

SUMMARY
Stem cells balance cellular fates through asymmetric and symmetric divisions in order to self-renew or to generate downstream progenitors. Symmetric commitment divisions in stem cells are required for rapid regeneration during tissue damage and stress. The control of symmetric commitment remains poorly defined. Using single-cell RNA sequencing (scRNA-seq) in combination with transcriptomic profiling of HSPCs (hematopoietic stem and progenitor cells) from control and m6A methyltransferase Mettl3 conditional knockout mice, we found that m6A-deficient hematopoietic stem cells (HSCs) fail to symmetrically differentiate. Dividing HSCs are expanded and are blocked in an intermediate state that molecularly and functionally resembles multipotent progenitors. Mechanistically, RNA methylation controls Myc mRNA abundance in differentiating HSCs. We identified MYC as a marker for HSC asymmetric and symmetric commitment. Overall, our results indicate that RNA methylation controls symmetric commitment and cell identity of HSCs and may provide a general mechanism for how stem cells regulate differentiation fate choice.
In Brief
Cheng et al. uncover RNA methylation as a guardian in hematopoietic stem cell (HSC) fate decisions. m6A maintains hematopoietic stem cell symmetric commitment and identity. This study may provide a general mechanism for how RNA methylation controls cellular fate.
Graphical Abstract
INTRODUCTION
Hematopoietic stem cells (HSCs) balance their long-lived regenerative capacity with the ability to maintain myeloid, lymphoid, and erythroid lineage output in the blood. This balance is mediated through cell fate decisions that occur during cellular division. When they divide, HSCs either self-renew or undergo differentiation toward a multipotent progenitor cell (MPP) fate, where the cells are metabolically more active than HSCs and retain multi-lineage potency but lack HSC-long-term engraftment activity. The choice between these distinct cellular outcomes is controlled by the ability to alternate between a symmetric or asymmetric fate choice (Knoblich, 2008; Morrison and Kimble, 2006). It remains unclear what signals can determine whether a cellular division leads to cellular commitment (differentiation) or self-renewal. Mechanistic insights into the regulation of cell fate decisions may inform approaches to bone marrow failure syndromes, differentiation therapy of hematopoietic malignancies, and stem cell expansion for therapeutic benefits.
A key controller of cellular fate is mRNA methylation. The most common reversible posttranscriptional mRNA modification on mRNA is N6-methyladenosine (m6A). The presence of m6A alters mRNA metabolism, including mRNA stability, mRNA splicing, and translation efficiency (Coots et al., 2017; Meyer and Jaffrey, 2017; Meyer et al., 2015; Ping et al., 2014; Roundtree et al., 2017; Schwartz et al., 2014; Slobodin et al., 2017; Wang et al., 2015; Xiao et al., 2016). The deposition of transcriptome-wide m6A marks depends on the METTL3-METTL14-WTAP-VIRMA-ZC3H13-RBM15 protein complex, in which METTL3 serves as the catalytic subunit. m6A controls cell state maintenance in a cellular-context-dependent manner. Embryonic stem cells (ESCs) with Mettl3 deficiency remain naive and fail to differentiate into primed ESCs (Batista et al., 2014; Geula et al., 2015) and specification of hematopoietic stem and progenitor cells (HSPCs) requires METTL3 in zebrafish and mouse embryos (Lv et al., 2018; Zhang et al., 2017).
A number of recent studies showed that m6A and METTL3 are important for survival and maintenance of the undifferentiated stages of myeloid leukemia cells (Barbieri et al., 2017; Vu et al., 2017a; Weng et al., 2018). However, as therapeutics toward METTL3 are being developed to target myeloid leukemia (Boriack-Sjodin et al., 2018), it is important to understand how loss of m6A affects normal blood development. Several studies have reported that disruption of m6A regulators impacts normal HSC function. Depletion of YTHDF2, a m6A reader protein, results in increased HSCs that are capable of normal engraftment, while loss of writer protein METTL3 leads to an accumulation of HSCs with impaired differentiation capacity and normal self-renewal (Lee et al., 2019; Li et al., 2018; Yao et al., 2018). However, the mechanism by which m6A affects HSC expansion remains unknown. Additionally, MYC was reported as a major target of m6A that contributes to the effects of m6A in myeloid leukemia and in HSCs (Lee et al., 2019; Vu et al., 2017a)However, it remains unclear if m6A simply alters MYC expression, or if m6A has other regulatory roles that mediate MYC’s effects in HSC accumulation.
To understand how m6A shapes the early differentiation decisions during hematopoietic differentiation, we performed singlecell RNA sequencing (RNA-seq) in wild-type (WT) and METTL3 knockout hematopoietic progenitor cells. In contrast to the HSC accumulation phenotype that has been described upon METTL3 depletion previously, we report here that HSCs are instead depleted. We show that the expanded population is not in the HSC pool but, instead, comprises a HSC-like intermediate state that molecularly and functionally resembles multipotent progenitors. Mechanistically, we show that m6A is required for HSCs’ symmetric commitment step in hematopoietic differentiation, with normal asymmetric commitment upon METTL3 depletion. We find that m6A controls Myc RNA stability and this m6A-regulated expression of Myc controls HSC symmetric commitment. The HSC-like intermediate population that is metabolically activated but fails to symmetrically commit has uncoupled the role for MYC in HSC activation and cellular commitment. Our data advance the concepts that m6A is essential for HSC identity maintenance and it tightly controls HSCs entry toward commitment. Overall, we find that the major role for m6A in hematopoietic differentiation is due to its ability to regulate symmetric commitment via controlling Myc mRNA stability.
RESULTS
Mettl3 Is Required for Normal Hematopoiesis
To study the role of m6A in normal hematopoiesis and cellular fate, we crossed the Mettl3 flox/flox (Mettl3 f/f) mice with the Mx1-Cre mice to generate Mettl3 f/f, Mx1-Cre+ conditional knockout (cKO) mice (Figure 1A). Mettl3 depletion was then induced by injection with polyinosinic-polycytidylic acid (pIpC) (Figure S1A). Analysis of pIpC-treated Mettl3 f/f, Mx1-Cre+ (Mettl3 cKO) and control Mettl3 f/f, Mx1-Cre− (Mettl3 f/f) confirmed efficient Mettl3 genomic deletion (Figure 1B) and ablation of METTL3 protein in HSPCs (LSK, Lineage− cKit+Sca-1+) (Figure S1B). We further confirmed that the loss of METTL3 reduced global m6A levels in bone marrow cells (Figure 1C).
Figure 1. Expansion of HSCs in Mettl3 cKO Mice.

(A) Target scheme for Mettl3 conditional knockout (cKO) mice. Primers were designed for genotyping by targeting Mettl3 exon 4.
(B) Successful deletion of Mettl3 in the cKO bone marrow 3 weeks post-pIpC injections. PCR by using genomic DNA from pIpC-treated mice bone marrow cells to validate Mettl3 exon4 deletion and Ore expression. Primers used were indicated in (A).
(C) Decreased m6A in cKO mice 3 weeks post-pIpC. Global m6A levels in Mettl3 f/f and Mettl3 cKO BM cells were measured by two-dimensional thin-layer chromatography (TLC). n = 3.
(D) Mettl3 cKO mice developed a pancytopenia phenotype 3 weeks post-pIpC. Whole blood counts of white blood cell (WBCs), red blood cells (RBCs), and platelets (PLT) of Mettl3 f/f and Mettl3 cKO mice. n = 11.
(E) Reduction in bone marrow cellularity in cKO mice 3 weeks post-pIpC. Bone marrow cellularity of Mettl3 f/f and Mettl3 cKO mice was determined. n = 11.
(F) Left: representative flow cytometry plots for gating strategies of hematopoietic stem and progenitor cell (HSPC) compartments. An expansion in HSPC population and a reduction in myeloid progenitors upon Mettl3 depletion. Right top: frequency of LSK (Lin−cKit+Sca-1+) and MP (Myeloid progenitor, Lin−cKit+Sca-1−) in BM cells. Right bottom: percentage of HSC (LSK, CD150+CD48−), MPP1 (LSK, CD150−CD48−), MPP2 (LSK, CD150+CD48+), and MPP4 (LSK, CD150−CD48+) in LSK population. n = 11.
(G) Absolute cell numbers of LSKs, HSCs, and MPPs in Mettl3 f/f and Mettl3 cKO mice at 3weeks post-pIpC injection. n = 11. Mean and SEM are shown (*p<0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). n represents number of mice.
We then evaluated the hematopoietic system upon depletion of Mettl3 and global m6A. We observed in Mettl3 cKO mice a 40% reduction in bone marrow cellularity and pancytopenia (Figures 1D and 1E). Flow analysis of bone marrow cells from Mettl3 cKO mice revealed a marked reduction in Gr1+Mac1+ mature granulocyte and Ter119+CD71med /Ter119+CD71low (Ery3/Ery4) erythroid cells (Figures S1C and S1D), as well as an increase in CD41+ megakaryocytes (Figure S1C) without major defects in lymphoid lineages compared to control mice (Figure S1E). Analysis of bone marrow sections confirmed that Mettl3 cKO mice had reduced mature granulocytes and increased immature myeloid cells (Figure S1F). Histopathological analysis in Mettl3 cKO mice further showed dysplasia in megakaryocytes characterized by abnormal micromegakaryocytes with a large fraction characterized with monolobated megakaryocytes, a feature associated with altered differentiation (Figure S1F). Moreover, METTL3-depleted bone marrow cells demonstrated a significant reduction in the ability to proliferate and differentiate to form myeloid colonies in vitro (Figure S1G). Additionally, Mettl3 cKO mice exhibited enlarged spleens with extensively infiltrated red pulp zones and perturbed splenic architecture, which is associated with an accumulation of immature erythroblasts at the expense of B and T cells (Figures S1H–S1K). Taken together, these data indicated an impairment in maturation and terminal differentiation of myeloid and erythroid lineages upon METTL3 ablation and reduction of m6A.
Given the broad, multi-lineage defects of METTL3 loss in the hematopoietic system, we hypothesized that the defects originate from the hematopoietic stem and progenitor compartments. The HSPC compartment can be further defined using cell surface markers (i.e., SLAM/CD150 and CD48) into HSCs (LSK, CD150+CD48−) and MPPs. MPPs, which all have short-term stem cell activity, can be subdivided into three populations: MPP1s, which are considered unbiased (LSK, CD150-CD48); MPP2s, which have a bias toward megakaryocytic and erythroid lineages (LSK, CD150+CD48+); and MPP4s, with lymphoid lineage bias (LSK, CD150−CD48+) (Kiel et al., 2005; Oguro et al., 2013; Rodriguez-Fraticelli et al., 2018). We examined all four populations and observed a 5-fold increase in the frequency of HSPCs (LSKs) and a nearly 10-fold expansion in immunophenotypic HSCs, as well as MPP2s and MPP4s in the bone marrow of Mettl3 cKO mice as compared to control Mettl3 f/f mice (Figures 1F and 1G). Notably, the MPP1 subset of MPPs was not increased in Mettl3 cKO mice. However, the downstream myeloid progenitors (MPs, Lin−ckit+Sca-1−) comprising common myeloid progenitors (CMP; Lin−cKit+Sca-1−CD34+FcrR−) and granulocyte myeloid progenitors (GMP; Lin−cKit+Sca-1−CD34+FcrR+) were significantly reduced in Mettl3 cKO mice (Figure S1L). Overall, these data suggested that loss of m6A imposed a blockage in transition of HSCs to MPs, thereby hindering commitment of HSCs toward a myeloid fate.
To explore the dosage effect of Mettl3 loss on hematopoiesis, we evaluated heterozygous Mettl3 f/−, Mx1-Cre+ (Het, Cre+) mice. Similar to the control mice, heterozygous Mettl3 deletion had equivalent bone marrow (BM) cellularity and peripheral blood counts (white blood cells [WBCs], red blood cells [RBCs], and platelets) 3 weeks post-deletion (Figures S2A and S2B). However, we found that the Mettl3 het mice also exhibited splenomegaly, which was associated with an accumulation of erythroid blasts and reduction of T cells (Figures S2C and S2D). Moreover, HSCs were also expanded in the Mettl3 het mice while the downstream lymphoid, myeloid, and erythroid populations were equivalent to controls except for the reduction in the pre-pro B cell population in the Mettl3 het mice (Figures S2E–S2H). These data suggest an intermediate dose requirement for Mettl3 in normal hematopoiesis.
Mettl3-Depleted HSCs Are More Activated and Functionally Defective in Reconstitution
As we observed defects in progenitor pools and mature lineages with an expansion of HSCs upon METTL3 depletion, we hypothesized that m6A is critical to maintain HSC function. We first examined whether loss of m6A results in the exit of quiescence in HSCs. Under normal homeostatic conditions, a majority of HSCs remain dormant in G0 while a fraction of HSCs enter cell cycle to divide and generate progenitor cells (Attar and Scadden, 2004). Loss of Mettl3 results in an increased frequency of cells that have exited G0 and increased percentage of cells that are actively dividing (Figures 2A, 2B, and S2I). These proliferative HSCs with depleted Mettl3 are also metabolically activated, with increased mitochondrial mass and activity (Figures 2C, 2D, and S2J). Consistent with previous findings that loss of quiescence in HSCs is attributed to reduced engrafting potential (Cheng et al., 2000; Hock et al., 2004), Mettl3 cKO bone marrow cells exhibited a significant defect in in vivo repopulating capacity, characterized by reduced chimerism of all HSPC compartments and mature lineage populations in recipient mice (Figures 2E, 2F, S2K, and S2L). These results indicated that m6A is functionally required for maintaining HSC quiescence and self-renewal potential. The data also suggested that the expansion of HSCs is possibly due to the accumulation of dividing HSCs, incapable of properly differentiating.
Figure 2. Mettl3 cKO HSCs Are Less Quiescent and Functionally Defective.

(A) Mettl3 cKO HSCs are less quiescent. Representative flow cytometry plots for assessing cell cycle status of Mettl3 f/f and Mettl3 cKO HSC by Pyronin Y staining.
(B) Quantification of cell cycle analysis. n = 5.
(C) Increased mitochondrial mass in Mettl3 cKO HSCs. Representative histograms of Mitotracker Green staining in Mettl3 f/f and Mettl3 cKO HSCs.
(D) Mitochondrial mass of different HSPC population was evaluated by Mitotracker Green staining quantified by flow cytometry. n = 5.
(E) Scheme of transplant strategy. Non-competitive reconstitution assay in which 106 donor BM cells from Mettl3 f/f or Mettl3 cKO mice at 3 weeks post-plpC were transplanted into CD45.1 congenic recipient mice.
(F) Mettl3 cKO bone marrow fails to reconstitute hematopoietic compartments in recipient mice. CD45.2 chimerisms of HSCs, MPPs and progenitor compartments from (E) were analyzed by flow cytometry at 16 weeks post-transplant. n = 10.
(G) Scheme of the experimental procedure in (H) and (I). CD45.1 recipient mice were transplanted with Mettl3 flox/flox Cre− or Cre+ bone marrow cells (pre-plpC). At 6 weeks post-transplantation, CD45.1 congenic recipient mice were then injected with plpC to deplete METTL3.
(H) The reconstitution defect in Mettl3 cKO bone marrow is cell-autonomous. CD45.2 chimerism analysis of HSCs, MPPs, and progenitor compartments from experiment (G) at 3 weeks post-pIpC. n = 5.
(I) Frequencies of HSCs and MPPs in the donor CD45.2 population from experiment (G). n = 5. Mean and SEM are shown (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). n represents number of mice.
To determine if the expansion of Mettl3-depleted HSCs and their defective ability to reconstitute the hematopoietic system was cell autonomous and not due to loss of RNA methylation in the heterogeneous niche environment, we engrafted bone marrow cells from both Mettl3 f/f, Mx1-Cre− and Mettl3 f/f, Mx1-Cre+ mice (Figure 2G), and then depleted Mettl3 following engraftment through pIpC administration. Donor reconstitution was comparable between Mettl3 f/f, Mx1-Cre− and Mettl3 f/f, Mx1-Cre+ mice before pIpC treatment (Figure S2M). However, the chimerism of donor engraftment was significantly decreased in the HSPC compartments and in mature lineages 3 weeks after METTL3 depletion (Figures 2H and S2N). Importantly, the expansion of HSCs following METTL3 depletion was recapitulated within the donor population (Figure 2I). These results strongly supported a cell-intrinsic role of METTL3 and m6A in regulating HSC numbers and function.
Next, we sought to determine if alteration in function of Mettl3 cKO HSCs is sustained after long-term engraftment (16 weeks post-pIpC), in which MPPs and other differentiated cell types are replenished from their precursors. Similar to acute deletion, the METTL3-depleted HSPCs remained expanded (HSC, MPP2, and MPP4) and demonstrated reduced RBCs in the primary Mettl3 cKO mice (Figures S3A–S3C). However, defects in downstream lineages were normalized in Mettl3 cKO mice, as characterized by normal bone marrow cellularity, WBCs, and mature lineages (Figures S3D–S3H). Splenomegaly was also observed in Mettl3 cKO mice with increased immature erythroblasts, even though the reductions of B and T cells were recovered (Figures S3I–S3K). These data were explained by genomic PCR analysis of bone marrow cells, demonstrating an emergence of a WT Mettl3 population (Figure S3L). Furthermore, these data suggest a strong selective pressure against the loss of RNA methylation during myeloid and lymphoid lineage restriction and the sustained increase in the numbers of METTL3-depleted HSCs suggested a continuous block in the ability of HSCs to differentiate to more committed lineage restricted cells.
Acute Depletion of Mettl3 Leads to a Reversible Defect in HSC Repopulating Capacity
Targeting METTL3 is a potential therapeutic strategy in acute myeloid leukemia (AML) (Barbieri et al., 2017; Vu et al., 2017a). Prolonged genetic ablation of METTL3 disrupts adult hematopoiesis in vivo in a dose-dependent manner. However, it remains unclear whether the effects of METTL3 inhibition on normal HSCs are reversible. Thus, we examined the effect of acute Mettl3 inhibition on hematopoiesis by transient small interfering RNA (siRNA) knockdown (Figure 3A). Sorted LSK cells were transfected with control or Mettl3 siRNA and then injected into recipient mice (Figure 3B). Pan hematopoietic donor reconstitution was significantly but modestly reduced in the Mettl3 knockdown group at a short-term time point (4 weeks) (Figure 3C). However, the engraftment defect induced by acute Mettl3 loss was normalized in the HSPC compartments at 12 weeks post-transplant (Figure 3D). The chimerism of donor engraftment of the Mettl3 knockdown group was also comparable to the control group in myeloid, T cell, and megakaryocytes lineages with modest reductions on the erythroid and B cell compartment (Figure 3E). These data suggest that acute inhibition of Mettl3 could induce a reversible defect in normal hematopoiesis. Together with the modest hematopoietic defect observed in Mettl3 heterozygous mice, these results highlight a wide therapeutic window to target METTL3 in AML.
Figure 3. Acute Depletion of Mettl3 Leads to a Reversible Defect in HSC Repopulating Capacity.

(A) Experimental scheme for acute knockdown of Mettl3 in LSK cells by siRNA followed by transplantation.
(B) qRT-PCR of Mettl3 to confirm the knockdown efficiency in LSK cells from (A).
(C) Donor engraftments ofCD45.2 in different lineage compartments were analyzed by flow cytometry in peripheral blood at 4 weeks post-transplant from (A). ctrl n = 10; siMettl3 n = 5.
(D) CD45.2 chimerism analysis of HSCs, MPPs, and progenitor compartments in bone marrow from experiment (A) at 12 weeks post-transplant. ctrl n = 10; siMettl3 n = 5.
(E) CD45.2 chimerism analysis of different lineage populations in bone marrow from experiment (A) at 12weeks post-transplant. ctrl n = 10; siMettl3 n = 5. Mean and SEM are shown (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). n represents number of mice.
Single-Cell Profiling Identifies Additional Cell Populations in Mettl3 cKO Mice
To decipher the effect of Mettl3 deletion on lineage commitment, identity, and priming of the hematopoietic hierarchy, we performed single-cell RNA-seq (scRNA-seq) using 6000 sorted LK cells (lineage−/cKit+ cells) from three independent control Mettl3 f/f and Mettl3 cKO mice (Figures S4A–S4C), at 3 weeks post-deletion. Single-cell t-Distributed Stochastic Neighbor Embedding (tSNE) analysis of Mettl3 f/f LK cells revealed the expected stem and progenitor clusters, including HSC, MPP, and erythroid, myeloid, and lymphoid progenitors, described previously in scRNA-seq studies (Giladi et al., 2018; Jaitin et al., 2014; Paul et al., 2015) (Figures 4A and S4D; Table S1). However, in Mettl3 cKO, the frequencies of basophils (Ba), megakaryocyte-erythroid progenitors (MEPs), megakaryocyte primed populations, and HSC clusters were significantly reduced (Figures 4A–4C). These data suggest that the reduction in myeloid and erythroid mature lineages is already manifested in lineage primed progenitors.
Figure 4. Mettl3 Deletion Results in a Defect in Lineage Commitment and the Development of Two Distinct HSC-like Populations.

(A) Identification of different hematopoietic clusters in Mettl3 f/f and Mettl3 cKO Lineage−cKit+ cells base on tSNE analysis from slngle-cell RNA sequencing (scRNA-seq).
(B) Two specific clusters in Mettl3 cKO Lin−cKit+ cells. tSNEs analysis of scRNA-seq. KO-specific clusters were labeled as red. n = 3.
(C) Quantification of cell frequencies of different populations in cKO Lin−cKit+ cells compared to Mettl3 f/f base on scRNA-seq.
(D) Specific clusters in Mettl3 cKO are HSC-like. Maximum likelihood of the KO-specific cells into the WT expression models was performed base on the transcriptome profile from scRNA-seq.
(E) Heatmap of differentially expressed genes between HSC cluster and KO SP clusters from scRNA-seq.
(F) Pseudo-time reconstruction of the hierarchy of cell differentiation by Monocle analysis base on scRNA-seq. Left branch indicated HSC commitment to MPPs. Right branch indicated trajectory from HSC to Mettl3 cKO specific clusters. n represents number of mice.
Importantly, projection of distinct cell types by tSNE revealed two populations in the Mettl3 cKO samples that were not present in control samples. The two Mettl3 cKO-specific populations (KOsp1 and KOsp2) accounted for nearly 30% of all immature LK cells in Mettl3 cKO bone marrow (Figures 4A–4C). To interrogate the identity of these cKO-specific clusters, we performed a maximum likelihood projection of KOsp1 and KOsp2 onto the annotated HSPC clusters in control mice. KOsp1 cells predominately clustered with HSCs (50%) and to a lesser degree with MPPs (25%). In contrast, the expression profile of KOsp2 cells was most similar to megakaryocyte progenitors (70%) and had a 20% similarity to HSCs (Figure 4D). A differential expression analysis comparing KOsp1 and KOsp2 cells to WT control HSCs demonstrated a higher expression of cell cycle genes, including Ki67 and Cdk1, and lower expression of key HSC self-renewal regulators and transcription factors, including Msi2, Hoxa9, Satb1, and Fos (Figures 4E and S4E). KOsp2 differed from normal megakaryocytic progenitors by their low expression of megakaryocyte lineage-regulating genes, including Pf4, Gp1bb, Myb, and Gata1, and increased expression of myeloid and lymphoid genes, including Cd63, Gimap1, and Gimap9 (Figure S4F). These gene expression changes are in line with the defects in HSC self-renewal and megakaryocyte maturation that are observed in these cells. These data indicated that the loss of METTL3 resulted in the emergence of actively dividing “HSC like” populations: one (KOsp1) with reduced self-renewal and the other (cKOsp2) acquiring lineage priming toward a megakaryocyte fate.
To better understand the origin of the cKO-specific clusters, we performed pseudo-time analysis and hierarchy reconstruction to capture early commitment decisions in HSCs. Both the KOsp clusters and normal MPP clusters were connected to the HSC cluster (Figure 4F). These data suggested that KOsp clusters represent an intermediate state of HSC that branches away from the normal HSC to MPP transition. While many Mettl3 cKO HSCs were still capable of normal HSC-to-MPP transition, KOsp1 and KOsp2 ascended from HSCs but the lack of m6A prevented them from properly committing to normal MPPs. These data strongly suggest that RNA methylation plays a key role in dictating cell identity and lineage commitment in HSCs.
Mettl3 cKO HSCs Lose HSC Identity and Function as MPPs
Since the single-cell sequencing data identified the HSC-like intermediate populations, we next want to delineate the relationship between immunophenotypic HSCs and MPPs and the molecularly defined HSC-like populations. We performed RNA-seq transcriptome profiling of classically defined HSCs and progenitor cells, i.e., HSCs, MPP1s, MPP2s, and MPP4s, whose frequencies were previously found to be altered (Figure S4A). Unsupervised clustering of the differentially expressed genes across all samples distinguished the Mettl3 cKO HSPC populations from the controls (by ANOVA test; padj < 0.01) (Figure S5A). The data confirmed that HSPCs from Mettl3 cKO mice are distinct from their Mettl3 f/f counterparts with a different transcriptome profile. We then examined enrichment of KOsp cluster-specific genes from scRNA-seq with the transcriptional profiles of Mettl3 f/f and Mettl3 cKO HSCs and MPPs in bulk RNA-seq. Gene set enrichment analysis (GSEA) analysis using the top expressed genes from each spleen (SP) cluster in scRNA-seq (KOsp1/KOsp2 gene sets) revealed that KOsp1 and KOsp2 cells were enriched with Mettl3 f/f immunophenotypic HSC signatures rather than MPPs (Figure S5B). Additionally, both KOsp1 and KOsp2 gene sets were enriched in Mettl3 cKO HSC cells compared to Mettl3 cKO MPPs (Figure S5C). These results indicated that despite the lineage priming features, the scRNA-seq cKO-specific populations still resemble the phenotypic HSCs. It further supports that ablation of m6A initially alters the cell fate decision of HSCs, and not downstream MPPs.
Next, we further evaluated the altered gene expression program within the HSPC compartment. We performed GSEA of 866 genes that are significantly upregulated in Mettl3 cKO HSCs (false discovery rate [FDR] < 0.05), compared with WT HSCs (Table S2). A gene set obtained from the Mettl3 KO mouse embryonic stem cells (mESCs) (Figures 5A, 5B, and S5D; http://amp.pharm.mssm.edu/Enrichr/) was the top-ranked gene set. These results suggest that loss of METTL3 in HSCs induces a gene expression program that is found in METTL3-deficient naive pluripotent ESCs. Interestingly, this was specific to HSCs, as this analysis in MPPs failed to match with METTL3-deficient ESCs. Thus, the analysis supported that Mettl3-deleted HSCs have a lineage priming defect that traps them in a more primitive state. The Rbm15 KO HSPC signature was also highly enriched in the Mettl3 cKO HSC gene expression program (Figures 5B and S5E). RBM15 is an RNA binding protein that recruits the m6A methyltransferase complex to target mRNAs (Patil et al., 2016). Moreover, the defects observed in Rbm15 KO mice closely match with many of the phenotypes observed post-METTL3 depletion, suggesting that the phenotypes that have been observed with RBM15 KO mice may be in part attributable to alteration in the m6A-dependent differentiation program (Patil et al., 2016). These data demonstrate an upregulation of a core m6A-dependent transcriptional program upon Mettl3 depletion in HSCs that is shared among models of RNA methylation depletion.
Figure 5. Mettl3-Deleted HSCs Are More Functionally and Molecularly MPP-like.

(A) Significant differentially expressed genes (padj < 0.05) in cKO HSCs compared to Mettl3 f/f were shown as heatmap.
(B) Differentially expressed genes in Mettl3 cKO HSCs were enriched with Mettl3 KO ESC and RBM15 KO LSK expression signatures. Enrichr analysis using upregulated genes in METTL3-depleted HSCs from bulk RNA-seq.
(C) Unbiased gene set enrichment analysis using 4,733 curated gene sets against the rank list of differential expressed genes between Mettl3 f/f and cKO HSCs. Self-renewal signature was negatively enriched in Mettl3 cKO HSCs. Translation and ribosome gene sets were positively enriched in Mettl3 cKO HSCs.
(D) Increased global translation in Mettl3 cKO HSCs. Representative histograms (top) and quantification (bottom) of OP-Puro incorporation into sorted Mettl3 f/f and cKO HSCs.
(E) Scheme of transplant strategy in (F). Sorted HSCs and MPP1s from Mettl3 f/f and Mettl3 cKO mice were injected into CD45.1 recipient mice with CD45.1 competitor BM cells.
(F) Mettl3 cKO HSCs function as MPP1s in competitive transplant assays. Engraftment of CD45.2 donor cells was analyzed in HSPCs from recipient mice at 6 weeks. n = 10.
(G) Engraftment of CD45.2 donor cells was analyzed in HSPCs from recipient mice at 40 weeks. n = 10.
(H) Spearman correlation among HSC and MPP1 populations from Mettl3 f/f and Mettl3 cKO mice.
(I) Gene set enrichment analysis of up-or downregulated genes in Mettl3 cKO HSC identified by bulk RNA-seq, against the rank list of differentially expressed genes between Mettl3f/f HSC and Mettl3f/f MPP1. Mean and SEM are shown (*p<0.05, **p<0.01, ***p<0.001, and ****p < 0.0001). n represents number of mice.
We next performed unbiased GSEA analysis on all 4,733 curated gene sets in the Molecular Signature Database (MSigDB), combined with 92 relevant gene sets from our experimentally derived or published hematopoietic self-renewal and differentiation signatures using HSC and MPP transcriptome profiles from Mettl3 f/f and cKO mice. We observed a negative enrichment of HSC self-renewal signature and a positive enrichment of ribosome and protein translation gene sets in the Mettl3 cKO HSC gene expression profile (Figure 5C). Moreover, protein translation-related ribosome signatures were also enriched in Mettl3 f/f MPPs compared to Mettl3 f/f HSCs (Figure S5F). We confirmed the increased protein synthesis in Mettl3 cKO HSCs based on O-Propargyl-puromycin (OP-Puro) (Figure 5D). These data suggest that there is an alteration in identity in Mettl3 cKO HSCs.
As our molecular profiling strongly suggests alterations in cell identity and commitment in METTL3-depleted HSCs, we examined whether these HSCs are functionally defective. To test whether there is an altered identity in Mettl3 cKO HSCs, we performed competitive transplantation using sorted HSCs and MPP1s from Mettl3 f/f and cKO mice (Figure 5E). As observed previously with total bone marrow cells, Mettl3 cKO HSCs and MPP1s had low reconstitution within all peripheral cellular compartments (Figure S5G). Within the bone marrow, the control HSCs engrafted normally and chimerism could be measured in all HSPC compartments: HSCs, MPP1s, MPP2s, and MPP4s. The control MPP1s also correctly reconstituted the MPP1 compartment, thus validating our sorting strategy in this functional assay (Figures 5F and S5H). However, the Mettl3 cKO HSCs demonstrated chimerism within the phenotypic MPP1 compartment but not within the HSC compartment (Figure 5F). Additionally, these transplanted HSCs in the MPP compartment behaved as MPP1s, as they were no longer detected at long-term analysis (40 weeks post-transplant) (Figures 5G and S5I). This altered fate was also validated by Spearman correlation analysis using gene expression profiles of Mettl3 f/f and Mettl3 cKO HSCs and MPPIs. Gene expressions between cKO HSCs and control HSCs were less correlated, and the cKO HSCs’ gene profile correlated similarly to control HSCs or MPPIs (Figure 5H). Furthermore, GSEA analysis also showed that Mettl3 cKO HSCs were indeed enriched for Mettl3 f/f MPP1 and MPP2 gene expression signatures (Figures 5I and S5J). These data indicated that m6A-ablated HSCs are more molecularly and functionally similar to MPP1 cells.
m6A Regulates HSC Asymmetric and Symmetric Division by Modulating Myc mRNA
We next sought to understand the molecular basis for abnormal cell fate decision and differentiation upon loss of m6A in HSCs. Given that m6A ablation traps HSCs in a more primitive state during HSC-to-MPP transition, we hypothesized that genes essential for this transition would differentially increase their m6A abundance in MPPs compared to HSCs. Thus, we reanalyzed previously obtained m6A profiles in HSCs and MPPs and selected genes with higher m6A marks in MPPs (Li et al., 2018). To identify m6Aand METTL3 direct targets, we then intersected the list of increased m6A methylated mRNAs in MPPs with the differentially expressed genes in HSCs upon Mettl3 deletion, resulting in eight genes (Figures 6A and S6A; Table S3). MYC was among these genes known to be a critical target of mRNA methylation (Vu et al., 2017a). Moreover, Mettl3 deletion partially phenocopied the expanded HSPC compartment found in the Myc-deficient mice (Bahr et al., 2018; Wilson et al., 2004). To further determine if MYC is a direct functional target of m6A in HSCs, we performed methylated RNA immune-precipitation (MeRIP)-qPCR and found reduced enrichment of m6A in Myc transcripts in Mettl3 cKO HSPCs (Figure 6B). Overall, these data suggest that Myc could be a functional downstream target of m6A and METTL3 in HSCs.
Figure 6. Mettl3 Is Required for HSC Symmetric Commitment by Regulating mRNA Stability of Myc.

(A) Overlap of the 52 genes that have increased m6A abundance in MPPs than HSCs with differentially expressed genes between Mettl3 f/f and cKO HSCs, as depicted in a Venn diagram. Myc is highlighted in red.
(B) m6A enrichment in the Myc transcript in Mettl3 f/f and cKO HSPCs was assessed by MeRIP-qPCR. Two primers were used to indicate different m6A sites on Myc RNA.
(C) MYC protein level is reduced in Mettl3 cKO HSCs upon cell division. MYC expression in Mettl3 cKO HSCs compared to Mettl3 f/f HSCs as quantified by immunofluorescence.
(D) Mettl3 cKO HSCs fail to undergo symmetric commitment compared to control HSCs. Left: representative immunofluorescence images of paired daughter cells stained with DAPI (blue), NUMB (green), MYC (red), and brightfield. Right: percentages of doublet cells in each type of cell division. Number of daughter pairs assessed: Mettl3 f/f n = 223; cKO n = 245. n represents number of paired HSCs measured.
(E) MYC expression correlates with NUMB expression during HSC division.
(F) Myc mRNA can be segregated into daughter cells asymmetrically, symmetrically high, and symmetrically low. Representative images of fluorescence in situ hybridization (FISH) of Myc mRNA (green) and fluorescence immunostaining of NUMB (red) and DAPI (blue) in HSCs.
(G) Myc mRNA is significantly decreased in Mettl3 cKO HSCs. Representative images of FISH of Myc mRNA (green) and DAPI (blue) in Mettl3 f/f and Mettl3 cKO HSCs.
(H) Quantification of Myc mRNA in HSCs from Mettl3 f/f and Mettl3 cKO mice.
(I) The mRNA half-life (t1/2) of Myc transcripts in Mettl3 f/f and Mettl3 cKO HSCs. Mean and SEM are shown (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).
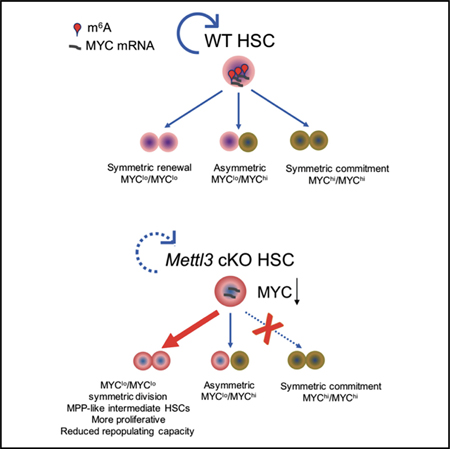
To directly assess HSC fate commitment and to evaluate MYC expression after Mettl3 deletion, we performed paired daughter cell division assays based on intracellular staining of NUMB and MYC proteins. Previous studies showed that HSCs can undergo three fate choices: symmetric renewal, symmetric commitment, and asymmetric cell division. Cellular commitment toward a differentiation cell fate is measured by increased NUMB staining in the post-dividing HSCs (Kharas et al., 2010; Park et al., 2014; Wu et al., 2007). Mettl3 deletion results in a significant reduction in symmetric commitment (25% to 7%; multiple t test; p = 0.000287) with an increase in symmetric renewal division and no effect on asymmetric cell division (Figures 6C and 6D). Surprisingly, we observed that MYC can be segregated in the dividing HSCs asymmetrically, symmetrically “low,” and symmetrically “high” in a manner correlated closely with the pattern of NUMB (Figures 6D and 6E). This indicated that MYC abundance could also be used as a marker for cellular commitment (Kharas et al., 2010; Park et al., 2014; Wu et al., 2007). As observed in NUMB staining, we found fewer daughter cells with high MYC staining (20% to 7%; multiple t test; p = 0.0307) (Figure 6D). These data suggest that METTL3 controlled the abundance of MYC in HSCs at a decision point that dictates the choice between symmetric self-renewal versus symmetric commitment.
To determine if asymmetric segregation of MYC was already determined at the level of mRNA, we performed fluorescence in situ hybridization (FISH) of Myc mRNA and fluorescence immunostaining of NUMB. Interestingly, Myc mRNA also can be segregated to daughter cells asymmetrically, symmetrically “low,” and symmetrically “high” in a manner correlating to NUMB and MYC (Figure 6F). Furthermore, consistent with our scRNA-seq and RNA-seq results, the Myc mRNA was dramatically reduced in Mettl3 cKO HSCs (Figures 6G and 6H). These data suggest that m6A RNA methylation controls Myc mRNA abundance that is required for HSC commitment. Given that m6A modification affects mRNA stability, we hypothesized that the reduced Myc mRNA level in Mettl3 cKO HSCs is due to decreased Myc mRNA stability. Indeed, the half-time of Myc mRNA was significantly decreased in Mettl3 cKO HSCs (Figures 6I and S6B). Reintroduction of MYC in Mettl3 cKO HSCs partially rescued the HSC commitment defect (Figure S6C and S6D). Overall, these data suggest that m6A controls HSC symmetric commitment through MYC regulation.
Next, to determine whether the failure of HSCs to properly differentiate is functionally driven by reduced MYC abundance during cell division, we overexpressed MYC in sorted Mettl3 cKO LSK cells and performed competitive bone marrow transplants. MYC overexpression resulted in a rescue in the peripheral blood and bone marrow HSPC compartment chimerism in mice transplanted with Mettl3 cKO HSPCs (Figures 7A–7C, S7A, and S7B). Furthermore, this increased chimerism could be observed within the HSC compartments in 5 out of the 10 mice (Figure S7B). A lethal myeloproliferative and AML-like disease was observed in the MYC overexpression group as previously reported (Figure S7C) (Luo et al., 2005), suggesting that highly expressed MYC could bypass the requirement for METTL3 in myeloid leukemia. These in vitro and in vivo data suggest that restoring MYC can at least partially override defects in HSC commitment and drive normal HSC differentiation.
Figure 7. MYC Overexpression Can Partially Rescue Reconstitution Defect in Mettl3 cKO HSPCs.

(A) Scheme of experiment strategy. LSK cells were sorted from Mettl3 f/f and Mettl3 cKO mice and transduced with control and MYC overexpression retrovirus. Donor cells were then transplanted into CD45.1 recipient mice with CD45.1 competitor BM cells.
(B) Quantification of the frequency of donor-derived cells was shown in LSK and MP populations, n = 9. n represents number of mice.
(C) MYC overexpression rescues the repopulating defect of Mettl3 cKO LSKs. Representative flow cytometry plots of engraftment of donor-derived CD45.2 cells.
(D) METTL3 overexpression rescue MYC expression defect in Mettl3 cKO HSCs as quantified by immunofluorescence.
(E and F) Paired daughter cell assay using sorted HSCs from Mettl3 f/f and Mettl3 cKO mice transfected with vectors expressing METTL3 or METTL3-CD or empty vector as control to indicate MYC (E) and NUMB (F) expression. Pie chart shows different types of cell divisions in HSCs as indicated. Number of daughter pairs assessed: Mettl3 f/f +Vec, n = 141; cKO+Vec, n = 35; cKO+METTL3 WT, n = 45; and cKO+METTL3 CD, n = 65.n represents number of paired HSCs measured. Mean and SEM are shown (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).
Altogether, these data suggest that MYC mRNA and protein is a marker for commitment and m6A-deficient HSCs are partially blocked in their ability to differentiate.
METTL3 Catalytic Function Controls MYC Abundance and HSC Commitment In Vivo
As METTL3 may have additional functions beyond its methyl-transferase activity, we then assessed if METTL3’s catalytic function is directly responsible for alterations in MYC abundance and HSC commitment. To this end, we overexpressed METTL3 or a catalytically dead METTL3 mutant (METTL3-CD) and found that it rescued the reduced overall MYC abundance in either Mettl3-siRNA transfected HSCs or in cKO HSCs (Figures 7D and S7D). Additionally, overexpression of METTL3, but not METTL3-CD, partially rescued HSC symmetric commitment in the paired daughter cell division assay and Mettl3 cKO HSC engraftment (Figures 7E, 7F, and S7E). Moreover, METTL3, but not METTL3-CD, significantly enhanced normal HSPC reconstitution capacity in vivo, suggesting that increased methyltransferase activity can enhance HSPC engraftment (Figure S7F). Overall, these data suggest that METTL3 catalytic activity is essential for MYC regulation, HSC commitment, and HSC function.
DISCUSSION
Here, by integrating transcriptome profiling with single-cell RNA sequencing and single-cell assays, we show that m6Ais required for the early cell fate decision in adult HSCs. Our studies find that m6A controls HSC identity by affecting the symmetric commitment step, while asymmetric divisions are less affected. m6A is a known regulator of Myc expression in HSCs. We show that Myc functions to promote HSC symmetric commitment, and its loss in m6A-deficient cells results in the formation of a cell type with MPP-like identity, rather than an accumulation in the HSC population, as is currently thought in the field (Lee et al., 2019; Yao et al., 2018). The current thinking about m6Ain normal hematopoiesis is that m6A controls HSC differentiation without affecting self-renewal or proliferation. Here, we show that the accumulated immunophenotypic “HSCs” that were identified in previous studies are instead a blocked HSC-MPP-like cell population. These cells behave as MPP-like cells as they exhaust and thus have reduced self-renewal in vivo. These HSC-like intermediate cells are also metabolically active and highly proliferative, but they fail to symmetrically commit and differentiate. Our study shows that m6A controls cell identity by regulating the symmetric commitment of HSCs during division at the early cell fate determination point. These intermediate populations could also be used to further understand the relationship between HSC expansion and commitment toward MPPs.
Interestingly, while m6A reader YTHDF2-deficient mice also exhibited increased HSCs number, Ythdf2 knockout HSCs demonstrated improved functional and regenerative capacity in contrast to what we observed in Mettl3-deficient HSCs (Li et al., 2018). These data suggest that YTHDF2 depletion may promote HSC self-renewal without compromising commitment, resulting in functional HSC expansion.
MYC was reported as a target of m6A in regulating myeloid leukemia and HSCs (Lee et al., 2019; Vu et al., 2017a). However, it is unclear how m6A controls MYC and in which step MYC affects HSCs. We identified MYC as a functional target of m6A that facilitates the transition from HSCs to MPPs during the early steps of cell fate determination and its RNA stability is controlled by m6A. Surprisingly, we demonstrated that MYC mRNA and protein could be segregated either asymmetrically or symmetrically during HSC division. An asymmetric partition of MYC was observed during T cell activation (Pollizzi et al., 2016; Verbist et al., 2016). Thus, the insight of m6A in controlling symmetric commitment by modulating MYC could be a general mechanism for m6A regulation of cell fate. Several known m6A-dependent cell fate regulations are consistent with our model. For example, in mouse ESCs and in naive T cells, loss of m6A locks the cells in its naive state and prevents differentiation (Geula et al., 2015; Li et al., 2017).
We previously reported that depletion of METTL3 in human AML cells results in increased MYC mRNA abundance and reduced translational efficiency, which finally leads to decreased MYC protein (Vu et al., 2017a). In contrast, METTL3 depletion in normal murine HSCs results in a decrease in Myc mRNA and protein levels, suggesting a cellular-context-dependent regulation of m6A targets or differential effects after transient versus prolonged depletion of m6A.
Previous studies revealed that m6A is required for leukemogenesis and AML cell survival, prompting the search for inhibitors to block this pathway as a potential therapeutic strategy in myeloid malignancies and cancer (Barbieri et al., 2017; Vu et al., 2017a; Weng et al., 2018). Our work describes the consequences of METTL3 depletion in the blood system and indicates the importance of this pathway in normal hematopoietic development at various stages including in more mature lineages. Importantly, our data also demonstrate that transient siRNA depletion of METTL3 results in reversible and modest effects on hematopoiesis. However, the true therapeutic window for METTL3 inhibitors will only be known once potent and selective inhibitors are developed.
Overall, our findings demonstrate that m6A is essential for the maintenance of HSC identity and symmetric commitment. As the control of symmetric commitment in adult stem cells is critical for rapidly replenishing tissue compartments after stress or damage, our study highlights the role of RNA methylation in adult stem cell commitment and fate determination.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Michael G Kharas (kharasm@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mettl3 flox/flox mice were generated by inserting loxP sites spanning the fourth exon of Mettl3 via homologous recombination base on construction taken from Knockout Mouse Project Repository (KOMP). We next crossed Mettl3 flox/flox mice with Mx1-Cre mice to obtain induced hematopoietic conditional knockout mice. Deletion of Mettl3 was initiated using pIpC HMW (InVivogen, Vac-Pic) by intraperitoneal injections at a dose of 10 mg/kg on 2 consecutive days as showed previously (Liu et al., 2015). Mice used for this experiment were 8 to 12 weeks of age. Both male and female mice were used. All mice were housed on a 12:12 hr light:dark cycle at 25°Cand received water and chow ad libitum. All of the animal experiments were approved under the Institutional Animal Care and Use Committee.
METHOD DETAILS
Noncompetitive Transplant
Noncompetitive transplants were performed as previously described(Park et al., 2014). Briefly, in the primary transplant, 106 whole bone marrow cells from Mettl3 cKO mice or littermate Mettl3 f/f mice (3 weeks post pIpC) were injected into lethally irradiated B6SJL congenic CD45.1 recipients. In the cell-autonomous transplants, we transplant 106 whole BONE MARROW cells from 8–12 weeks old Mettl3 flox/flox, Mx1-Cre+ or Mx1-Cre-mice into lethally irradiated B6SJL congenic CD45.1 recipients. After 6 weeks of engraftment, these transplanted mice were treated with pIpC as shown previously. All recipient mice used were 6–8 weeks old. All animals were randomly assigned to the experimental groups. For chimerism, either peripheral blood or bone marrow cells was extracted and subjected to flow cytometry at different time points.
PCR and Quantitative RT-PCR
Genomic DNA from Mettl3 f/f and Mettl3 cKO whole bone marrow cells was isolated by using genomic DNA extraction kit (QIAGEN) and used as template for PCR to genotype. Primers are Forward: ACCACAACAGCCAAGGAACA; Reverse: CCGGAGCTCT GAAACCTTGT. For qRT-PCR, total RNA was extracted from cells using TRIZOL (Life Technologies) following the standard manual. Equal amount of RNA from samples was reverse transcribed into cDNA with Verso cDNA Synthesis Kit (Thermo), and qPCR was performed using an ABI7500 sequence detection system using primers together with SYBR green master mix (ABI systems). Primers are listed below:
Myc: Forward: GCTGTTTGAAGGCTGGATTTC
Reverse: GATGAAATAGGGCTGTACGGAG
Mettl3: Forward: AAGGAGCCGGCTAAGAAGTC
Reverse: TCACTGGCTTTCATGCACTC
Actin: Forward: ACCAACTGGGACGACATGGAGAAG
Reverse: TACGACCAGAGGCATACAGGGACA
Flow Cytometry and Cell Sorting
Bone marrow or spleen cells were harvested and subjected to red blood cell lysis. To measure HSPC compartments, cells were stained with following cocktail: Linage marker (CD3 (15–0031-83), CD4(15–0041-83), CD8(15–0081-83), Gr1(15–5931-82), B220(15–0452-83), CD19(15–0193-83) and Ter119(15–5921-82))-PE-Cy5(Invitrogen, eBioscience), cKit-APC-Cy7 (BioLegend, 105826), Sca-1-Pacific Blue (BioLegend, 122520), CD150-APC (BioLegend, 115910), CD48-PE (BD Bioscience, 557485), CD34-FITC (eBioscience, 11–0341-85), CD16/32-PE-Cy7 (BD Bioscience, 560829). To monitor linage cells differentiation cells were stained with cocktail including Gr1-APC (Invitrogen, 17–5931-82), Mac1-Pacific Blue (BioLegend, 101224), Ter119-PE-Cy5 (Invitrogen, 15–5921-82), CD71-FITC (Invitrogen, 11–0711-81), CD41-PE (BD Bioscience, 558040) or cocktail containing CD3-Pacific Blue (eBioscience, 48–0031-80), CD4-PE (BD Bioscience, 557308), B220-PE-Cy7 (BD Bioscience, 552772), CD19-PE-Cy5 (Invitrogen, 15–0193-83), IgM-APC (BioLegend, 406509), CD43-FITC (BD Bioscience, 553270). For transplanted mice, we added CD45.1-PE-Txase Red (BD Bioscience, 562452) and CD45.2-A700 (Invitrogen, 56–0454-82) to distinguish donor and recipient cells. For cell cycle analyses, Ki67/Hoechst staining was performed according to manufacturer’s recommendation (BD Biosciences). For PY/Hoechst staining, bone marrow cells stained with HSC surface markers (Linage marker (CD3, CD4, CD8, Gr1, B220, CD19 and Ter119)-PE-Cy5, cKit-APC-Cy7, Sca-1-PECy7 (BioLegend, 122514), CD150-APC, CD48-FITC (BioLegend, 103404)) were resuspended in 500ul NASS buffer (1 phosphate citrate tablet (sigma), 0.9g EDTA, 0.45 g NaCl, 0.25 g BSA in 50ml ddH2O), 3.3ul diluted Hoechst (1:10 in NASS) was added followed by 30mins in room temperature. Samples were then incubated on ice for 5 mins to stop reaction. 2.5ul of diluted PY (1:100 of 10mg/ml PY stock in NASS) was added to samples. Incubate for 10mins at 4 degree. Wash the samples before analysis. For mitochondrial mass and activity, BMCs were incubated with 100nM MitoTracker Green (Invitrogen) or 50nM DilC5 (Thermo Fisher) for 30 min at 37°C in the dark after cell surface staining (Linage marker (CD3, CD4, CD8, Gr1, B220, CD19 and Ter119)-PE-Cy5, cKit-APC-Cy7, Sca-1-Pacific Blue, CD150-APC/CD150 FITC, CD48-PE). Cells were analyzed on a BD FACS LSR or Fortessa instrument. For HSPC(LSK) or HSC (LSK, CD150+CD48-) cell sorting, bone marrow cells were harvested and incubated with MACS beads (CD117, Miltenyi Biotec, 130–091-224). Then enriched c-Kit+ cells were collected by running AutoMACS (Miltenyi Biotec) according to the manufacturer’s instructions. The cells were then stained with cocktail: Linage marker (CD3, CD4, CD8, Gr1, B220, CD19 and Ter119)-PE-Cy5, cKit-APC-Cy7, Sca-1-Pacific Blue, CD150-APC, CD48-PE. Specific cell population was sorted on BD Aria machine.
Peripheral Blood Analysis
Peripheral blood was collected from the retroorbital cavity using a heparinized glass capillary tube. Complete peripheral blood count analysis including a differential blood count was obtained by using Hemavet (Drew Scientific). For flow cytometry, peripheral blood was treated by red blood cell lysis to remove red blood cells and then applied to flow as showed earlier.
Western-Blot
LSK cells were sorted from Mettl3 f/f and Mettl3 cKO bone marrow cells following protocol described before. 50,000 cells were then lysed in 30 μL1 × Laemmli protein loading buffer and boiled for 5 min. Whole-cell lysates were run on a 4%–15% gradient SDS–PAGE and transferred to a nitrocellulose membrane. Membranes were probed with the METTL3 (Proteintech, 150731–1-AP) and ACTIN (Sigma, A3854) antibody.
Determination of Relative m6A Levels
Relative levels of global m6A was determined by thin-layer chromatography (TLC), as described previously(Vu et al., 2017a). Briefly, poly(A)-purified RNA was digested with 2 U ribonuclease T1 for 2 h at 37°C in the presence of RNaseOUT (Invitrogen) first. Next, we labeled digested RNA at 5′ with [γ−32P] ATP. After removal of γ phosphate of ATP by apyrase (NEB), labeled RNA was purified by phenol-chloroform extraction and ethanol precipitation and digested to single nucleotides nuclease. 1 mL of released 5′ monophosphates was analyzed on glass-backed PEI-cellulose plates (Merck-Millipore) and exposed to a storage phosphor screen (GE Healthcare Life Sciences), as described previously. The relative m6A was calculated as a percentage of the total of A, C and U spots which is quantified with ImageJ, as described previously (Mauer et al., 2017).
Colony Assay
10,000 whole Mettl3 f/f or Mettl3 cKO bone marrow cells were plated in in M3434 methylcellulose media (STEMCELL Technologies, 03434), and colonies were scored at 10 days post plating, respectively.
Competitive Transplant
HSC and MPP1 cells from Mettl3 f/f or Mettl3 cKO mice were sorted following protocol described above. 500 sorted HSC and MPP1 cells plus 500K CD45.1 whole bone marrow cells as support cells were injected into lethally irradiated B6SJL congenic CD45.1 recipients directly after cell sorting. For chimerism, either peripheral blood or bone marrow cells was extracted and subjected to flow cytometry at different time points.
HSPC Cell Sorting, Cell Culture, Retroviral Infection, and Transplant
HSPC(LSK) cells from Mettl3 f/f or Mettl3 cKO mice were sorted following protocol described above. Sorted LSK cells were cultured in the SFEM medium (STEMCELL Technologies, NC9753895) supplemented with murine cytokines (50 ng/ml SCF, 10 ng/ml IL-3, and 10 ng/ml IL-6,10ng/ml TPO and 20 ng/ml FLT3L, PeproTec). Cells were then transduced with high-titer concentrated retroviral suspensions in the presence of 4 μg/ml polybrene and followed with spin infection for 1.5 hr. Next day, second round of transduction was performed as described. For transplant, 10,000 LSK cells plus500KCD45.1 whole bone marrow cells were injected into lethally irradiated B6SJL congenic CD45.1 recipients. For chimerism, either peripheral blood or bone marrow cells was extracted and subjected to flow cytometry at different time points.
O-Propargyl-puromycin (OP-Puro) Flow Analysis
Protein synthesis rate was assessed by O-Propargyl-puromycin (OP-Puro) as described previously(Vu et al., 2017b). Mettl3 f/f and Mettl3 cKO HSC cells were sorted and treated with 50 μM O-propargyl-puromycin (OP-Puro; NU-931–05, Jena Bioscience) for 1 hour. Cells were washed once, collected, and then subjected to processing using the Click-iT Flow Cytometry Assay kit (C10418, Invitrogen) following the manufacturer’s instructions. Labeled cells were analyzed using a BD Fortessa instrument.
Immunofluorescence
HSCs were sorted from Mettl3 f/f and Mettl3 cKO mice following protocol shown before. We cultured the sorted HSCs with SFEM media (STEMCELL Technologies, NC9753895) containing 10 ng/ml heparin, 10 ng/ml SCF, 20 ng/ml TPO, 20 ng/ml IGFII, and 10 ng/ml FGF, PeproTec) in 96 round-bottom wells for 16 h and then treated cells with 10 nM Nocodazole for 24 h. After incubation, cells were fixed with 4% paraformaldehyde 15 mins at room temperature and permeabilized with cold methanol. Fixed HSCs were then cytospun onto glass slides and were blocked for 1h followed with staining on slides with anti-NUMB (Abcam, ab4147), anti-MYC (Cell Signaling, 5605) and secondary Ab (donkey anti–goat Alexa fluor 488, Invitrogen, A11055; donkey anti–rabbit Alexa fluor 568, Invitrogen, A10042; Molecular Probes) with DAPI counterstaining. We evaluate symmetric and asymmetric percentages based on the fluorescence signal intensity of each cell acquired by Axio Imager M2 microscope (Carl Zeiss) and quantified by FIJI. Thresholds to determine NUMB high/low/asymmetric were set for experimental replicates. Briefly, both of daughter cells with high NUMB staining or average staining intensity is above NUMB high threshold and there is less than 2-fold difference in the daughter pairs, this condition was counted as symmetric commitment. Daughter pair cells were scored as a symmetric renewal division when both of them were low or no staining or average staining intensity is below NUMB high threshold and there is less than 2-fold difference in the daughter pairs. If else, the division was considered an asymmetric division(Kharas et al., 2010).
RNA-FISH in Conjugation with Fluorescent Immunostaining
Sorted HSCs were cultured in SFEM media and treated with Nocodazole as shown above. HSC cells were then fixed with 4% paraformaldehyde and permeabilized with cold methanol. Fixed HSCs were then cytospun onto glass slides. RNA in situ hybridization was performed using RNAscope multiplex fluorescent detection kit according to the manufacturer’s instructions (Advanced Cell Diagnostics). RNAscope probes targeting mouse Myc was designed and produced by ACDbio. After the in situ hybridization was completed, slides were washed twice and subjected to immunostaining as shown above.
RNA Stability Assay
Sorted HSCs were cultured in SFEM media supplemented with cytokines as shown above. After overnight culture, HSCs were treated with 5 μM actinomycin D (Sigma) for inhibition of mRNA transcription. Cells were collected at 0 min, 10 min, 20 min or 40 min post treatment, and total RNA was extracted and used for qRT-PCR.
Electroporation Transfection
Neon nucleofection of HSPCs was performed asdescribed(Gundry etal., 2016). HSCs were sorted and cultured in the SFEM medium for 1–3 hours before transfection. 1ug Plasmids or 100pmol siRNAs were electroporated into 100,000 HSCs or LSKs by using Neon transfection system (Thermo Fisher Scientific) under a condition:1700V, 20ms, 1 pulse. If higher numbers of cells were needed, multiple electroporations were performed (e.g., 6 electroporations of 100,000 LSKs for 600,000 total cells).
m6A-seq Peak Enrichment Analysis
Previously published data of m6A-seq and input RNA-seq from mouse LT-HSCs, ST-HSCs, and MPPs were downloaded from NCBI GEO (GSE107957). PCR duplicates were removed using the pyDuplicateRemover.py script of the pyCRAC tool suite (version 1.2.2.3). Deduplicated sequences were then aligned to the mm10 mouse genome build using bowtie2 (version 2.3.4.2) (bowtie2-x mm10-N 0–trim5 3). Significantly enriched m6A peaks were determined using the R/Bioconductor package exomePeak.
MeRIP-qPCR
MeRIP-qPCR was performed as described previously(Vu et al., 2017a). Briefly, total RNA was isolated from sorted LSKs cells (from 3 Mettl3 cKO or control mice) by Trizol and poly(A)+ RNA was isolated using Dynabeads Oligo-(dT)25 magnetic beads (ThermoFisher Scientific). 5 μg anti-m6A antibody (Abcam, ab151230) was pre-bound to Protein A/G magnetic beads (Pierce) in IP buffer (20-mM Tris pH 7.5, 140-mM NaCl, 1% NP-40, 2-mM EDTA) for 1h. 1 μg Poly(A)+ RNA was mixed with 400 μl of IP buffer and added to the Protein A/G beads for 2 hours at 4°C. Samples were washed twice in low-salt-wash buffer (10-mM Tris pH 7.5, 5 mM EDTA), twice with high-salt-wash buffer (20-mM Tris pH 7.5,1-M NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1-mM EDTA) and twice with RIPA buffer (20-mM Tris pH 7.5, 150-mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1-mM EDTA). RNA was eluted from the beads by incubating with 200 μl 0.5 mg/ml N6-methyladenosine 5-monophosphate sodium salt (Sigma Aldrich) for 1h at 4°C. Following ethanol precipitation, input and eluted poly(A)+ RNA was reverse-transcribed using Superscript III with random hexamers, and enrichment of m6A-containing transcripts was determined by quantitative PCR.
RNA-Seq
3 replicates (from 3 different mice) of HSC, MPP1, MPP2 and MPP4 cells were sorted from Mettl3 f/f and Mettl3 cKO mice base on cell surface marker. RNA was extracted from bulk cells using the SMARTer RNA extraction method for minimal cells RNA extraction. After amplification, cDNA was subjected to automated Illumina paired-end library construction. Libraries were sequenced on Illumina HiSeq2000 instruments with paired reads of 50 bp in length per sample. Sequence data were aligned using the tophat software to the GRCh37-lite human reference genome. Fragments Per Kilobase of transcript per Million mapped reads were calculated using the cufflink software. Differentially expressed genes were identified as those with FPKM greater than 1 for Mettl3 f/f or Mettl3 cKO showing differential expression greater than twofold (up or down) with a Benjamini–Hochberg corrected p value less than 0.05.
Single-Cell RNA-Seq
Single-cell RNA-seq data processing, alignment, cell type classification and clustering: 10× data were processed using Cell Ranger 2.1.0 with default parameters. Reads were aligned to the mouse reference mm10. The Seurat package(Butler et al., 2018) was used to perform unbiased clustering of the cells bone marrow Lin-, c-Kit+ sorted hematopoietic progenitors. Briefly, cells with < 200 detected genes or percentage of mitochondrial reads > 20% were filtered out. In addition, cells that showed lower number of genes than expected from the number of UMIs detected (Figure S4B) were filtered out as low-quality cells. The data were log normalized using a scale factor of 10,000. Potential confounders (e.g., number of UMI per cell, the proportion of mitochondrial genes and proportion of ribosomal genes) were regressed out of the data before principle component analysis (PCA) was performed using variable genes. JackStraw method was used to determine the statistically significant PCs to be used for graph-based clustering. t-SNE was used to visualize the clusters. Cell types were classified according to expression of previously reported marker genes(Paul et al., 2015). Pseudotime ordering was performed using Monocle(Trapnell et al., 2014), setting the origin of pseudotime in the cell state containing the highest fraction of cells from the HSC cluster.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were processed using GraphPad Prism v.7. All analyses were performed using two-tailed Student’s t tests, except where stated otherwise. Graphs and error bars reflect means ± s.e.m., except where stated otherwise. In all corresponding figures, * represents p < 0.05. ** represents p < 0.01. *** represents p < 0.001. ns represents p > 0.05. Replicate information is indicated in the figures.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| METTL3 Ab | Proteintech | Cat# 150731–1-AP; RRID: AB_2142033 |
| MYC Ab | Cell signalingTechnology | Cat# 5605S; RRID:AB_1903938 |
| Actin Ab | Sigma Aldrich | Cat# A3854; RRID:AB_262011 |
| Gr1-APC cloneRB6–8C5 | Thermo Fisher Scientific | Cat# 17–5931-82; RRID:AB_469476 |
| Mac1-PB clone M1/70 | Biolegend | Cat# 101224; RRID:AB_755986 |
| c-Kit APCCy7 clone2B8 | Biolegend | Cat# 105826; RRID:AB_1626278 |
| B220-PeCy7 clone RA3–6B2 | BD Biosciences | Cat# 552772; RRID:AB_394458 |
| CD19-PeCy5 clone eBio1D3 | Thermo Fisher Scientific | Cat# 15–0193-83; RRID:AB_657673 |
| IgM APC clone RMM-1 | Biolegend | Cat# 406509; RRID:AB_315059 |
| CD3e-PB clone 145–2C11 | Thermo Fisher Scientific | Cat# 48–0031-80; RRID:AB_10733280 |
| CD4 PE clone GK1.5 | BD Biosciences | Cat# 557308; RRID:AB_396634 |
| CD8 FITC clone 53–6.7 | BD Biosciences | Cat# 553031; RRID:AB_394569 |
| CD34-FITC clone RAM34 | eBioscience | Cat# 11–0341-85; RRID:AB_465022 |
| Sca1-PB clone E13–161.7 | Biolegend | Cat# 122520; RRID:AB_2143237 |
| CD150-APC clone TC15–12F12.2 | Biolegend | Cat# 115910; RRID:AB_493460 |
| CD48-PE clone HM48–1 | BD Biosciences | Cat# 557485 RRID:AB_396725 |
| FcgRIIb-PeCy7 clone 2.4G2 | BD Biosciences | Cat# 560829 RRID:AB_10563207 |
| CD41-PE clone MWReg30 | BD Biosciences | Cat# 558040 RRID:AB_397004 |
| Ter119-PeCy5 clone TER-119 | Thermo Fisher Scientific | Cat# 15–5921-82 RRID:AB_468810 |
| CD71-FITC clone R17217 | Thermo Fisher Scientific | Cat# 11–0711-81 RRID:AB_465123 |
| CD45.1 Pe-CF594 clone A20 | BD Biosciences | Cat# 562452 RRID:AB_11152958 |
| CD45.2 Alexa 700 clone 104 | Thermo Fisher Scientific | Cat# 56–0454-82 RRID:AB_657752 |
| CD3-PeCy5 | Invitrogen | Cat# 15–0031-83 RRID:AB_468691 |
| CD4-PeCy5 | Invitrogen | Cat# 15–0041-83 RRID:AB_468696 |
| CD8-PeCy5 | Invitrogen | Cat# 15–0081-83 RRID:AB_468767 |
| Gr1-PeCy5 | Invitrogen | Cat# 15–5931-82 RRID:AB_468813 |
| B220-PeCy5 | Invitrogen | Cat# 15–0452-83 RRID:AB_468756 |
| CD19-PeCy5 | Invitrogen | Cat# 15–0193-83 RRID:AB_657673 |
| Ki67 −FITC | Biolegend | Cat# 652410 RRID: AB_2562141 |
| NUMB Ab | Abcam | Cat# Ab4147 RRID:AB_304320 |
| m6A Ab | Abcam | Cat# Ab151230 RRID:AB_2753144 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| pIpC HMW | InVivogen | Cat# Vac-Pic |
| MitoTracker Green | Invitrogen | Cat# M7514 |
| Pyronin Y | Sigma | Cat# 213519 |
| Murine SCF | Peprotech | Cat# 250–03 |
| Murine IL-3 | Peprotech | Cat# 213–13 |
| Murine IL-6 | Peprotech | Cat# 216–16 |
| Murine TPO | Peprotech | Cat# 315–14 |
| Murine FLT3L | Peprotech | Cat# 250–31L |
| MethoCult M3434 | Stem Cell Technologies | Cat# 03434 |
| SFEM medium | Stem Cell Technologies | Cat# NC9753895 |
| O-Propargyl-puromycin (OP-Puro) | Jena Bioscience | Cat# NU–931–05 |
| Actinomycin D | Sigma | Cat# 50–76–0 |
| Critical commercial assays | ||
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| Murine CD117 MicroBeads Kit | Miltenyi Biotec | Cat# 130091224 |
| Verso cDNA Synthesis Kit | Thermo Fisher Scientific | Cat# AB1453-B |
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat# 4367659 |
| Click-iT Flow Cytometry Assay kit | Thermo Fisher Scientific | Cat# C10418 |
| RNAscope multiplex fluorescent detection kit | Advanced Cell Diagnostics | Cat# 320851 |
| Deposited Data | ||
| Single cell RNA-sequencing data and bulk RNA-sequencing data |
National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) |
GSE132357 |
| Experimental Models: Strains | ||
| Mouse: Mettl3 f/f Mx1-Cre mice | This paper | N/A |
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| B6.SJL-Ptprc < a > /Boy | Taconic Biosciences | Stock No: 4007 |
| Oligonucleotides | ||
| Genotyping primers-Forward | ACCACAACAGCCAAGGAACA | N/A |
| Genotyping primers-Reverse | CCGGAGCTCTGAAACCTTGT | N/A |
| Myc-Forward | GCTGTTTGAAGGCTGGATTTC | N/A |
| Myc-Reverse | GATGAAATAGGGCTGTACGGAG | N/A |
| Mettl3-Forward | AAGGAGCCGGCTAAGAAGTC | N/A |
| Mettl3-Reverse | TCACTGGCTTTCATGCACTC | N/A |
| Actin-Forward | ACCAACTGGGACGACATGGAGAAG | N/A |
| Actin-Reverse | TACGACCAGAGGCATACAGGGACA | N/A |
| Recombinant DNA | ||
| MSCV-MYC-IRES-GFP | This paper | N/A |
| MSCV-FLAG-METTL3-IRES-GFP | This paper | N/A |
| MSCV-FLAG-METTL3-CD-IRES-GFP | This paper | N/A |
| Software and Algorithms | ||
| ImageJ version 2.0.0 | ImageJ | N/A |
| FlowJo software (version10.2) | FlowJo | N/A |
| GraphPad Prism 7 | GraphPad Software | N/A |
| R version 3.5.2 (2018–12-20) | R development core team, 2018 | N/A |
| Rstudio 1.1.456 | Rstudio, Inc. | N/A |
| cellrangerRkit 2.0.0 | 10× Genomics | https://www.r-project.org |
| Seurat 2.3.4 | Raul Satija laboratory at New York Genome Center | https://www.rstudio.com/ |
| dplyr 0.8.1 | Romain François, Lionel Henry and Kirill Müller | software@10xgenomics.com |
Highlights.
Single-cell RNA-seq uncovers an HSC-MPP intermediate population after m6A depletion
m6A-depleted HSC-MPPs molecularly and functionally resemble normal MPPs
m6A controls HSC symmetric commitment and identity through MYC mRNA stability
Asymmetric segregation of MYC during HSC division provides a marker for commitment
ACKNOWLEDGMENTS
We would like to thank members of the Kharas, Jaffrey, and Landau laboratories and colleagues for helpful advice and suggestions. We thank Dr. Linheng Li for providing the m6A-seq information in LT/ST HSCs and MPPs. We thank Brooke Greenstein from Basepair for bioinformatics analysis. We would like to thank the Weill Cornell Medicine Epigenomics Core for their assistance with single-cell RNA sequencing. We thank the Integrated Genomics Operation (IGO) core in MSKCC for their help with our bulk RNA sequencing. M.G.K. is a Scholar of the Leukemia and Lymphoma Society and supported by the US NIH National Institute of Diabetes Digestive and Kidney Diseases Career Development Award; NIDDK NIH R01-DK101989–01A1; NCI 1R01CA193842–01, R01HL135564, and R01CA225231–01; the Kimmel Scholar Award; the V-Scholar Award; the Geoffrey Beene Award; the Starr Cancer Consortium; the Alex’s Lemonade Stand A Award; the LLS Translation Research Program; the Susan and Peter Solomon Fund; and the Tri-Institutional Stem Cell Initiative 2016–014. D.A.L. is supported by the Burroughs Wellcome Fund Career Award for Medical Scientists; the ASH Scholar Award; Pershing Square Sohn Prize for Young Investigators in Cancer Research, the NIH Director’s New Innovator Award (DP2-CA239065); the National Heart, Lung, and Blood Institute (R01HL145283–01); a Stand Up to Cancer Innovative Research Grant (SU2C-AACR-IRG-0616); and the Leukemia Lymphoma Society Translational Research Program. S.R.J. is supported by R01CA186702. H.L. is supported by NYSTEM training award contract C32599GG. B.F.P. is supported by F32CA22104. L.P.V. is supported by K99 CA229993 and the LLS Career Development Award. This work was also supported by P30CA008748.
Footnotes
DECLARATION OF INTERESTS
D.A.L. received consulting honoraria from Pharmacyclics. S.R.J. is a scientific founder of Gotham Therapeutics and has equity in this company.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.032.
DATA AND CODE AVAILABILITY
The single cell RNA-seq and bulk RNA-seq data have been deposited in the Gene Expression Omnibus with GEO: GSE132357.
REFERENCES
- Attar EC, and Scadden DT (2004). Regulation of hematopoietic stem cell growth. Leukemia 18, 1760–1768. [DOI] [PubMed] [Google Scholar]
- Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, Murison A, Langenfeld K, Petretich M, Scognamiglio R, et al. (2018). A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553, 515–520. [DOI] [PubMed] [Google Scholar]
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. (2017). Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 552, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. (2014). m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boriack-Sjodin PA, Ribich S, and Copeland RA (2018). RNA-modifying proteins as anticancer drug targets. Nat. Rev. Drug Discov. 17, 435–453. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, and Scadden DT (2000). Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287, 1804–1808. [DOI] [PubMed] [Google Scholar]
- Coots RA, Liu XM, Mao Y, Dong L, Zhou J, Wan J, Zhang X, and Qian SB (2017). m(6)A Facilitates eIF4F-Independent mRNA Translation. Mol. Cell 68, 504–514.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Giladi A, Paul F, Herzog Y, Lubling Y, Weiner A, Yofe I, Jaitin D, Cabezas-Wallscheid N, Dress R, Ginhoux F, et al. (2018). Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat. Cell Biol. 20, 836–846. [DOI] [PubMed] [Google Scholar]
- Gundry MC, Brunetti L, Lin A, Mayle AE, Kitano A, Wagner D, Hsu JI, Hoegenauer KA, Rooney CM, Goodell MA, and Nakada D (2016). Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Rep. 17, 1453–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, and Orkin SH (2004). Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431, 1002–1007. [DOI] [PubMed] [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, and Amit I (2014). Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharas MG, Lengner CJ, Al-Shahrour F, Bullinger L, Ball B, Zaidi S, Morgan K, Tam W, Paktinat M, Okabe R, et al. (2010). Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat. Med. 16, 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121,1109–1121. [DOI] [PubMed] [Google Scholar]
- Knoblich JA (2008). Mechanisms of asymmetric stem cell division. Cell 132, 583–597. [DOI] [PubMed] [Google Scholar]
- Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, Hanna JH, and Ding L (2019). Stage-specific requirement for Mettl3-dependent m(6)A mRNA methylation during haematopoietic stem cell differentiation. Nat. Cell Biol. 21, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, Bailis W, Cao G, Kroehling L, Chen Y, et al. (2017). m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548,338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, Yu Z, Wang Y, Qi M, Zhu Y, et al. (2018). Suppression of m(6)A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 28, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Cheng G, Hamard PJ, Greenblatt S, Wang L, Man N, Perna F, Xu H, Tadi M, Luciani L, and Nimer SD (2015). Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J. Clin. Invest. 125, 3532–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Li Q, O’Neal J, Kreisel F, Le Beau MM, and Tomasson MH (2005). c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood 106, 2452–2461. [DOI] [PubMed] [Google Scholar]
- Lv J, Zhang Y, Gao S, Zhang C, Chen Y, Li W, Yang YG, Zhou Q, and Liu F (2018). Endothelial-specific m6A modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res. 28, 249–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, et al. (2017). Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 541, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, and Jaffrey SR (2017). Rethinking m6A Readers, Writers, and Erasers. Annu. Rev. Cell Dev. Biol. 33, 319–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, and Jaffrey SR (2015). 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell 163, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, and Kimble J (2006). Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441, 1068–1074. [DOI] [PubMed] [Google Scholar]
- Oguro H, Ding L, and Morrison SJ (2013). SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Deering RP, Lu Y, Tivnan P, Lianoglou S, Al-Shahrour F, Ebert BL, Hacohen N, Leslie C, Daley GQ, et al. (2014). Musashi-2 controls cell fate, lineage bias, and TGF-β signaling in HSCs. J. Exp. Med. 211, 71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, and Jaffrey SR (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D,Gury M, Weiner A, et al. (2015). Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 163, 1663–1677. [DOI] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollizzi KN, Sun IH, Patel CH, Lo YC, Oh MH, Waickman AT, Tam AJ, Blosser RL, Wen J, Delgoffe GM, and Powell JD (2016). Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat. Immunol. 17, 704–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, Sun J, Calogero RA, Klein AM, and Camargo FD (2018). Clonal analysis of lineage fate in native haematopoiesis. Nature 553,212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, and He C (2017). Dynamic RNA Modifications in Gene Expression Regulation. Cell 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. (2014). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 8, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobodin B, Han R, Calderone V, Vrielink J, Loayza-Puch F, Elkon R, and Agami R (2017). Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-adenosine Methylation. Cell 169, 326–337.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbist KC, Guy CS, Milasta S, Liedmann S, Kamiński MM, Wang R, and Green DR (2016). Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 532, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. (2017a). The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu LP, Prieto C, Amin EM, Chhangawala S, Krivtsov A, Calvo-Vidal MN, Chou T, Chow A, Minuesa G, Park SM, et al. (2017b). Functional screen of MSI2 interactors identifies an essential role for SYNCRIP in myeloid leukemia stem cells. Nat. Genet. 49, 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al. (2018). METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 22, 191–205.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, Macdonald HR, and Trumpp A (2004). c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 18, 2747–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Kwon HY, Rattis F, Blum J, Zhao C, Ashkenazi R, Jackson TL, Gaiano N, Oliver T, and Reya T (2007). Imaging hematopoietic precursor division in real time. Cell Stem Cell 1, 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al. (2016). Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 61, 507–519. [DOI] [PubMed] [Google Scholar]
- Yao QJ, Sang L, Lin M, Yin X, Dong W, Gong Y, and Zhou BO (2018). Mettl3-Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res. 28, 952–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, Lv J, Heng J, Ding Y, Xue Y, et al. (2017). m6A modulates haematopoietic stem and progenitor cell specification. Nature 549, 273–276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.