged after initiation of statin or placebo therapy were not durable even though the total cholesterol and LDL‐cholesterol reductions were sustained over the 18‐month study period. Our data robustly suggest that the significant differences at 12 months were not due to changes in central or peripheral blood pressure or differences in LDL cholesterol concentration. The changes also were not accompanied by a reduction in inflammation, measured by hsCRP, or improvements vascular reactivity, measured by FMD. Because this study was designed to evaluate the impact of statin therapy on dementia‐related biomarkers, the transient differences in AIx at 12 months could be mediated through unmeasured variables, which may be more prominent in offspring of those with AD. There could also be gene‐mediated effects of statin therapy on AIx resulting from changes in extracellular matrix genes in vascular smooth muscle cells that help maintain elasticity and could lead to a reduction in AIx. The transient nature of the AIx improvements observed could be due to the impact of APOE on differences in unmeasured lipoprotein particles, such as apoE‐containing HDL particles, rather than measurable LDL cholesterol.16, 17, 34 There may be other short‐lived, unmeasured changes at the molecular and cellular level that could, alone or in combination, account for our observations. Changes in matrix metalloproteinase expression have been shown to have plaque‐stabilizing effects.35, 36 Alterations in the expression of matrix metalloproteinase alters the elastin‐to‐collagen ratio in the vessel wall. Last, differences in unmeasured inflammatory markers or inflammatory cytokines or changes in T‐cell activation may also transiently alter AIx following statin therapy.10, 11, 35, 37 Additionally, statins have been shown to dampen the sympathetic nervous system, which could also play into the transient impact on arterial stiffness.38
Most previous randomized controlled trials that have evaluated the effects of statin therapy on AIx and arterial stiffness with statin therapy have been small (ranging from 5 to 891 subjects), and most had less than 18 months of follow‐up (ranging from 5 days to 3.5 years).18, 19, 20, 21, 22 There might have been a regression toward less difference in AIx in the treatment group if there had been longer follow‐up. The larger randomized controlled trials with more prolonged follow‐up also differed from the current study in that they evaluated patients with known CVD risk factors, including hypertension, which is a well‐established driver of arterial stiffness.39, 40, 41 To our knowledge there are only 4 randomized controlled trials with over 1 year of follow‐up, and all recruited participants with hypertension25, 42 or other proinflammatory states such as rheumatoid arthritis43 or chronic kidney disease.44 The largest randomized controlled trial with the longest follow‐up to date (CAFE‐LLA [Conduit Artery Function Evaluation‐Lipid‐Lowering Arm] study; n=891; 3.5 years of follow‐up) recruited hypertensive subjects who also randomized to specific antihypertensive regimens in addition to statin therapy.25 Although blood pressure and blood pressure control robustly impact arterial stiffness and AIx,39, 41 in the CAFE‐LLA study, statin therapy did not influence central aortic blood pressure measurements or AIx.25 Older patients may have too advanced arterial damage to reveal an impact of statin therapy, which may be more apparent in younger subjects, although even in a relatively younger patient population, like those recruited in the current study, we did not see a durable difference in arterial stiffness at the 18‐month time point between the 2 groups. Additionally, atorvastatin was used in CAFE‐LLA, rather than simvastatin. The non‐LDL effects on AIx may be impacted by the statin subtypes due to the differences in lipophilicity, which varies the effects on matrix metalloproteinase expression.35
Limitations
Although this study was a well‐designed, double‐blind, randomized controlled trial, there were still several important limitations that should be pointed out. First, this was a substudy of a randomized controlled trial looking at the effects of statin therapy on markers of AD. Those recruited were predominantly younger, white subjects free from known CVD without a guideline‐based indication for lipid‐lowering therapy. Although there was limited ethnic diversity, this is 1 of the first randomized controlled trials to evaluate the effects of statin therapy on arterial stiffness measures and central pressures in a younger, healthier US patient population free from hypertension. Because the outcomes measured here were meant to be exploratory, the sample size and power calculations were not based on arterial stiffness measures, central pressures, or FMD. However only 1 other randomized controlled trial (the CAFE‐LLA study) that had more than 1 year of follow‐up enrolled more patients than the current study.25 There are other unmeasured variables at both the cellular and molecular levels that could impact the arterial wall as well as ventricular contractility and systemic vascular resistance, which also could contribute the impact of the effects of statin therapy on AIx. Unlike pulse‐wave velocity, AIx is affected by the heart rate. We reported AIx data that were corrected to a heart rate of 75 bpm and showed that heart rate data were similar between groups at each time point (P>0.4), although similar findings were also found using AIx that was not corrected for heart rate (Table S1). Arterial stiffness was not directly measured; instead, we evaluated AIx, a well‐established surrogate marker of pulse wave velocity, which utilizes a generalized transfer function that is assumed to be constant across the study visits, for both the placebo‐ and statin‐treated groups. Specifically, there is a possibility that if there are changes to the physical properties of the conduit arteries with statin therapy, and they may make the use of the same transfer function less valid, this assumption could be a confounding limitation to the absolute changes of AIx observed with statin therapy, potentially underestimating their absolute delta. To explore this further, we also analyzed changes in the peripheral augmentation index of the directly sampled radial artery, avoiding any mathematical assumptions, and found no differences between treatment groups at any visit (Table S2). Although the differences in the treatment effects on central and peripheral augmentation could be related to the general transfer function, they could also be due to inherent differences between central and peripheral pressure augmentation and the properties of central (more elastic) and peripheral (more muscular) arteries.
Conclusions
In low‐risk, middle‐aged individuals free of CVD, statin therapy had no impact on central aortic blood pressures or brachial artery FMD. There was a significant but transient reduction in AIx after 12 months of statin treatment compared with placebo, but between‐group differences in AIx did not persist after 18 months despite a durable reduction in LDL cholesterol levels.
Sources of Funding
This work was supported by the National Institutes of Health (R01AG031790‐01A1), NIH CTSA at UW 1UL1TR002373, and NIH P50 AG033514.
Disclosures
None.
Supporting information
Table S1. Effect of Simvastatin Treatment on Unadjusted Augmentation Index
Table S2. Effects of Simvastatin Treatment on Peripheral Augmentation Index Using Mixed Models
Figure S1. Scatter plots of the effect of simvastatin treatment or LDL on augmentation index. LDL indicates low‐density lipoprotein.
(J Am Heart Assoc. 2019;8:e009792 DOI: 10.1161/JAHA.118.009792.)
This work was presented as a poster at the American Heart Association Scientific Sessions, November 11 to 15, 2017, in Anaheim, CA.
References
- 1. Ben‐Shlomo Y, Spears M, Boustred C, May M, Anderson SG, Benjamin EJ, Boutouyrie P, Cameron J, Chen CH, Cruickshank JK, Hwang SJ, Lakatta EG, Laurent S, Maldonado J, Mitchell GF, Najjar SS, Newman AB, Ohishi M, Pannier B, Pereira T, Vasan RS, Shokawa T, Sutton‐Tyrell K, Verbeke F, Wang KL, Webb DJ, Willum Hansen T, Zoungas S, McEniery CM, Cockcroft JR, Wilkinson IB. Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta‐analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol. 2014;63:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Sloten TT, Sedaghat S, Laurent S, London GM, Pannier B, Ikram MA, Kavousi M, Mattace‐Raso F, Franco OH, Boutouyrie P, Stehouwer CD. Carotid stiffness is associated with incident stroke: a systematic review and individual participant data meta‐analysis. J Am Coll Cardiol. 2015;66:2116–2125. [DOI] [PubMed] [Google Scholar]
- 3. Gepner AD, Korcarz CE, Colangelo LA, Hom EK, Tattersall MC, Astor BC, Kaufman JD, Liu K, Stein JH. Longitudinal effects of a decade of aging on carotid artery stiffness: the multiethnic study of atherosclerosis. Stroke. 2014;45:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all‐cause mortality with arterial stiffness: a systematic review and meta‐analysis. J Am Coll Cardiol. 2010;55:1318–1327. [DOI] [PubMed] [Google Scholar]
- 5. Hughes TM, Wagenknecht LE, Craft S, Mintz A, Heiss G, Palta P, Wong D, Zhou Y, Knopman D, Mosley TH, Gottesman RF. Arterial stiffness and dementia pathology: Atherosclerosis Risk in Communities (ARIC)‐PET study. Neurology. 2018;90:e1248–e1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hughes TM, Craft S, Lopez OL. Review of “the potential role of arterial stiffness in the pathogenesis of Alzheimer's disease.” Neurodegener Dis Manag. 2015;5:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hazzouri AZ, Newman AB, Simonsick E, Sink KM, Tyrrell KS, Watson N, Satterfield S, Harris T, Yaffe K. Pulse wave velocity and cognitive decline in elders: the health, aging, and body composition study. Stroke. 2013;44:388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cui C, Sekikawa A, Kuller LH, Lopez OL, Newman AB, Kuipers AL, Mackey RH. Aortic Stiffness is Associated with Increased Risk of Incident Dementia in Older Adults. J Alzheimers Dis. 2018;66:297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta‐analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Catapano AL, Pirillo A, Norata GD. Vascular inflammation and low‐density lipoproteins: is cholesterol the link? A lesson from the clinical trials. Br J Pharmacol. 2017;174:3973–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenson RS. Pluripotential mechanisms of cardioprotection with HMG‐CoA reductase inhibitor therapy. Am J Cardiovasc Drugs. 2001;1:411–420. [DOI] [PubMed] [Google Scholar]
- 12. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 13. Pena JM, MacFadyen J, Glynn RJ, Ridker PM. High‐sensitivity C‐reactive protein, statin therapy, and risks of atrial fibrillation: an exploratory analysis of the JUPITER trial. Eur Heart J. 2012;33:531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Silverman MG, Ference BA, Im K, Wiviott SD, Giugliano RP, Grundy SM, Braunwald E, Sabatine MS. Association between lowering LDL‐C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta‐analysis. JAMA. 2016;316:1289–1297. [DOI] [PubMed] [Google Scholar]
- 15. Pedersen TR, Cater NB, Faergeman O, Kastelein JJ, Olsson AG, Tikkanen MJ, Holme I, Larsen ML, Lindahl C, Szarek M. Comparison of atorvastatin 80 mg/day versus simvastatin 20 to 40 mg/day on frequency of cardiovascular events late (five years) after acute myocardial infarction (from the Incremental Decrease in End Points through Aggressive Lipid Lowering [IDEAL] trial). Am J Cardiol. 2010;106:354–359. [DOI] [PubMed] [Google Scholar]
- 16. Kothapalli D, Liu SL, Bae YH, Monslow J, Xu T, Hawthorne EA, Byfield FJ, Castagnino P, Rao S, Rader DJ, Pure E, Phillips MC, Lund‐Katz S, Janmey PA, Assoian RK. Cardiovascular protection by ApoE and ApoE‐HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep. 2012;2:1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma Y, Malbon CC, Williams DL, Thorngate FE. Altered gene expression in early atherosclerosis is blocked by low level apolipoprotein E. PLoS One. 2008;3:e2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castejon R, Castaneda A, Sollet A, Mellor‐Pita S, Tutor‐Ureta P, Jimenez‐Ortiz C, Yebra‐Bango M. Short‐term atorvastatin therapy improves arterial stiffness of middle‐aged systemic lupus erythematosus patients with pathological pulse wave velocity. Lupus. 2017;26:355–364. [DOI] [PubMed] [Google Scholar]
- 19. Davenport C, Ashley DT, O'Sullivan EP, McHenry CM, Agha A, Thompson CJ, O'Gorman DJ, Smith D. The effects of atorvastatin on arterial stiffness in male patients with type 2 diabetes. J Diabetes Res. 2015;2015:846807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oh PC, Han SH, Koh KK, Lee K, Seo JG, Suh SY, Ahn T, Choi IS, Shin EK. Rosuvastatin treatment improves arterial stiffness with lowering blood pressure in healthy hypercholesterolemic patients. Int J Cardiol. 2014;176:1284–1287. [DOI] [PubMed] [Google Scholar]
- 21. Sahebkar A, Pecin I, Tedeschi‐Reiner E, Derosa G, Maffioli P, Reiner Z. Effects of statin therapy on augmentation index as a measure of arterial stiffness: a systematic review and meta‐analysis. Int J Cardiol. 2016;212:160–168. [DOI] [PubMed] [Google Scholar]
- 22. Upala S, Wirunsawanya K, Jaruvongvanich V, Sanguankeo A. Effects of statin therapy on arterial stiffness: a systematic review and meta‐analysis of randomized controlled trial. Int J Cardiol. 2017;227:338–341. [DOI] [PubMed] [Google Scholar]
- 23. Cooper LL, Palmisano JN, Benjamin EJ, Larson MG, Vasan RS, Mitchell GF, Hamburg NM. Microvascular function contributes to the relation between aortic stiffness and cardiovascular events: the Framingham Heart Study. Circ Cardiovasc Imaging. 2016;9:e004979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wiviott SD, de Lemos JA, Cannon CP, Blazing M, Murphy SA, McCabe CH, Califf R, Braunwald E. A tale of two trials: a comparison of the post‐acute coronary syndrome lipid‐lowering trials A to Z and PROVE IT‐TIMI 22. Circulation. 2006;113:1406–1414. [DOI] [PubMed] [Google Scholar]
- 25. Williams B, Lacy PS, Cruickshank JK, Collier D, Hughes AD, Stanton A, Thom S, Thurston H. Impact of statin therapy on central aortic pressures and hemodynamics: principal results of the Conduit Artery Function Evaluation‐Lipid‐Lowering Arm (CAFE‐LLA) Study. Circulation. 2009;119:53–61. [DOI] [PubMed] [Google Scholar]
- 26. Wechsler D. Wechsler Memory Scale. 3rd ed. San Antonio, TX: Psychological Corp; 1997.
- 27. Anderson TJ, Charbonneau F, Title LM, Buithieu J, Rose MS, Conradson H, Hildebrand K, Fung M, Verma S, Lonn EM. Microvascular function predicts cardiovascular events in primary prevention: long‐term results from the Firefighters and Their Endothelium (FATE) study. Circulation. 2011;123:163–169. [DOI] [PubMed] [Google Scholar]
- 28. Gottdiener JS, Bednarz J, Devereux R, Gardin J, Klein A, Manning WJ, Morehead A, Kitzman D, Oh J, Quinones M, Schiller NB, Stein JH, Weissman NJ. American Society of Echocardiography recommendations for use of echocardiography in clinical trials. J Am Soc Echocardiogr. 2004;17:1086–1119. [DOI] [PubMed] [Google Scholar]
- 29. Stein JH, Brown TT, Ribaudo HJ, Chen Y, Yan M, Lauer‐Brodell E, McComsey GA, Dube MP, Murphy RL, Hodis HN, Currier JS. Ultrasonographic measures of cardiovascular disease risk in antiretroviral treatment‐naive individuals with HIV infection. Aids. 2013;27:929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilkinson IB, Mohammad NH, Tyrrell S, Hall IR, Webb DJ, Paul VE, Levy T, Cockcroft JR. Heart rate dependency of pulse pressure amplification and arterial stiffness. Am J Hypertens. 2002;15:24–30. [DOI] [PubMed] [Google Scholar]
- 31. Wilkinson IB, MacCallum H, Flint L, Cockcroft JR, Newby DE, Webb DJ. The influence of heart rate on augmentation index and central arterial pressure in humans. J Physiol. 2000;525(Pt 1):263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Du R, Cai J, Zhao XQ, Wang QJ, Liu DQ, Leng WX, Gao P, Wu HM, Ma L, Ye P. Early decrease in carotid plaque lipid content as assessed by magnetic resonance imaging during treatment of rosuvastatin. BMC Cardiovasc Disord. 2014;14:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nissen SE, Nicholls SJ, Sipahi I, Libby P, Raichlen JS, Ballantyne CM, Davignon J, Erbel R, Fruchart JC, Tardif JC, Schoenhagen P, Crowe T, Cain V, Wolski K, Goormastic M, Tuzcu EM. Effect of very high‐intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–1565. [DOI] [PubMed] [Google Scholar]
- 34. Yang H, Zhang N, Okoro EU, Guo Z. Transport of apolipoprotein B‐containing lipoproteins through endothelial cells is associated with apolipoprotein e‐carrying HDL‐like particle formation. Int J Mol Sci. 2018;19:e3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Izidoro‐Toledo TC, Guimaraes DA, Belo VA, Gerlach RF, Tanus‐Santos JE. Effects of statins on matrix metalloproteinases and their endogenous inhibitors in human endothelial cells. Naunyn Schmiedebergs Arch Pharmacol. 2011;383:547–554. [DOI] [PubMed] [Google Scholar]
- 36. Lijnen HR. Extracellular proteolysis in the development and progression of atherosclerosis. Biochem Soc Trans. 2002;30:163–167. [DOI] [PubMed] [Google Scholar]
- 37. Tziakas DN, Chalikias GK, Stakos DA, Papanas N, Chatzikyriakou SV, Mitrousi K, Maltezos E, Boudoulas H. Effect of statins on collagen type I degradation in patients with coronary artery disease and atrial fibrillation. Am J Cardiol. 2008;101:199–202. [DOI] [PubMed] [Google Scholar]
- 38. Lewandowski J, Symonides B, Gaciong Z, Sinski M. The effect of statins on sympathetic activity: a meta‐analysis. Clin Auton Res. 2015;25:125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gepner AD, Tedla Y, Colangelo LA, Tattersall MC, Korcarz CE, Kaufman JD, Liu K, Burke GL, Shea S, Greenland P, Stein JH. Progression of carotid arterial stiffness with treatment of hypertension over 10 years. Hypertension. 2017;69:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA. 2012;308:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tedla YG, Gepner AD, Vaidya D, Colangelo L, Stein JH, Liu K, Greenland P. Association between long‐term blood pressure control and ten‐year progression in carotid arterial stiffness among hypertensive individuals: the multiethnic study of atherosclerosis. J Hypertens. 2017;35:862–869. [DOI] [PubMed] [Google Scholar]
- 42. Manisty C, Mayet J, Tapp RJ, Sever PS, Poulter N, Thom SAMcG, Hughes AD. Atorvastatin treatment is associated with less augmentation of the carotid pressure waveform in hypertension: a substudy of the Anglo‐Scandinavian Cardiac Outcome Trial (ASCOT). Hypertension. 2009;54:1009–1013. [DOI] [PubMed] [Google Scholar]
- 43. Tam LS, Li EK, Shang Q, Tomlinson B, Lee VW, Lee KK, Li M, Kuan WP, Li TK, Tseung L, Yip GW, Freedman B, Yu CM. Effects of rosuvastatin on subclinical atherosclerosis and arterial stiffness in rheumatoid arthritis: a randomized controlled pilot trial. Scand J Rheumatol. 2011;40:411–421. [DOI] [PubMed] [Google Scholar]
- 44. Fassett RG, Robertson IK, Ball MJ, Geraghty DP, Sharman JE, Coombes JS. Effects of atorvastatin on arterial stiffness in chronic kidney disease: a randomised controlled trial. J Atheroscler Thromb. 2010;17:235–241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Effect of Simvastatin Treatment on Unadjusted Augmentation Index
Table S2. Effects of Simvastatin Treatment on Peripheral Augmentation Index Using Mixed Models
Figure S1. Scatter plots of the effect of simvastatin treatment or LDL on augmentation index. LDL indicates low‐density lipoprotein.