

Abstract
Motivation
Transcriptional networks are models that allow the biological state of cells or tumours to be described. Such networks consist of connected regulatory units known as regulons, each comprised of a regulator and its targets. Inferring a transcriptional network can be a helpful initial step in characterizing the different phenotypes within a cohort. While the network itself provides no information on molecular differences between samples, the per-sample state of each regulon, i.e. the regulon activity, can be used for describing subtypes in a cohort. Integrating regulon activities with clinical data and outcomes would extend this characterization of differences between subtypes.
Results
We describe RTNsurvival, an R/Bioconductor package that calculates regulon activity profiles using transcriptional networks reconstructed by the RTN package, gene expression data, and a two-tailed Gene Set Enrichment Analysis. Given regulon activity profiles across a cohort, RTNsurvival can perform Kaplan-Meier analyses and Cox Proportional Hazards regressions, while also considering confounding variables. The Supplementary Information provides two case studies that use data from breast and liver cancer cohorts and features uni- and multivariate regulon survival analysis.
Availability and implementation
RTNsurvival is written in the R language, and is available from the Bioconductor project at http://bioconductor.org/packages/RTNsurvival/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Transcriptional networks are useful in integrating and interpreting information generated by large-cohort genomics studies. Solutions like RTN (reconstruction of transcriptional networks) (Castro et al., 2016) reconstruct these networks, which consist of units made up of a regulatory element and its targets, called regulons. Regulons provide functional annotations on regulatory associations, and serve as inputs to calculate regulon activity profiles (RAPs) across a cohort. Two recent studies calculated RAPs with the RTN package: Castro et al. (2016) associated regulon activity with disease-specific survival in breast cancer, and Robertson et al. (2017) used regulon status to inform on differences between tumour subtypes in muscle-invasive bladder cancer. While the RTN package supports determining regulon activity, it offers no way to integrate this information with clinical variables. To facilitate such integration, RTNsurvival extends RTN by combining RAPs with clinical and molecular covariates, and performing uni- or multivariate outcomes analysis, aiding in interpreting differences between subtypes in a cohort.
2 Regulon activity inference and survival analyses
Figure 1a gives an overview of the RTNsurvival analysis pipeline. The first step in the pipeline is to infer regulon activity from a transcriptional network. Regulon activity is calculated separately for each sample and regulon, using a two-tailed Gene-Set Enrichment Analysis (GSEA-2T), a modified version of the GSEA-2T approach developed to assess enrichment of two sets of genes (Lamb et al., 2006). The Supplementary Information gives a thorough walkthrough of the GSEA-2T metric, and compares it to the related three-tailed analytic Rank-based Enrichment Analysis (aREA-3T) metric (Alvarez et al., 2016). Figure 1b shows the estimation of ESR1 regulon activity for a breast cancer tumour sample from the METABRIC study (Curtis et al., 2012). For each regulon, a cohort can be stratified by activity in order to fit a survival function and generate Kaplan-Meier curves (Kaplan and Meier, 1958). For example, Figure 1c shows three panels for breast cancer tumours from the METABRIC cohort 1. The first panel shows a ranking of cohort’s tumours based on GSEA-2T ESR1 regulon activity, with the ER-/basal-like samples at the bottom and the ER+/luminal samples at the top. The samples are divided into three groups based on their regulon status (positive dES, undetermined, and negative dES). The second panel shows selected covariates for each tumour, ordered according to the ESR1 regulon activity. The third panel shows a Kaplan-Meier analysis for samples stratified by ESR1 regulon activity. A second survival analysis available in RTNsurvival is a Cox Proportional Hazards Model (Cox et al., 1992), which is fit for selected regulons and covariates. Figure 1d shows a forest plot for 10 breast cancer regulons, with covariates age and tumour grade.
Fig. 1.
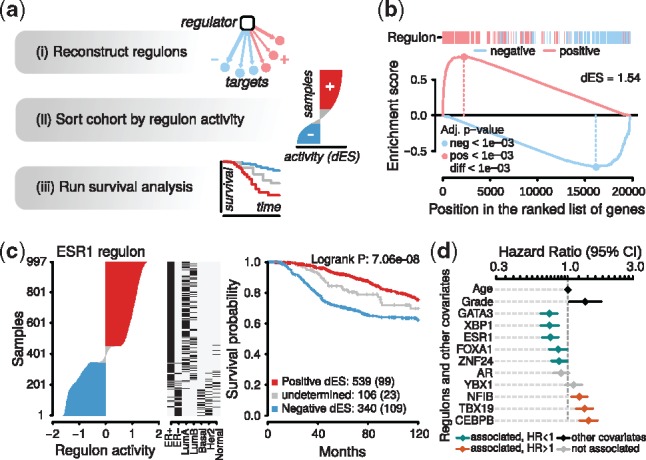
RTNsurvival pipeline and results. (a) Overview of the pipeline. Given a regulatory network from RTN, RTNsurvival calculates RAPs, which are used to stratify samples for a Kaplan-Meier analysis, or to fit a Cox Proportional Hazards model, including confounding variables. In (b), we show the GSEA-2T regulon activity calculation for sample MB-5365, the luminal A tumour in the METABRIC cohort that has the most-activated ESR1 regulon. The MB-5365 transcriptome is enriched with induced ESR1-positive targets and enriched with repressed ESR1-negative targets. The ‘dES’ score quantifies regulon activity in this sample; GSEA-2T returns one dES per regulon per sample. (c) A covariate and survival analysis for the ESR1 regulon. In the left panel, samples are ranked and stratified according to ESR1 regulon activity. The centre panel adds covariates, and shows that samples with higher ESR1 activity were also found to be ER+ in immunohistochemical assays; such patients were more likely to receive hormone therapy. In the right panel, Kaplan-Meier curves are plotted for the 3 strata. (d) A forest plot generated by a multivariate RTNsurvival analysis showing hazard ratios derived from regulon activity for selected regulons, with age and tumour grade
3 Case studies
In Section 1 of the Supplementary Information, we apply RTNsurvival to explore RAPs and perform survival analysis using the clinical variables from the METABRIC study (Curtis et al., 2012). We calculate RAPs for 997 tumour samples and 36 risk-associated transcription factor regulons, describe the association between regulon activity and subtyping, and report survival results from a Kaplan-Meier analysis and a multivariate Cox regression. In Section 2 of Supplementary Information, for the TCGA hepatocellular cancer (LIHC) cohort (The Cancer Genome Atlas Research Network, 2017), we walk through a similar analysis that uses GRch38/hg38 harmonized RNA-seq data from the NCI Genomic Data Commons (GDC).
Funding
This work was supported by the National Council for Scientific and Technological Development (CNPq) (407090/2016-9); and the Cancer Research UK (CRUK), the Breast Cancer Research Foundation (BCRF) (BCRF-17-127). C.S.G. and V.S.C. are funded by the Coordination for the Improvement of Higher Education Personnel (CAPES). S.J.M.J. and A.G.R. are funded by the National Cancer Institute of the National Institutes of Health (U24CA210952). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest: none declared.
Supplementary Material
References
- Alvarez M.J. et al. (2016) Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet., 48, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro M.A.A. et al. (2016) Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat. Genet., 48, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C. et al. (2012) The genomic and transcriptomic architecture of 2, 000 breast tumours reveals novel subgroups. Nature, 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D.R. et al. (1992) Regression models and life-tables In: Kotz S., Johnson N.L. (eds) Breakthroughs in Statistics. Springer Series in Statistics (Perspectives in Statistics). Springer, New York, NY. [Google Scholar]
- Kaplan E.L., Meier P. (1958) Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc., 53, 457–481. [Google Scholar]
- Lamb J. et al. (2006) The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science, 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Robertson A.G. et al. (2017) Comprehensive molecular characterization of muscle-Invasive bladder cancer. Cell, 171, 540–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. (2017) Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell, 169, 1327–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.