


Abstract
The NF-κB family of dimeric transcription factors regulates transcription by selectively binding to DNA response elements present within promoters or enhancers of target genes. The DNA response elements, collectively known as κB sites or κB DNA, share the consensus 5′-GGGRNNNYCC-3′ (where R, Y and N are purine, pyrimidine and any nucleotide base, respectively). In addition, several DNA sequences that deviate significantly from the consensus have been shown to accommodate binding by NF-κB dimers. X-ray crystal structures of NF-κB in complex with diverse κB DNA have helped elucidate the chemical principles that underlie target selection in vitro. However, NF-κB dimers encounter additional impediments to selective DNA binding in vivo. Work carried out during the past decades has identified some of the barriers to sequence selective DNA target binding within the context of chromatin and suggests possible mechanisms by which NF-κB might overcome these obstacles. In this review, we first highlight structural features of NF-κB:DNA complexes and how distinctive features of NF-κB proteins and DNA sequences contribute to specific complex formation. We then discuss how native NF-κB dimers identify DNA binding targets in the nucleus with support from additional factors and how post-translational modifications enable NF-κB to selectively bind κB sites in vivo.
INTRODUCTION
Nuclear factor κB (NF-κB) is a family of dimeric DNA binding transcription factors that modulate diverse biological responses to cellular stress by regulating the expression of hundreds of effector genes controlling these processes (1). Under the typical conditions of a resting cell, most NF-κB is rendered inactive through its noncovalent association with a class of inhibitor proteins known collectively as IκB. Even under these steady state conditions, however, a small pool of NF-κB remains free of IκB and directs expression of genes that help maintain cellular homeostasis (2,3). Although NF-κB:IκB complexes localize predominantly to the cytoplasm of resting cells, a small fraction can be detected within the nucleus where, by virtue of their inability to bind DNA, the IκB-bound NF-κB are mostly inactive. There are exceptions to this rule since certain NF-κB:IκB complexes do, in fact, exhibit DNA binding activity once the complex-associated IκB is modified post-translationally in response to a specific stimulus (discussed later). The pool of free, active nuclear NF-κB dimers becomes amplified by several-fold, however, upon cell stimulation by a vast number of diverse chemical, biological, or environmental factors.
Five related polypeptide subunits constitute the mammalian NF-κB family: p50/NF-κB1, p52/NF-κB2, p65/RelA, c-Rel and RelB (Figure 1). Each shares high amino acid sequence conservation throughout a large portion of roughly 300 residues in length located near their N-termini and referred to as the Rel Homology Region (RHR). This RHR is responsible for sequence-specific DNA binding, protein dimerization and IκB binding. These proteins can be further divided into two sub-classes: the NF-κB p50 and p52 subunits belong to class I by virtue of their not possessing a transcriptional activation domain (TAD). The other three family members, RelA, c-Rel and RelB (class II), each contain a TAD within their respective C-terminal portions. The NF-κB p50 and p52 subunits are generated by partial proteolytic processing of the inhibitory p105 and p100 proteins, respectively. The p50 and p52 subunits terminate with a short ∼70 residues long segment rich in glycine residues.
Figure 1.

Domain organization of the NF-κB family. (A) The upper panel contains domain schematic diagrams for NF-κB family members. The p50 and p52 subunits (Class I) are generated from larger precursors (p105 and p100, respectively). The precursors, which contain an Ankyrin repeat domain (ARD) in their C-termini, function as inhibitors of NF-κB. The precursors also contain a glycine rich region (GRR) which connects the RHR and ARD and a death domain (DeD). The Rel homology region (RHR) is folded into two distinct domains, N-terminal domain (NTD) and dimerization domain (DD). All NF-κB proteins contain a nuclear localization signal (NLS). NF-κB RelA, RelB and c-Rel subunit proteins (Class II) contain a transactivation domain (TAD). RelB has an N-terminal leucine zipper (LZ) domain. Depicted within the lower left panel is a ribbon diagram representation of the X-ray crystal structure of NF-κB p50 homodimer bound to a κB DNA in two different orientations (PDB ID: 1NFK). The complex displays its characteristic ‘butterfly-like’ shape with the DNA in the center and the two NF-κB monomers embracing it. The three panels to the right deptict solution structures of RelA TAD (TA1 and TA2 in red) bound to different proteins (TFIIH and CBP in grey) (PDB ID: 2LWW, 5URN and 5U4K). (B) Ribbon diagram (above left) and topology map (below left) of the NF-κB DD immunoglobulin (Ig)-like fold (PDB ID: 1MY5). Right, sequence alignment of the DD of the NF-κB subunits. Secondary structures and connecting loops are drawn above the sequences. The NLS is highlighted in purple. Residues critical for dimer formation are colored (invariant residues are blue and conserved residues are red or green).
Since its original discovery in 1986 as a nuclear factor with binding specificity for a DNA element within an intronic enhancer of the kappa light chain gene in B cells (4), NF-κB has served as a treasure trove for the discovery of novel biochemical mechanisms in intracellular signal transduction. As examples, the study of NF-κB regulation has contributed directly to our current understanding of effector modulation and subcellular localization via protein-protein interaction, protein kinase activation and substrate specificity, phosphorylation-dependent ubiquitylation, partial and complete processing of signaling molecules via proteasomes, and the use of alternative ubiquitin linkages as components of innate immune signaling. Many excellent reviews have been written that highlight the molecular mechanisms of these critical processes (5–9). However, fundamental questions remain surrounding the mechanisms by which active NF-κB functions within the nucleus to elevate the expression of select target genes. In this review, we focus on mechanistic details of DNA binding by NF-κB dimers with emphasis on correlating current genome-wide and cellular observations to in vitro biochemical as well as structural and biophysical studies.
NF-κB PROTEIN STRUCTURE
The 3D structures for the RHR of all five mammalian NF-κB subunits have each been determined by X-ray crystallography (10–16). As predicted from their high degree of primary amino acid sequence similarity, their folded structures are also highly conserved (Figure 1A). The RHR contains two separately folded domains: an N-terminal domain (NTD), which is primarily responsible for sequence-specific DNA binding, and a C-terminal dimerization domain (DD). A short, ∼10 amino acids long linker connects the two folded domains. A final conserved stretch of ∼20 amino acids C-terminal to the DD, which contains the type I nuclear localization signal (NLS), is disordered in free or DNA-bound NF-κB but converts to an alpha-helical secondary structure upon IκB binding (17–19).
Each DD adopts a rendition of the common immunoglobulin (Ig)-like fold in which one four-stranded anti-parallel β-sheet packs against another three-stranded anti-parallel β-sheet (Figure 1B) (20). Unlike the Ig domains of antibodies, no cysteinyl disulfide bonds stabilize the folded DD. NF-κB dimerization results from the juxtaposition of two Ig-like dimerization domains with C2 symmetry (20,21). Although assembly of fifteen distinct NF-κB homo- and heterodimers is possible through the pairwise combination of five NF-κB subunits, not every potential dimer is observed in vivo (Figure 2). Sequence variations at the dimer interface support a mechanism for preferential homo- and heterodimer formation through amino acid side chain complementarity (Figure 1B) (22–24). But residues outside of the subunit interface also play import roles in modulating dimer stability and dictating preferred dimer combinations (21,23,25,26). The precise mechanism by which these distal residues function through space to control NF-κB subunit dimerization remains unclear. In addition to the direct and indirect effects of specific amino acids within the DD, regulated synthesis/degradation of individual subunits and preferential binding by IκB inhibitors also serve to bias the assembly of particular NF-κB dimers within the context of the cell (27).
Figure 2.

NF-κB dimers. Cartoon representations of all possible NF-κB homo- and heterodimer combinations. Dimeric NF-κB proteins involved in canonical signaling are colored red with the ubiquitous p50:RelA heterodimer bolded for emphasis (29). Dimers that function in response to non-canonical (alternative) NF-κB signaling are colored in green with p52:RelB, the predominant NF-κB dimer of this pathway, darkened for emphasis (38). NF-κB dimers not observed in cells are colored in light grey (26).
Of the NF-κB dimers with potential to activate gene transcription, the p50:RelA heterodimer is ubiquitous and is involved in most biological activities associated with NF-κB (28,29) (Figure 2). The RelA:RelA homodimer is less abundant, but also plays critical roles. Indeed, RelA:RelA can compensate for the loss of p50:RelA heterodimer in p50 null mice. Consequently, despite their increased susceptibility to infection, p50 subunit knockout mice survive into adulthood (30). Like RelA, c-Rel preferentially forms two dimers: p50:c-Rel heterodimer and the c-Rel:c-Rel homodimer (31). c-Rel protein expression has been analyzed via the Human Protein Atlas Database and was shown to be expressed in hematopoietic cells as well as in cardiac tissue, hepatocytes and keratinocytes (www.proteinatlas.org/ENSG00000162924-REL/tissue) (32). RelB also preferentially forms two dimers, though both p50:RelB and p52:RelB are heterodimers (16,23,33). Whereas p50:RelA is the principal end product of induced NF-κB transcriptional activity via canonical signaling, the p52:RelB heterodimer becomes active as a result of signaling through the ‘non-canonical’ (also known as ‘alternative’) NF-κB pathway (34–38). Finally, though they lack C-terminal TADs and, consequently, inherent transcriptional activation potential, both p50:p50 and p52:p52 homodimers can activate or inhibit expression of select target genes through their association with IκB proteins Bcl-3, IκBζ and IκBNS. Gene knockout studies in mice indicate complicated, context dependent in vivo roles for these IκB proteins in modulating NF-κB-dependent gene expression (39–41). Though structurally similar to prototypical IκB inhibitors, Bcl-3, IκBζ and IκBNS interact only weakly with RelA, c-Rel or RelB (42–44). On account of their propensity to accumulate in the nucleus, Bcl-3, IκBζ and IκBNS are referred to collectively as ‘nuclear IκB’.
A STRUCTURAL PERSPECTIVE ON DNA RECOGNITION BY NF-κB
κB DNA in vitro and in vivo
NF-κB dimers bind with sequence specificity to double-stranded DNA elements that vary in length from between 5 and 11 base pairs (bp). Early comparisons of the first DNA sequences demonstrated that NF-κB binds to the consensus sequence: 5′-GGGRNWYYCC-3′, where R = A or G; N = A, C, G, or T; W = A or T; and Y = C or T (Figure 3) (45). The subsequent identification of new NF-κB DNA binding sites has broadened the consensus to 5′-GGGNNNNNCC-3′ (46). DNA from within gene enhancer regions that meet this later consensus and that can be shown experimentally to drive NF-κB-dependent reporter gene expression are termed ‘κB DNA’ or ‘κB sites.’
Figure 3.

Classification of different types of κB DNA sequences. Top panel, the κB DNA sequence, as defined during the late 1980s, is labeled as ‘Early Consensus.’ This consensus was ‘Broadened’ in the late 1990s after the discovery of NF-κB binding to novel sequences. The central base pair is highlighted in red. Conserved flanking residues are underlined. Variable central portion residues are boxed in black. Middle panel, κB DNA sequences can be sorted into three classes with respect to the nature of the nucleotide sequences within the central portion. The central base pair is highlighted in red. Bottom panel, two examples of NF-κB target genes display that only half consensus κB DNA (48). Nucleotides that do not match with the κB DNA consensus sequence are boxed in red.
The critical feature of the consensus κB DNA sequence is the presence of a series of G and C nucleotide bases at the 5′ and 3′ ends, respectively, while the bases at the central portion display greater variation. Assuming the flanking G/C regions remain constant, the observed sequence variation within the central portion leads to three broad classifications of sequences: (i) sequences rich in AT, wherein all five central bp are A or T, (ii) sequences rich in GC and (iii) sequences with mixed AT and GC. Hundreds of such sequences belonging to all three classes have been identified by sequencing and confirmed experimentally, and the total unconfirmed κB sites detected by computational methods alone number in the thousands (47).
Many of these hypothetical κB sites exhibit significantly greater sequence variation than allowed by the original consensus κB DNA. Of particular interest among these predicted sequences are a class of κB DNA that display only partial (∼5 bp) consensus such as those present in the promoters of CCND1 (GGGGACTTTT) and CCR7 (GGGGCTTTTT) genes (48). Taking these κB DNA into consideration, an alternative κB DNA ‘half site’ consensus is 5′-GGGRNNYNNN-3′ (Figure 3).
The critical issue to consider is whether the NF-κB DNA response elements in vivo follow the same general features of κB sites characterized in vitro. The widely used genome-wide method for motif identification is chromatin immunoprecipitation coupled to high throughput sequencing (ChIP-Seq). Over the past 15 years, ChIP-Seq studies, notably by Encyclopedia of DNA Elements (ENCODE) Project Consortium, have identified DNA motifs bound by nearly all transcription factors including NF-κB (49–51). Although this method is technically limited in its ability to determine DNA binding motifs with absolute precision (52), it has generated a wealth of valuable data. As expected, the NF-κB dimers associate with consensus κB sites in addition to binding at sites that display only half-site consensus (53–56). Some of these κB sites (GGGGATTTT or GGGGGTTTT) appear to be strong binders on the basis of the z-score of ChIP peaks (56), but they are weaker binding sites for NF-κB p50:RelA heterodimer in vitro. Conversely, several consensus sites are only weakly recognized by NF-κB in vivo. The mechanism by which NF-κB binds with either high or low affinity to ‘partial’ κB DNA sequences vs. consensus κB DNA, respectively, in vivo remains unclear. Intriguingly, however, NF-κB also binds to sites that possess no κB consensus (53–55). Therefore, in vitro data do not fully capture the complexity of DNA recognition by the NF-κB dimers in vivo. We discuss these aspects of NF-κB-mediated gene regulation in vivo later in this review.
NF-κB:DNA complexes
Successful determination of 3D structures of several NF-κB RHR dimers in complex with diverse κB DNA by X-ray crystallography has helped establish the chemical principles for sequence specific DNA recognition by NF-κB (10,11,13–16,33,57–62). In general, κB DNA is pseudo-symmetric and the RHR of each NF-κB monomer binds to one κB DNA half site. In its DNA-bound conformation, the NF-κB subunit NTD, which also adopts a derivative Ig-like fold, positions itself such that an extensive basic (positively charged) surface counteracts the acidic phosphate-ribose DNA backbone. Movement of the NTD relative to the DD is afforded by virtue of the short stretch of 10 or so amino acids that link the two domains. This potential for independent domain motion allows for the RelA NTD to move ∼40 Å and to rotate by nearly 180° upon binding the IκBα inhibitor protein, as evidenced by NF-κB:IκBα complex X-ray crystal structures (17,18).
Amino acid side chains from the immunoglobulin (Ig)-like NTD and the interdomain linker (loop L3) of each NF-κB RHR mediate all of the direct contacts to specific DNA bases (Figure 4). The NF-κB dimer interface remains unchanged upon κB DNA binding, and several additional DNA backbone interactions involve residues from both the NTD and DD. The mode of DNA binding by the NF-κB RHR was unique at the time of its determination because all of the DNA contacts were mediated by amino acids on loops that connect β-strand elements of secondary structure. The arrangement of the NF-κB dimer within the major groove of one entire turn of DNA gives rise to a global structure that is reminiscent of a butterfly with a DNA ‘body’ and a pair of RHR ‘wings.’ The short, conserved NLS-containing portion of the RHR that is immediately C-terminal to the DD is disordered when present in NF-κB constructs used for X-ray crystal structure determination in complex with DNA (14).
Figure 4.

NF-κB residues that participate in sequence-specific DNA contacts. Left, ribbon diagram representation of RelA homodimer (PDB ID: 2RAM) highlighting loops L1 (cyan) and L3 (green). These two loops contribute all amino acid side chains that contact DNA bases. Right, the amino acid sequences and numbering for loops L1 and L3 are shown for all five murine NF-κB subunits. Filled circles above mark positions of identically conserved DNA base-contacting residues. Red letters correspond to nonidentical residues that either participate in DNA base-contacts in all cases (denoted by circle) or in specific circumstances, as described in the text.
NF-κB recognition of κB DNA at the 5′-end
A set of conserved amino acid residues that are present within a large loop that connects the first two β-strands of the NTD (referred to as loop L1) directly contact bases within κB DNA (63). In the p50 subunit these residues are Arg54, Arg56, Tyr57, Glu60 and His64 (murine amino acid numbering will be used throughout this report) (Figures 4 and 5A). The two Arg, the Tyr and the Glu are each found in all NF-κB subunits. His64, which is conserved in p52 (His62 of human p52) interacts with the 5′-G of the consensus κB DNA sequence. Substitution at this position with Ala in c-Rel, RelA and RelB allows for variance in the identity of this base position and explains why the consensus DNA half-site preferred by these NF-κB subunits is one base shorter than for p50 or p52 (Figure 5B). Consequently, within the context of the biologically relevant NF-κB heterodimers, p50 and p52 subunits preferentially bind a 5 bp long 5′-half site beginning with 5′-GGG while RelA, RelB or c-Rel subunits select for 4 bp half sites that begin 5′-GG. As discussed later, the RelA:RelA homodimer and possibly c-Rel:c-Rel can accommodate 5′-GGGRR if presented with κB DNA containing such a half site. A central bp, which is nearly always A:T, is not contacted by either subunit of the NF-κB dimer. This suggests that p50:p50 or p52:p52 homodimers bind preferentially to an 11 bp κB DNA composed of two 5 bp half sites separated by a central A:T bp while RelA, RelB and c-Rel select for 9 bp κB DNA (two 4 bp half sites and a central A:T bp). This model agrees perfectly with the original observation that NF-κB p50:RelA heterodimer binds to a 10 bp κB DNA from the enhancer of the immunoglobulin kappa light chain gene (4). The non-contacted central A:T bp provides a convenient reference point for analyzing base-specific interactions by NF-κB subunits on κB DNA half sites. For example, the extreme 5′-G that is contacted by the p50 subunit His64 occupies positions at ±5 bp from this central A:T origin. The G:C bp that occupy positions ±4 and ±3 are contacted similarly by identical loop L1 residues in each of the NF-κB subunits. The two signature arginines (Arg56 and Arg54 in p50) directly contact the two G bases and the invariant glutamate (Glu60 in p50) contacts the paired C base from the complementary strand at the ±3 positions. Recognition of both nucleotide bases of the G:C bp at the ±3 position indicates that it plays a more critical role than either of the G:C bp at the ±5 and ±4 positions (Figure 5B).
Figure 5.

Variable modes of NF-κB:DNA recognition. (A) NF-κB binds to κB DNA as a dimer and each monomer recognizes one half site. (B) Standard recognition of κB DNA at the 5′-end by NF-κB. The conserved NF-κB subunit loop L1 residues involved in direct contacts with DNA bases are indicated. Arg35 (murine RelA numbering) contacts the G base at the -4 position and Arg33 contacts the G at -3. His64 or His62 are specific only to p50 and p52 subunits, respectively. (C) NF-κB recognition of κB DNA at the central bases. Left, different modes of DNA binding (for RelA and c-Rel vs. p50 and p52) are shown. Right, table summarizing how the presence of specific nucleotides at certain central positions affect NF-κB:DNA binding. (D) κB DNA sequence induces NF-κB binding with distinct modes. R41/R125 of RelA/RelB binds the G:C bp at –5 position of a 5 bp half-site. R187 of RelA cannot bind the –2 position of the same half-site.
NF-κB binding to the central bases of κB DNA
The nucleotide bases at positions ±2 and ±1 of κB DNA vary to a considerably greater degree than the flanking bases at positions ±3 to ±5. In the X-ray crystal structure of NF-κB p50:RelA heterodimer in complex with κB DNA from the immunoglobulin kappa light chain gene (14), an arginine residue (Arg187) contained within the flexible loop L3 region of the RelA subunit crosses over to the homologous complementary strand and contacts the T base of an A:T bp at position ±2 (Figure 5C). The c-Rel subunit also contains Arg at this position (15). The corresponding residue is a lysine in p50 (Lys241 of murine p50), p52 and RelB. The lysine residue of p50 and p52 can interact either with the complementary T of a ±2 A:T bp or with a G base from a G:C bp at the same location (16,33). Thus, p50 and p52 can accommodate either A:T or G:C bp at this position. Interestingly, the corresponding lysine in RelB (Lys274 in murine RelB) does not contact DNA. Rather, it engages in an ion pair interaction with the side chain of nearby Asp272. This suggests that RelB is more tolerant than other NF-κB subunits to DNA base sequence diversity at the ±2 position of its κB DNA targets (33,64). This result agrees well with a study reported by Britanova, et al., in which optimal binding sites for RelB:p52 were identified by a random site selection method. The investigators concluded that the preferred RelB:p52 binding sequence mirrors that of the classical p50:RelA consensus, bringing into question the existence of unique RelB:p52 specific binding sites (65).
Base pairs at κB DNA position ±1 are not contacted by either NF-κB RelA or c-Rel subunits (Figure 5C). Lys241 from p50 and Lys221 of murine p52 can contact the ±1 bp with a preference for A bases (10,11,13). An invariant Tyr on loop L1 (Tyr57 in murine p50 and Tyr36 in murine RelA) crosses over and stacks against the paired bases on complementary strand at both ±1 and ±2 positions (63). This mode of interaction is favored by the presence of two successive T bases through contact with their exocyclic 5-methyl groups. Although a Phe at the same position could maintain these stacking interactions, the loop L1 Tyr also mediates hydrogen bonding through its hydroxyl group to a DNA backbone phosphate making Tyr the preferred residue for recognition and binding of the central bases. Two C bases or a T and C can also accommodate binding by the loop L1 Tyr, but an A or G base at either position is unfavorable.
The importance of this Tyr in κB DNA sequence selection is illustrated by the popularity of the sequence AAATT or AATTT (the central bp is underlined) at the central 5 positions in κB DNA targeted by RelA and c-Rel homodimers. It seems likely that the favorable interaction between Tyr and neighboring pyrimidine bases near the center of the 9 bp κB sites compensate for the fact that NF-κB RelA and c-Rel subunits each contact fewer flanking GC bp than either p50 or p52. The importance of two neighboring pyrimidines at the complementary strand ±1 and ±2 positions is further reinforced by the observation that they are present in at least one half site of all functional κB sites known to date, even if the second half site exhibits significant degeneracy. By contrast, otherwise ideal κB DNA sequences that lack neighboring pyrimidine bases at these positions, such as GGGATAATCC and GGGATTATCC (central bp is underlined), fail as functional κB sites.
It is not clearly evident from the structural studies why position 0 is dominated by AT bp. It seems likely that this might be necessary to convey the proper DNA bending and/or dynamic characteristics (discussed later) required for optimum NF-κB:DNA complex formation. Interestingly, despite the pseudo-symmetry in κB sites with identical half sites, NF-κB homodimers bind DNA asymmetrically. This is clearly demonstrated in the co-crystal structure of p52:p52 homodimer bound to a κB DNA of sequence GGGGATTCCCC (13). This DNA contains two identical half sites and only the central base-pair (underlined) breaks the otherwise perfect symmetry. However, one p52 subunit from the homodimer participates in many more DNA ribose-phosphate backbone contacts than the other. Non-uniformity in DNA bending and resultant asymmetrical positioning of the NTD on different DNA half sites might explain the asymmetry of the two half complexes.
κB DNA sequence induces distinct NF-κB binding modes
While the initial X-ray crystal structures together with accompanying in vitro and cellular biochemical studies supported DNA binding according to the rules described above, subsequent structural analyses revealed that alternate binding modes are available to RelA and RelB (33,58,66). As previously discussed, both RelA and RelB bind preferentially to 4 bp 5′-GGAA-3′ type half-sites. However, slight rearrangement of side chain geometries together with modest movement of the entire, folded N-terminal domain allows both of these NF-κB subunits to also bind a 5 bp 5′-GGGAA-3′ half-site (Figure 5D). In other words, like the p50 and p52 subunits, RelA and RelB subunits are capable of contacting the ±5 G in spite of their lack of a histidine at the position equivalent to His64 of p50 (16,33). This is accomplished when a nearby arginine (Arg41 of RelA and Arg125 of RelB) alters its conformation to contact the guanine at position ±5. However, this does not occur without disrupting the ability of the interdomain linker arginine residue (Arg187 of RelA) to contact the A:T bp at ±2 positions in all 4 bp half sites. The switch by RelA and RelB to binding outer versus central bp in a 5 bp half site is probably the result of three factors: the differential strength of the contact between Arg41/125 and G-5 and between Arg187/Lys274 and T-2 of A:T, structural difference between 5′-SGGAN-3′ (S = C, T or A) versus 5′-GGGAN-3′ half sites on account of relative dynamic behavior as discussed later, and the preferences for the adoption of preferred conformations by the protein side chains involved. Therefore, NF-κB proteins have evolved alternative stable conformations that allow for binding of a variety of DNA sites by accommodating DNA sequence and conformational variations. Although it has not yet been captured in X-ray crystal structures, we expect c-Rel to be capable of similar alterations in conformation in order to accommodate binding to different classes of κB DNA.
Stabilization of NF-κB:DNA complexes through NF-κB subunit inter-domain interactions
It is well established that amino acid residues located far from the interaction surface of a protein can contribute indirectly to modulate a binding interface and NF-κB is not an exception (67). This becomes clear upon comparison of DNA binding by homologous pairs of NF-κB homodimers such as p50:p50 and p52:p52 or RelA:RelA and c-Rel:c-Rel. In both pairs, all DNA contacting residues are identical. However, each dimer binds DNA in a unique manner. In vitro selection experiments have revealed a preference for each NF-κB homodimer toward a subset of κB sites (68). It is difficult, however, to pinpoint precisely which distal residues affect binding or by what mechanisms they exert their influence over DNA sequence selectivity.
Additional cellular factors that interact with NF-κB can have a profound effect on DNA recognition and binding, even when the site of interaction with NF-κB is far from its DNA binding surfaces. The effect is mutual, as NF-κB binding to DNA can also influence distal NF-κB:protein interactions. This suggests that the assembly of NF-κB into large multiprotein enhanceosome complexes can be either facilitated or inhibited by subtle changes in DNA conformation. One mechanism by which co-factor interaction could directly influence DNA binding affinity is illustrated by the interplay of two loops within the DNA binding RHR of NF-κB subunits: one from the DD and the other from the NTD. The loop that connects β-strands f and g in the NF-κB DD (the ‘βf-βg loop’) projects toward κB DNA but does not directly contact it (63). Two conserved acidic residues (Asp267 and Glu269 in chicken c-Rel) are located within this loop and reside near the DNA in the complex between c-Rel:c-Rel homodimer and the IL2-CD28RE κB DNA complex. Although these residues do not directly contact DNA, their proximity and combined electrostatic surface potential likely repel DNA and weaken binding (15). These negative charges are neutralized, however, by one of the Arg residues from loop L1. Loop L1 is the same loop that contributes five of the six base contacting residues. Loop L1 can be divided into three parts: N-terminal front, N-terminal back, and the C-terminal portion. The C-terminal portion of loop L1 is flexible and can contact the DNA backbone of nucleotides flanking the κB sequence. The N-terminal front and back, on the other hand, adopt stable structures that remain unchanged both in DNA bound and unbound states (15). Surface residues from the front portion contribute the DNA base-contacting residues. It is an Arg from the back surface of this ordered N-terminal portion of loop L1 that contacts the acidic residues of the βf-βg loop. Interestingly, although these residues are conserved across NF-κB family subunit proteins, not all NF-κB:DNA complex crystal structures exhibit this interaction. We suggest that DNA conformational differences play a role in dictating RHR inter-domain interactions that serve either to augment or weaken DNA binding affinity. In the case of oncogenic v-Rel, a viral homologue of c-Rel from the avian Reticuloendotheliosis virus, two core residues within the rigid part of loop L1 are mutated. Mutation of these two residues is at least partly responsible for the observed altered DNA binding profiles by v-Rel as compared to c-Rel (69). Finally, the βf-βg and L1 loops contain sites for post-translational modification, which also regulates NF-κB:DNA recognition as discussed below. Taken together, these observations suggest that subtle changes to the interactions between domains of the RHR, either as a direct result of binding to proteins or indirectly via factors that modulate target DNA conformation, serve to influence NF-κB:DNA complex stability.
The development and implementation of high throughput techniques to study how transcription factors bind to target DNA sequences have been extraordinarily useful in providing new insights to the field of DNA binding by NF-κB. Some of these approaches include SELEX (systematic evolution of ligands by exponential enrichment), PBM (protein-binding microarrays), and EMSA-Seq. Such genome-wide analyses have led to the identification of a plethora of new non-consensus NF-κB DNA binding motifs as well as serving to classify NF-κB binding sites, including those that contain only a single consensus κB half-site, revealing the intrinsic plasticity of NF-κB dimers (64,70). In general, results from these approaches agree very well with data obtained through structural and in vitro biochemical methods.
DNA CONFORMATION AND NF-κB BINDING
Sequence-dependent DNA conformations
DNA duplexes are not static entities that simply present themselves to proteins to be read and then seed assembly of multiprotein complexes at specific sequences. Rather, in solution DNA is intrinsically dynamic on many levels and time scales. The movement of DNA through its different conformational states is continuous and is influenced by, but not completely dependent upon, its nucleotide sequence. Nuclear magnetic resonance (NMR) and X-ray crystal structures of different κB DNA (21,71–72) together with molecular dynamics simulations of DNA in its unbound state (73) have revealed insights that highlight the complexity of the NF-κB:DNA recognition process.
NMR solution studies of free κB DNA from the long terminal repeat (LTR) of proviral DNA encoding HIV-1 (HIV-κB DNA) as well as two different mutant versions of the same sequence revealed that phosphate backbone dynamics vary not only between different contiguous base-pairs but also between the same contiguous base-pairs within different contexts (71,72). This complicated backbone dynamic dictates local major and minor groove depth and width as well subtle differences in base functional group location and geometry. For example, it has been observed that the sequence of nucleotides that flank a κB site impact the overall groove chemistry of the κB site in ways that affect p50:RelA heterodimer binding (44,74). The significance of flanking sequences in modulating DNA target site structure and transcription factor (TF) binding has also been reported for other systems such as the glucocorticoid receptor (GR) (75).
Microsecond time-scale molecular dynamics (MD) simulations of a 20 bp long DNA fragment of the IL-2 promoter containing the IL-2 κB site revealed two striking features: the –2 position A nucleotide of the IL-2 κB site (AGAAATTCC) begins intercalative stacking with the cross-strand bases, disrupting the base pairing of the central bases and consistently leading to thymine of the central A:T base pair flipping out of the DNA helix (73) (Figure 6). In light of what is known about interactions that stabilize NF-κB:DNA complexes, these massive structural variations in a κB site must affect NF-κB binding. Although we do not know precisely how DNA conformations impact NF-κB binding, it is likely that the AT-rich sequence typical within central portion of κB DNA facilitates both cross-stand base stacking and base-flipping processes. Many κB sites that favor binding to RelA:RelA or c-Rel:c-Rel contain four or five A:T bp at their center. It is tempting to speculate that these DNA rely upon the dynamic inherent to such sequences either to facilitate initial binding by these dimers or to hinder binding by other dimers.
Figure 6.
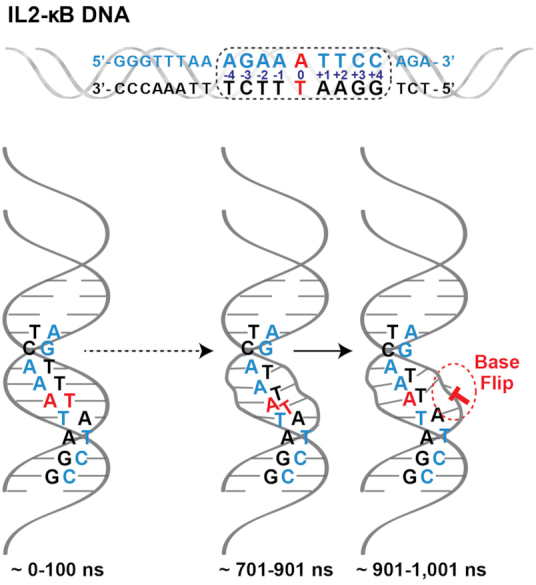
Molecular dynamics (MD) of IL-2 κB DNA in its unbound state. Upper panel, the 20 bp DNA sequence from the promoter for IL-2 is depicted with its consensus κB DNA site enclosed in the dashed outline box. Bottom panel, a cartoon representation of changes that occur consistently during the time course of the MD simulation within the central A:T portion the IL-2 κB DNA that lead to flipping of the 0 position T base. Time ranges are denoted in nanoseconds (ns).
Single nucleotide changes in κB DNA can alter gene expression
One of the fascinating observations involving gene regulation by NF-κB is the ability of a particular NF-κB dimer to initiate different gene expression programs upon binding κB sites that differ at only a single bp. This phenomenon was first described by Leung et al. in the Baltimore Laboratory where the authors showed that the RelA:RelA homodimer activates transcription of genes within a promoter containing the κB DNA sequence GGGAATTTCC but represses gene transcription when the κB DNA sequence is GGGAAATTCC (76). These two κB DNA differ only by a single bp (A:T to T:A) at the 0 position. RelA:RelA homodimer is recruited to the two sites and binds to both sequences with similar affinity both in vivo and in vitro. As previously discussed, the central bp that separates the half-sites has not been observed to participate in contacts with protein in any NF-κB:DNA complex structure studied. How such a small, seemingly inconsequential change in sequence might lead to such severe functional differences remains unclear. However, the importance of seemingly minor variations in κB DNA sequence to transcriptional regulation has since been observed in other NF-κB dimers. In 2012, researchers observed that p52:p52 homodimer could differentiate between A:T and G:C bp at the central position (48). In both instances, the p52:p52 binds its DNA target but only in the case of the central G:C bp can the homodimer also associate with its protein co-regulator Bcl-3 and activate transcription by recruiting histone acetyltransferases. When the central bp is A:T, the same p52:p52:Bcl-3 complex represses gene transcription via recruitment of histone deacetylases. Since most κB sites include A/T-centric κB sites, the p52:p52:Bcl-3 complex down regulates most genes that are preferentially activated by RelA dimers. On the other hand, the p52:p52:Bcl-3 complex appears to be the primary NF-κB activator for a smaller group of genes that contain G/C-centric κB sites within their promoters. RelA-containing dimers bind poorly to these G/C-centric κB sites. Since most NF-κB responsive promoters include multiple κB sites, selection of these κB sites will be guided generally by the relative availability of specific NF-κB dimers and the affinities of the respective NF-κB:DNA complexes. But, as these studies on the functional consequences of single nucleotide changes attest, select κB DNA sequences can specialize in the recruitment of particular NF-κB dimers with specific transcriptional outcomes.
NF-κB binds to κB DNA with only one half-site consensus
The mutually semi-independent, two-domain modular architecture of the NF-κB subunit RHR endows it with potential to accommodate expanded target DNA sequences. As described previously, both the NTD and DD participate in contacting DNA, although the DD does so nonspecifically through contacts with the DNA phosphate-ribose backbone only. The two domains are linked by a short stretch of roughly 10 residues. Due to the fact that NF-κB dimerization is mediated by relatively stable association between the DD from two subunits, the resultant NF-κB dimers possess three stable structural entities: two NTDs and one platform composed of two DDs. The flexibility of the NTDs relative to the DD platform allows them to move upon encountering non-ideal binding half-sites in order to locate optimal binding surfaces as first shown by Chen et al. (57). If necessary, the NTD can even move so far on a half-site as to interact only with the DNA phosphate-ribose backbone, abandoning any base-specific interactions (57,77) (Figure 7). Consequently, κB DNA that contain only one half-site consensus can serve as NF-κB dimer binding targets of reasonable affinity (61). In these cases, the NTD from one subunit recognizes one consensus DNA half-site with specificity making all possible base contacts while the other contributes positively to complex stability by moving into a position that fails to contact any nucleotide bases directly but allows for non-specific interactions with the DNA backbone. Recent in vivo and in vitro results have confirmed the existence of κB sites with only half-site specificity (56,64,77).
Figure 7.

NF-κB dimer binding to κB sites with only half-site consensus. Left, one monomer makes all specific possible contacts whereas the other monomer fails to specifically contact any nucleotide base. Right, when the non-consensus κB DNA half site includes either GGGG or AAAA, the NF-κB:DNA complex formation is strongly disfavored.
Interestingly, in these cases of NF-κB binding to κB DNA that bear only one consensus half-site the non-specific half-site sequence cannot be random. As discussed previously, the stacking of a Tyr against preferred TT bases on that complementary DNA strand at both ±1 and ±2 positions contributes both specificity and affinity to NF-κB:DNA complexes. Therefore, a non-specific half-site with TT in positions +1 and +2, as in GGGAA A/T TTNN, is favored. Moreover, non-specific half-site sequences with stretches of G or A (for example, GGGAA A/T GGGG or GGGAA A/T AAAA) are not preferred (Figure 7). As discussed in a previous section, DNA base sequences outside the border of the κB DNA consensus can indirectly influence NF-κB binding by altering the conformation and dynamics of the non-specific half-site. A similar mode of binding is observed in tetrameric p53 where the tetramerization domain is flexibly linked to the DNA binding domain. In that system it is observed that one p53 dimer binds DNA with sequence specificity whereas the other dimer binds non-specifically with alternative conformation (78,79). It seems likely that many, if not most, multimeric TFs with multidomain modular structures can accommodate expanded DNA sequences through optimized half-site binding.
NF-κB dimers bind to tandem κB sites
All gene promoters contain binding sites for many transcription factors and often promoters contain multiple binding sites for a single transcription factor family. It was initially proposed that multiple transcription factors bind to their respective sites cooperatively and activate transcription synergistically (80–82). In support of this notion, researchers measured increased levels of reporter gene expression by multiple transcription factors in overexpression systems. In fact, synergistic transcriptional activation through cooperative interactions of transcription factors has been found to be the case in only some instances. In such cases, although cooperative interaction between two transcription factors bound to tandem sites occurs, it is mostly, if not exclusively, the result of two monomeric transcription factors or else a dimer and a monomer binding to a composite DNA response element (83–85). Other studies have also highlighted that the DNA sequence has an active role in determining how TFs bind and act cooperatively to regulate gene expression (86,87). Along these lines, it has been suggested that when two transcription factor binding sites are within sufficiently close proximity, their corresponding flanking regions can overlap and, as a consequence, their final sequences might differ considerably from the expected consensus. Such composite sites would be expected to display relatively low affinity toward their respective transcription factors individually, suggestive of the crucial role of the DNA sequence in transcription factor cooperativity. NF-κB dimers have never been shown conclusively to interact cooperatively with other transcription factors bound to tandem sites. One recent report described an approach to determine cooperative interaction between a large number of TF pairs through the use of selected DNA (86). When the NF-κB p50 subunit was used in this in vitro selection-based method, only p50 itself could be identified as a partner protein.
The most extensively studied system for measuring cooperativity in NF-κB-driven gene expression is the interferon beta (IFN-β) gene promoter, which contains a high affinity κB site adjacent to two IFN-stimulated response element (ISRE) binding sites for transcription factor IRF3/7 (88). Although both TFs bind independently to their respective sites, direct protein-protein interaction between NF-κB the RelA homodimer or p50:RelA heterodimer and IRF3 that facilitates their DNA binding has never been convincingly shown (89). Using solution-based experiments, however, Dragan, et al., were able to observe cooperative binding interactions between the p50 homodimer, IRF3, and DNA (90). In addition to κB and ISRE, the IFN-β promoter also contains a binding site for the AP1 transcription factor family member ATF2/c-Jun. Although attempts have been made to assemble all these transcription factors on an IFN-β promoter fragment in vitro, a stable assembled complex containing all proteins could not be formed (62).
Several NF-κB response genes contain two or more κB sites within their promoters or enhancers where the arrangements of the κB sites do not follow any specific spacing or orientation. The most extensively studied of these promoters is the HIV LTR where two identical κB sites are present with an intervening spacing of 4 bp. It was shown nearly 30 years ago that NF-κB dimers do not bind these sites cooperatively. Yet, they do synergistically activate reporter gene transcription (91,92). That is to say, together the two κB sites drive reporter gene expression at levels higher than the sum of each individual site. Indeed, one study suggested a cooperative binding model based on their observation that two dimers can occupy both sites simultaneously (92). In vitro biochemical experiments suggested an anti-cooperative relationship of binding between the two NF-κB dimers for tandem κB sites (61). It was further showed that expansion beyond two κB sites can further increase levels of reporter gene expression and that the maximum transcriptional threshold is achieved by four tandemly arranged κB sites (92). A relatively recent report examined the relationship between NF-κB concentration, gene expression, and number of κB sites within the regulatory element. They found gene expression was gradual and increased as a function of number of κB sites (93).
The apparent lack of cooperativity between NF-κB dimers in binding to tandem κB sites makes sense since, due to the variability in spacing between pairs of κB sites observed in gene promoters, oriented physical interaction between the dimers bound to neighboring sites is likely not possible. There have been two X-ray crystal structures reported that capture a pair of NF-κB dimers bound to DNA containing tandem κB sites. One contains two NF-κB p50:RelA heterodimers bound to tandem HIV-κB sites and the other includes two NF-κB RelA:RelA homodimers bound to tandem E-Selectin κB sites (66,94). Biochemical experiments revealed that binding of one dimer does not facilitate binding of the second dimer (61). Rather, the in vitro study indicates that binding affinity of a second NF-κB dimer is weakened by the presence of a first bound dimer. In other words, assembly of the dimers on tandem κB sites is anti-cooperative (Figure 8). This is in spite of the fact that there are direct contacts between the two dimers bound to DNA (66,94). Interestingly, although the two κB sites in each DNA fragment are identical in sequence, the complexes form asymmetrically. For example, the RelA:RelA homodimer:E-Selectin κB DNA complex reveals a unique binding mode in which one of the dimers binds its respective κB site suboptimally with several DNA contacts missing. The authors speculate that this mode of binding represents an early step in the process of NF-κB dissociation from DNA (66).
Figure 8.

NF-κB dimers binding to tandem κB sites. Anti-cooperative binding of two NF-κB dimers to tandem κB sites. Only one dimer can stably bind to one site at any given time.
If NF-κB dimers on promoters with close tandem κB sites do not cooperate with one another during the assembly process, and if this is even less likely to be the case when the sites are separated by tens of bp, then why do so many target genes possess multiple NF-κB binding sites? We envision two possible consequences of multiple κB sites. First, a promoter with multiple κB sites increases its probability of successfully recruiting NF-κB. While this at first seems obvious, it raises the intriguing possibility that the binding event, and not necessarily the stable complex, is key to transcriptional activation. Were this the case, then a mechanism that raises the number individual binding events over time would serve to increase overall transcriptional output (Figure 8). The second advantage is that the different κB sites could be NF-κB family member-specific. As explained previously, RelA:RelA homodimer binds poorly to a G/C-centric DNA but with high affinity to A/T-centric DNA, whereas the p52:p52 homodimer in complex with Bcl-3 binds both classes of DNA but with opposite transcriptional outcomes. Thus by simply exchanging dimers, transcriptional output can be prolonged or tempered (95). Under this model, the addition of a particular κB site within a target gene promoter would expand inducible expression of that gene to the specific class of NF-κB that functions through that site and the signaling pathway that mobilizes the corresponding NF-κB.
ALLOSTERIC REGULATION OF NF-κB BINDING TO DNA
Over the past few years, genome-wide experiments have raised some intriguing observations surrounding DNA binding by transcriptional regulatory factors. Among these, that in addition to binding their own consensus sites, most transcriptional activator proteins also bind at places in which there is no consensus site present (96,97). In separate studies, transcription factors have been studied by introducing mutations that target their DNA interacting residues and render them incapable of binding to DNA response elements (98). Surprisingly, when such mutants are then tested for their transcriptional potential, many are still able to engage in promoter-specific binding events and elevate gene expression (99,100). Overall, observations from these studies suggest that a typical transcription factor is able to regulate gene expression in two ways: by directly binding to their cognate DNA response elements, and by associating with DNA indirectly via interaction with another DNA-bound factor. Due to this second mode of action, DNA binding by the transcription factor is not required and, hence, mutations that abolish its DNA binding or even the deletion of the DNA binding domain entirely do not necessarily affect its ability to regulate gene expression (76,101).
As is the case with other transcription factors, NF-κB RelA-containing dimers have been shown to regulate gene expression through both direct and indirect interaction with promoter DNA. The observation that NF-κB could regulate gene expression through an indirect mechanism arose from a genome-wide study reported 15 years ago (54). This study found that nearly one third of RelA genome reading events could not result from direct interaction with DNA since κB sites were absent from those genes. Several subsequent reports have validated this earlier observation and found that NF-κB associates with many promoter/enhancer elements that lack any κB site (55). Consensus binding sites for other transcription factors are often present within the DNA where NF-κB is bound in these cases. The most well studied example came from a RelA ChIP experiment targeting a promoter that contains ISRE (interferon-sensitive response element) sites but not κB sites. The ISRE recruits IRF3/7 transcription factors through direct binding, while NF-κB is tethered indirectly to the DNA via its interaction with IRF3/7. The reciprocal case has also been reported wherein NF-κB was identified bound to a κB site and IRF3 associated with the same DNA indirectly via binding through the NF-κB RelA subunit (101,102). The following sections discuss how additional factors can tether themselves to DNA via NF-κB and why this is an essential component of NF-κB-dependent gene expression.
DNA binding by NF-κB is influenced by protein co-factors
In addition to IRF3, many other factors have been reported to interact with the RelA subunit when NF-κB is bound directly to κB DNA. Many of these ‘other factors’ are themselves DNA binding transcription factors, such as p53, E2F1, FOXM1 and KLF-6 (53,55,103–105). However, several additional factors that are not recognized as TFs, such as the DNA repair protein OGG1, architectural DNA binding protein HMGA1 (formerly HMGI(Y)), RNA binding proteins such as RPS3 and Sam68, and the histone chaperone protein nucleophosmin/NPM1, have also been reported to engage with NF-κB on promoters of target genes (106–109). Gene knockout or knockdown studies have revealed that each of these proteins plays critical roles in gene expression via RelA. However, their effect is selective and only a subset of RelA-regulated genes is influenced by each of these proteins. In vitro experiments have shown that p53, FOXM1, RPS3, Sam68, and NPM1 facilitate DNA binding of RelA dimers (103,106,108,110). These factors interact directly with RelA, often through its DNA binding RHR segment. Although never quantitatively measured, the affinity of interaction between NF-κB and these factors appears to be weak, since their complexes have not been successfully isolated by size exclusion chromatography though their interaction in vitro or in cells has been confirmed by co-immunoprecipitation or other affinity-based pull-down methods. Such protein:protein interactions are not unprecedented in the literature, where it has been reported that many micromolar and even some nanomolar affinity complexes that are readily by identifiable via immunodetection fail to co-purify in the time scale requirements of size exclusion chromatography (111,112). We refer to these protein factors as ‘co-factors’ of NF-κB. The increased DNA binding affinity exhibited by NF-κB in the presence of protein co-factors has been described as site-specific since ChIP showed that NF-κB and co-factors assemble on specific promoters in response to specific stimuli (113). In vitro binding studies concurred with in vivo experiments for many of these proteins and in all cases tested it was shown that the co-factors strengthened NF-κB binding to κB sites (53,108,114,115). Interaction of p53 and FOXM1 with p50:RelA heterodimer was confirmed both in cells and in vitro (103).
It has long been observed that NF-κB dimers do not efficiently bind to κB DNA under physiological ionic strength (116–119). High ion concentrations effectively shield charged/polar groups on the surface of RelA, p50, and DNA and decrease the strength of their interactions. However, in the presence of RPS3 and, to a lesser extent, p53, RelA dimers recover the ability to binding to κB DNA with high affinity (119). Although the precise mechanism for how RPS3 and other co-factors accomplish this is unclear, we can speculate that RPS3 mobilizes or otherwise occupies ions, mitigating their shielding effect near the NF-κB DNA binding surfaces. Furthermore, low affinity but direct contacts between RPS3 and RelA could induce long-range allosteric changes in DNA contacting residues of RelA such that these residues can participate in base-specific contacts with DNA (Figure 9). Protein flexibility test has shown that the segments of both p50 and RelA far away from the DNA binding surface become more flexible upon binding to a κB DNA (120). These distal allosteric sites might be the targets of co-factors affecting DNA binding. Whatever the mechanism, it is clear that diverse protein co-factors function to negate the detrimental ion effect and facilitate formation of NF-κB RelA:DNA complexes. Since many, if not all DNA binding transcription factors exhibit decreased DNA binding as a result of increased ion concentration, as a general rule they all likely require co-factors to facilitate their binding.
Figure 9.

A model of how co-factor stabilizes NF-κB:DNA complex. At physiological ionic strength, ions shield DNA contacting residues along their variable conformational flexibility. Upon binding to a co-factor, dissolved ions may lose some of their ability to shield DNA and/or RelA side-chain conformations might alter themselves to a mode primed to bind DNA.
The observation that many different proteins can function in the nucleus as co-factors to modulate transcriptional activity sheds light on some previously studied interactions between NF-κB and other transcription factors. For example, nuclear hormone receptors, in particular glucocorticoid receptor (GR), are known to inhibit NF-κB activity, consequently behaving as anti-inflammatory agents. Although several mechanisms have been invoked to explain the anti-inflammatory activity of GR, the most critical is likely its ability to block transcriptional activity of RelA-containing NF-κB dimers on DNA (99,101). Upon binding to glucocorticoids, GR can inhibit the effect of co-factor interaction with RelA by removing it from its DNA binding transcription factor partner IRF3, as GR and IRF3 appear to bind RelA through a common surface. In its glucocorticoid-bound state, GR can remove RelA from IRF3 at ISRE sites. Furthermore, GR can suppress inflammation by binding RelA upon κB sites within the promoters of pro-inflammatory target genes. In contrast to its removal of RelA from DNA-bound IRF3, GR inhibits NF-κB-dependent target gene expression without disrupting the RelA:DNA complex. Exactly how GR association inhibits the transcriptional activation potential of RelA even when a RelA-containing NF-κB dimer remains bound to DNA is at present unknown and open to speculation. As mentioned previously, merely binding to DNA does not necessarily give a transcription factor license to activate gene transcription. It is possible that GR binding alters the kinetic rates with which RelA-containing NF-κB dimers associate with and release DNA. If particular DNA binding kinetics were linked to downstream gene regulatory events such as co-activator or co-repressor recruitment, then one could envision how disruption of this timing could alter the transcriptional output without necessarily removing the DNA binding protein. Such kinetic control over enzyme catalytic mechanisms has been proven an effective mechanism employed by many binding protein enzyme inhibitors, such as INK proteins toward cyclin-dependent kinases (Cdk) in cell cycle regulation (121). Alternatively, GR can directly block RelA recruitment of activating components such as p300/CBP or other co-activators independent of its effect on DNA binding kinetics.
Co-factors can influence DNA conformation
Not all co-factors that affect NF-κB binding to DNA do so through direct interactions with NF-κB. The DNA repair protein OGG1, for example, binds to a damaged site (O6-methyl guanine) somewhat distal to the κB site, which is essential for NF-κB recruitment (107). It is likely that upon binding to the damaged DNA, OGG1 alters the structure of the κB site (Figure 10A). In the process, the location and/or context of the modified base are critical. In vitro experiments suggest that modification of G residues in specific positions lead to more efficient recruitment of NF-κB (107). This is another area in which mechanistic details remain to be worked out, but it is possible that both linear distance and relative orientation of the modified base influence its ability to regulate recruitment of NF-κB to κB sites.
Figure 10.

Co-factors affecting NF-κB:DNA complex altering DNA structural conformation. (A) DNA repair protein OGG1 enhances the recruitment of RelA dimers to the κB sites through binding to an O6-methyl guanine damaged site (O6-MeG). Upon OGG1:O6-MeG binding, the κB site structure is altered favoring NF-κB binding. (B) Architectural transcription factor HMGA1 enhances the recruitment of RelA dimer to the IFNβ-κB site. The proposed model shows how HMGA1 transiently bind the DNA minor groove affecting the conformation of the DNA major groove facilitating NF-κB binding.
Architectural transcription factors can also enhance recruitment of RelA dimer to κB sites (Figure 10B). HMGA1, an AT hook binding protein, was shown to increase promoter activity of IFN-β (109). The IFN-β promoter contains multiple HMGA1 binding sites, one of which overlaps with the κB site (IFNβ-κB). IFNβ-κB site contains five A:T bp in the central region, which is also the binding site for HMGA1. Interestingly, both HMGA1 and NF-κB are proposed to bind the same κB site with HMGA1 lining the minor groove side while NF-κB contacts DNA bases through one turn of the major groove. However, on the basis of X-ray crystal structure analysis, it is not possible for both proteins bind to a single κB site simultaneously without significant structural rearrangement. Although the positive role of HMGA1 in NF-κB-mediated gene regulation in vivo is unquestioned, the mechanism of how it facilitates the formation of NF-κB:DNA complexes remains unresolved.
The role of HMGA1 in activation of the immunoglobulin heavy chain enhancer μ during B cell development has been investigated thoroughly (122). HMGA1 displays strong interactions with PU.1 and Ets-1 transcription factors and binds weakly with DNA. These interactions have been proven critical for μ chain gene enhancer activation. Based on these and other experiments, we propose the following model for HMGA1 enhancement of NF-κB dimer binding to DNA. HMGA1 transiently binds through the minor groove at AT-rich sites altering the conformation of the DNA major groove and allowing NF-κB dimers to initiate binding from major groove side. HMGA1 then dissociates upon binding of NF-κB dimers to κB DNA (Figure 10B). In this model the HMGA1 protein acts more as a ‘catalyst’ to accelerate proper NF-κB:DNA complex formation. Several reports also suggest that HMG proteins compete with Histone H1 protein and that, through competitive removal of H1, HMG proteins cause chromatin decompaction facilitating transcription factor access to their DNA response elements (123). Thus, HMGA1 can facilitate DNA binding of NF-κB in at least two ways: by improving access to DNA binding sites and by priming the κB DNA conformation for more efficient NF-κB binding.
The transcription activation domain influences RelA binding to DNA
All structural and most other biophysical characterization of NF-κB:DNA complexes has been performed using the DNA binding RHR. It was believed that the acidic, C-terminal transcription activation domain (TAD) of RelA functions independently in solution and, perhaps, might negatively affect DNA binding through non-specific charge-change interactions between the basic RHR and acidic TAD. Interestingly, early experiments revealed full length RelA that was produced in an in vitro transcription-translation system failed to bind κB DNA. However, deletion of the TAD resulted in enhanced DNA binding by the RelA RHR (124). We have subsequently reproduced those original results and further demonstrated that an independently expressed RelA TAD could act as an inhibitor preventing the RHR from binding to κB DNA (66).
The biophysical and biochemical properties of the RelA TAD have been studied extensively. The RelA TAD can be divided into two parts, TA1 and TA2, based on their ability to interact with partner proteins. The more C-terminal TA1 interacts with Mediator and with components of the basal transcription machinery (125–127). The more N-terminal TA2 binds the CBP/p300 co-activator (Figure 1A). In addition, the KIX domain of CBP/p300 was shown to contact the RelA RHR through a binding interaction that is dependent upon phosphorylation at RelA residue Ser276 (128). Overall, these observations suggested a general mechanism in which CBP/p300 facilitates RelA binding to DNA by neutralizing the inhibitory effect of the highly acidic TAD. However, more recent solution-based experiments reveal that the TAD does not prevent DNA binding by RelA under conditions that approximate physiological cellular environment, even in the absence of co-factors (119). Furthermore, recombinant full length RelA retains its ability to bind DNA, albeit with lower affinity, under high salt conditions that fail to support DNA binding by the RelA RHR alone. The weak binding exhibited by full length RelA increases significantly in the presence of protein co-factors while, surprisingly, most co-factors tested, RPS3 and p53, fail to influence DNA binding by the same RelA in the absence of its TAD (119) (Figure 9). Therefore, the RelA TAD appears to play a fundamental role in DNA binding under physiological conditions. Precisely, how the interplay between the well characterized DNA binding RHR, the complicated regulatory potential of the TAD, and the subtle facilitative nature of protein co-factors results in κB DNA binding and target gene expression remains unclear at this time. It is possible that it influences phase separation and formation of nuclear condensates as has been described recently for other transcription factors such as OCT4 and GCN4 (129). However, this is an intriguing area of investigation that is expected to unravel critical insights into the mechanism of NF-κB:DNA complex formation.
Post-translational modifications affect DNA binding by NF-κB
Several recent reports have shown that methylation and acetylation of arginine and lysine residues within or immediately after RelA RHR modulate gene expression by altering the DNA binding properties of RelA. Residues that have been observed to become modified in response to treatment of cells with pro-inflammatory cytokines include Arg30, Lys37, Lys218, Lys221 and Lys310 (130–133) (Figure 11). Both Arg30 and Lys37 are located within loop L1, which has been discussed previously and is critical for mediating base-specific contacts with κB DNA. Interestingly, neither Arg30 nor Lys 37 are directly involved in DNA contacts (132). Although a detailed mechanism that explains how modification of these residues affects DNA binding remains to be deduced, their positions within of loop L1 suggest that the effect is likely indirect through altering the conformation of nearby loop L1 residues that directly contact DNA. In support of this hypothesis, it is worth pointing out that some amino acid residues from the DNA base contacting loop L1 interact with one another to help stabilize the loop L1 conformation and allow it to contact DNA as a stable, prearranged unit. In the NF-κB p50 subunit, for example, Glu60 bridges Arg54 and Arg56 as together they contact DNA as a structured module. The stability and utility of this folded polypeptide structure were illustrated when it was found to be exploited by RNA in selection binding experiments (134,135). In RelA and c-Rel, a similar Glu brings together one of the two loop L1 Arg residues and the Arg from the interdomain linker. Residues Lys218 and Lys221 have been identified as sites of acetylation upon RelA activation (130,131). These residues are positioned directly above the central base pairs. Although they do not directly contact DNA, acetylation of these lysine residues could extend them closely enough to influence DNA bending at the center of the complex or even participate in direct contacts with DNA bases that prevent efficient binding. Lys310 is present within the flexible, NLS-containing region at the RHR C-terminal end, which affects subcellular localization of RelA and could influence function of the C-terminal TAD. Therefore, it is expected that post-translational modifications of RelA within the RHR will impact DNA binding affinity and their impact could be DNA sequence dependent.
Figure 11.

Post-translational modifications affect NF-κB DNA binding. Acetylation and methylation (left) and phosphorylation (right) of different NF-κB subunits affecting NF-κB dependent transcription through altered DNA binding.
The most extensively studied RelA post-translational modification is the previously mentioned phosphorylation at Ser276. This residue is targeted for phosphorylation by two different kinases, MSK1/2 and protein kinase A (PKA), and its phosphorylation appears to be essential for RelA transcriptional activity (136–139). Mice expressing a version of RelA in which Ser276 is mutated to Ala demonstrate embryonic lethality, a phenotype that is similar to complete knockout of the gene, and exhibit severe developmental defects, most notably in eye development (137). Surprisingly, the Ser276Ala mutant affects expression of only a subset of NF-κB target genes and the mutant RelA binds DNA with near equal affinity as the wild type protein. This suggests that phosphorylated Ser276 (p-Ser276) provides additional functionality to RelA that is required for expression of a select subset of genes including some that are critical for development. One likely possibility is that p-Ser276 facilitates CBP/p300 recruitment, as discussed previously. Therefore, phosphorylation may play a role in changing chromatin dynamics through acetylation activity of CBP/p300. It deserves mentioning that replacement of Ser276 with a phosphomimetic Glu residue results in a RelA mutant that completely fails to bind DNA in vitro (unpublished observation). Together, these results suggest a complicated and incompletely understood role for p-Ser276 in RelA function. One possibility is that only a subset of total cellular RelA undergoes phosphorylation at Ser276 and the two forms of the protein, phosphorylated and unphosphorylated, are involved in two distinct functions. Under this model, the unphosphorylated protein might be directly responsible for transcriptional activation by binding to κB sites while the phosphorylated form could be involved indirectly in chromatin regulation and/or act as co-factors to regulate binding of other TF to their DNA sites discussed above. In addition to Ser276, RelA is a substrate for phosphorylation at multiple sites within its TAD, but none of these modifications has any impact on DNA binding.
The NF-κB p50 subunit also undergoes phosphorylation at various locations (140). One of these sites is Ser329 (141). This DNA damage-induced cytotoxic phosphorylation primes the p50:RelA heterodimer to avoid binding κB sites with C at the –1 position, such as GGGACTYYCC, without affecting binding to GGGAATYYCC type of sequences (–1 bp is underlined). Phosphorylation-dependent κB site binding selectivity prevents expression of anti-apoptotic factors and tips the balance of cell stress signaling toward cell death. Surprisingly, the phosphorylation site of p50 is located near the tip of the dimerization domain, far from the protein-DNA interface (Figure 11). Cells that express a phosphorylation-resistant version of p50 (Ser329Ala) function properly, as do cells that lack a p50 subunit entirely (141). This suggests that other NF-κB dimers can substitute the function of p50:RelA heterodimers. It is possible that p-Ser329 allosterically affects DNA binding of p50. Without further structural and biochemical data, it is difficult to predict how structural changes at a distal site propagate to residues that directly contact DNA in a manner that alters binding specificity. It is also possible that a co-factor specific to the p50 subunit is critical for guiding p50 to bind DNA. Phosphorylation at Ser329 could block binding to such a co-factor and lead to reduced DNA binding. Ser337 and Ser20 of p50 have also been shown to undergo phosphorylation (142) (Figure 11). Phosphorylation at Ser20, which is not conserved in murine p50, affects p50:RelA heterodimer binding to VCAM-κB sites. There are two κB sites within the VCAM promoter, both of which deviate from the consensus at positions -1 and -2 (143). Ser20 is located within an apparently flexible N-terminal segment, which is not directly involved in DNA contacts. How this phosphorylation might alter DNA binding specificity of p50 requires further study. Ser337 is homologous to Ser276 of RelA and, similar to RelA Ser276, Ser337 of p50 also undergoes phosphorylation. Whereas phosphorylation at this site by protein kinase A (PKA) was shown to augment DNA binding by the p50 homodimer in cells (144), both homodimerization and DNA binding are negatively affected by the phosphomimetic Ser337Asp in vitro (145). These observations suggest that p-Ser276 of RelA and p-Ser337 of p50 might be targeted by other factors to enhance DNA binding in vivo.
IκB proteins influence DNA binding
The inhibitor of NF-κB (IκB) proteins employ a centrally located ankyrin repeat domain (ARD) to associate noncovalently with select NF-κB dimers. IκB proteins are classified into three groups: the classical IκB group includes IκBα, IκBβ and IκBϵ; the ‘precursor’ IκB proteins p100/IκBδ and p105/IκBγ give rise to the mature NF-κB p50 and p52 subunits, respectively; and the nuclear IκB group includes Bcl-3, IκBζ and IκBNS (63,146). IκB binding typically renders NF-κB incapable of DNA binding and partitions the transcription factor out of the nucleus.
Nuclear IκB proteins do not act as typical inhibitors that form inhibitory NF-κB:IκB complexes in the cytoplasm. Rather, these proteins regulate NF-κB function in the nucleus by acting as signaling and target gene-specific ‘co-activators’ of transcription (147) (Figure 12). The expression of all three nuclear IκB proteins is regulated by NF-κB, primarily through RelA and c-Rel dimers. Upon translation, nuclear IκB proteins localize to the nucleus and associate with NF-κB p50 and p52 homodimers on DNA as ternary complexes. Ternary complex formation by Bcl-3 requires that it first be phosphorylated, as unphosphorylated Bcl-3 favors formation of binary complexes with p50 and p52 (148). However, the phosphorylation pattern of Bcl-3 is highly complex and several Bcl-3 phospho-isoforms coexist in cells, each of which can potentially regulate a distinct subset of genes. Two of the essential phosphorylation sites are located within a structurally flexible region C-terminal to the ARD. Since the C-terminus is not involved in binary complex formation with p50 or p52, it is reasonable to suppose that phosphorylation within this region enables Bcl-3 to make additional contacts with p50/p52 allowing them to associate with DNA (148). (Figure 12). In the absence of structural information, it is unclear why unmodified Bcl-3 cannot form a ternary complex with κB DNA and NF-κB and how phosphorylation enables ternary complex formation.
Figure 12.

Nuclear IκB proteins affect transcription by forming NF-κB:IκB:DNA complexes. Wild type IκBα forms stable binary complexes with NF-κB (top left). Mutations on the acidic IκBα acidic PEST C-terminal domain partially favor ternary complexes with NF-κB and DNA. Such ternary complexes have not been seen in vivo (boxed). Nuclear IκBβ forms ternary complexes with NF-κB and DNA only in its hypo-phosphorylated form. These promoter specific complexes induce gene transcription. Bcl-3:NF-κB:DNA ternary complexes occur only when Bcl-3 is phosphorylated. Unphosphorylated Bcl-3 acts like classical IκB proteins in vitro. It is possible that specific phosphatases exist in vivo that convert Bcl-3 into a classical IκB protein. Finally, IκBζ is known as a transcriptional coactivator by forming ternary complexes. It remains to be determined whether it is regulated similarly to the other nuclear IκB proteins.
IκBζ selectively binds the p50 homodimer and, as part of a ternary complex on DNA, IκBζ makes direct specific contact with the promoter DNA. This explains how IκBζ:p50 recognizes specific promoters to regulate downstream genes (44,149). Interestingly, although IκBβ does not associate with NF-κB and DNA forming a ternary complex under normal conditions where it is hyperphosphorylated (pp-IκBβ), a specific partially dephosphorylated state of IκBβ, referred to as its hypo-phosphorylated form (p-IκBβ), can form ternary complexes at specific promoters similar to the Bcl-3:NF-κB:DNA complexes (Figure 12). Unlike Bcl-3, which associates with p50 or p52 and DNA on a large number of promoters and affects the expression of hundreds of target genes, promoter association of p-IκBβ is highly selective. Only expression of IL-1β and TNF-α has been shown to be regulated by p-IκBβ (150–153). IκBα also can bind DNA as a ternary complex with NF-κB. But this ternary complex is extremely transient and was observed only as a rapid first step in removal of NF-κB from an active promoter to down-regulate gene expression (154,155). Mutations within the C-terminal acidic residues of IκBα alter its NF-κB ‘stripping’ function resulting in a more stable ternary complex (156). Thus, it appears as though the gene regulatory activity of IκB proteins at promoters is a function of the amount of time they typically spend bound to NF-κB on DNA. If the half-life is very short, the IκB function as repressors by catalyzing removal of NF-κB from the promoter DNA. If, on the other hand, IκB binding extends the amount of time an NF-κB dimer remains bound to DNA, then the IκB takes on the function of transcriptional ‘co-activator.’ Modifications, primarily by phosphorylation, enable different IκB proteins to act as transcriptional regulators of specific genes through promoter recruitment. Interestingly, multiple phosphorylation events on Bcl-3 convert it into a transcriptional co-activator, while hyperphosphorylation renders IκBα and IκBβ more effective inhibitors of transcription. The simple temporal model presented here suggests that IκBα and IκBβ can fulfill roles as transcriptional co-activation if negative charges of their C-terminal segments are reduced or shielded (Figure 12).
With regards to the influence on transcriptional regulation, IκB proteins can be viewed as a special class of co-factors. Unlike the previously described co-factors, which interact with NF-κB weakly and transiently to facilitate DNA binding, p-Bcl-3, IκBζ, and p-IκBβ bind NF-κB with considerably higher affinity (44). The stable interaction with NF-κB is essential for Bcl-3 and IκBζ since they regulate the transcriptional activity of p50 and p52 homodimers, which lack their own transcriptional activation domains. The more transient interacting protein co-factors, on the other hand, only regulate DNA binding by RelA, c-Rel, and possibly RelB. In addition to stabilizing DNA binding by NF-κB p50 and p52 homodimers, both Bcl-3 and IκBζ regulate selective promoter DNA binding and the events downstream of DNA binding such as histone acetyltransferase and deacetylase recruitment (149,157,158).
Chromatin influences NF-κB binding to DNA
Unlike the pioneering transcription factors such as FOXA and GATA, among others (159,160), NF-κB has no inherent ability to free target DNA from its association with chromatin. Indeed, the nucleosome acts as a barrier for NF-κB to induce gene expression. Consequently, several NF-κB target genes exhibit delayed expression since their target κB sites are masked by nucleosomes and they must await remodeling factors to free the sites (161–163). It has been reported that NF-κB facilitates RNA Pol II elongation and it is known to interact with transcriptional co-activators such as CBP/p300 (164–166). Presumably, the interaction between NF-κB and co-activators is in synchrony with its DNA binding. Whether NF-κB simultaneously interacts with a neighboring nucleosome is not known. Over a period of nearly ten years, a series of studies were carried out that seemed to suggest that NF-κB could bind to κB sites hidden within the nucleosome DNA in vitro (167,168). However these experiments were performed with DNA sequences that were not optimum and failed to form nucleosomes akin to the established 601 nucleosome positioning sequence (169). Further analysis revealed that under dilute concentrations the non-ideal nucleosome assemblies could possibly lose H2A and H2B dimers, thus allowing NF-κB to bind the unstable half-nucleosomes with κB sites exposed. NF-κB can, however, competitively displace the linker histone H1 (170).
These experiments seem to suggest that NF-κB is only capable of binding to DNA sequences that are completely free of nucleosomes or, at most, to half nucleosomes where H2A/H2B dimers are removed (170). However, neither the precise extent to which a κB site needs to be cleared of histone proteins to enable NF-κB binding nor the effect of one bound NF-κB on movement of nearby by nucleosomes are well known. These are critical measures as they could provide support for a mechanism for NF-κB-dependent effects on PolII elongation. We speculate that protein co-factor facilitated DNA binding by NF-κB is a critical early event that, followed by regulated binding by CPB/p300, nuclear IκB, or possibly other co-activators, initiates the process of moving histone octamers and clearing more of the promoter. This process then facilitates binding of additional transcription factors to neighboring sites, including additional NF-κB subunits on promoters with multiple κB sites or as non-DNA binding co-factors. Interestingly, many NF-κB target genes contain κB sites ∼12–500 bp from the TSS. This fairly well conserved length of DNA might give some clue as to the distances covered during the steps of proper nucleosome sliding/eviction from the chromatin and subsequent assembly of complexes formed around NF-κB as well as other factors. Transcriptional co-repressors could function by interacting with the nucleosome to halt its progress or by targeting NF-κB by competing for co-activator binding. A neighboring nucleosome thus could act as facilitator of cooperative binding between NF-κB and coactivator/corepressor as predicted previously (Figure 13) (171). The relative frequency of interaction of co-activators or co-repressors with DNA-bound NF-κB and/or nucleosomes might then dictate the efficiency of mediator/Pol II binding and hence transcriptional output. Interference by classical IκB proteins that can actively strip NF-κB from DNA would serve to decrease the half-life of NF-κB:DNA complexes disrupting the entire process. Under this model, the interplay between factors that support or block NF-κB binding to DNA combined with additional NF-κB-dependent factors that control clearance of DNA from nucleosomes might be sufficient to ensure that all subsequent downstream events leading to transcription initiation or repression ensue.
Figure 13.
A model of NF-κB and the transcriptional coactivator CBP/p300 bound to a κB site in a chromatin context. The NF-κB RHR binds the κB site while its TAD contact CBP/p300 and/or nearby nucleosomes simultaneously. Similar ternary complexes with co-repressors and nucleosome could also be possible.
CONCLUDING THOUGHTS
X-ray crystal structures of an array of NF-κB:DNA complexes detail the stereochemical foundation of the thermodynamically and kinetically stable endpoint of the protein-DNA interaction. Intermediates along the path of recognition and binding, however, are unknown. This is the true for almost every TF:DNA complex studied to date. A vast body of experimental results from genetics, biochemical, and cell-based experiments revealed that numerous factors affect this binding event. As discussed in this report, these regulators of DNA binding can be divided into three categories: (i) the composition and concentrations of ions that can shield the interacting functional groups on key NF-κB residues from making direct contact with DNA thus acting as a negative regulator; (ii) the DNA sequence itself both within its core recognition part (κB sites) and flanking segments, which together determine both local and overall geometry and dynamics of the DNA and (iii) co-factor proteins and covalent modifications to NF-κB, which not only negate the shielding effect of ions but also induce the interacting protein side chains to adopt specific conformations to facilitate DNA contacts. While the impact of the first two regulators can be easily verified by biophysical and biochemical experiments, assessing the relative contributions to DNA binding by NF-κB modifications and co-factors is considerably more complicated. Although very different in chemistry, both covalent modification, at least in some instances, and co-factors, in all cases, regulate DNA binding through allosteric mechanisms in which through bond and through space effects lead to modulation of the DNA binding surfaces on NF-κB. The mechanism of co-factor protein facilitation of DNA binding by NF-κB is at least partially worked out. Diverse co-factor proteins contact NF-κB directly and specifically around a surface on the NTD that is distal to the DNA binding surface. This interaction is then relayed to the residues that directly contact DNA. In addition to priming DNA binding residues, these allosteric changes may also serve to alter the shielding effect by ions. The combined effects of various extrinsic (other factors) and intrinsic (protein and DNA sequences) factors determine the kinetics of binding, which is converted into transcriptional output.
One challenge with which we are currently faced is to identify new modifications of specific residues on NF-κB that influence its DNA binding and transcriptional activation functions. Another is to identify new co-factors that regulate DNA binding. Together with these discoveries, efforts are needed to determine mechanistically how these modifications and co-factors regulate NF-κB binding broadly to κB DNA in general as well as to specific κB sites. Finally, in this review, we have discussed DNA binding by NF-κB under equilibrium conditions, despite the fact that transcriptional regulation is a process that is wholly dependent upon timing. We have briefly touched upon kinetic control by discussing the consequences on transcriptional activity of Bcl-3 and IκBβ in their different phosphorylation states. We also speculated that the observed one bp change that switches the potential of a κB DNA from one that activates transcription to one that represses could be due to its effect on NF-κB binding kinetics. More and better experiments are required that measure and correlate NF-κB:DNA binding kinetics and transcriptional output under physiological conditions.
FUNDING
Funding for this work and for open access charges: National Institutes of Health [GM085490 to G.G.]; Science and Technology Development Fund, Macau SAR (FDCT) [File no. 023/2016/A1 to V.Y.-F.W.]; Multi-Year Research Grant from University of Macau [MYRG2018-00093-FHS to V.Y.-F.W.]; California Metabolic Research Foundation to San Diego State University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Zhang Q., Lenardo M.J., Baltimore D.. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017; 168:37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mathes E., O’Dea E.L., Hoffmann A., Ghosh G.. NF-κB dictates the degradation pathway of IκBα. EMBO J. 2008; 27:1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O’Dea E.L., Barken D., Peralta R.Q., Tran K.T., Werner S.L., Kearns J.D., Levchenko A., Hoffmann A.. A homeostatic model of IκB metabolism to control constitutive NF-κB activity. Mol. Syst. Biol. 2007; 3:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sen R., Baltimore D.. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986; 46:705–716. [PubMed] [Google Scholar]
- 5. Hayden M.S., Ghosh S.. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014; 26:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huxford T., Ghosh G.. A structural guide to proteins of the NF-κB signaling module. Cold Spring Harb. Perspec. Biol. 2009; 1:a000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Iwai K. Diverse ubiquitin signaling in NF-κB activation. Trends Cell Biol. 2012; 22:355–364. [DOI] [PubMed] [Google Scholar]
- 8. Mitchell S., Vargas J., Hoffmann A.. Signaling via the NF-κB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016; 8:227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Napetschnig J., Wu H.. Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 2013; 42:443–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Müller C.W., Rey F.A., Sodeoka M., Verdine G.L., Harrison S.C.. Structure of the NF-κB p50 homodimer bound to DNA. Nature. 1995; 373:311–317. [DOI] [PubMed] [Google Scholar]
- 11. Ghosh G., van Duyne G., Ghosh S., Sigler P.B.. Structure of NF-κB p50 homodimer bound to a κB site. Nature. 1995; 373:303–310. [DOI] [PubMed] [Google Scholar]
- 12. Ghosh G., Wang V.Y., Huang D.B., Fusco A.. NF-κB regulation: lessons from structures. Immunol. Rev. 2012; 246:36–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cramer P., Larson C.J., Verdine G.L., Müller C.W.. Structure of the human NF-κB p52 homodimer-DNA complex at 2.1 Å resolution. EMBO J. 1997; 16:7078–7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen F.E., Huang D.B., Chen Y.Q., Ghosh G.. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature. 1998; 391:410–413. [DOI] [PubMed] [Google Scholar]
- 15. Huang D.B., Chen Y.Q., Ruetsche M., Phelps C.B., Ghosh G.. X-ray crystal structure of proto-oncogene product c-Rel bound to the CD28 response element of IL-2. Structure. 2001; 9:669–678. [DOI] [PubMed] [Google Scholar]
- 16. Moorthy A.K., Huang D.B., Wang V.Y., Vu D., Ghosh G.. X-ray structure of a NF-κB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem κB sites. J. Mol. Biol. 2007; 373:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huxford T., Huang D.B., Malek S., Ghosh G.. The crystal structure of the IκBα/NF-κB complex reveals mechanisms of NF-κB inactivation. Cell. 1998; 95:759–770. [DOI] [PubMed] [Google Scholar]
- 18. Jacobs M.D., Harrison S.C.. Structure of an IκBα/NF-κB complex. Cell. 1998; 95:749–758. [DOI] [PubMed] [Google Scholar]
- 19. Malek S., Huang D.B., Huxford T., Ghosh S., Ghosh G.. X-ray crystal structure of an IκBβ:NF-κB p65 homodimer complex. J. Biol. Chem. 2003; 278:23094–23100. [DOI] [PubMed] [Google Scholar]
- 20. Huang D.B., Huxford T., Chen Y.Q., Ghosh G.. The role of DNA in the mechanism of NFκB dimer formation: crystal structures of the dimerization domains of the p50 and p65 subunits. Structure. 1997; 5:1427–1436. [DOI] [PubMed] [Google Scholar]
- 21. Huang D.B., Vu D., Ghosh G.. NF-κB RelB forms an intertwined homodimer. Structure. 2005; 13:1365–1373. [DOI] [PubMed] [Google Scholar]
- 22. Sengchanthalangsy L.L., Datta S., Huang D.B., Anderson E., Braswell E.H., Ghosh G.. Characterization of the dimer interface of transcription factor NFκB p50 homodimer. J. Mol. Biol. 1999; 289:1029–1040. [DOI] [PubMed] [Google Scholar]
- 23. Hart D.J., Speight R.E., Sutherland J.D., Blackburn J.M.. Analysis of the NF-κB p50 dimer interface by diversity screening. J. Mol. BIol. 2001; 310:563–575. [DOI] [PubMed] [Google Scholar]
- 24. Chirgadze D.Y., Demydchuk M., Becker M., Moran S., Paoli M.. Snapshot of protein structure evolution reveals conservation of functional dimerization through intertwined folding. Structure. 2004; 12:1489–1494. [DOI] [PubMed] [Google Scholar]
- 25. Ryseck R.P., Novotny J., Bravo R.. Characterization of elements determining the dimerization properties of RelB and p50. Mol. Cell Biol. 1995; 15:3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vu D., Huang D.B., Vemu A., Ghosh G.. A structural basis for selective dimerization by NF-κB RelB. J. Mol. Biol. 2013; 425:1934–1945. [DOI] [PubMed] [Google Scholar]
- 27. Tsui R., Kearns J.D., Lynch C., Vu D., Ngo K.A., Basak S., Ghosh G., Hoffmann A.. IκBβ enhances the generation of the low-affinity NFκB/RelA homodimer. Nat. Commun. 2015; 6:7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smale S.T. Dimer-specific regulatory mechanisms within the NF-κB family of transcription factors. Immunol. Rev. 2012; 246:193–204. [DOI] [PubMed] [Google Scholar]
- 29. Hayden M.S., Ghosh S.. Signaling to NF-κB. Genes Dev. 2004; 18:2195–2224. [DOI] [PubMed] [Google Scholar]
- 30. Sha W.C., Liou H.C., Tuomanen E.I., Baltimore D.. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995; 80:321–330. [DOI] [PubMed] [Google Scholar]
- 31. Kontgen F., Grumont R.J., Strasser A., Metcalf D., Li R., Tarlinton D., Gerondakis S.. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995; 9:1965–1977. [DOI] [PubMed] [Google Scholar]
- 32. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A. et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 33. Fusco A.J., Huang D.B., Miller D., Wang V.Y., Vu D., Ghosh G.. NF-κB p52:RelB heterodimer recognizes two classes of κB sites with two distinct modes. EMBO Rep. 2009; 10:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ghosh S., Karin M.. Missing pieces in the NF-κB puzzle. Cell. 2002; 109:S81–S96. [DOI] [PubMed] [Google Scholar]
- 35. Senftleben U., Cao Y., Xiao G., Greten F.R., Krahn G., Bonizzi G., Chen Y., Hu Y., Fong A., Sun S.C. et al.. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 2001; 293:1495–1499. [DOI] [PubMed] [Google Scholar]
- 36. Xiao G., Harhaj E.W., Sun S.C.. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell. 2001; 7:401–409. [DOI] [PubMed] [Google Scholar]
- 37. Bonizzi G., Bebien M., Otero D.C., Johnson-Vroom K.E., Cao Y., Vu D., Jegga A.G., Aronow B.J., Ghosh G., Rickert R.C. et al.. Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers. EMBO J. 2004; 23:4202–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011; 21:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwarz E.M., Krimpenfort P., Berns A., Verma I.M.. Immunological defects in mice with a targeted disruption in Bcl-3. Genes Dev. 1997; 11:187–197. [DOI] [PubMed] [Google Scholar]
- 40. Yamamoto M., Yamazaki S., Uematsu S., Sato S., Hemmi H., Hoshino K., Kaisho T., Kuwata H., Takeuchi O., Takeshige K. et al.. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IκBζ. Nature. 2004; 430:218–222. [DOI] [PubMed] [Google Scholar]
- 41. Touma M., Antonini V., Kumar M., Osborn S.L., Bobenchik A.M., Keskin D.B., Connolly J.E., Grusby M.J., Reinherz E.L., Clayton L.K.. Functional role for IκBNS in T cell cytokine regulation as revealed by targeted gene disruption. J. Immunol. 2007; 179:1681–1692. [DOI] [PubMed] [Google Scholar]
- 42. Hatada E.N., Nieters A., Wulczyn F.G., Naumann M., Meyer R., Nucifora G., McKeithan T.W., Scheidereit C.. The ankyrin repeat domains of the NF-κB precursor p105 and the protooncogene Bcl-3 act as specific inhibitors of NF-κB DNA binding. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:2489–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamazaki S., Muta T., Takeshige K.. A novel IκB protein, IκBζ, induced by proinflammatory stimuli, negatively regulates nuclear factor-κB in the nuclei. J. Biol. Chem. 2001; 276:27657–27662. [DOI] [PubMed] [Google Scholar]
- 44. Trinh D.V., Zhu N., Farhang G., Kim B.J., Huxford T.. The nuclear IκB protein IκBζ specifically binds NF-κB p50 homodimers and forms a ternary complex on κB DNA. J. Mol. Biol. 2008; 379:122–135. [DOI] [PubMed] [Google Scholar]
- 45. Lenardo M.J., Baltimore D.. NF-κB: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989; 58:227–229. [DOI] [PubMed] [Google Scholar]
- 46. Chen F.E., Ghosh G.. Regulation of DNA binding by Rel/NF-κB transcription factors: structural views. Oncogene. 1999; 18:6845–6852. [DOI] [PubMed] [Google Scholar]
- 47. Ghosh G., Huang D.-B., Huxford T.. Williams MC, Maher LJ. Biophysics of DNA-Protein Interactions: From Single Molecules to Biological Systems. 2011; NY: Springer; 85–106. [Google Scholar]
- 48. Wang V.Y., Huang W., Asagiri M., Spann N., Hoffmann A., Glass C., Ghosh G.. The transcriptional specificity of NF-κB dimers is coded within the κB DNA response elements. Cell Rep. 2012; 2:824–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R. et al.. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012; 489:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y. et al.. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012; 22:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Inukai S., Kock K.H., Bulyk M.L.. Transcription factor-DNA binding: beyond binding site motifs. Curr. Opin. Genet. Dev. 2017; 43:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lim C.A., Yao F., Wong J.J., George J., Xu H., Chiu K.P., Sung W.K., Lipovich L., Vega V.B., Chen J. et al.. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-κB upon TLR4 activation. Mol. Cell. 2007; 27:622–635. [DOI] [PubMed] [Google Scholar]
- 54. Martone R., Euskirchen G., Bertone P., Hartman S., Royce T.E., Luscombe N.M., Rinn J.L., Nelson F.K., Miller P., Gerstein M. et al.. Distribution of NF-κB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:12247–12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao B., Barrera L.A., Ersing I., Willox B., Schmidt S.C., Greenfeld H., Zhou H., Mollo S.B., Shi T.T., Takasaki K. et al.. The NF-κB genomic landscape in lymphoblastoid B cells. Cell Rep. 2014; 8:1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tong A.J., Liu X., Thomas B.J., Lissner M.M., Baker M.R., Senagolage M.D., Allred A.L., Barish G.D., Smale S.T.. A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell. 2016; 165:165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen Y.Q., Ghosh S., Ghosh G.. A novel DNA recognition mode by the NF-κB p65 homodimer. Nat. Struct. Biol. 1998; 5:67–73. [DOI] [PubMed] [Google Scholar]
- 58. Chen Y.Q., Sengchanthalangsy L.L., Hackett A., Ghosh G.. NF-κB p65 (RelA) homodimer uses distinct mechanisms to recognize DNA targets. Structure. 2000; 8:419–428. [DOI] [PubMed] [Google Scholar]
- 59. Escalante C.R., Shen L., Thanos D., Aggarwal A.K.. Structure of NF-κB p50/p65 heterodimer bound to the PRDII DNA element from the interferon-β promoter. Structure. 2002; 10:383–391. [DOI] [PubMed] [Google Scholar]
- 60. Berkowitz B., Huang D.B., Chen-Park F.E., Sigler P.B., Ghosh G.. The x-ray crystal structure of the NF-κB p50:p65 heterodimer bound to the interferon-β-κB site. J. Biol. Chem. 2002; 277:24694–24700. [DOI] [PubMed] [Google Scholar]
- 61. Chen-Park F.E., Huang D.B., Noro B., Thanos D., Ghosh G.. The κB DNA sequence from the HIV long terminal repeat functions as an allosteric regulator of HIV transcription. J. Biol. Chem. 2002; 277:24701–24708. [DOI] [PubMed] [Google Scholar]
- 62. Panne D., Maniatis T., Harrison S.C.. An atomic model of the interferon-β enhanceosome. Cell. 2007; 129:1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huxford T., Hoffmann A., Ghosh G.. Understanding the logic of IκB:NF-κB regulation in structural terms. Curr. Top. Microbiol. Immunol. 2011; 349:1–24. [DOI] [PubMed] [Google Scholar]
- 64. Siggers T., Chang A.B., Teixeira A., Wong D., Williams K.J., Ahmed B., Ragoussis J., Udalova I.A., Smale S.T., Bulyk M.L.. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol. 2011; 13:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Britanova L.V., Makeev V.J., Kuprash D.V.. In vitro selection of optimal RelB/p52 DNA-binding motifs. Biochem. Biophys. Res. Commun. 2008; 365:583–588. [DOI] [PubMed] [Google Scholar]
- 66. Mulero M.C., Huang D.B., Nguyen H.T., Wang V.Y., Li Y., Biswas T., Ghosh G.. DNA-binding affinity and transcriptional activity of the RelA homodimer of nuclear factor κB are not correlated. J. Biol. Chem. 2017; 292:18821–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huxford T., Mishler D., Phelps C.B., Huang D.B., Sengchanthalangsy L.L., Reeves R., Hughes C.A., Komives E.A., Ghosh G.. Solvent exposed non-contacting amino acids play a critical role in NF-κB/IκBα complex formation. J. Mol. Biol. 2002; 324:587–597. [DOI] [PubMed] [Google Scholar]
- 68. Kunsch C., Ruben S.M., Rosen C.A.. Selection of optimal κB/Rel DNA-binding motifs: interaction of both subunits of NF-κB with DNA is required for transcriptional activation. Mol. Cell Biol. 1992; 12:4412–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Phelps C.B., Ghosh G.. Discreet mutations from c-Rel to v-Rel alter κB DNA recognition, IκBα binding, and dimerization: implications for v-Rel oncogenicity. Oncogene. 2004; 23:1229–1238. [DOI] [PubMed] [Google Scholar]
- 70. Wong D., Teixeira A., Oikonomopoulos S., Humburg P., Lone I.N., Saliba D., Siggers T., Bulyk M., Angelov D., Dimitrov S. et al.. Extensive characterization of NF-κB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 2011; 12:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tisne C., Delepierre M., Hartmann B.. How NF-κB can be attracted by its cognate DNA. J. Mol. Biol. 1999; 293:139–150. [DOI] [PubMed] [Google Scholar]
- 72. Tisne C., Hartmann B., Delepierre M.. NF-κB binding mechanism: a nuclear magnetic resonance and modeling study of a GGG → CTC mutation. Biochemistry. 1999; 38:3883–3894. [DOI] [PubMed] [Google Scholar]
- 73. Mura C., McCammon J.A.. Molecular dynamics of a κB DNA element: base flipping via cross-strand intercalative stacking in a microsecond-scale simulation. Nucleic Acids Res. 2008; 36:4941–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huang D.B., Phelps C.B., Fusco A.J., Ghosh G.. Crystal structure of a free κB DNA: insights into DNA recognition by transcription factor NF-κB. J. Mol. Biol. 2005; 346:147–160. [DOI] [PubMed] [Google Scholar]
- 75. Schone S., Jurk M., Helabad M.B., Dror I., Lebars I., Kieffer B., Imhof P., Rohs R., Vingron M., Thomas-Chollier M. et al.. Sequences flanking the core-binding site modulate glucocorticoid receptor structure and activity. Nat. Commun. 2016; 7:12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Leung T.H., Hoffmann A., Baltimore D.. One nucleotide in a κB site can determine cofactor specificity for NF-κB dimers. Cell. 2004; 118:453–464. [DOI] [PubMed] [Google Scholar]
- 77. Cheng C.S., Feldman K.E., Lee J., Verma S., Huang D.B., Huynh K., Chang M., Ponomarenko J.V., Sun S.C., Benedict C.A. et al.. The specificity of innate immune responses is enforced by repression of interferon response elements by NF-κB p50. Sci. Signal. 2011; 4:ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kitayner M., Rozenberg H., Kessler N., Rabinovich D., Shaulov L., Haran T.E., Shakked Z.. Structural basis of DNA recognition by p53 tetramers. Mol. Cell. 2006; 22:741–753. [DOI] [PubMed] [Google Scholar]
- 79. McLure K.G., Lee P.W.. How p53 binds DNA as a tetramer. EMBO J. 1998; 17:3342–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ellwood K., Huang W., Johnson R., Carey M.. Multiple layers of cooperativity regulate enhanceosome-responsive RNA polymerase II transcription complex assembly. Mol. Cell Biol. 1999; 19:2613–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kim T.K., Maniatis T.. The mechanism of transcriptional synergy of an in vitro assembled interferon-β enhanceosome. Mol. Cell. 1997; 1:119–129. [DOI] [PubMed] [Google Scholar]
- 82. Perkins N.D., Edwards N.L., Duckett C.S., Agranoff A.B., Schmid R.M., Nabel G.J.. A cooperative interaction between NF-κB and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993; 12:3551–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bandukwala H.S., Wu Y., Feuerer M., Chen Y., Barboza B., Ghosh S., Stroud J.C., Benoist C., Mathis D., Rao A. et al.. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity. 2011; 34:479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chen L., Glover J.N., Hogan P.G., Rao A., Harrison S.C.. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998; 392:42–48. [DOI] [PubMed] [Google Scholar]
- 85. Tahirov T.H., Inoue-Bungo T., Morii H., Fujikawa A., Sasaki M., Kimura K., Shiina M., Sato K., Kumasaka T., Yamamoto M. et al.. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFbeta. Cell. 2001; 104:755–767. [DOI] [PubMed] [Google Scholar]
- 86. Jolma A., Yin Y., Nitta K.R., Dave K., Popov A., Taipale M., Enge M., Kivioja T., Morgunova E., Taipale J.. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015; 527:384–388. [DOI] [PubMed] [Google Scholar]
- 87. Stampfel G., Kazmar T., Frank O., Wienerroither S., Reiter F., Stark A.. Transcriptional regulators form diverse groups with context-dependent regulatory functions. Nature. 2015; 528:147–151. [DOI] [PubMed] [Google Scholar]
- 88. Maniatis T., Falvo J.V., Kim T.H., Kim T.K., Lin C.H., Parekh B.S., Wathelet M.G.. Structure and function of the interferon-β enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 1998; 63:609–620. [DOI] [PubMed] [Google Scholar]
- 89. Panne D., Maniatis T., Harrison S.C.. Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-β enhancer. EMBO J. 2004; 23:4384–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dragan A.I., Carrillo R., Gerasimova T.I., Privalov P.L.. Assembling the human IFN-β enhanceosome in solution. J. Mol. Biol. 2008; 384:335–348. [DOI] [PubMed] [Google Scholar]
- 91. Gaynor R.B., Kuwabara M.D., Wu F.K., Garcia J.A., Harrich D., Briskin M., Wall R., Sigman D.S.. Repeated B motifs in the human immunodeficiency virus type I long terminal repeat enhancer region do not exhibit cooperative factor binding. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:9406–9410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pettersson M., Schaffner W.. Synergistic activation of transcription by multiple binding sites for NF-κB even in absence of co-operative factor binding to DNA. J. Mol. Biol. 1990; 214:373–380. [DOI] [PubMed] [Google Scholar]
- 93. Giorgetti L., Siggers T., Tiana G., Caprara G., Notarbartolo S., Corona T., Pasparakis M., Milani P., Bulyk M.L., Natoli G.. Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol. Cell. 2010; 37:418–428. [DOI] [PubMed] [Google Scholar]
- 94. Stroud J.C., Oltman A., Han A., Bates D.L., Chen L.. Structural basis of HIV-1 activation by NF-κB–a higher-order complex of p50:RelA bound to the HIV-1 LTR. J. Mol. Biol. 2009; 393:98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Saccani S., Pantano S., Natoli G.. Modulation of NF-κB activity by exchange of dimers. Mol. Cell. 2003; 11:1563–1574. [DOI] [PubMed] [Google Scholar]
- 96. Heinz S., Romanoski C.E., Benner C., Allison K.A., Kaikkonen M.U., Orozco L.D., Glass C.K.. Effect of natural genetic variation on enhancer selection and function. Nature. 2013; 503:487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Jin F., Li Y., Dixon J.R., Selvaraj S., Ye Z., Lee A.Y., Yen C.A., Schmitt A.D., Espinoza C.A., Ren B.. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013; 503:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Reichardt H.M., Kaestner K.H., Tuckermann J., Kretz O., Wessely O., Bock R., Gass P., Schmid W., Herrlich P., Angel P. et al.. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998; 93:531–541. [DOI] [PubMed] [Google Scholar]
- 99. De Bosscher K., Vanden Berghe W., Haegeman G.. The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr. Rev. 2003; 24:488–522. [DOI] [PubMed] [Google Scholar]
- 100. Ratman D., Vanden Berghe W., Dejager L., Libert C., Tavernier J., Beck I.M., De Bosscher K.. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol. Cell Endocrinol. 2013; 380:41–54. [DOI] [PubMed] [Google Scholar]
- 101. Ogawa S., Lozach J., Benner C., Pascual G., Tangirala R.K., Westin S., Hoffmann A., Subramaniam S., David M., Rosenfeld M.G. et al.. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005; 122:707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Freaney J.E., Kim R., Mandhana R., Horvath C.M.. Extensive cooperation of immune master regulators IRF3 and NFκB in RNA Pol II recruitment and pause release in human innate antiviral transcription. Cell Rep. 2013; 4:959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cooks T., Pateras I.S., Tarcic O., Solomon H., Schetter A.J., Wilder S., Lozano G., Pikarsky E., Forshew T., Rosenfeld N. et al.. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell. 2013; 23:634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Rahnamoun H., Lu H., Duttke S.H., Benner C., Glass C.K., Lauberth S.M.. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat. Commun. 2017; 8:754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang Y., Lei C.Q., Hu Y.H., Xia T., Li M., Zhong B., Shu H.B.. Kruppel-like factor 6 is a co-activator of NF-κB that mediates p65-dependent transcription of selected downstream genes. J. Biol. Chem. 2014; 289:12876–12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lin J., Kato M., Nagata K., Okuwaki M.. Efficient DNA binding of NF-κB requires the chaperone-like function of NPM1. Nucleic Acids Res. 2017; 45:3707–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pan L., Zhu B., Hao W., Zeng X., Vlahopoulos S.A., Hazra T.K., Hegde M.L., Radak Z., Bacsi A., Brasier A.R. et al.. Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase-1-mediated epigenetic regulation of Nuclear Factor κB-driven gene expression. J. Biol. Chem. 2016; 291:25553–25566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wan F., Anderson D.E., Barnitz R.A., Snow A., Bidere N., Zheng L., Hegde V., Lam L.T., Staudt L.M., Levens D. et al.. Ribosomal protein S3: a KH domain subunit in NF-κB complexes that mediates selective gene regulation. Cell. 2007; 131:927–939. [DOI] [PubMed] [Google Scholar]
- 109. Yie J., Liang S., Merika M., Thanos D.. Intra- and intermolecular cooperative binding of high-mobility-group protein I(Y) to the β-interferon promoter. Mol. Cell Biol. 1997; 17:3649–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fu K., Sun X., Zheng W., Wier E.M., Hodgson A., Tran D.Q., Richard S., Wan F.. Sam68 modulates the promoter specificity of NF-κB and mediates expression of CD25 in activated T cells. Nat. Commun. 2013; 4:1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Stubenrauch K., Wessels U., Essig U., Kowalewsky F., Vogel R., Heinrich J.. Characterization of murine anti-human Fab antibodies for use in an immunoassay for generic quantification of human Fab fragments in non-human serum samples including cynomolgus monkey samples. J. Pharm. Biomed. Anal. 2013; 72:208–215. [DOI] [PubMed] [Google Scholar]
- 112. Ren C., Bailey A.O., VanderPorten E., Oh A., Phung W., Mulvihill M.M., Harris S.F., Liu Y., Han G., Sandoval W.. Quantitative determination of protein-ligand affinity by size exclusion chromatography directly coupled to high-resolution native mass spectrometry. Anal. Chem. 2019; 91:903–911. [DOI] [PubMed] [Google Scholar]
- 113. Ren Z., Kang W., Wang L., Sun B., Ma J., Zheng C., Sun J., Tian Z., Yang X., Xiao W.. E2F1 renders prostate cancer cell resistant to ICAM-1 mediated antitumor immunity by NF-κB modulation. Mol. Cancer. 2014; 13:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Choy M.K., Movassagh M., Siggens L., Vujic A., Goddard M., Sanchez A., Perkins N., Figg N., Bennett M., Carroll J. et al.. High-throughput sequencing identifies STAT3 as the DNA-associated factor for p53-NF-κB-complex-dependent gene expression in human heart failure. Genome Med. 2010; 2:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kawauchi K., Araki K., Tobiume K., Tanaka N.. Activated p53 induces NF-κB DNA binding but suppresses its transcriptional activation. Biochem. Biophys. Res. Commun. 2008; 372:137–141. [DOI] [PubMed] [Google Scholar]
- 116. Hart D.J., Speight R.E., Cooper M.A., Sutherland J.D., Blackburn J.M.. The salt dependence of DNA recognition by NF-κB p50: a detailed kinetic analysis of the effects on affinityand specificity. Nucleic Acids Res. 1999; 27:1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Menetski J.P. The structure of the nuclear factor-κB protein-DNA complex varies with DNA-binding site sequence. J. Biol. Chem. 2000; 275:7619–7625. [DOI] [PubMed] [Google Scholar]
- 118. Phelps C.B., Sengchanthalangsy L.L., Malek S., Ghosh G.. Mechanism of κB DNA binding by Rel/NF-κB dimers. J. Biol. Chem. 2000; 275:24392–24399. [DOI] [PubMed] [Google Scholar]
- 119. Mulero M.C., Shahabi S., Ko M.S., Schiffer J.M., Huang D.B., Wang V.Y., Amaro R.E., Huxford T., Ghosh G.. Protein cofactors are essential for high-affinity DNA binding by the Nuclear Factor κB RelA subunit. Biochemistry. 2018; 57:2943–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ramsey K.M., Dembinski H.E., Chen W., Ricci C.G., Komives E.A.. DNA and IκBα both induce long-range conformational changes in NFκB. J. Mol. Biol. 2017; 429:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Morgan D.O. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997; 13:261–291. [DOI] [PubMed] [Google Scholar]
- 122. McCarthy K.M., McDevit D., Andreucci A., Reeves R., Nikolajczyk B.S.. HMGA1 co-activates transcription in B cells through indirect association with DNA. J. Biol. Chem. 2003; 278:42106–42114. [DOI] [PubMed] [Google Scholar]
- 123. Zhao K., Kas E., Gonzalez E., Laemmli U.K.. SAR-dependent mobilization of histone H1 by HMG-I/Y in vitro: HMG-I/Y is enriched in H1-depleted chromatin. EMBO J. 1993; 12:3237–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nolan G.P., Ghosh S., Liou H.C., Tempst P., Baltimore D.. DNA binding and IκB inhibition of the cloned p65 subunit of NF-κB, a rel-related polypeptide. Cell. 1991; 64:961–969. [DOI] [PubMed] [Google Scholar]
- 125. Guermah M., Malik S., Roeder R.G.. Involvement of TFIID and USA components in transcriptional activation of the human immunodeficiency virus promoter by NF-κB and Sp1. Mol. Cell Biol. 1998; 18:3234–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Naar A.M., Beaurang P.A., Zhou S., Abraham S., Solomon W., Tjian R.. Composite co-activator ARC mediates chromatin-directed transcriptional activation. Nature. 1999; 398:828–832. [DOI] [PubMed] [Google Scholar]
- 127. Yamit-Hezi A., Dikstein R.. TAFII105 mediates activation of anti-apoptotic genes by NF-κB. EMBO J. 1998; 17:5161–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mukherjee S.P., Behar M., Birnbaum H.A., Hoffmann A., Wright P.E., Ghosh G.. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcription. PLoS Biol. 2013; 11:e1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Boija A., Klein I.A., Sabari B.R., Dall'Agnese K., Coffey E.L., Zamudio A.V., Li C.H., Shrinivas K., Manteiga J.C., Hannett N.M. et al.. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell. 2018; 175:1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Chen L.F., Williams S.A., Mu Y., Nakano H., Duerr J.M., Buckbinder L., Greene W.C.. NF-κB RelA phosphorylation regulates RelA acetylation. Mol. Cell Biol. 2005; 25:7966–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Chen L.F., Mu Y., Greene W.C.. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J. 2002; 21:6539–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ea C.K., Baltimore D.. Regulation of NF-κB activity through lysine monomethylation of p65. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:18972–18977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lanzillotta A., Sarnico I., Ingrassia R., Boroni F., Branca C., Benarese M., Faraco G., Blasi F., Chiarugi A., Spano P. et al.. The acetylation of RelA in Lys310 dictates the NF-κB-dependent response in post-ischemic injury. Cell Death Dis. 2010; 1:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ghosh G., Huang D.B., Huxford T.. Molecular mimicry of the NF-κB DNA target site by a selected RNA aptamer. Curr. Opin. Struct. Biol. 2004; 14:21–27. [DOI] [PubMed] [Google Scholar]
- 135. Huang D.B., Vu D., Cassiday L.A., Zimmerman J.M., Maher L.J. 3rd, Ghosh G.. Crystal structure of NF-κB (p50)2 complexed to a high-affinity RNA aptamer. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:9268–9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zhong H., Voll R.E., Ghosh S.. Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell. 1998; 1:661–671. [DOI] [PubMed] [Google Scholar]
- 137. Dong J., Jimi E., Zhong H., Hayden M.S., Ghosh S.. Repression of gene expression by unphosphorylated NF-κB p65 through epigenetic mechanisms. Genes Dev. 2008; 22:1159–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Reber L., Vermeulen L., Haegeman G., Frossard N.. Ser276 phosphorylation of NF-κB p65 by MSK1 controls SCF expression in inflammation. PLoS One. 2009; 4:e4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Vermeulen L., De Wilde G., Van Damme P., Vanden Berghe W., Haegeman G.. Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 2003; 22:1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hou S., Guan H., Ricciardi R.P.. Phosphorylation of serine 337 of NF-κB p50 is critical for DNA binding. J. Biol. Chem. 2003; 278:45994–45998. [DOI] [PubMed] [Google Scholar]
- 141. Crawley C.D., Raleigh D.R., Kang S., Voce D.J., Schmitt A.M., Weichselbaum R.R., Yamini B.. DNA damage-induced cytotoxicity is mediated by the cooperative interaction of phospho-NF-κB p50 and a single nucleotide in the κB-site. Nucleic Acids Res. 2013; 41:764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Ju J., Naura A.S., Errami Y., Zerfaoui M., Kim H., Kim J.G., Abd Elmageed Z.Y., Abdel-Mageed A.B., Giardina C., Beg A.A. et al.. Phosphorylation of p50 NF-κB at a single serine residue by DNA-dependent protein kinase is critical for VCAM-1 expression upon TNF treatment. J. Biol. Chem. 2010; 285:41152–41160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Iademarco M.F., McQuillan J.J., Dean D.C.. Vascular cell adhesion molecule 1: contrasting transcriptional control mechanisms in muscle and endothelium. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:3943–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Guan H., Hou S., Ricciardi R.P.. DNA binding of repressor nuclear factor-κB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit. J. Biol. Chem. 2005; 280:9957–9962. [DOI] [PubMed] [Google Scholar]
- 145. Vonderach M., Byrne D.P., Barran P.E., Eyers P.A., Eyers C.E.. DNA binding and phosphorylation regulate the core structure of the NF-κB p50 transcription factor. J. Am. Soc. Mass Spectrom. 2019; 30:128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Basak S., Kim H., Kearns J.D., Tergaonkar V., O’Dea E., Werner S.L., Benedict C.A., Ware C.F., Ghosh G., Verma I.M. et al.. A fourth IκB protein within the NF-κB signaling module. Cell. 2007; 128:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bates P.W., Miyamoto S.. Expanded nuclear roles for IκBs. Sci. STKE. 2004; 2004:pe48. [DOI] [PubMed] [Google Scholar]
- 148. Wang V.Y., Li Y., Kim D., Zhong X., Du Q., Ghassemian M., Ghosh G.. Bcl3 phosphorylation by Akt, Erk2, and IKK is required for its transcriptional activity. Mol. Cell. 2017; 67:484–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kohda A., Yamazaki S., Sumimoto H.. The nuclear protein IκBζ forms a transcriptionally active complex with nuclear factor-κB (NF-κB) p50 and the Lcn2 promoter via the N- and C-terminal ankyrin repeat motifs. J. Biol. Chem. 2016; 291:20739–20752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Rao P., Hayden M.S., Long M., Scott M.L., West A.P., Zhang D., Oeckinghaus A., Lynch C., Hoffmann A., Baltimore D. et al.. IκBβ acts to inhibit and activate gene expression during the inflammatory response. Nature. 2010; 466:1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Scheibel M., Klein B., Merkle H., Schulz M., Fritsch R., Greten F.R., Arkan M.C., Schneider G., Schmid R.M.. IκBβ is an essential co-activator for LPS-induced IL-1β transcription in vivo. J. Exp. Med. 2010; 207:2621–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Suyang H., Phillips R., Douglas I., Ghosh S.. Role of unphosphorylated, newly synthesized IκBβ in persistent activation of NF-κB. Mol. Cell Biol. 1996; 16:5444–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Yamauchi S., Ito H., Miyajima A.. IκBβ, a nuclear IκB protein, positively regulates the NF-κB-mediated expression of proinflammatory cytokines. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:11924–11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Alverdi V., Hetrick B., Joseph S., Komives E.A.. Direct observation of a transient ternary complex during IκBα-mediated dissociation of NF-κB from DNA. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Sue S.C., Alverdi V., Komives E.A., Dyson H.J.. Detection of a ternary complex of NF-κB and IκBα with DNA provides insights into how IκBα removes NF-κB from transcription sites. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:1367–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Dembinski H.E., Wismer K., Vargas J.D., Suryawanshi G.W., Kern N., Kroon G., Dyson H.J., Hoffmann A., Komives E.A.. Functional importance of stripping in NFκB signaling revealed by a stripping-impaired IκBα mutant. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:1916–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Tartey S., Matsushita K., Vandenbon A., Ori D., Imamura T., Mino T., Standley D.M., Hoffmann J.A., Reichhart J.M., Akira S. et al.. Akirin2 is critical for inducing inflammatory genes by bridging IκBζ and the SWI/SNF complex. EMBO J. 2014; 33:2332–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Horber S., Hildebrand D.G., Lieb W.S., Lorscheid S., Hailfinger S., Schulze-Osthoff K., Essmann F.. The atypical inhibitor of NF-κB, IκBζ, controls macrophage Interleukin-10 expression. J. Biol. Chem. 2016; 291:12851–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Cirillo L.A., Lin F.R., Cuesta I., Friedman D., Jarnik M., Zaret K.S.. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell. 2002; 9:279–289. [DOI] [PubMed] [Google Scholar]
- 160. Zaret K.S., Carroll J.S.. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011; 25:2227–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Natoli G., Saccani S., Bosisio D., Marazzi I.. Interactions of NF-κB with chromatin: the art of being at the right place at the right time. Nat. Immunol. 2005; 6:439–445. [DOI] [PubMed] [Google Scholar]
- 162. Weinmann A.S., Mitchell D.M., Sanjabi S., Bradley M.N., Hoffmann A., Liou H.C., Smale S.T.. Nucleosome remodeling at the IL-12 p40 promoter is a TLR-dependent, Rel-independent event. Nat. Immunol. 2001; 2:51–57. [DOI] [PubMed] [Google Scholar]
- 163. Weinmann A.S., Plevy S.E., Smale S.T.. Rapid and selective remodeling of a positioned nucleosome during the induction of IL-12 p40 transcription. Immunity. 1999; 11:665–675. [DOI] [PubMed] [Google Scholar]
- 164. Barboric M., Nissen R.M., Kanazawa S., Jabrane-Ferrat N., Peterlin B.M.. NF-κB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell. 2001; 8:327–337. [DOI] [PubMed] [Google Scholar]
- 165. Bhatt D., Ghosh S.. Regulation of the NF-κB-mediated transcription of inflammatory genes. Front. Immunol. 2014; 5:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Merika M., Williams A.J., Chen G., Collins T., Thanos D.. Recruitment of CBP/p300 by the IFN-β enhanceosome is required for synergistic activation of transcription. Mol. Cell. 1998; 1:277–287. [DOI] [PubMed] [Google Scholar]
- 167. Angelov D., Charra M., Seve M., Cote J., Khochbin S., Dimitrov S.. Differential remodeling of the HIV-1 nucleosome upon transcription activators and SWI/SNF complex binding. J. Mol. Biol. 2000; 302:315–326. [DOI] [PubMed] [Google Scholar]
- 168. Steger D.J., Workman J.L.. Stable co-occupancy of transcription factors and histones at the HIV-1 enhancer. EMBO J. 1997; 16:2463–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Lowary P.T., Widom J.. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998; 276:19–42. [DOI] [PubMed] [Google Scholar]
- 170. Lone I.N., Shukla M.S., Charles Richard J.L., Peshev Z.Y., Dimitrov S., Angelov D.. Binding of NF-κB to nucleosomes: effect of translational positioning, nucleosome remodeling and linker histone H1. PLoS Genet. 2013; 9:e1003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Mirny L.A. Nucleosome-mediated cooperativity between transcription factors. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:22534–22539. [DOI] [PMC free article] [PubMed] [Google Scholar]