

Abstract
Non-alcoholic fatty liver disease (NAFLD) enhances the growth and recurrence of colorectal cancer (CRC) liver metastasis. With the rising prevalence of NAFLD, a better understanding of the molecular mechanism underlying NAFLD-associated liver metastasis is crucial. Tumor-associated macrophages (TAMs) constitute a large portion of tumor microenvironment that promotes tumor growth. NOD-like receptor C4 (NLRC4), a component of an inflammasome complex, plays a role in macrophage activation and IL-1β processing. We aimed to investigate whether NLRC4-mediated TAM polarization contributes to metastatic liver tumor growth in NAFLD. Wild type (WT) and NLRC4−/− mice were fed low-fat or high-fat diet (HFD) for 6 weeks followed by splenic injection of mouse CRC MC38 cells. The tumors were analyzed 2 weeks after CRC cell injection. HFD-induced NAFLD significantly increased the number and size of CRC liver metastasis. TAMs and CD206-expressing M2 macrophages accumulated markedly in tumors in the presence of NAFLD. NAFLD upregulated the expression of IL-1β, NLRC4, and M2 markers in tumors. In NAFLD, but not normal livers, deletion of NLRC4 decreased liver tumor growth accompanied by decreased M2 TAMs and IL-1β expression in tumors. WT mice showed increased vascularity and VEGF expression in tumors with NAFLD, but these were reduced in NLRC4−/− mice. When IL-1 signaling was blocked by recombinant IL-1 receptor antagonist, liver tumor formation and M2-type macrophages were reduced, suggesting IL-1 signaling contributes to M2 polarization and tumor growth in NAFLD. Finally, we found that TAMs, but not liver macrophages, produced more IL-1β and VEGF following palmitate challenge.
Conclusions
In NAFLD, NLRC4 contributes to M2 polarization, IL-1β, and VEGF production in TAMs, which promote metastatic liver tumor growth.
Liver is the most frequent site for metastasis of visceral cancers, such as gastric, colorectal (CRC) and pancreatic ductal adenocarcinoma (PDAC)(1). Metastasis in the liver is a major determinant of the survival of patients with these cancers(1). The prevalence of obesity has been increasing, and non-alcoholic fatty liver disease (NAFLD), a hepatic manifestation of metabolic syndrome, is becoming a serious health concern worldwide(2). Importantly, obesity and NAFLD increase the risk of primary cancers, including liver, pancreas, colon, prostate, and breast in both humans and rodents(3–5). Previous animal studies have demonstrated that high-fat diet (HFD)-induced NAFLD promotes the tumor growth of CRC and PDAC liver metastasis(6–8). Large prospective and retrospective human cohorts have demonstrated that cancer patients with NAFLD have increased risk of liver metastasis and for recurrence after resection of liver metastasis compared with patients without NAFLD(9–17). The increased prevalence of obesity and NAFLD will likely increase the occurrence of liver metastasis. However, the mechanism(s) by which fatty liver mediates and enhances cancer liver metastasis is poorly understood.
Innate immune cells, including macrophages, dendritic cells (DCs), and natural killer (NK) cells, in conjunction with adaptive immune T and B cells, monitor newly transformed or metastasized tumor cells. The immune surveillance system against tumors can eliminate early stage of metastatic tumor cells in the liver. However, immune cells also have capacity to promote tumor growth by producing tumor promoting cytokines, such as IL-6, IL-1, and IL-17(4,18,19). Immune cells constitute a part of the tumor microenvironment (TME), along with myofibroblasts and extracellular matrix, which create a favorable condition for the engraftment and growth of metastatic tumors in the liver(20,21). Macrophages are the major producer of cytokines and growth factors to enhance the tumor promoting role of TME. Macrophages can be classified into tumor-suppressing M1 macrophages and tumor-promoting M2 macrophages(22–24). IL-4, IL-13, and TGF-β are associated with M2 macrophage polarization and suppress anti-tumor immunity, supporting metastatic tumor growth. In the early stage of liver metastasis, liver macrophages might play a role in the prevention of the initial engraftment and growth of metastatic tumor cells in the liver through their tumoricidal activity, likely by M1-type macrophages. However, liver macrophages as well as tumor-associated macrophages (TAMs) could promote a pro-metastatic niche formation in the liver, likely by polarizing to M2 macrophage, to enhance the growth of metastatic tumor cells that survived the initial tumoricidal attack. Inflammatory, fibrotic, and fatty liver microenvironment enhances liver tumor growth(3–17). Indeed, in a NAFLD condition, CD4+T cells are depleted and tumor surveillance is impaired, which promotes hepatocellular carcinoma (HCC) growth(25). Moreover, NAFLD enhances the infiltration of CD8+T cells and NKT cells, promoting NAFLD-associated HCC growth(26). These studies suggest that NAFLD modulates the hepatic immune response that enhances liver tumor growth. Here, we hypothesize that the altered liver microenvironment associated with NAFLD modulates TAM polarization in the TME, promoting metastatic liver tumor growth.
IL-1β is a pleiotropic cytokine and has dual roles in tumorigenesis. It has been reported to promote tumorigenesis in multiple cancers, including HCC, gastric cancer, and melanoma(27–29). IL-1β has also been reported to play a role in the development of NAFLD as well as NAFLD-associated HCC(18,30). Alternatively, IL-1β also contributes to anti-tumor surveillance as an M1-type cytokine(31,32). IL-1β is initially produced as a pro-form that is processed through the inflammasome complex to convert to the active form of IL-1β(32). IL-18 is also converted from a pro-form to the active form through the inflammasome. The inflammasome complex consists of caspase-1, ASC, and AIM2, NLRP3, or NLRC4(32). A previous study has investigated the role of NLRP3 and IL-18 in CRC liver metastasis(33). IL-18 regulated the maturation of NK cells, which prevented metastatic liver tumor growth. This study determined that the IL-18 necessary for appropriate control of metastasis was processed by the NLRP3 inflammasome(33). NLRP3−/− mice displayed similar phenotypes as IL-18−/− mice, showing augmented CRC liver metastasis. However, the role of IL-1 and NLRC4 inflammasome activation in metastatic tumor growth enhanced by fatty liver has not been examined.
The present study aimed to investigate the role of NLRC4 and IL-1 signaling and their roles in the polarity of TAM in the progression of metastatic liver tumor growth in the unique context of NAFLD. The present study determined that NLRC4 and IL-1 signaling play a role in M2 polarization and angiogenesis in metastatic CRC tumors in a fatty liver condition. This pathophysiology is distinct from tumor growth in a non-fatty liver microenvironment. The present study provides insight into the new molecular mechanism underlying metastatic liver tumor growth in different liver conditions, which prompts us to consider different therapeutic strategies for liver metastasis in patients with or without NAFLD.
MATERIALS and METHODS
Animals
C57BL/6 wild type (WT) mice were purchased from Jackson Laboratories (Bar Harbor, ME). The generation of NLRC4−/− mice and NLRC4-Flag knock-in mice were described(34,35). All mice were bred in the Cedars-Sinai Medical Center vivarium. All genetically modified mice were back-crossed at least 10 generations onto the C57BL/6 background. Female mice were used for in vivo experiments. All studies were done in accordance with National Institutes of Health recommendations outlined in the Guide for the Care and Use of Laboratory Animals. All animal experiment protocols were approved by the Cedars-Sinai Medical Center Institutional Animal Care and Use Committee.
Fatty liver-liver metastasis splenic injection model
Eight to 12-week-old female mice were fed a low-fat diet (LFD; 12% fat calories) or HFD (60% fat calories) for 6 weeks. Mice were then anesthetized. After a laparotomy, MC38 cells (2×105 cells; C57BL/6 background CRC cell line) were injected via the spleen(33). Two weeks following injection (8 total weeks of HFD feeding), tumors were harvested and analyzed. For the treatment of IL-1 receptor antagonist, 10 mg/kg of recombinant IL-1 receptor antagonist (Anakinra®, Amgen) was injected subcutaneously (s.c.) daily, starting on the second day after the intrasplenic MC38 cell injection. To assess early engraftment of metastatic cancer cells, MC38 cells were labeled with Vybrant Dil Cell-Labeling system (Thermo Fisher) and livers were harvested three days after cancer cell injection.
Tumor analysis
We evaluated metastatic liver tumors macroscopically and histologically (H&E staining) by counting visible tumor number and measuring maximal size of tumors, area, and whole liver weight. Tumor and non-tumor liver tissues were separated macroscopically followed by mRNA and protein analyses. To assess early engraftment of cancer cells, liver sections were analyzed using fluorescent microscopy and tumor foci labeled by Vybrant Dil Cell-Labeling system were counted from randomly selected 10 fields of x100 magnification per slide. Liver parameters were measured using alanine transaminase (ALT) levels in blood, and triglyceride levels in the blood and liver.
Histological Analysis
Formalin-fixed, paraffin-embedded liver tissues were used. After being deparaffinized and rehydrated, endogenous peroxidase blocking and antigen retrieval were applied. After blocking, sections were incubated with primary antibody against F4/80 (clone BM8; #14–4801, eBioscience) or CD206 (#AF2535, Novus). Secondary antibodies were used. F4/80 or CD206-positive area was quantified from randomly selected 8–10 fields of x100 magnification per slide by NIH ImageJ software (https://imagej.nih.gov/ij). Neutral lipids were analyzed by Oil Red O staining. Oil Red O-positive area was evaluated from randomly selected 10 fields of x200 magnification per slide.
Immunofluorescence staining
For NLRC4-Flag and F4/80 double staining, liver sections were incubated with anti-Flag antibody Cell Signaling) and anti-F4/80 antibody, followed by Cy3- and AlexaFluor® 488-conjugated secondary antibodies. For CD31 staining, anti-CD31 antibody (Invitrogen) and FITC-conjugated secondary antibody were used. Imaging analyses were performed using a Zeiss LSM800 inverted stand and Axio Imager Z2m confocal system.
RNA Isolation and quantitative RT-PCR Analysis
RNA was extracted using TRIzol (Life Technologies) plus NucleoSpin® (Clontech). Extracted RNA was converted to cDNA using reverse transcription kit (Applied Biosystems). Quantitative real-time PCR was performed using SYBR Green. Quantification was performed by comparing Ct values of each sample with normalization to 18S RNA. Sequences of primers were summarized in Supplemental Table 1.
Western blots
Protein extracts from liver and tumor tissues were electrophoresed and then blotted. Blots were incubated with antibody for IL-1β (Cell Signaling Technologies), mouse caspase-1 p20 (Adipogen), and β-actin (Sigma).
Measurement of cytokines
The levels of IL-4, IL-13, and IFN-γ in liver and tumor tissues as well as IL-1β in the culture supernatant were analyzed by ELISA kit (R&D).
Cell Culture Experiments
Liver macrophages and TAMs were isolated from mice by in situ liver perfusion with Pronase E (Sigma-Aldrich) and Collagenase D (Sigma-Aldrich), followed by density gradient centrifugation with Percoll (GE Healthcare Life Science). Magnetic antibody sorting (MACS; Miltenyi Biotec) using CD11b (Miltenyi Biotec) were used to positively select macrophages. Liver macrophages were cultured in RPMI 1640. Palmitate (200 μM, Sigma) was used to stimulate liver macrophages or TAMs. TAK-242 (CLI-095, 1μM, Inviviogen) was used to inhibit TLR4.
Flow cytometry
The single-cell suspension of mouse tumor tissue was prepared by enzymatic digestion, followed by Percoll isolation. To obtain the population of M1 or M2 macrophages, F4/80 and iNOS, or F4/80 and CD206 antibodies were used and detected by BD flow cytometry. The data were analyzed by the FlowJo software.
Statistical Analysis
Statistical significance was assessed by using GraphPad Prism 8.1.0 software (GraphPad Software, Inc). Differences between the two groups were compared using a two-tailed unpaired Student’s t-test. Differences between multiple groups were compared using one-way ANOVA, followed by Tukey’s post hoc analysis. P values <0.05 were considered significant.
RESULTS
Fatty liver condition enhances the growth of CRC liver metastasis
After feeding mice 6 weeks of HFD that resulted in the development of fatty liver, we injected syngeneic CRC MC38 cells via the spleen to establish a CRC liver metastasis model. In comparison to LFD feeding, HFD feeding augmented the growth of metastatic liver tumors as demonstrated by increased number of visible tumors and histological tumor area in the liver (Figure 1A–D). We confirmed the duration of HFD feeding that we used was sufficient to develop fatty liver via histology as well as through increased serum triglyceride levels (Figure 1E,F).
FIGURE 1. High-fat diet (HFD)-induced fatty liver enhances metastatic tumor growth in the liver.
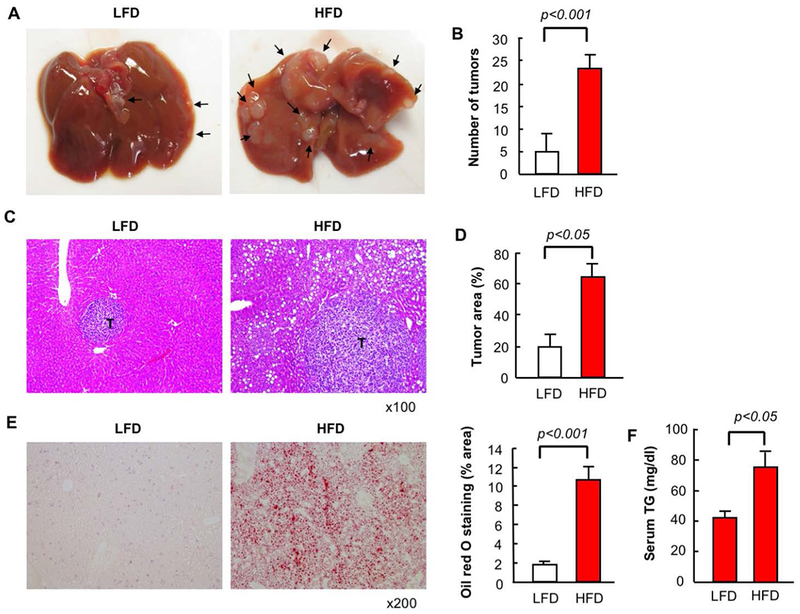
After 6 weeks of low-fat diet (LFD) or HFD, mice had splenic injection of MC38 colorectal cancer (CRC) cells and continued on LFD or HFD for an additional 2 weeks. (A) Macroscopic appearance of the liver. Arrow, tumors. (B) Number of visible liver tumors. (C) H&E staining. T, tumor. (D) Measurement of tumor area based on H&E staining (E) Oil red O staining and its quantification. (F) Serum triglyceride levels. n=8, each group. Representative pictures are shown. Data are shown as mean ± S.D. per group.
Fatty liver augments TAM infiltration in the tumor area of CRC liver metastasis
Next, we investigated macrophage infiltration in the tumors. Macrophages infiltrated in the tumor were referred to as TAMs. We observed infiltration of F4/80-positive TAMs in mice fed with a LFD versus HFD, and found TAM infiltration was augmented by the fatty liver condition (Figure 2A). Because M2-polarized macrophages are known to promote tumor growth(24), we evaluated TAMs for expression of CD206, a M2 marker. The number of the infiltrating CD206-expressing TAMs were also significantly increased when mice were fed HFD (Figure 2B). Next, we measured expression of IFN-γ and NOS2 as M1 macrophage markers and IL-4, IL-13, and CD206 as M2 macrophage markers in the non-tumorous liver and tumors. IFN-γ mRNA and protein expression was significantly upregulated, but NOS2 mRNA expression was unchanged, in tumors in fatty liver compared with tumors without fatty liver (Figure 2C,D). IL-4 and IL-13 mRNA and protein expression and CD206 mRNA expression were significantly upregulated in tumors with fatty liver compared with tumors without fatty liver (Figure 2E,F). These results suggest that TAMs and M2-polarized TAMs accumulated at higher levels in metastatic liver tumors with fatty liver.
FIGURE 2. The infiltration of tumor-associated macrophages (TAMs) in tumors of CRC liver metastasis is enhanced in non-alcoholic fatty liver disease (NAFLD).
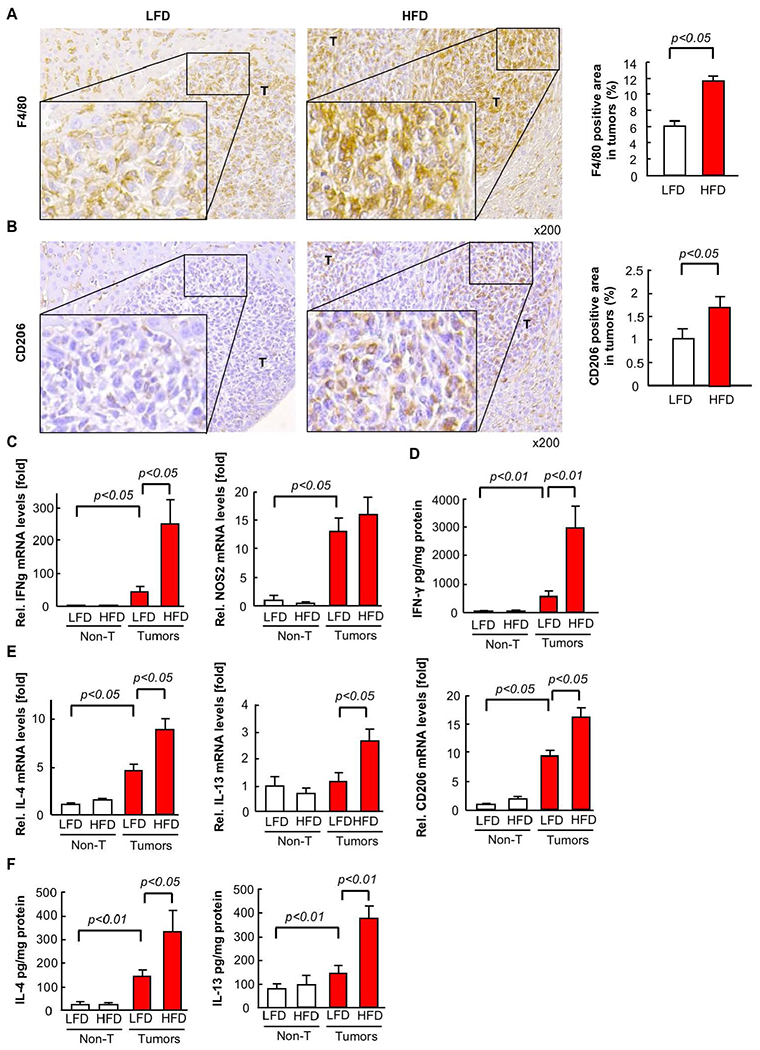
Mice had splenic injection of MC38 colorectal cancer (CRC) cells after 6 weeks of low-fat diet (LFD) or high-fat diet (HFD) which was continued for an additional 2 weeks. (A, B) TAMs and M2 macrophages were examined by immunohistochemistry (IHC) for F4/80 and CD206, respectively (left). The F4/80- or CD206-positive area in tumors was quantified by ImageJ (right). n=8, each group. T, tumor. (C,E) The mRNA expression of M1 (C, IFN-γ, NOS2) and M2 (E, IL-4, IL-13, CD206) macrophage markers in tumors was examined by quantitative real-time PCR. (D,F) The protein expression of M1 (D, IFN-γ) and M2 (F, IL-4, IL-13) macrophage markers in tumors was examined by ELISA. n=6, each group. Data are shown as mean ± SEM per group.
Fatty liver enhanced the expression of IL-1β and inflammasome components in metastatic tumors
Because our data show that IL-1β mRNA as well as mature IL-1β protein expression were higher in tumors with fatty liver than those without fatty liver (Figure 3A,B), it is possible that inflammasome activation contributes to tumor progression in the fatty liver condition. Thus, we measured the expression of inflammasome components in tumors. The expression of caspase-1, ASC, NLRP3, and NLRC4 was increased in tumors with fatty liver compared with those without fatty liver (Figure 3A). Consistent with the increased mature IL-1β protein expression, the active form of caspase-1 was increased in tumors with fatty liver (Figure 3C), suggesting that the fatty liver condition enhanced caspase-1-dependent IL-1β processing in metastatic tumors. A previous study using MC38 CRC cells demonstrated that NLRP3−/− mice had exacerbated liver associated with the loss of inflammasome-dependent IL-18 (33). This previous study(33) and our results as shown in Figure 1–3 suggest that IL-1β and inflammasome components other than NLRP3 could be associated with increased liver metastasis in the fatty liver condition.
FIGURE 3. Expression of inflammasome components is increased in tumors enhanced by NAFLD.
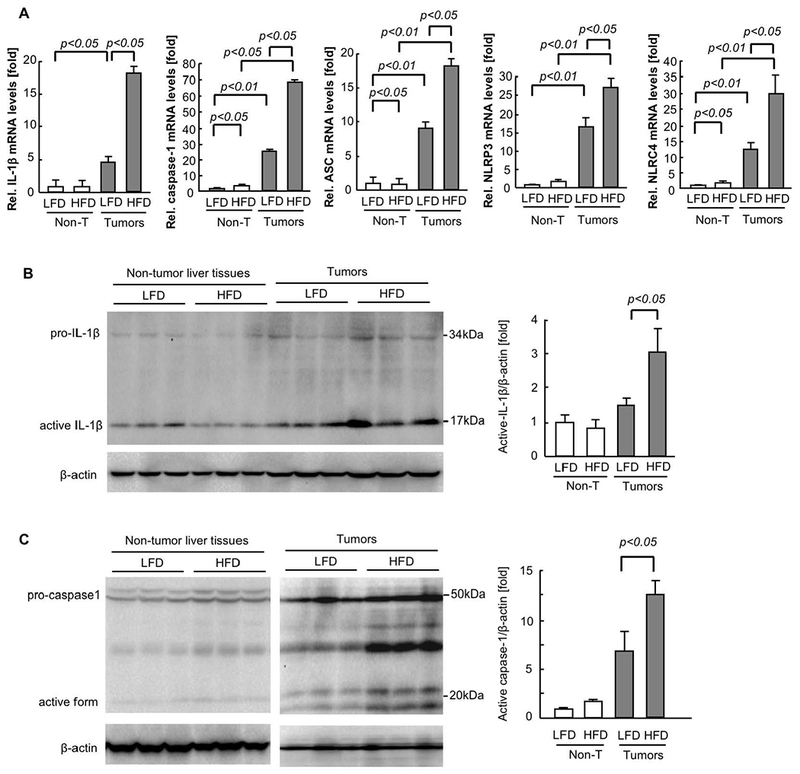
After 6 weeks of low-fat diet (LFD) or high-fat diet (HFD), mice had splenic injection of MC38 colorectal cancer (CRC) cells and continued on LFD or HFD for 2 additional weeks. (A) The mRNA expression of inflammasome components (IL-1β, caspase-1, ASC, NLRP3, NLRC4) in tumors was examined by quantitative real-time PCR. n=6, each group. Data are shown as mean ± SEM per group. (B,C) Western blot analyses for IL-1β and caspase-1. Representative images are shown. The quantifications were analyzed by Image J. n=5, each group. Data are shown as mean ± SEM per group.
Loss of NLRC4 reduces the fatty liver-enhanced metastatic liver tumor growth
NLRC4 is another inflammasome-forming NLR family member that had enhanced expression in tumors that was further upregulated in tumors of HFD-fed mice. Accordingly, we hypothesized that NLRC4 may contribute to the enhanced liver tumor growth in the fatty liver condition. Both WT and NLRC4−/− mice received either HFD or LFD for 6 weeks, followed by splenic injection of MC38 cells. After an additional 2 weeks of continued LFD or HFD, liver and tumor samples were harvested. The liver weight, visible tumor number, and maximal tumor size were similar in WT and NLRC4−/− mice fed a LFD (Figure 4A–E). Although NLRC4 deficiency did not affect HFD-induced fatty liver development (Figure 4F), NLRC4 deficiency inhibited liver tumor growth in the mice fed HFD (Figure 4A–E). We also investigated the role of NLRC4 in the early engraftment of metastatic tumors in the liver. The early engraftment was assessed by counting tumor foci histologically 3 days after injection of MC38 cells. Interestingly, HFD feeding enhanced the engraftment whereas NLRC4 deficiency did not affect the engraftment (Supplementary Figure 1). These results suggest that NLRC4 plays a role in the metastatic tumor growth in the presence of fatty liver but NLRC4 is involved in neither early engraftment nor fatty liver development.
FIGURE 4. Metastatic liver tumor growth enhanced by NAFLD is suppressed by NLRC4 deficiency.
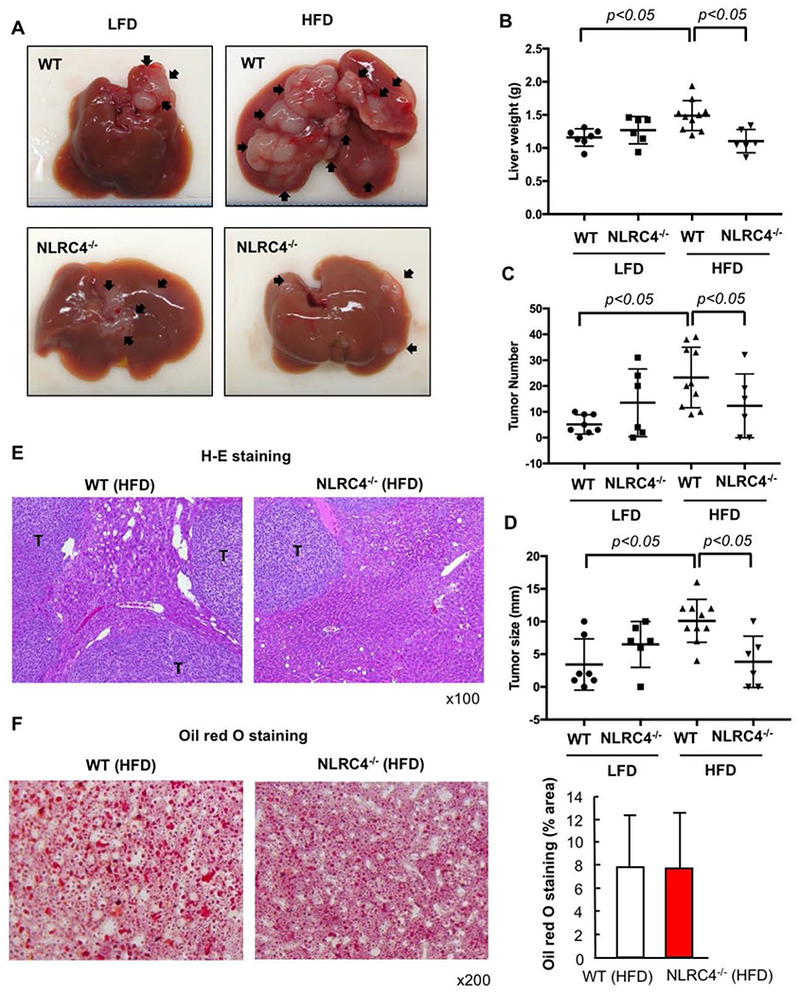
WT and NLRC4−/− mice were injected via the spleen with MC38 colorectal cancer (CRC) cells after 6 weeks of being fed low-fat diet (LFD) or high-fat diet (HFD) which was continued for an additional 2 weeks. (A) Macroscopic appearance of the liver. Arrow, tumors. (B) Liver weight. (C) Number of visible liver tumors. (D) Maximal size of tumors. (E) H&E staining. T, tumor. (F) Oil red O staining and its quantification. n=6–10, each group. Representative pictures are shown. Data are shown as mean ± S.D.
NLRC4 mediates TAM infiltration and polarization in the CRC liver metastasis with fatty liver
We next investigated the role of NLRC4 in TAMs. The infiltration of F4/80-positive TAMs in HFD-fed WT mice was reduced in mice deficient in NLRC4 (Figure 5A). Next, we examined M2-polarized macrophages in tumors with fatty liver. The number of CD206-expressing M2-polarized TAMs was significantly reduced in NLRC4−/− mice compared to WT mice (Figure 5A). While M1 macrophage markers IFN-γ mRNA and protein expression and NOS2 mRNA expression trended towards being increased in HFD-fed NLRC4−/− mice compared to WT mice, this finding did not reach statistical significance (Figure 5B,C). In contrast, M2 macrophage markers IL-4 and IL-13 mRNA and protein expression and CD206 mRNA expression were significantly reduced in HFD-fed NLRC4−/− mice compared to WT mice (Figure 5D,E). Moreover, FACS analysis showed that the proportion of iNOS-expressing M1 TAMs was decreased in both WT and NLRC4−/− mice when fed with HFD (Figure 5F). The proportion of CD206-expressing M2 TAMs was markedly increased in WT mice after HFD feeding but this increase was lost in the NLRC4-deficient mice despite being fed the HFD (Figure 5F). These results demonstrate that NLRC4 is required for the infiltration of tumors by CD206-expressing M2 TAMs, suggesting that the mechanism by which NLRC4 regulates enhanced tumor growth in the fatty liver condition is through modification of M2 TAM infiltration or polarization.
FIGURE 5. NLRC4 regulates the recruitment and polarization of M2 TAMs in the NAFLD-enhanced tumors.
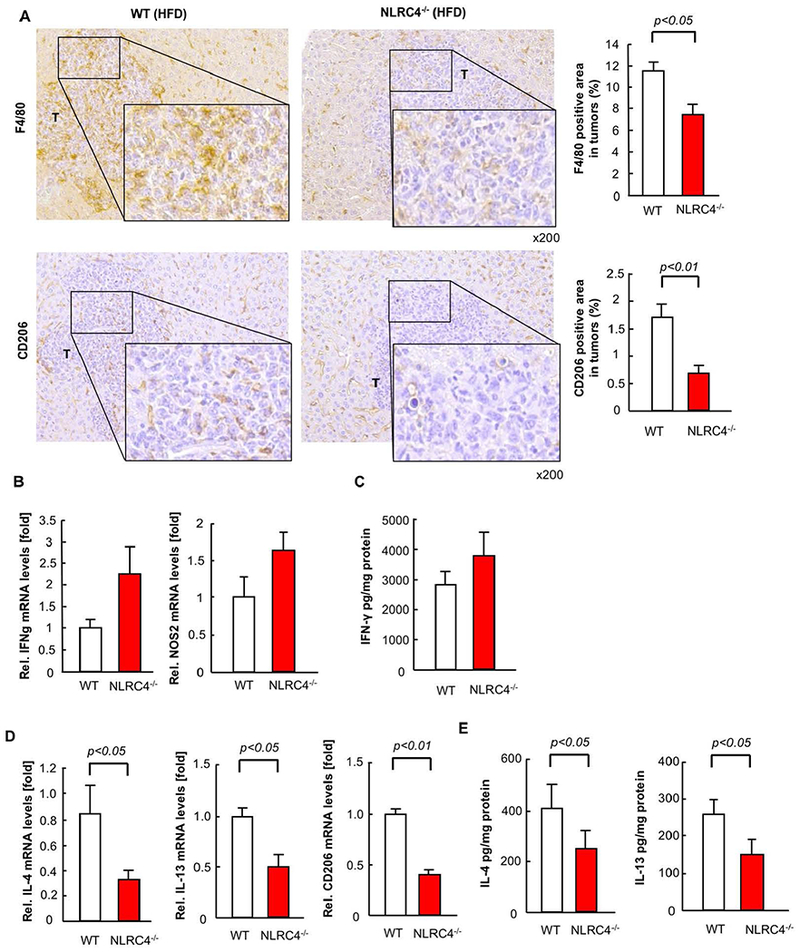
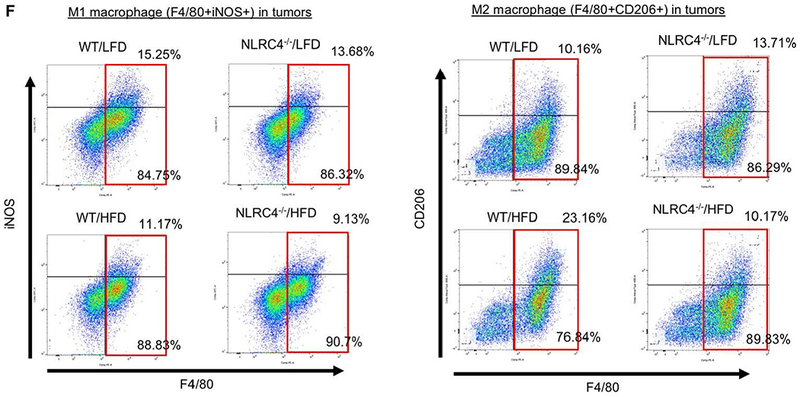
WT and NLRC4−/− mice were fed low-fat diet (LFD) or high-fat diet (HFD) for 6 weeks followed by splenic injection of MC38 colorectal cancer (CRC) cells and continued LFD or HFD for an additional 2 weeks. (A) TAMs and M2 macrophages were examined by immunohistochemistry (IHC) with F4/80 and CD206, respectively. The F4/80- or CD206-positive area in tumors was quantified by ImageJ. n=8, each group. T, tumor. (B,D) The mRNA expression of M1 (IFN-γ, NOS2) and M2 (IL-4, IL-13, CD206) macrophage markers in tumors from HFD-fed mice was examined by quantitative real-time PCR. n=6–10, each group. Data are shown as mean ± SEM per group. (C,E) The protein expression of M1 (C, IFN-γ) and M2 (E, IL-4, IL-13) macrophage markers in tumors was examined by ELISA. (F) FACS analysis was performed for M1 and M2 TAMs. TAMs were isolated from liver tumors of LFD- and HFD-fed mice. F4/80, iNOS, and CD206 were used to define TAMs, M1 (F4/80+iNOS+), and M2 (F4/80+CD206+) macrophages, respectively. Representative results of 3 independent experiments are shown.
TAMs are the responsible cells expressing NLRC4 in HFD-enhanced CRC liver metastasis
To determine the role of NLRC4 in the expression of the inflammasome components in tumors with fatty livers, we examined expression of caspase-1, ASC, IL-1β, IL-18, and NLRC4. The loss of NLRC4 significantly reduced the mRNA expression of caspase-1, ASC, and IL-1β, but not IL-18 (Figure 6A). NLRC4 deficiency also decreased the active form of caspase-1 and mature IL-1β in tumors (Figure 6B,C). These results suggest that decreased caspase-1 activation and IL-1β maturation are associated with the reduced tumor growth in NLRC4−/− mice. Of note, NLRC4 expression was blunted in tumors of NLRC4−/− mice compared to WT mice (Figure 6A), suggesting that implanted tumor cells do not express NLRC4 and that NLRC4-expressing cells in the liver are host-derived and are components of the TME in the liver. To further determine the responsible cell types expressing NLRC4, we stained TAMs with F4/80 in liver tumors using the mice with knock-in of NLRC4-Flag fusion protein. NLRC4-Flag protein was nicely co-localized with F4/80-expressing TAMs (Figure 6D). The HFD feeding condition increased NLRC4-Flag-expressing F4/80 positive cells in tumors (Figure 6D). These data indicate that TAMs are the NLRC4-expressing cells in the metastatic tumors.
FIGURE 6. NLRC4 is expressed in TAMs.
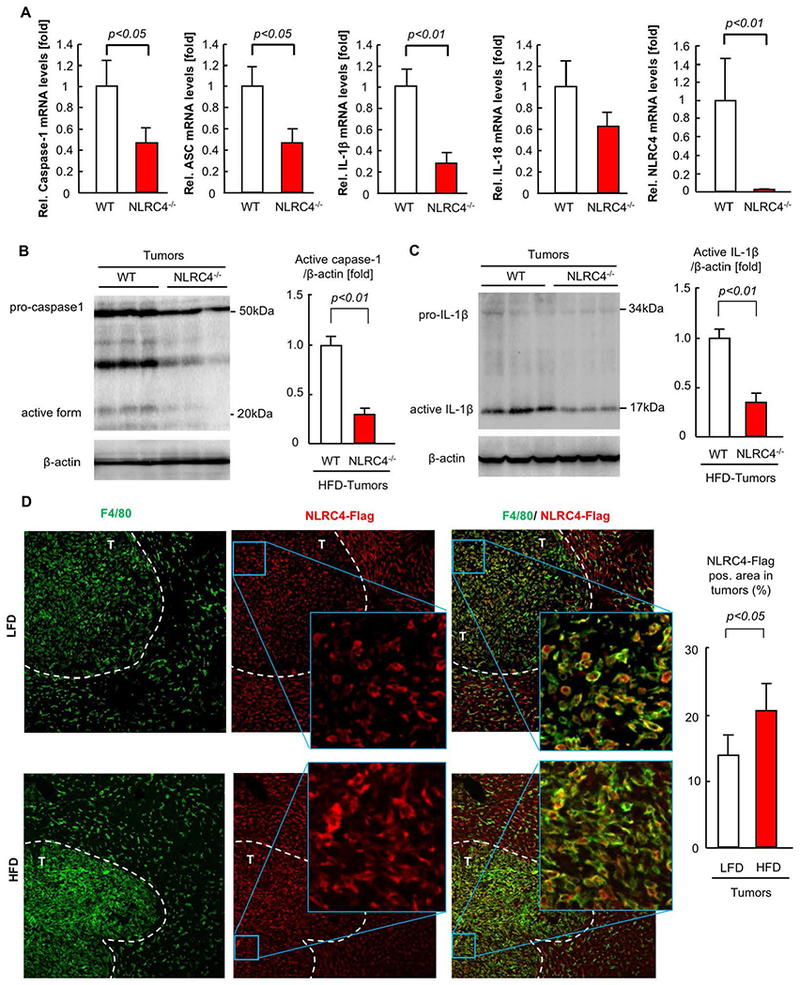
After 6 weeks of high-fat diet (HFD), WT and NLRC4−/− mice injected via the spleen with MC38 colorectal cancer (CRC) cells and continued on HFD for an additional 2 weeks. (A) The mRNA expression of inflammasome components (caspase-1, ASC, IL-1β, IL-18, NLRC4) in tumors was examined by quantitative real-time PCR. n=6–10, each group. Data are shown as mean ± SEM per group. (B,C) Western blot analyses for IL-1β and caspase-1. Representative images are shown. The quantifications were evaluated by Image J. n=5, each group. Data are shown as mean ± SEM per group. (D) NLRC4-Flag knock-in mice had splenic injection of CRC cells after 6 weeks of low-fat diet (LFD) or HFD which was continued for an additional 2 weeks. Immunofluorescence for F4/80 was performed. Green, F4/80. Red, NLRC4-Flag. White dotted line indicates tumors. T, tumor. Representative results of 3 independent experiments are shown.
IL-1 receptor signaling regulates metastatic liver tumor progression and M2 TAM infiltration in NAFLD
Because mature IL-1β expression was reduced in tumors of NLRC4−/− mice, IL-1 signaling might play a role in NLRC4-mediated metastatic tumor growth enhanced in the fatty liver. To investigate the role of IL-1 in tumor growth enhanced in NAFLD, we inhibited IL-1 receptor signaling using a recombinant IL-1 receptor antagonist. Two weeks of inhibition of IL-1 receptor signaling did not affect metastatic liver tumor growth in mice fed a LFD (Figure 7A,B), consistent with a previous study showing that IL-1R−/− mice were not protected against metastatic liver tumor growth(33). In contrast, in mice fed a HFD, the blockade of IL-1 receptor signaling by IL-1 receptor antagonist significantly suppressed the growth of liver tumors as shown by reduced liver weight, visible tumor number, and maximal tumor size (Figure 7A,B). Notably, two weeks of IL-1 inhibition did not affect the degree of fatty liver (Figure 7C). The increased F4/80 and CD206-positive TAM infiltration induced in tumors of mice fed a HFD was reduced by inhibition of IL-1 signaling (Figure 7D,E). These results suggest that IL-1 receptor signaling contributes to metastatic liver tumor growth specifically in the fatty liver microenvironment.
FIGURE 7. IL-1 receptor signaling contributes to metastatic liver tumor growth in the NAFLD condition.
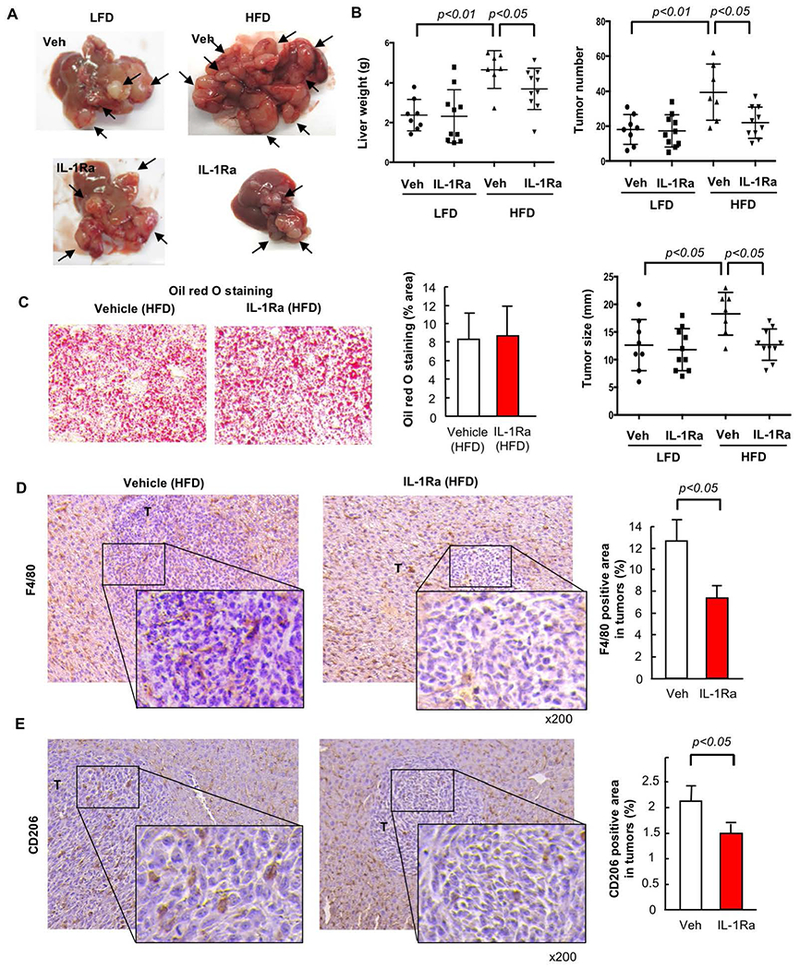
WT mice were injected via the spleen with MC38 colorectal cancer (CRC) cells after 6 weeks of low-fat diet (LFD) or high-fat diet (HFD) which was continued for an additional 2 weeks. A subset of mice were treated with recombinant IL-1 receptor antagonist (IL-1Ra, 10 mg/kg body weight) s.c. daily. (A, left) Macroscopic appearance of the liver. Arrow, tumors. (A, right) Liver weight. (B, left) Number of visible liver tumors. (B, right) Maximal size of tumors. (C) Oil red O staining and its quantification. n=7–10, each group. Representative pictures are shown. Data are shown as mean ± S.D. per group. (D,E) TAMs (D) and M2 macrophages (E) were examined by immunohistochemistry (IHC) for F4/80 and CD206, respectively. The F4/80- or CD206-positive area in tumors was quantified by ImageJ. n=7–10, each group. T, tumor. Representative pictures are shown. Data are shown as mean ± S.D. per group.
NLRC4 contributes to tumor angiogenesis through vascular endothelial growth factor (VEGF) production in TAMs
Endothelial cell expansion and angiogenesis can promote metastatic tumor growth. VEGF is a potent angiogenic factor and we found its expression in tumors was markedly increased when mice were fed HFD compared to LFD (Figure 8A). Consistently, the number of cells positive for CD31, a marker for vascular endothelial cells, was significantly increased in tumors of HFD-fed mice compared to LFD-fed mice (Figure 8B). Of note, the increase in CD31-positive cells and VEGF expression induced by HFD feeding were reduced in tumors from NLRC4−/− mice (Figure 8B,C). These results suggest that endothelial cell expansion and VEGF expression enhanced by HFD feeding required NLRC4. Because NLRC4 is solely expressed in macrophages (Figure 6D), we investigated whether macrophages contribute to VEGF expression. We isolated liver macrophages from normal mouse livers and TAMs from MC38-implanted livers. To investigate whether free fatty acid found in the TME regulates the different TAM phenotypes found in metastatic tumors from HFD-fed mice versus LFD-fed mice, we treated normal liver macrophages and TAMs with palmitate. Palmitate treatment significantly increased NLRC4 in TAMs, but not in liver macrophages (Figure 8D). Palmitate treatment increased IL-1β expression by up to 50-fold in TAMs, but 15-fold in liver macrophages (Figure 8D). Because TLR4 is involved in palmitate-mediated inflammatory response(36,37), we examined whether palmitate induces NLRC4 and IL-1β expression via TLR4 in TAMs. TLR4 inhibition almost abolished palmitate-induced NLRC4 mRNA upregulation and dramatically suppressed secretion of active IL-1β but it did not dramatically reduce pro-IL-1β mRNA expression (Figure 8D, Supplementary Figure 2). This result suggests that palmitate-mediated NLRC4 upregulation largely depends on TLR4 but molecule(s) other than TLR4 might be more important for palmitate-mediated proIL-1β mRNA induction. Then, we investigated the role of NLRC4 and IL-1β in VEGF expression in TAMs. Palmitate treatment increased IL-1β protein secretion and VEGF expression in TAMs but NLCR4 deficiency abolished IL-1β production and VEGF induction (Figure 8E). Recombinant IL-1β treatment increased VEGF expression in TAMs, but not in liver macrophages (Figure 8E). These results demonstrate that NLCR4-mediated IL-1β contributes to VEGF expression in TAMs. Taken together, it is suggested that a fatty liver condition could promote TAM activation to produce IL-1β and VEGF through overexpression of NLRC4, which contributes to angiogenesis and tumor growth in the liver.
FIGURE 8. NLRC4 regulates tumor angiogenesis through VEGF production.
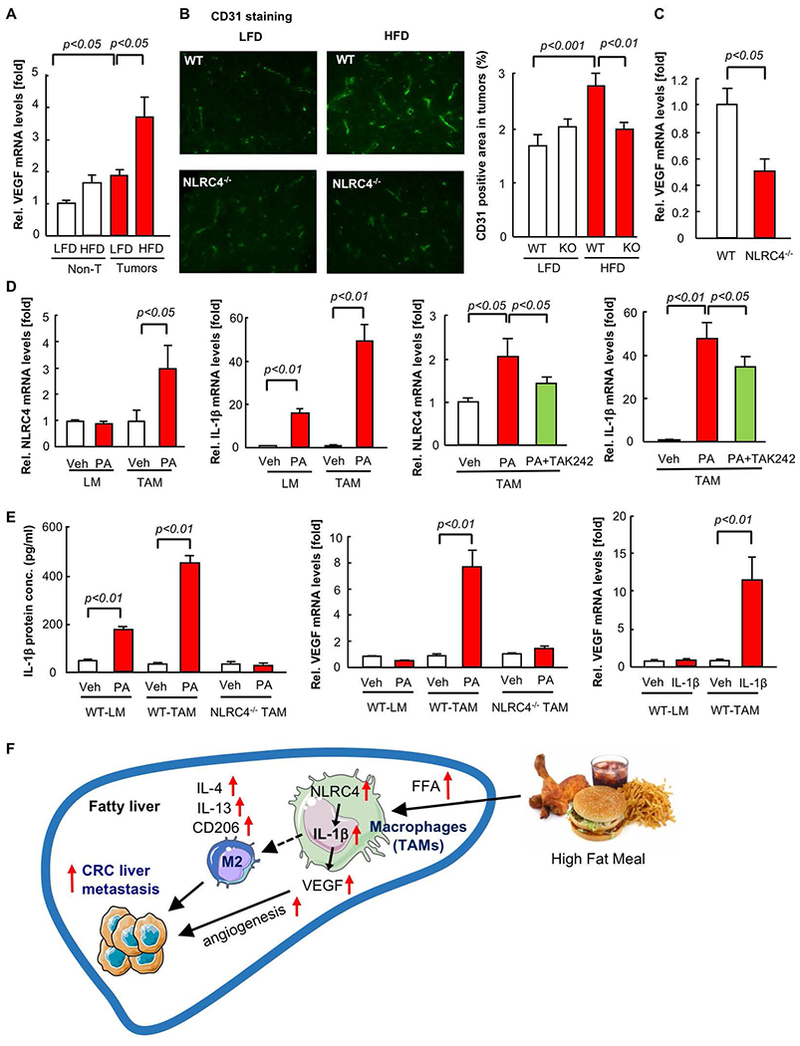
(A-C) WT and NLRC4−/− mice were fed low-fat diet (LFD) or high-fat diet (HFD) for 6 weeks after which they underwent splenic injection of MC38 colorectal cancer (CRC) cells and continued on LFD or HFD for an additional 2 weeks. (A) VEGF mRNA was examined by quantitative real-time PCR. (B) Immunofluorescence for CD31 in liver sections is shown. Representative pictures are shown. Quantification of CD31-positive area in tumors. (C) VEGF mRNA expression in tumors from HFD-fed mice. n=6–10, each group. Data are shown as mean ± SEM per group. (D,E) Liver macrophages and TAMs were isolated from WT and NLRC4−/− mice. Cells were treated with 200μM palmitate for 24 hours, with or without pretreatment of 1μM TAK-242 for 1 hour. Cells were also treated with 10ng IL-1β for 24 hours, mRNA expression of NLRC4, IL-1β, and VEGF was determined by quantitative real-time PCR. IL-1β protein concentrations in culture supernatant were measure by ELISA. Data are shown as mean ± S.E.M. from 3 independent experiments. (F) Summary of the proposed model.
DISCUSSION
The major cause of death by CRC is due to cancer dissemination and distant metastasis. Liver is the primary organ for CRC metastasis, in part due to the anatomical relationship between the liver and intestines through the portal vein. Hepatic innate immune cells, including Kupffer cells, NK cells, and DCs patrol hepatic sinusoids and prevent the initial engraftment of migrated CRC cells to the liver and also inhibit the proliferation of tumor cells in the liver microenvironment. Today, NAFLD is becoming the leading cause of chronic liver disease and afflicts 25% of adults in the United States. Although liver metastasis can occur in CRC patients without NAFLD, a recent large prospective human cohort analyzing 2,715 CRC patients with liver metastasis as well as other retrospective studies have suggested that the patients with NAFLD have an increased risk of liver metastasis and for the recurrence of liver metastasis after resection, when compared with patients without NAFLD(9–13). Moreover, higher recurrence rate of liver metastasis in patients with fibrosis have also been reported in recent studies(12,38–41). However, previous studies underestimated the risk of liver metastasis or have reported contradictory results in patients with underlying fatty liver and fibrosis(42–44). Numerous animal studies suggested both fatty liver and fibrotic liver conditions create a TME that promotes tumor growth, and that this is mediated through tumor-promoting immune cells including TAMs, regulatory T cells, and myeloid-derived suppressor cells (MDSCs), and hepatic stellate cells, and extracellular matrix. In the present study, we have demonstrated the HFD-induced fatty liver condition enhanced metastatic liver tumor growth. We found that the NAFLD condition dramatically increased infiltration of TAMs, in which CD206-expressing M2 macrophage population was increased. Although M1 marker IFN-γ was relatively increased in tumors with NAFLD compared to that without NAFLD, iNOS expression and iNOS-expressing M1 TAMs were not increased by NAFLD. This suggests that M2 TAM infiltration contributes to enhanced tumor growth in NAFLD. Indeed, a recent human study demonstrated that an increased M2/M1 ratio in resected livers is correlated with CRC liver metastasis, suggesting the relevance of M2 macrophages in this clinical setting(45). In addition, in HCC, the NAFLD condition decreased the number of anti-tumorigenic CD4+T cells, contributing to tumor growth(25). These studies strongly suggest that the modulated hepatic immune microenvironment is associated with enhanced tumor growth in NAFLD.
Altered polarization of hepatic TAMs by NAFLD could affect the tumor-promoting conditions in TME. In the fatty liver TME, the expression of mature IL-1β, the active form of caspase-1, and the other inflammasome components were increased. An inflammasome is a multiprotein complex, in which the components include NLRP3, NLRC4, or AIM2 with ASC and caspase-1. Inflammasome activation results in activation of caspase-1 which then cleaves IL-1β and IL-18 to their mature and active forms. Previous studies demonstrated that mice lacking NLRP3 were more susceptible to experimental colitis and colitis-associated CRC models, and that NLRP3−/− mice had increased CRC liver metastases(46–49). NLRP3-dependent IL-18 activation was shown to stimulate NK cells, which enhanced their tumoricidal activity against CRC liver metastasis(33). In that study, consistent with our data, NLRC4 did not play a significant role in tumor growth in a normal chow-diet feeding condition(33).
In contrast to the protective role of the other NLRs, our study demonstrated that NLRC4 promoted CRC metastatic liver tumor growth specifically in the NAFLD state, but not in the absence of NAFLD. Interestingly, we did not find any role played by NLRC4 in the early engraftment of tumor cells to the liver. Our data show that NLRC4−/− mice had reduced TAM infiltration, particularly CD206-expressing M2 TAMs, along with reduced expression of Th2 cytokines IL-4 and IL-13. This could enhance tumoricidal M1 macrophage functions. Moreover, our data show reduced expression of mature IL-1β and the active form of caspase-1 in NLRC4−/− mice and that blockade of IL-1 signaling reduced tumor growth in NAFLD, suggesting that NLRC4-mediated IL-1β promotes tumor growth in NAFLD (Figure 8F). IL-1β is involved in the development of gastric cancer, melanoma, lung metastasis, and HCC(27–29). In gastric cancer, IL-1β contributes to increased infiltration of MDSC to the stomach(29). Inflammation mediated by inflammasomes and IL-1 results in the suppression of anti-tumor immunity associated with NK cells and T cells, which is involved in the development of melanoma, gastric carcinoma, and lung metastasis. It has also been reported that activation of the NLRP3 inflammasome by the anti-cancer drugs gemcitabine and 5-fluorouracil promoted IL-1β production in MDSCs that promote cancer growth(50). MDSCs are the immature myeloid cell population that express both monocyte and granulocyte markers along with both M1 and M2 markers. Although MDSC infiltration mediated by IL-1β could inhibit anti-tumor immunity, promoting tumor growth, few reports show the involvement of IL-1β in M2 macrophage infiltration and/or polarization for cancer development. However, our data show that NLRC4−/− mice had a significant reduction of M2 markers, but not M1 markers, suggesting that NLRC4 and IL-1β are associated with TAM M2 infiltration and/or polarization (Figure 8F).
Although IL-1 signaling is not involved in metastatic liver tumor growth in a non-fatty liver condition(33), IL-1 signaling promoted tumor growth in the context of NAFLD. In fact, the expression of mature IL-1β and the active form of caspase-1 were increased in tumors with NAFLD. A related finding has also been demonstrated in NAFLD-associated HCC progression, in which IL-1 signaling plays a crucial role(18). Using NLRC4-Flag knock-in mice, we determined that NLRC4 is predominantly expressed in macrophages whereas MC38 cells and hepatocytes do not express NLRC4. Our data show that TAMs, but not normal liver macrophages, increased NLRC4 and IL-1β expression in response to palmitate, which mimics the conditions of the fatty liver microenvironment in vitro. Our data demonstrate that palmitate-mediated NLRC4 upregulation was mediated mainly through TLR4, but TLR4 is likely less important for palmitate-mediated pro-IL-1β expression in TAMs. This suggests that the regulatory mechanisms of NLRC4 and pro-IL-1β by palmitate could be different. Nevertheless, our study suggests that NLRC4 is crucial for activating caspase-1 and IL-1β maturation in tumors with NAFLD. Our data also show that palmitate induced VEGF expression through NLRC4 and that IL-1β stimulation upregulated VEGF expression in TAMs. Together with the data showing that decreased VEGFA expression and endothelial cell expansion in NLRC4−/− mice, tumor growth enhanced by NAFLD could be mediated by NLRC4- as well as IL-1β-mediated VEGFA production and angiogenesis, an observation that is very similar to our previous study(35). Consistent with the current study, NLRC4 did not play a role in the breast cancer growth in mice fed a normal chow diet, but NLRC4 played a significant role in breast cancer growth enhanced by HFD feeding(35). Moreover, that study demonstrated that the HFD-promoted breast cancer growth is dependent on IL-1β and IL-1β-induced VEGFA production and angiogenesis(35). Because we have also shown that NLRC4 inhibited tumor growth by enhancing the function of anti-tumor CD4 and CD8 T cells in melanoma(34), the role of NLRC4 in tumor growth is context- and tumor type-dependent.
In summary, NLRC4 contributes to metastatic tumor growth specifically in NAFLD. These data suggest the underlying molecular mechanism of metastatic tumor growth may be different between normal and fatty liver conditions. Because cancer patients with underlying fatty liver are increasing, we need to understand the precise molecular mechanisms of metastatic tumor growth in both normal and NAFLD livers in order to treat these patients accurately.
Supplementary Material
Acknowledgements
We thank Ms. Fiona Miao (Department of Medicine at Cedars-Sinai Medical Center) for the technical assistance.
Grant Support: This study was supported by NIH grant R01AA027036 (E.S), R01DK085252 (E.S), 3R01DK107288–02S1 (S.C.L., M.P.), R21AA025841 (E.S, S.C.L.), R01AI118719 (F.S.S) and T32HL134637 (Y.M.Y), American Liver Foundation Irwin M. Arias, MD Postdoctoral Research Fellowship (Y.M.Y), AASLD The Leonard B. Seeff Award for Outstanding Research by Young Investigator (Y.M.Y.), Winnick Research award from Cedars-Sinai Medical Center (E.S.), and the Center for Integrated Research in Cancer and Lifestyle Award by the Samuel Oschin Comprehensive Cancer Institute at Cedars-Sinai Medical Center (E.S.).
Abbreviations
- CRC
colorectal cancer
- DCs
dendritic cells
- HCC
hepatocellular carcinoma
- HFD
high-fat diet
- IL-1Ra
IL-1 receptor antagonist
- LFD
low-fat diet
- MDSCs
myeloid-derived suppressor cells
- NAFLD
non-alcoholic fatty liver disease
- NK
natural killer
- NLRC4
NOD-like receptor C4
- PDAC
pancreatic ductal adenocarcinoma
- TAM
tumor-associated macrophages
- TME
tumor microenvironment
- WT
wild-type
Footnotes
Disclosures: The authors declare that they have no conflict of interest to declare.
REFERENCES
- 1.Zarour LR, Anand S, Billingsley KG, Bisson WH, Cercek A, Clarke MF, Coussens LM, et al. Colorectal Cancer Liver Metastasis: Evolving Paradigms and Future Directions. Cell Mol Gastroenterol Hepatol 2017;3:163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 2003;348:1625–1638. [DOI] [PubMed] [Google Scholar]
- 4.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim GA, Lee HC, Choe J, Kim MJ, Lee MJ, Chang HS, Bae IY, et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. J Hepatol 2017. [DOI] [PubMed] [Google Scholar]
- 6.VanSaun MN, Lee IK, Washington MK, Matrisian L, Gorden DL. High fat diet induced hepatic steatosis establishes a permissive microenvironment for colorectal metastases and promotes primary dysplasia in a murine model. Am J Pathol 2009;175:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Earl TM, Nicoud IB, Pierce JM, Wright JP, Majoras NE, Rubin JE, Pierre KP, et al. Silencing of TLR4 decreases liver tumor burden in a murine model of colorectal metastasis and hepatic steatosis. Ann Surg Oncol 2009;16:1043–1050. [DOI] [PubMed] [Google Scholar]
- 8.Dawson DW, Hertzer K, Moro A, Donald G, Chang HH, Go VL, Pandol SJ, et al. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev Res (Phila) 2013;6:1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulz PO, Ferreira FG, Nascimento Mde F, Vieira A, Ribeiro MA, David AI, Szutan LA. Association of nonalcoholic fatty liver disease and liver cancer. World J Gastroenterol 2015;21:913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamady ZZ, Rees M, Welsh FK, Toogood GJ, Prasad KR, John TK, Lodge JP. Fatty liver disease as a predictor of local recurrence following resection of colorectal liver metastases. Br J Surg 2013;100:820–826. [DOI] [PubMed] [Google Scholar]
- 11.Brouquet A, Nordlinger B. Metastatic colorectal cancer outcome and fatty liver disease. Nat Rev Gastroenterol Hepatol 2013;10:266–267. [DOI] [PubMed] [Google Scholar]
- 12.Kondo T, Okabayashi K, Hasegawa H, Tsuruta M, Shigeta K, Kitagawa Y. The impact of hepatic fibrosis on the incidence of liver metastasis from colorectal cancer. Br J Cancer 2016;115:34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duran Ocak A, Yildirim A, Inanc M, Karaca H, Berk V, Bozkurt O, Ozaslan E, et al. Hepatic steatosis is associated with higher incidence of liver metastasis in patients with metastatic breast cancer; an observational clinical study. J BUON 2015;20:963–969. [PubMed] [Google Scholar]
- 14.Molla NW, Hassanain MM, Fadel Z, Boucher LM, Madkhali A, Altahan RM, Alrijraji EA, et al. Effect of non-alcoholic liver disease on recurrence rate and liver regeneration after liver resection for colorectal liver metastases. Curr Oncol 2017;24:e233–e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramos E, Torras J, Llado L, Rafecas A, Serrano T, Lopez-Gordo S, Busquets J, et al. The influence of steatosis on the short- and long-term results of resection of liver metastases from colorectal carcinoma. HPB (Oxford) 2016;18:389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amptoulach S, Gross G, Kalaitzakis E. Differential impact of obesity and diabetes mellitus on survival after liver resection for colorectal cancer metastases. J Surg Res 2015;199:378–385. [DOI] [PubMed] [Google Scholar]
- 17.Shen Z, Ye Y, Bin L, Yin M, Yang X, Jiang K, Wang S. Metabolic syndrome is an important factor for the evolution of prognosis of colorectal cancer: survival, recurrence, and liver metastasis. Am J Surg 2010;200:59–63. [DOI] [PubMed] [Google Scholar]
- 18.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013;499:97–101. [DOI] [PubMed] [Google Scholar]
- 19.Gomes AL, Teijeiro A, Buren S, Tummala KS, Yilmaz M, Waisman A, Theurillat JP, et al. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016;30:161–175. [DOI] [PubMed] [Google Scholar]
- 20.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer 2009;9:239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013;144:512–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maina V, Sutti S, Locatelli I, Vidali M, Mombello C, Bozzola C, Albano E. Bias in macrophage activation pattern influences non-alcoholic steatohepatitis (NASH) in mice. Clin Sci (Lond) 2012;122:545–553. [DOI] [PubMed] [Google Scholar]
- 23.Dey A, Allen J, Hankey-Giblin PA. Ontogeny and polarization of macrophages in inflammation: blood monocytes versus tissue macrophages. Front Immunol 2014;5:683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014;59:2034–2042. [DOI] [PubMed] [Google Scholar]
- 25.Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, Stroncek DF, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014;26:549–564. [DOI] [PubMed] [Google Scholar]
- 27.Okamoto K, Ishida C, Ikebuchi Y, Mandai M, Mimura K, Murawaki Y, Yuasa I. The genotypes of IL-1 beta and MMP-3 are associated with the prognosis of HCV-related hepatocellular carcinoma. Intern Med 2010;49:887–895. [DOI] [PubMed] [Google Scholar]
- 28.Qin Y, Ekmekcioglu S, Liu P, Duncan LM, Lizee G, Poindexter N, Grimm EA. Constitutive aberrant endogenous interleukin-1 facilitates inflammation and growth in human melanoma. Mol Cancer Res 2011;9:1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008;14:408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010;139:323–334 e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009;15:1170–1178. [DOI] [PubMed] [Google Scholar]
- 32.Eisenbarth SC, Flavell RA. Innate instruction of adaptive immunity revisited: the inflammasome. EMBO Mol Med 2009;1:92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dupaul-Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A, Rodrigue-Gervais IG, et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 2015;43:751–763. [DOI] [PubMed] [Google Scholar]
- 34.Janowski AM, Colegio OR, Hornick EE, McNiff JM, Martin MD, Badovinac VP, Norian LA, et al. NLRC4 suppresses melanoma tumor progression independently of inflammasome activation. J Clin Invest 2016;126:3917–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, Xie Q, et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun 2016;7:13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006;116:3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, Yoo W, et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun 2017;8:2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huo T, Cao J, Tian Y, Shi X, Wu L, Zhang M, Wong LL, et al. Effect of Concomitant Positive Hepatitis B Surface Antigen on the Risk of Liver Metastasis: A Retrospective Clinical Study of 4033 Consecutive Cases of Newly Diagnosed Colorectal Cancer. Clin Infect Dis 2018;66:1948–1952. [DOI] [PubMed] [Google Scholar]
- 39.Han EC, Ryoo SB, Park JW, Yi JW, Oh HK, Choe EK, Ha HK, et al. Oncologic and surgical outcomes in colorectal cancer patients with liver cirrhosis: A propensity-matched study. PLoS One 2017;12:e0178920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiou WY, Chang CM, Tseng KC, Hung SK, Lin HY, Chen YC, Su YC, et al. Effect of liver cirrhosis on metastasis in colorectal cancer patients: a nationwide population-based cohort study. Jpn J Clin Oncol 2015;45:160–168. [DOI] [PubMed] [Google Scholar]
- 41.Augustin G, Bruketa T, Korolija D, Milosevic M. Lower incidence of hepatic metastases of colorectal cancer in patients with chronic liver diseases: meta-analysis. Hepatogastroenterology 2013;60:1164–1168. [DOI] [PubMed] [Google Scholar]
- 42.Murono K, Kitayama J, Tsuno NH, Nozawa H, Kawai K, Sunami E, Akahane M, et al. Hepatic steatosis is associated with lower incidence of liver metastasis from colorectal cancer. Int J Colorectal Dis 2013;28:1065–1072. [DOI] [PubMed] [Google Scholar]
- 43.Hayashi S, Masuda H, Shigematsu M. Liver metastasis rare in colorectal cancer patients with fatty liver. Hepatogastroenterology 1997;44:1069–1075. [PubMed] [Google Scholar]
- 44.Karube H, Masuda H, Hayashi S, Ishii Y, Nemoto N. Fatty liver suppressed the angiogenesis in liver metastatic lesions. Hepatogastroenterology 2000;47:1541–1545. [PubMed] [Google Scholar]
- 45.Cui YL, Li HK, Zhou HY, Zhang T, Li Q. Correlations of tumor-associated macrophage subtypes with liver metastases of colorectal cancer. Asian Pac J Cancer Prev 2013;14:1003–1007. [DOI] [PubMed] [Google Scholar]
- 46.Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol 2010;185:4912–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010;32:379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 2010;207:1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, Lam V, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis 2011;17:1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med 2013;19:57–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.