

Abstract
Summary
For the analysis of high-throughput genomic data produced by next-generation sequencing (NGS) technologies, researchers need to identify linkage disequilibrium (LD) structure in the genome. In this work, we developed an R package gpart which provides clustering algorithms to define LD blocks or analysis units consisting of SNPs. The visualization tool in gpart can display the LD structure and gene positions for up to 20 000 SNPs in one image. The gpart functions facilitate construction of LD blocks and SNP partitions for vast amounts of genome sequencing data within reasonable time and memory limits in personal computing environments.
Availability and implementation
The R package is available at https://bioconductor.org/packages/gpart.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
In recent genome wide association studies (GWAS) and population genetic studies, researchers increasingly investigate dense single nucleotide polymorphism (SNP) data produced by new sequencing technologies (Kilpinen and Barrett, 2013). To reduce the dimension of high-throughput genomic data for genetic association analysis or to find evidence for population genetic phenomenon, one can utilize genomic linkage disequilibrium (LD) structure, especially LD blocks (or haplotype blocks).
The development of algorithms and software to identify the LD blocks from SNP genotype data mostly occurred before the era of deep sequencing technology. To determine the LD blocks, Gabriel et al. (2002) proposed a method based on estimation of the confidence interval of . Zhang et al. (2002, 2003) developed a dynamic programming algorithm to detect common haplotypes in a block. Wang et al. (2002) proposed an approach using a four-gamete test. Barrett (2005) proposed the Solid Spine method which finds blocks based on the strong LD with markers at the block boundary, and Pattaro et al. (2008) developed a method based on an MCMC algorithm. As reported in Kim et al., (2018), the previous methods and definitions for LD blocks (Gabriel et al., 2002; Pattaro et al, 2008; Wang et al, 2002) do not serve well to identify long range LD blocks in sequencing data such as available in the 1000 Genomes Project. We previously proposed a new method of LD block construction called Big-LD (Kim et al. 2018), using graph-based clustering techniques. We showed that Big-LD produces larger size blocks, achieves better optimization in terms of LD strength within and across LD blocks, and agrees better with recombination hotspots, compared to existing approaches such as methods implemented in Haploview (Barrett, 2005; Gabriel et al., 2002; Wang et al, 2002) or related methods (Pattaro et al, 2008; Taliun et al, 2014, 2016).
In this R/Bioconductor implementation gpart, we provide a new SNP partitioning method based on not only LD block structures but also on gene positions, together with a visualization tool to display a LD heatmap with LD block partitioning information and gene positions. The algorithm GPART uses an updated version of Big-LD which can deal with both r2 and LD measures and has improved speed and memory efficiency for construction of LD blocks by means of a new heuristic algorithm.
2 Implementation and main functions
The R package gpart provides three main functions, BigLD, GPART, LDblockHeatmap; and is available at the Bioconductor repository (https://bioconductor.org/packages/gpart). The package contains a vignette with detailed explanation about the functions and their options, illustrated by various examples and figures.
2.1 Updated version of Big-LD
Big-LD is a method to identify LD blocks using SNPs (Kim et al., 2018). The results of the Big-LD algorithm can be obtained using the BigLD function in the gpart package. In gpart, the Big-LD algorithm adopts an updated version of the published CLQ algorithm (Kim et al., 2018) that finds LD bins using the newly added heuristic algorithm (near-nonhrst algorithm, detailed in Supplementary Methods) which has been extended to account for both LD measures (r2 and ). Although the new heuristic algorithm is not as fast as the existing heuristic CLQ algorithm (fast algorithm), it returns results more similar to those obtained by the non-heuristic CLQ algorithm in a reasonable time (Supplementary Table S1). Users can choose a CLQ mode (maximal/density) and heuristic algorithm (nonhrst/fast/near-nonhrst) depending on their research aim or computational environment (see Supplementary Results, Supplementary Tables S2 and S3). We apply BigLD to 1000 Genomes Project phase 3 data for MAF > 5% (Supplementary Table S4) and to a GWAS dataset (Supplementary Table S5) (Roshandel et al., 2018).
2.2 GPART: SNP partitioning method
We developed a SNP partitioning algorithm, GPART, which partitions sets of contiguous SNPs into blocks using the Big-LD results combined with gene position information. Big-LD considers only LD structure within the given data; therefore depending on the LD, the results can include a large number of singleton SNPs or extremely large LD blocks. According to the purpose of downstream analysis, it can be appropriate to limit the number of SNPs in each block to increase analytical effectiveness. The GPART algorithm partitions an entire set of SNPs in a specified region so that all blocks satisfy specified minimum and maximum size limits, where size refers to a number of SNPs.
The function GPART provides two different method types, a gene-based method (geneBased) and an LDblock-based method (LDblockBased). The gene-based method first fuses gene position information and Big-LD blocks, then splits or merges blocks that do not meet pre-defined size criteria. The LDblock-based method splits large LD blocks to satisfy the pre-defined size criteria and first takes them as new blocks. Then it merges the remaining consecutive small-sized LD blocks into new blocks of at least the minimum size. In this merging stage, as many small LD blocks as possible can be merged if the small blocks overlap with a gene region. Depending on whether the gene position information is used when combining small blocks, the LDblock-based method is divided into two methods: the only-block method (onlyBlocks) and the use-gene-region method (useGeneRegions). The algorithm is detailed in Supplementary methods. Application of GPART to 1000 Genomes Project phase 3 data and a GWAS dataset (Roshandel et al., 2018) is reported in Supplementary results (Supplementary Tables S6 and S7).
2.3 LDblockheatmap: visualization function to show LD structure and gene positions
The LDblockheatmap function provides plotting capabilities to visualize the LD heatmap, LD block boundaries of Big-LD results or genomic sequence partitioning results of GPART, and physical location of LD blocks and genes (Fig. 1). The function displays gene regions when gene positions are provided and can draw a figure including up to 20 000 SNPs. See Supplementary Figures S1–S3 for examples using various number of SNPs. For datasets with less than 200 SNPs, the LD bin structure obtained by the CLQ algorithm can be visualized (Fig. 1 and Supplementary Fig. S1). The LD heatmap can also be visualized without Big-LD results or gene positions.
Fig. 1.
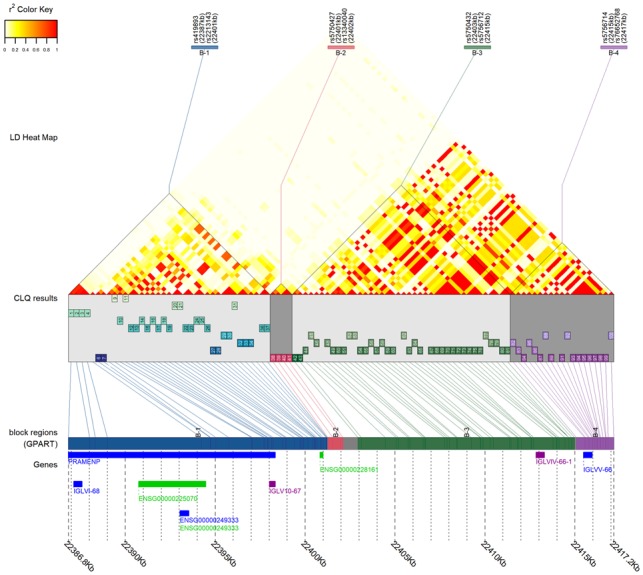
Example plot produced by LDblockHeatmap function with GPART result (see Supplementary Fig. S1 for detailed explanation of each component of the plot)
For various examples plotted by LDblockHeatmap, see the vignette of the package gpart.
3 Conclusion
In this paper, we introduce an R package, called gpart, which provides novel functions to cluster and partition a given genomic region by modeling the underlying LD structures of the SNPs as graphs. In addition, the package offers an efficient visualization function to display the obtained results with genomic information. The package gpart is available at Bioconductor.
Funding
This work was supported by the National Research Foundation of Korea (NRF) [NRF-2018R1A2B6008016]; the Canadian Institutes of Health Research (CIHR) [Operating/Projects MOP-84287 & PJT 159463]; the Canadian Statistical Sciences Institute; and the Canadian Institutes of Health Research Strategic Training for Advanced Genetic Epidemiology (CIHR STAGE) [GET-101831].
Conflict of Interest: none declared.
Supplementary Material
References
- Barrett J.C. (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics, 21, 263–265. [DOI] [PubMed] [Google Scholar]
- Gabriel S.B. et al. (2002) The structure of haplotype blocks in the human genome. Science, 296, 2225–2229. [DOI] [PubMed] [Google Scholar]
- Kilpinen H., Barrett J.C. (2013) How next-generation sequencing is transforming complex disease genetics. Trends Genet., 29, 23–30. [DOI] [PubMed] [Google Scholar]
- Kim S.A. et al. (2018) A new haplotype block detection method for dense genome sequencing data based on interval graph modeling of clusters of highly correlated SNPs. Bioinformatics, 34, 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattaro C. et al. (2008) Haplotype block partitioning as a tool for dimensionality reduction in SNP association studies. BMC Genomics, 9, 405.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshandel D. et al. (2018) Meta-genome-wide association studies identify a locus on chromosome 1 and multiple variants in the MHC region for serum C-peptide in type 1 diabetes. Diabetologia, 61, 1098–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliun D. et al. (2014) Efficient haplotype block recognition of very long and dense genetic sequences. BMC Bioinf., 15, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliun D. et al. (2016) Fast sampling-based whole-genome haplotype block recognition. IEEE/ACM Trans. Comput. Biol. Bioinf., 13, 315–325. [DOI] [PubMed] [Google Scholar]
- Wang N. et al. (2002) Distribution of recombination crossovers and the origin of haplotype blocks: the interplay of population history, recombination, and mutation. Am. J. Hum. Genet., 71, 1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K. et al. (2002) A dynamic programming algorithm for haplotype block partitioning. Proc. Natl Acad. Sci. USA, 99, 7335–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Jin L. (2003) HaploBlockFinder: haplotype block analyses. Bioinformatics, 19, 1300–1301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.