

Trib1 controls atherosclerotic plaque macrophage function by up-regulating OLR1, promoting foam cell formation and atherosclerosis.
Abstract
Macrophages drive atherosclerotic plaque progression and rupture; hence, attenuating their atherosclerosis-inducing properties holds promise for reducing coronary heart disease (CHD). Recent studies in mouse models have demonstrated that Tribbles 1 (Trib1) regulates macrophage phenotype and shows that Trib1 deficiency increases plasma cholesterol and triglyceride levels, suggesting that reduced TRIB1 expression mediates the strong genetic association between the TRIB1 locus and increased CHD risk in man. However, we report here that myeloid-specific Trib1 (mTrib1) deficiency reduces early atheroma formation and that mTrib1 transgene expression increases atherogenesis. Mechanistically, mTrib1 increased macrophage lipid accumulation and the expression of a critical receptor (OLR1), promoting oxidized low-density lipoprotein uptake and the formation of lipid-laden foam cells. As TRIB1 and OLR1 RNA levels were also strongly correlated in human macrophages, we suggest that a conserved, TRIB1-mediated mechanism drives foam cell formation in atherosclerotic plaque and that inhibiting mTRIB1 could be used therapeutically to reduce CHD.
INTRODUCTION
Atherosclerosis, a progressive disease of arterial blood vessels and the main underlying cause of stroke, myocardial infarction, and cardiac death (1), is initiated by the conversion of plaque macrophages to cholesterol-laden foam cells (2) in the arterial intima (3). In the early-stage atherosclerotic plaque, this transformation is induced by the uptake of both low density lipoprotein-cholesterol (LDL-C) and oxidized LDL (oxLDL) (2, 4), which may serve a beneficial purpose (3); but unrestrained, the crucial function of plaque macrophages in resolving local inflammation is compromised, and the development of unstable, advanced lesions ensues (3). It has been shown that foamy macrophages are not only less effective in clearing apoptotic cells (5), they are also more prone to apoptosis (6), thus increasing secondary necrosis and the release of cellular components and lipids that ultimately form the necrotic core of advanced plaques. Hence, there have been investigations into the identities of macrophage-specific proteins that induce lipid accumulation. Thus, myeloid–lipoprotein lipase (LPL), for example, has been shown to enhance the retention of LDL-C and triglyceride-rich remnant particles within the artery wall (7) and induce foam cell formation (8), while the scavenger receptor, oxidized low-density lipoprotein receptor 1 (OLR1) has been found to internalize oxLDL (9), promoting not only lipid accumulation and growth but also the survival of macrophage foam cells (10). Conversely, myeloid-ApoE expression has been shown to promote high-density lipoprotein (HDL)–mediated cholesterol efflux (11) and macrophage switching from a proinflammatory (M1) to an alternatively (M2) activated phenotype (12). However, substantial advances in the development of cardiovascular disease (CVD) therapeutics await the identification of an apical regulator(s) that acts in a coordinated manner on the multiple downstream processes governing lipid accumulation, as well as atherogenicity of plaque-resident macrophages.
Tribbles 1 (Trib1) has been detected in murine plaque-resident macrophages (13), and variants at the TRIB1 locus have been associated with increased risk of hyperlipidemia and atherosclerotic disease in multiple populations (14, 15). However, no study had examined the macrophage-specific cellular processes dependent on myeloid-specific Trib1 (mTrib1) expression and how these tally with the assumed atheroprotective properties of this pseudokinase. At the whole-body level, one study has shown that Trib1-deficient mice have markedly reduced numbers of M2-like (F4/80+ MR+) macrophages in multiple organs, including adipose tissue (16). Hence, these studies strongly implicated that loss of macrophage-Trib1 expression within the arterial wall would lead to excessive atherosclerotic plaque inflammation and/or impair inflammation resolution and promote atheroma formation. Moreover, in hepatocytes, Trib1 suppresses very-low-density lipoprotein production and de novo lipogenesis (15), indicating that the association between variants at the TRIB1 locus and atherosclerotic disease (14, 15) relates to loss of TRIB1 activity.
In the current study, we found that contrary to expectations, myeloid-specific knockout (KO) of Trib1 is atheroprotective, while mTrib1 expression is detrimental. In brief, Trib1 increased OLR1 RNA and protein expression, oxLDL uptake, foamy macrophage formation, and atherosclerotic burden in two distinct mouse models of human disease. The expression of these two genes, as well as those of LPL and SCARB1 (which mediates selective HDL-cholesterol uptake (17)), is also tightly linked in human macrophages. Collectively, our studies reveal an unexpected beneficial effect for selectively silencing Trib1 in arterial plaque macrophages.
RESULTS
Myeloid Trib1 increases atherosclerosis burden
Immunostaining of human coronary atheromas from patients undergoing endarterectomies detected Trib1 in the arterial wall, including in 42.79 (± 2.31)% of CD68+ macrophages (Fig. 1A). We therefore examined the impact of macrophage Trib1 expression on atherogenesis by creating mice expressing low, wild-type (WT), and elevated levels of myeloid Trib1 as outlined in Fig. 1 (B to F). Although previous studies have demonstrated that global Trib1 KO significantly increases perinatal lethality (16), Trib1-floxed mice and myeloid-specific Trib1 KO (Trib1mKO) mice were fully viable and bred normally (fig. S1, A to D). mTrib1 transgenic (overexpressing, Trib1mTg) mice were also fully viable and bred normally. mTrib1 RNA levels were substantially lower in Trib1mKO than in floxed WT littermates, as judged by reverse transcription–quantitative polymerase chain reaction (RT-qPCR) assays performed on bone marrow–derived macrophages (BMDMs) prepared from these animals (Fig. 1G). As judged by enhanced green fluorescent protein (eGFP) expression, the bicistronic Trib1 transgene was expressed in 78.43 ± 2.33% and 65.58 ± 0.92% of blood monocytes and peritoneal macrophages, respectively (Fig. 1G), and overall, the transgene increased BMDM Trib1 RNA levels by 2.49 ± 0.43 (SEM) fold (Fig. 1G, bottom right). Consistent with previous findings (18), the transgene was also expressed in neutrophils, which form a minor component of the immune cell population within very early-stage atherosclerotic lesions (19). Thus, we detected eGFP in 53.88 ± 2.41% and 34.93 ± 2.96% of Trib1mTg blood and bone marrow CD11b+/Ly6C−/Ly6G+ cells, respectively, compared to 25.95 ± 3.16% and 12.42 ± 2.01% in their CD11b+/Ly6C+/Ly6G− monocyte counterparts (fig. S1, E to I). However, in marked contrast to the reported full-body Trib1 KO mouse (16), Trib1mKO mice were not afflicted by reduced numbers of total, or individual, white blood cells (Fig. 1H) or by reduced macrophage numbers in their adipose tissue (F4/80+, fig. S2A), liver (F4/80+, fig. S2B), or spleen (F4/80+ and CD206+, fig. S2C). Similar to Trib1mKO mice, Trib1mTg mice displayed no gross abnormalities and had WT numbers of white blood cells (Fig. 1H). In addition, the sizes of their adipocytes (fig. S2A), liver (fig. S2B), and splenic (fig. S2C) macrophage populations were unaltered.
Fig. 1. Generation and characterization of myeloid-specific Trib1 mouse strains.
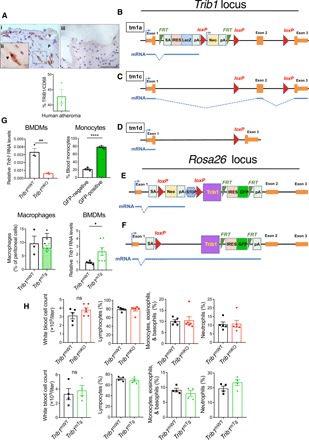
(A) Representative immunohistochemistry image of human atherosclerotic plaque (P). Red, TRIB1; brown, CD68+. (ii) Magnification (×40) of boxed area. Arrowhead highlights a double-positive cell. Quantification (mean ± SD) of three patient samples. (iii) Isotype control (scale bar, 50 μm). (B) Targeting construct used to produce the null, conditional-ready/floxed (tm1c) and conditional-null (tm1d) Trib1 alleles. Predicted transcripts below. FRT, flippase recognition target; SA, splice acceptor; pA, polyadenylation motif; IRES, internal ribosome entry site; LacZ, β-galactosidase; Neo, neomycin resistance gene. (C) Trib1mWT allele following removal of the “gene-trap” cassette. (D) Conditional-null Trib1 allele was produced by crossing tm1c and Cre-expressing mice. (E) Construct used to produce Trib1mWT and Trib1mTransgenic (Tg) mice. (F) Cre-mediated excision of the STOP cassette produces a bicistronic Trib1-eGFP transcript. Bent arrow, indicates transcription from endogenous Rosa26 promoter. (G) Trib1 RNA (relative to Actb) in BMDMs from homozygous tm1c (i.e., Trib1mWT) and Trib1mKO mice (n = 3 per group). eGFP expression in monocytes of three Trib1mTg mice and peritoneal macrophages from specified mice (n = 3 per group). Trib1 RNA levels (relative to Trib1mWT) in BMDMs of Trib1mTg (n = 5 to 7 per group). (H) Blood cell counts of mixed-gender Trib1mKO (top) and Trib1mTg (bottom) and their respective WT littermates (n = 5 to 6 per group). Data are means ± SEM. Significances were determined by Student’s t test, *P < 0.05, **P < 0.01, and ****P < 0.0001. ns, non-significant.
To address the contribution of mTrib1 in early atherosclerosis, we first transplanted bone marrow cells from the Trib1mKO and Trib1mTg mice and their respective controls (i.e., non-CRE, floxed KO, and Tg alleles) into 12- to 13-week-old lethally irradiated male ApoE−/− mice (Fig. 2A). Thus, all recipient mice received ApoE+/+–bone marrow cells to mitigate the previously described effects of total ablation of this apolipoprotein on both classical/proinflammatory (M1) and alternative/anti-inflammatory (M2) polarization (12) and to provide them with a physiologically important source of APOE to aid normalization of plasma cholesterol levels and of the lipoprotein profile in this otherwise extreme hyperlipidemic mouse model of human atherosclerosis (12, 20). In short, this experiment allowed modeling of early atherogenesis in a setting more akin to the human disease. Following a 7-week recovery period, the chimeric mice were fed a Western diet containing 0.2% cholesterol for 12 weeks. At sacrifice, and consistent with expectations of the study design, the chimeric mice had relatively low plasma cholesterol levels for a mouse model of human atherosclerosis (fig. S3A).
Fig. 2. Myeloid-specific Trib1 expression increases atherosclerosis burden in two murine models of human atherosclerosis.
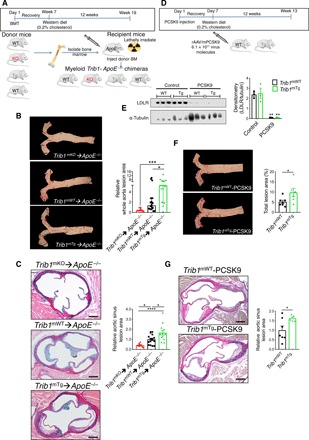
(A) Schematic of the bone marrow transplant experiment. Bone marrow cells from myeloid-specific Trib1 KO and transgenic (Tg) mice and their respective WT controls were transplanted into ApoE−/− recipients. (B) Representative en face Oil Red O staining of thoracic aortas (week 19) from specified chimeras. Lesion areas were calculated as percentages of the total surface area of the whole aorta and normalized (median, ±95% confidence interval) to Trib1mWT; n = 10 to 18 per group. (C) Representative images of Elastic van Gieson–stained aortic sinus lesions. Quantification relative to WT (n = 10 to 16 mice per group). (D) Second model of human atherosclerosis. rAAV/mPCSK9, recombinant adenovirus–produced murine proprotein convertase subtilisin/kexin 9. (E) LDLR protein in liver samples from specified mice was quantified by Western blotting (n = 3 per group). (F) Representative en face Oil Red O staining of thoracic aortas from specified mice. Lesion areas were calculated as percentages of the total surface areas of the whole aorta (n = 6 to 7 per group). (G) Representative images of Elastic van Gieson–stained aortic sinus lesions of specified mice and quantification, relative to WT (n = 5 to 7 per group). Scale bars, 200 μm (C and G). Data are means ± SEM. Significance was determined by one-way (B and C) or two-way analysis of variance (ANOVA) (E) or Student’s t test (F and G). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Unexpectedly, we found less atherosclerosis in the thoracic aorta of Trib1mKO➔ApoE−/− chimeras than in the control WT mice (Fig. 2B and fig. S4A). Conversely, there was a significantly higher atheroma burden in the Trib1mTg➔ApoE−/− mice (Fig. 2B and fig. S4A). Similarly, the lesions in the aortic sinus were, on average, smaller in the Trib1mKO➔ApoE−/− mice and larger in the Trib1mTg➔ApoE−/− mice (Fig. 2C and fig. S4B). However, the collagen contents of the Trib1mKO➔ApoE−/− and Trib1mTg➔ApoE−/− in these “early-stage” plaques and their clinical pathology were comparable to those of the chimeric Trib1mWT➔ApoE−/− mice (fig. S4C). In short, we found that mTrib1 expression increased the atherosclerotic burden of ApoE−/− mice, despite having little impact on plasma LDL-cholesterol levels (fig. S3A).
To confirm that mTrib1 accelerates the development of atherosclerosis (Fig. 2, B and C), we created an LDL receptor (Ldlr) knockdown model of human atherosclerosis (21) to induce hyperlipidemia and atherosclerosis in otherwise WT mice (21). Specifically, mice were injected with an adeno-associated virus (rAAV8) encoding for proprotein convertase subtilisin/kexin 9 (Fig. 2D), which previous studies have shown lowers both hepatic and extrahepatic surface cell expression of the Ldlr (22). Following feeding a Western diet for 12 weeks, this intervention produced comparable, highly significant reductions in LDLR protein levels in the Trib1mTg and Trib1mWT mice (Fig. 2E) and a similar degree of hyperlipidemia (fig. S3B). However, despite this and consistent with mTrib1 expression increasing atheroma formation in ApoE−/− mice, Trib1mTg injected with rAAV8-Pcsk9 developed a significantly higher atherosclerotic burden in their aorta and aortic sinus than their similarly injected Trib1mWT mice (Fig. 2, F and G).
Myeloid Trib1 increases macrophage/foam cell size in the atherosclerotic plaque
Next, we investigated the macrophage content and phenotype in the atherosclerotic plaque in each mouse model. This revealed that the aortic sinus lesions of Trib1mKO➔ApoE−/− mice contained a much smaller MAC-3+ immunoreactive area than the chimeric Trib1mWT➔ApoE−/− mice, while on average, the Trib1mTg➔ApoE−/− atheromas contained a marginally larger stained area (Fig. 3, A and B). However, there was no preferential loss of YM1+ macrophages in the Trib1mKO➔ApoE−/− lesions (Fig. 3B), consistent with the finding that M2 polarization of Trib1-deficient BMDMs isolated from whole-body Trib1mKO mice are compromised to a similar extent as M1 polarization (23). In addition, we could not attribute the proatherogenic activity of myeloid Trib1 expression to a preferential increase in the proinflammatory macrophage (NOS2+) content of Trib1mTg➔ApoE−/− plaque (Fig. 3B, right). Rather, the increased atherosclerosis in the Trib1mTg➔ApoE−/− chimeras was attributable to a doubling of foam cell numbers (cells with characteristic foamy appearance and MAC-3+ (fig. S4D), and on average, these cells were also larger (Fig. 3, A and C, and fig. S4D). While the Trib1mWT-Pcsk9 atheromas had a similar-sized macrophage population (Fig. 3D), the size of the plaque-resident foam cells in the transgenic animals was larger (Fig. 3D).
Fig. 3. Myeloid-Trib1 induces foam cell expansion.
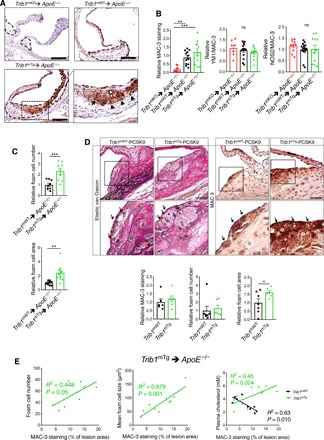
(A) MAC-3 staining (brown) of representative cross sections of the aortic sinus from specified mice (scale bars, 100 μm). Dashed lines indicate lesion boundaries. Arrows highlight foam cells in the boxed region (40-fold magnification). (B) Staining of aortic sinus lesions from specified chimeric mice with specified antibodies: MAC-3 (n = 9 to 12 per group) and double-positive YM1/MAC-3 (n = 7 to 14 per group) and NOS2/MAC-3 (n = 10 to 16 per group) cells. (C) Quantification of relative foam cell numbers (top) and size (bottom) in aortic sinus lesions of specified chimeras. n = 10 to 16 per group. (D) Representative images (scale bars, 30 μm) of Elastic van Gieson– and MAC-3–stained aortic sinus lesions from specified mice injected with rAAV/mPCSK9 (n = 9 to 11 per group). Arrows in magnified (×40) images of boxed area highlight foam cells. Quantification of MAC-3 staining and foam cell numbers and sizes (n = 6 to 7 per group). (E) Correlation between MAC-3 staining (x axis) in aortic sinus lesions of specified chimeric mice and foam cell numbers (left), foam cell size (middle), and plasma cholesterol levels (right). MAC-3 staining expressed as percentage (%) of total aortic sinus lesion area. R2, Pearson correlation coefficient. In (B) to (D), data (means ± SEM) are expressed relative to WT. Statistical differences were determined by one-way ANOVA (B) or Student’s t test (C and D). *P < 0.05, **P < 0.01, and ***P < 0.001.
In the Trib1mTg➔ApoE−/− chimeras, there was a stronger correlation between the mean foam cell size and the percentage of aortic sinus stained by MAC-3 than between MAC-3+ staining and foam cell numbers (Fig. 3E), and we could not ascribe the observed increase in plaque-foam cell numbers on the effects of mTrib1 expression on blood cholesterol levels (Fig. 3E), HDL-C levels, or the nonsignificant rise in LDL-C (fig. S3). While the Trib1mWT➔ApoE−/− chimeras with the highest plasma cholesterol, HDL-C, and LDL-C concentrations had the lowest amount of MAC-3+ staining in their aortic sinus lesions, the inverse was true for the Trib1mTg➔ApoE−/− chimeras (Fig. 3E, right, and fig. S3). Thus, collectively, these data indicate that increased macrophage lipid uptake/storage was the prominent driving force for the observed foam cell expansion besetting the early stage of the atherosclerotic process in these and the Trib1mTg-Pcsk9 mice.
Myeloid TRIB1 expression induces OLR1 expression in both mouse and man
To identify potential cellular mechanisms by which mTrib1 enhances foam cell expansion, we analyzed the gene expression characteristics of human TRIB1High monocytes and TRIB1High monocyte-derived macrophages (MDMs) using the microarray RNA data produced in the Cardiogenics Transcriptomic Study (CTS) (24). In this dataset, involving samples from 758 individuals, TRIB1 RNA levels were, on average, higher in monocytes than in MDMs (Fig. 4A, top) but, as is evident from the analyses of RNA levels in 596 paired samples, there was no correlation between TRIB1 RNA levels in these two cell types (Fig. 4A, bottom). Moreover, genes differentially expressed in TRIB1High versus TRIB1Low monocytes were enriched for different sets of “DAVID” Gene Ontology cluster terms (tables S1 and S2) than those characterizing the more lipid-based transcriptome of TRIB1High MDM (Fig. 4, B and C, and table S3).
Fig. 4. Myeloid TRIB1 induces reciprocal changes in oxLDL and HDL receptor expression in human and mouse macrophages.
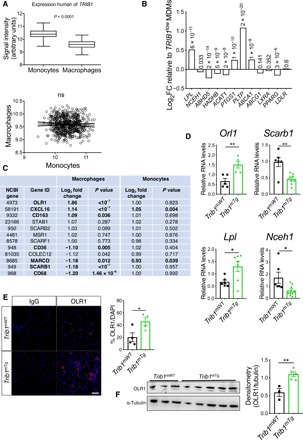
(A) TRIB1 RNA levels in monocytes and MDMs of CTS participants (24). Correlation (R2 < 0.001, P = 0.47) was performed on 596 paired monocyte and MDM samples. (B) MDM (n = 596) and monocytes (n = 758) were ranked according to TRIB1 RNA contents and gene expression values in the top and bottom quartiles compared. Log2 fold changes (FC) of specified RNAs in TRIB1High (n = 149) versus TRIB1Low (n = 149) MDM, with associated P values. (C) FC and P values for differential expression of RNAs encoding representative scavenger receptors, including CD36, which mediates (ox)-phospholipid and long-chain fatty acid uptake (39), the acetylated-LDL scavenging receptor (40), and macrophage scavenger receptor (40). Comparisons are between TRIB1High versus TRIB1Low MDMs and between TRIB1High (n = 191) versus TRIB1Low (n = 191) monocytes. (D) RT-qPCR quantification of RNA (mean ± SEM) in BMDM prepared from specified mice (n = 5 to 9 per group). (E) Immunocytochemistry of nonpolarized Trib1mWT and Trib1mTg BMDMs. OLR1 (red), nuclei counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 50 μm. Quantifications performed on BMDMs prepared from four to five mice per group. (F) Western blot analysis of OLR1 in Trib1mWT and Trib1mTg BMDMs (n = 3 to 5 per group). In (D) to (F), significance was determined by Student’s t test; *P < 0.05, and **P < 0.01.
The Cardiogenics Transcriptomic data strongly suggested that the mTRIB1-induced foam cell phenotype stemmed from increased oxLDL uptake rather than changes in LDLR and scavenger receptor class B type 1 [which mediates selective HDL-cholesterol uptake and efferocytosis (17)] expression (Fig. 4C) or reductions in ABCG1- and ABCA1-mediated cholesterol efflux (Fig. 4B). Notably, OLR1 was the fourth most differentially expressed gene in the TRIB1High human MDMs and the most highly altered scavenger receptor in these cells (Fig. 4C). We therefore examined the effect of mTrib1 transgene expression on the expression of this oxLDL receptor (9). This revealed that Trib1mTg BMDMs contained more Olr1 RNA but fewer Scarb1 transcripts (Fig. 4D) than their Trib1mWT counterparts, indicating that the increased numbers of OLR1 and reduced numbers of SCARB1 transcripts in human TRIB1High MDMs are causally related to the increased number of TRIB1 transcripts in these cells. The reciprocal relationship between OLR1 and SCARB1 RNA levels in TRIB1High MDMs was also recapitulated in interferon-γ (IFN-γ)/lipopolysaccharide (LPS), interleukin-4 (IL-4)–polarized (fig. S5A) and fatty acid–polarized MDM samples but not in HDL-polarized MDMs (fig. S5B and table S4). Rather, HDL-polarized MDMs contained OLR1 and SCARB1 RNA levels indistinguishable from those of nonpolarized MDMs (fig S5, A and B, and table S4), indicating that Trib1 induces Olr1 expression and foam cell formation via a non-HDL uptake–mediated mechanism. Last, to substantiate the evidence for causal TRIB1 involvement in OLR1 expression, we stained BMDMs for OLR1 protein. OLR1 was detected in twice as many Trib1mTg cells than their WT counterparts (Fig. 4E), consistent with the Western blotting analysis of whole BMDM cell lysates (Fig. 4F).
mTrib1-induced OLR1 expression in plaque macrophages increases atherosclerotic burden
To corroborate the evidence for causal mOLR1 involvement in plaque-resident macrophage foam cells expansion, we quantified OLR1 expression in the aortic sinus lesions of our mouse models using an OLR1 antibody that recognizes the cell surface–expressed form of this oxLDL receptor, as well as the proteolytically cleaved (soluble) extracellular form (9). The antibody detected OLR1 in MAC-3+ cells and in acellular areas of the mouse aortic sinus lesions, including in regions adjacent to plaque macrophages (Fig. 5A). Moreover, as expected from the known expression and regulation of this scavenger receptor in endothelial cells (9), significant amounts of OLR1 was also detected in the nonmacrophage (i.e., MAC-3−) cell population at the plaque surface of the more hyperlipidemic model of human atherosclerosis (Fig. 5A and fig. S3). Last, confirming the causal involvement of mTRIB1 in mOLR1 expression (Fig. 4, D to F), Fig. 5A shows that the anti-OLR1 antibody detected OLR1 in more of the plaque macrophages of Trib1mTg➔ApoE−/− mice than in those of the Trib1mWT➔ApoE−/− control animals (33.89 ± 6.56% versus 16.18 ± 4.05%); a result which was replicated in the Trib1mTg-Pcsk9 and Trib1mWT-Pcsk9 mice (29.35 ± 7.42% versus 10.38 ± 3.36%) (Fig. 5A).
Fig. 5. Myeloid-Trib1 increases ORL1 expression, cholesterol uptake, and neutral lipid accumulation.
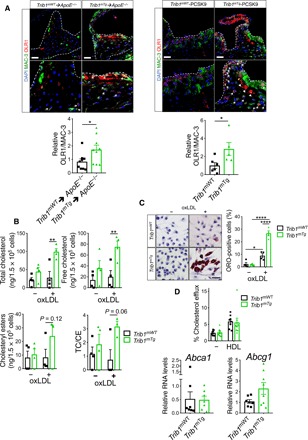
(A) Representative images (scale bars, 25 μm) of aortic sinus lesions from specified mice and enlarged images. Dashed lines indicate boundaries of lesions. MAC-3 (green), OLR1 (red), and nuclei counterstained with DAPI (blue). Arrows indicate OLR1-positive macrophages. Arrowheads indicate assumed acellular OLR1. Quantification: Trib1mWT➔ApoE−/− and Trib1mTg➔ApoE−/− chimeras, nine per group; mTrib1-PCSK9, five to six per group (mean ± SEM). (B) Intracellular total cholesterol, unesterified cholesterol, and cholesteryl ester contents of Trib1mTg and Trib1mWT bone marrow cells differentiated into macrophages and incubated with oxLDL (25 μg/ml) for 24 hours (n = 4 per group). (C) Representative images of BMDMs stained with Oil Red O (scale bar, 50 μm). Quantification was performed on three fields of view per sample. (D) Quantification of cholesterol efflux from cholesterol-loaded BMDMs to human HDL (n = 8 to 9 per group). RT-qPCR quantification of Abca1 and Abcg1 RNA in nonpolarized BMDMs prepared from specified mice (n = 7 to 8 per group). Data are means ± SEM. Significance determined by Student’s t test (A and D, bottom panels) or two-way ANOVA with Sidak’s multiple comparisons posttest (B to D) *P < 0.05, **P < 0.01, and ****P < 0.001.
To mechanistically validate the contribution of mTrib1-induced Olr1 expression to foam cell expansion, we incubated nonpolarized BMDMs with oxLDL for 24 hours. This led to marked rises in the intracellular levels of both total cholesterol (2.71 ± 0.24–fold, P = 0.0091) and unesterified cholesterol (5.81 ± 0.83–fold, P = 0.0049) in the Trib1mTg BMDMs but not in their WT counterparts (Fig. 5B). In addition, as judged by Oil Red O staining, oxLDL transformed nearly three times as many Trib1mTg BMDMs into foam cells than Trib1mWT cells (P < 0.0001, Fig. 5C), as evidenced by the very visible increase in neutral lipid accumulation in these cells upon exposure to oxLDL. In contrast to the profound effects of oxLDL on cholesterol accumulation in unpolarized Trib1mTg BMDMs, we observed no impairment of HDL-mediated cholesterol efflux (Fig. 5D), consistent with the observation that neither Abca1 nor Abcg1 RNA levels are reduced in these BMDMs (Fig. 5D) and that the Trib1mTg➔ApoE−/− chimeras have higher, rather than lower, HDL-C levels than their WT peers (fig. S3). Thus, collectively, our results suggest that Trib1-induced foam cell expansion in early-stage atherosclerotic plaque arises from increased cholesterol/neutral lipid uptake and retention rather than reduced HDL-mediated cholesterol efflux.
DISCUSSION
Despite the success in establishing that hepatic Trib1 expression affects the regulation of multiple cellular processes modulating blood cholesterol and triglyceride levels (15), the influence of global-KO of Trib1 on shaping the phenotype of macrophages (16), and the finding that variants at the TRIB1 locus are associated with and increased coronary heart disease (CHD) risk (14), the contribution of Trib1 on atherogenesis remains to be addressed. Here, we demonstrate that there is a wide distribution of TRIB1 RNA levels in human MDMs and that genetically engineered changes in mTrib1 expression in mouse models of early-stage human atherosclerosis markedly affect the size of developing plaques and the morphological and functional properties of plaque macrophages (Fig. 6). In summary, we have confirmed the proatherogenic impact of myeloid TRIB1 in two distinct in vivo models of human atherosclerosis.
Fig. 6. Model summarizing the proposed effects of differences in mTRIB1 expression on foam cell expansion in early-stage atherosclerosis.
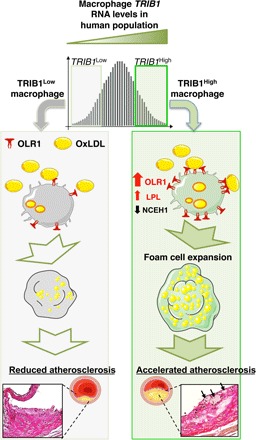
Factors up-regulating mTRIB1 expression in human MDMs and plaque macrophages simultaneously increase cholesterol and neutral lipid uptake, with no compensatory rise in cholesterol efflux. Schematic recognizes that increased OLR1 expression increases the probability of this scavenger receptor assembling as a hexamer made up of three homodimers on the macrophage cell surface and that this configuration leads to a marked increase in its affinity for oxLDL (9) and hence OLR1-mediated uptake of oxLDL lipids. In the setting of no compensatory rise in HDL-mediated cholesterol efflux, accelerated foam cell expansion and increased atheroma burden ensue, highlighting the therapeutic potential of inhibiting macrophage Trib1 expression to block the gene expression changes that both promote macrophage cholesterol uptake and cholesteryl ester formation accumulation and prevent the increased hydrolysis of the accumulated cholesteryl ester and thus the up-regulation of the reverse cholesterol transport pathway to mediate the removal of cholesterol from the arterial wall.
In the experiments reported here, we provide evidence that mTrib1 transgene expression may reduce vascular cell exposure to oxLDL given that it increased the size and lipid contents of plaque-resident foam cells in two independent models of early-stage atherosclerosis by increasing mOLR1 expression and oxLDL uptake. Notably, in the less hyperlipidemic of these two transgenic models, we could also discern a very strong positive correlation between plaque macrophage numbers and plasma cholesterol/LDL-C levels, implying that in early-stage atherosclerosis, mTrib1High macrophages (in marked contrast to Trib1-deficient macrophages) are uniquely equipped to increase plaque macrophage numbers in response to lipid excess and hence foam cell and fatty streak formation.
Recent studies have established that oxLDL accumulates steadily in both early-stage (growing) and mature-stage (yellow plaque without a necrotic core) human coronary plaques but that, in more advanced vulnerable plaques (yellow plaques with a necrotic core), this lipoprotein is removed either by metabolism or replacement with other substances, including cell debris (2). These in vivo data dovetail well with the early in vitro work, which showed that while oxLDL promotes macrophage growth and survival in a dose-dependent manner, beyond a certain lipid concentration, cell death ensues, albeit by an unknown mechanism (10). Thus, a critical question to consider is whether mTRIB1-induced OLR1 expression serves an (athero-) protective role in human atherosclerosis, for example, by reducing the exposure of plaque-resident vascular cells (where this disease is initiated) to oxLDL. This lipoprotein is a well-described activator of endothelial cell OLR1 expression with the totality of the data indicating that this activation culminates in arterial endothelium dysfunction (9). That said, the endothelium is activated by numerous factors, including blood flow, hypertension, and inflammation (25), which over the life span of an individual may attenuate (and even erase) the potential atheroprotective effect arising from an mTRIB1-mediated reduction in the exposure of endothelial cells to oxLDL. By contrast, the effects of an mTRIB1-induced increase in OLR1 expression will directly bear on atherosclerotic disease progression. Notably, it has been shown that mice overexpressing Olr1 on an ApoE KO background develop markedly more atherosclerosis than their nontransgenic littermates (26) and that Olr1 deficiency on an Ldlr KO background reduces the atherogenic burden conferred by 18 weeks of a 4% cholesterol/10% cocoa butter diet (27). With specific reference to the macrophage component of atheromas, Schaeffer and colleagues (28) have shown that cytokine-induced up-regulation of OLR1 increased OxLDL internalization by >40%, suggesting that (in line with our data presented in fig. S5), in inflamed microenvironments, where proinflammatory cytokines are relatively abundant (such as in atherosclerotic lesions), increased OLR1 expression could play a substantial role in up-regulating oxLDL uptake and atheroma formation. Supporting this contention, we show here that the TRIB1-induced increase in OLR1 expression increased the uptake of oxLDL and lipid accumulation in plaque macrophages, which in vitro (10) and in vivo (9) studies have indicated, all other things being equal, will accelerate the formation of foam cells, foam cell apoptosis/necrosis, and the development of more advanced, complex lesions.
On the basis of the strong evidence of a causal link between hyperlipidemia and CHD (29) and the demonstration that ablating hepatic TRIB1 expression increased plasma levels of cholesterol, LDL-C, and triglyceride (15), the implication was, which seemed to be entirely consistent with results from genome-wide association studies (GWAS) (14), that increasing TRIB1 would be atheroprotective. Silencing TRIB1 expression in macrophages, however, turns out to be atheroprotective, as judged by analyses of the aortas and aortic sinuses from Trib1mKO➔ApoE−/− chimeric mice, after a 12-week Western dietary regime. This counterintuitive result, which fits well with our in vitro and in vivo analysis of the consequences of mTrib1High expression on foam cell expansion, suggests moving forward that developing a therapy to specifically silence Trib1 expression in macrophages would provide clinical benefit beyond that of lipid-lowering medications, although full realization of this benefit may require its adoption at an early stage of atherogenesis. More generally, our study also demonstrates that mechanistic probing of GWAS signals is not only warranted, but also critical, to identify both the totality and directionality of disease risk factors, even when, as was the case for TRIB1, the association between a genetic variant(s), disease risk and a major disease-risk factor (plasma lipids) appeared congruent. Hence, we acknowledge that a limitation of the current study is that we have not addressed whether changes in vascular cell TRIB1 expression might affect early-stage atherosclerosis development and whether the GWAS-CHD signal at the TRIB1 locus reflects that, in these cells (and hepatocytes), TRIB1 serves an atheroprotective role, in contrast to the situation in plaque-resident macrophages, where it induces foam cell expansion.
One of the most notable outcomes arising from roughly doubling Trib1 expression in macrophages was the increase in foam cell size that developed in both of our models of human atherosclerosis. Our results indicate that this was driven by the failure of these cells to increase HDL-mediated cholesterol efflux in response to an up-regulation of cholesterol and fatty acid uptake. In addition, this up-regulation was attributable to increased OLR1 RNA and protein expression, consistent with the in vitro studies of Lazar and co-workers (30), which demonstrated that Rosiglitazone induction of Olr1 expression in adipocytes increased oxLDL and palmitate uptake and cellular cholesterol levels. Likewise, when Steinbrecher and colleagues (28) increased macrophage OLR1 expression via lysophosphatidylcholine stimulation, oxLDL uptake was also induced, prompting them to introduce the concept that macrophages within atheromas may be quiescent with respect to oxLDL uptake until Olr1 expression is induced (28). Here, we validate this concept by demonstrating overexpressing mTrib1 led directly to an increase in OLR1 protein in plaque-resident macrophages, alongside their increased lipid contents and size. Our data also show that by doubling mTrib1 expression, we removed the requirement for an external stimulus to up-regulate OLR1 expression and the consequent clinical sequelae. The phenotype observed in response to this rise in mTrib1 expression fits well with earlier computational modeling studies that indicated that relatively modest changes in TRIB1 would have a major impact on the activity of mitogen-activated protein kinase pathways; thus, defining the concentration of these proteins is critical for shaping cell function (31).
Thus, from the therapeutic standpoint, our results reveal that targeting TRIB1 expression could, in addition to beneficially affecting OLR1 expression, moderate CHD pathogenesis by simultaneously altering in favorable directions the expression of a number of other disease-promoting genes affected by changes in TRIB1 expression. These would include, for example, beneficially increasing NCEH1 expression to reduce the release of proinflammatory cytokines from plaque-resident macrophages (32), while reducing the expression of LPL to help reduce the retention of LDL in the artery wall during early-stage atherosclerosis (7), as well as excessive accumulation of cholesteryl ester and triglycerides within macrophage foam cells (8). Whether mTRIB1 modulates atheroma regression (33, 34) and late-stage atherosclerotic plaque stability by orchestrating a coordinated response to the lipid and inflammatory challenges encountered by plaque-resident macrophages/foam cells in advanced stage atherosclerosis now requires investigation.
MATERIALS AND METHODS
Human samples
Human tissue and blood samples were collected under protocols approved by the University of Sheffield Research Ethics Committee and Sheffield Teaching Hospitals Trust Review Board (ref. STH 16346 and SMBRER310) and in accordance with the Declaration of Helsinki. All participants gave written informed consent. Human atheromas were from the left anterior descending artery of explanted hearts from three patients with ischemic heart disease undergoing cardiac transplantation. Patients were male and had a mean age of 55.67 ± 2.51 (SD) years at the time of surgery. Lesions were classified as type V (American Heart Association). Specimens were fixed in 10% (v/v) formalin and embedded in paraffin wax. Antigen retrieval was performed with trypsin for 15 min at room temperature (MP-955-K6, A. Menarini Diagnostics, UK). Sections were incubated with mouse anti-human CD68 (M0814, Clone KP1, Dako) antibody (1:100 dilution) and rabbit anti-human TRIB1 (09-126, Millipore, UK) antibody (1:100) and the appropriate secondary antibodies, biotinylated horse anti-mouse secondary antibody and goat anti-rabbit secondary (BA-2000 and BA-1000, Vector Laboratories) antibody (both 1:200 dilution); and the detection reagents, Elite Mouse ABC horseradish peroxidase (HRP) (PK-6100, Vector Laboratories), 3, 3′-diaminobenzidine (SIGMAFAST, D4293, Sigma-Aldrich), and rabbit ABC-Alkaline phosphatase and a Vector Red Alkaline phosphatase substrate kit (AK-5000 and SK-5100, Vector Laboratories). Sections were counterstained with hematoxylin and mounted with DPX mountant (44581, Sigma-Aldrich).
Mice and creation of murine models of myeloid-specific Trib1 expression
Mice were handled in accordance with U.K. legislation (1986) Animals (Scientific Procedures) Act. Mouse experiments were approved by the University of Sheffield Project Review Committee and carried out under a U.K. Home Office Project Licence (70/7992). All mice used were congenic on a C57BL/6J background (N17) and were housed in a controlled environment with a 12-hour light/dark cycle, at 22°C in Optimice® individually ventilated cages (Animal Care Systems) and given free access to a standard chow diet (2918; Harlan Teklad) and water. A knockout mouse project (KOMP) repository embryonic stem (ES) cell clone containing loxP sites flanking exon 2 of Trib1 (EPD0099_5_D04) was used to generate a floxed Trib1 allele. The clone was genotyped to validate its authenticity, injected into C57BL/6J blastocysts, and transferred to pseudopregnant recipient females (Geneta). Resulting chimeras were mated with a flippase (FLP)–deleter strain maintained on a C57BL/6J background (Geneta), and floxed-Trib1 mice were generated. Trib1mKO mice were generated by crossing floxed mice with Lys2–cre recombinase transgenic mice (www.jax.org/strain/004781), excising all but the first 120 amino acids of TRIB1. Murine Trib1 complementary DNA (cDNA) was introduced into the previously described pROSA26, loxP-flanked STOP and Frt-flanked internal ribosomal entry site–eGFP targeting construct (35) and then inserted into the ubiquitously expressed Rosa26 locus of Bruce4 (C57/BL6J origin) mouse ES cells by homologous recombination. Correct integration was confirmed by Southern blotting. Trib1mTg mice were generated by crossing floxed mice with the Lys2–cre recombinase transgenic mice described above. Mice were genotyped by PCR amplification of ear-clip samples. Trib1 fl/fl × Lyz2Cre and Rosa26.Trib1 × Lyz2Cre were genotyped for the presence of Lyz2Cre and for either Trib1 fl/fl or Rosa26.Trib1 using three primer sets (table S5). For the atherosclerosis experiments, mice were fed a Western (21% fat and 0.2% cholesterol) diet (829100; Special Diet Services, Braintree, UK) for 12 weeks.
mTrib1 RNA quantification
BMDMs were cultured for 5 days in complete medium: Dulbecco’s modified Eagle’s medium (DMEM) (BE12-604F, Lonza) containing 10% (v/v) L929-conditioned medium, 10% (v/v) ultralow endotoxin fetal bovine serum (FBS) (S1860-500, Biowest, USA), and penicillin (100 U/ml) and streptomycin (100 ng/ml) (15140-122, Gibco). Total RNA was isolated using a ReliaPrep Kit (Z6011, Promega) and reverse transcribed into cDNA using an iScript cDNA Synthesis Kit (1708890, Bio-Rad). Trib1 was quantified using the TaqMan assay Mm00454875, which amplifies a 99-nucleotide amplicon comprising exon 2 and 3 sequences.
myeloid-green fluorescent protein (mGFP) quantification
GFP-positive cells in freshly purified peripheral blood monocytes were isolated by positive selection using magnetic MicroBeads conjugated with F4/80 (130-110-443, Miltenyi Biotec) and CD115 (130-096-354, Miltenyi Biotec) using a modified version of the protocol described by Houthuys et al. (36). Inflammation was induced by injecting phosphate-buffered saline (PBS) containing thioglycollate into the peritoneal cavity and isolating the infiltrating cells, as described (37). The percentages of GFP-positive cells in blood and bone marrow cells (from femurs and tibiae) were determined using mice euthanized humanely by cervical dislocation. Red blood cells (RBCs) were lysed using RBC lysis buffer (00-4300-54, eBioscience). Approximately 106 cells per sample were resuspended in PBS, and dead cells were removed using an amine-reactive dye, NIR Zombie (1:500 dilution in the antibody master mix; 423105, BioLegend). Live cells were stained with the following cell surface marker–specific antibodies, at 0.1 μg/ml each in 100-μl total volume: AF647-conjugated anti-human/mouse CD11b (101220, BioLegend), phycoerythrin (PE)–conjugated anti-mouse Ly6C (101220, BioLegend), and PE/Cy7-conjugated anti-mouse Ly6G (127617, BioLegend). Cells were sorted on an LSR II Cytometer (BD Bioscience) equipped with 355-, 405-, 488-, and 633-nm excitation lasers. Quantifications were performed with FlowJo software.
Blood counts
Blood counts of heparinized blood, obtained via cardiac puncture, were determined using a Sysmex KX-21 N quantitative automated hematology analyzer.
Histological analysis
Adipose tissue and liver samples were paraffin-embedded. Cross sections were stained with hemotoxylin and eosin or incubated with F4/80 [1:50; 565409 (clone T45-2342), BD Pharmingen], followed by biotinylated rabbit anti-rat antibody (1:200; Vector Laboratories, UK) and PE-streptavidin (1:20; 405203, BioLegend). They were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (P36931, Invitrogen) and mounted with ProLong Gold antifade mountant. Mean adipocyte sizes were determined using NIS-Elements software (Nikon Instruments, UK) by measuring at least 15 cells per field of view and three fields of view per mouse. Frozen spleen sections were stained with F4/80 [PE-rat anti-mouse 123019 (clone BM8, BioLegend)] or CD206− [Alexa Fluor 647 rat anti-mouse 321116 (clone 15-2, BioLegend)]–conjugated antibodies (1:200), counterstained with DAPI, and mounted with Aqua-Mount (Thermo Fisher Scientific). Fluorescent images were captured using an inverted wide-field fluorescence microscope (Leica AF6000).
Models of early-stage human atherosclerosis
For the bone marrow transplantation model, 12- to 13-week-old mixed-gender donor mice were euthanized humanely by cervical dislocation. Femur/tibae bone marrow cells were isolated and purified by standard methods and resuspended in Hank’s balanced salt solution (without phenol red, 14175053, Thermo Fisher Scientific) containing 10% (v/v) fetal calf serum. Donor cells (2 × 106 to 4 × 106) were transplanted via tail vein injection to randomly allocated 12- to 13-week-old male ApoE−/− recipient mice that were lethally irradiated with 11 grays (Gy) in two doses (5.5 Gy on two occasions separated by 4 hours) on the day of the transplantation. Bone marrow transplant experiments were undertaken in two waves, one for each mTrib1 model and respective WT control. During the 7-week posttransplant recovery period, the chimeras were fed a standard chow (2918; Harlan Teklad) diet and then switched to Western diet (21% fat and 0.2% cholesterol, 829100; Special Diet Services, Braintree, UK) for 12 weeks. Chimera mice were given sterile acidified drinking water [1.1% (v/v) HCl] until the end of the procedure.
PCSK9 model
An adeno-associated virus-based vector (rAAV8) that supports transport of the mPCSK9D377Y gene to the liver was purchased from UNC GTC Vector Core (Chapel Hill, NC). The mice received 6.1 × 1011 viral particles via a single tail vein injection. Following 7-day recovery, they were transferred to the Western diet (829100, Special Diet from Braintree, UK) for 12 weeks.
Lipid measurements
Fasting, plasma total cholesterol, HDL-C, triglycerides, and glucose levels were measured on a Roche Cobas 8000 modular analyzer. LDL-C was estimated by the Friedewald equation. In addition, colorimetric assays (Cholesterol Quantification Kit, MAK043, Sigma-Aldrich; Triglyceride Assay Kit, ab65336, Abcam) were used.
Atherosclerotic plaque assessment
Mice were perfused through the heart with PBS and then with 10% (w/v) neutral-buffered formalin. The aorta was segmented at the diaphragm and at the top of the aortic arch. Following careful removal of surrounding extraneous fat and connective tissue, it was dissected longitudinally and fixed in 4% (w/v) paraformaldehyde. Dissected aortas were stained with Oil Red O [60% (v/v) in isopropanol] and pinned onto wax. Images were taken with a macroscopic charge-coupled device camera and analyzed by computer-assisted image analysis (NIS-Elements software, Nikon Instruments, UK). Aortic sinus samples were obtained by excising the heart and transecting parallel to the atria. Following fixation in 10% formalin (v/v) buffered saline for at least 24 hours, samples were serially cut (at 7-μm intervals) from the valve leaflets until the beginning of the aorta.
Immunohistochemistry
Aortic sinus sections were dewaxed and rehydrated. Heat-mediated antigen retrieval was performed with 10 mM sodium citrate and nonspecific staining reduced by incubation in 5% (v/v) goat serum (G9023, Sigma-Aldrich) for 30 min at room temperature. Primary antibodies diluted as appropriate were as follows: rat anti-mouse MAC-3 antibody clone M3/84 (1:100 dilution, BD Pharmingen), rabbit polyclonal NOS2 antibody (1:100; ab15323; Abcam, UK), rabbit anti-mouse YM1 polyclonal antibody (1:100; ab93034; Abcam, UK) and or rabbit polyclonal OLR1 (1:100; ab203246; Abcam, UK). The secondary antibodies were biotinylated rabbit anti-rat secondary antibody (1:200, Vector Laboratories BA-4000), goat anti-rat DyLight 488 goat anti-rat DyLight 488 (GtxRt-003488NHSX, ImmunoReagents Inc.), and goat anti-rabbit DyLight 550 (GtxRb-003-D550NHSX, ImmunoReagents Inc.). Rabbit anti-rat–conjugated biotinylated antibody was visualized with the Vectastain ABC-HRP complex (PK-6100, Vector Laboratories). Sections were counterstained with Carazzi’s hematoxylin. The number and area of foam cells (MAC-3+ macrophages containing a vacuolated cytoplasm) were quantified using NIS-Elements software (Nikon, UK). Fluorescent images were captured using an inverted wide-field fluorescence microscope (Leica AF6000). Quantification of YM1, NOS2, and OLR1 in the aortic sinus lesions was confined to macrophage (MAC-3+) cells only and analyzed using ImageJ.
Clinical grading
The atherosclerotic burden of the atheroma in aortic sinus samples was graded according to the Stary system (e.g., 1 = presence of macrophage foam cells, 2 = presence of intracellular lipid accumulation, and 3 = presence of extracellular lipid pools) (38) using a light microscope (Zeiss Axiophot, Carl Zeiss, Jena, Germany) and Image Access. Slides were randomized, and the cardiac pathologist was fully blinded to sample origin.
Western blotting
Twenty micrograms of total protein lysates was size-fractionated on 4 to 12% NuPAGE Bis-Tris Gel (NP0321, Invitrogen). OLR1 was detected by incubation with a rabbit anti-mouse OLR1 antibody (1:500, ab203246; Abcam), followed by HRP-conjugated goat anti-rabbit immunoglobulin G (IgG) (1:1000; P0448, Dako). LDLR was detected by incubation with a rabbit polyclonal antibody (1:1000; 3839-100, BioVision, USA) at 4°C overnight followed by HRP-conjugated goat anti-rabbit IgG (1:1000; P0448, Dako). α-Tubulin was used as a housekeeping control (sc-32293, Santa Cruz Biotechnology). Detection of immunoreactive products was performed using 1:1 ECL reagent (RPN2235, GE Healthcare) and a C-DiGit Blot scanner (Model 3600, LI-COR). Densitometry was performed with Image Studio Lite software (LI-COR).
Gene expression studies
RNA from BMDM was prepared as described above. RT-qPCR was performed using primer sequences provided in table S5 and either SYBR-Green (PrecisionPLUS, Primer Design, UK) or a TaqMan (Invitrogen) assay (TRIB1, Hs00921832; OLR1 Hs01552593; SCARB1, Hs00969821, Thermo Fisher Scientific). Values were normalized to either Actb (Mm02619580, Invitrogen) for mouse samples or GAPDH for human samples (Hs02786624, Invitrogen). Fold changes were calculated using the ΔΔCt method.
Microarray analyses
Details of the CTS, which comprises transcriptomic data from isolated monocytes from 758 donors and matched MDM samples from 596 donors, have been published (24). In brief, monocytes were isolated from whole blood using CD14 immunobeads and cultured for 7 days with macrophage colony-stimulating factor (MCS-F) to generate macrophages. The top and bottom quartiles of TRIB1-expressing samples were defined respectively as TRIB1High and TRIB1Low. Comparable sizes of differentially expressed gene lists (n = 1842 monocytes; n = 2171, MDM) were obtained by using false discovery rate (FDR)- adjusted P values (i.e., q values) of <0.01 plus cutoff log2 fold changes of >0.071 (up-regulated) and >−0.071 (down-regulated) for the MDM dataset. The Database for Annotation, Visualization and Integrated Discovery version 6.8 was used to identify for enrichment of functionally related Gene Ontology terms.
Gene expression in polarized human macrophages
Healthy human monocytes were isolated from whole blood by Ficoll-Paque PLUS (17144003, GE Healthcare) density centrifugation followed by magnetic selection with CD14 Human MicroBeads (130-050-201, Miltenyi Biotec). Monocytes were incubated for 7 days with RPMI 1640 (Gibco) supplemented with MCS-F (100 ng/ml) (300-25-100, PeproTech, UK), 10% FBS (Biowest), 1 mM glutamine (Invitrogen), and 1% penicillin/streptomycin (Gibco), followed by polarization with either LPS (100 ng/ml) (581-007-L002, Enzo Life Sciences, USA) and IFN-γ (20 ng/ml) (300-02, PeproTech, UK), IL-4 (20 ng/ml) (200-04, PeproTech, UK), or IL-10 (20 ng/ml) (200-10, PeproTech, UK) for 24 hours. RNA and RT-qPCR were performed, as described above using the primer sets listed in table S4.
OxLDL uptake and HDL-mediated cholesterol efflux
Assays were performed on BMDMs cultured for 5 days as described above. OLR1 was detected as described above. For the uptake assays, cells were incubated at 37°C for a further 24 hours with human oxLDL (5685-3557, Bio-Rad) at a concentration of 0 and 25 μg/ml. Total cell lipids were extracted using 7:11:01 (v/v) chloroform: isopropanol:IGEPAL CA-630 (I8896, Sigma-Aldrich), dried at 50°C, and resuspended in 120 μl of cholesterol assay buffer (MAK043, Sigma-Aldrich). Total cholesterol, free cholesterol, and cholesteryl esters were measured using a cholesterol quantification kit (MAK043, Sigma-Aldrich), according to the manufacturer’s instructions. Foam cells were assessed by Oil Red O [60% (v/v) in isopropanol] staining as described above. For the efflux assays, BMDMs were incubated for 24 hours in DMEM (BE12-604F, Lonza) supplemented with 0.2% (w/v) fatty acid–free bovine serum albumin (BSA) (A8806, Sigma-Aldrich) and 2.5 μM TopFluor (Bodipy) cholesterol (Avanti Polar Lipids Inc. USA). The medium was removed, and the cells were washed with PBS and equilibrated for 18 hours in DMEM supplemented with 0.2% (w/v) fatty acid–free BSA. Efflux was measured after a 4-hour incubation period with human HDL (0 and 50 μg/ml) (5685-2004, Bio-Rad). Supernatants were collected, and the cells were lysed with 1% (w/v) cholic acid (C1129, Sigma-Aldrich) in ethanol. Cholesterol efflux was calculated as the percentage of fluorescence (excitation, 490 nm; emission, 520 nm) in the cell medium at the end of the incubation period divided by the total fluorescence in the medium and cells.
Statistics
All data are reported as means ± SEM unless stated otherwise in the figure legend. Graphs were produced and analyzed by GraphPad Prism software. Each data point represents a single mouse or human donor. For analysis of two groups, Student’s t test was performed. When three or more groups were compared, analysis of variance (ANOVA) was used, accompanied with the appropriate ad hoc posttest, as detailed in the specific figure legends. P values <0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank M. Ariaans, C. Wright, and the Biological Service Unit staff at the University of Sheffield for technical support during the in vivo experiments; F. Wright for tissue processing and histology; S. Haynes for mouse genotyping; and L. Martínez-Campesino for assistance and logistical support. Funding: This work was supported by a Transatlantic Network of Excellence grant 10CVD03 from the Fondation Leducq to E.K.-T. and D.J.R., funds from the British Heart Foundation (PG/16/44/32146 to E.K.-T., S.E.F., and C.C.S.) and BBSRC (BB/J00152X/1 and BB/J004472/1 to M.T.). The CTS data were provided by the Cardiogenics project, which was supported by the European Union Sixth Framework Programme (LSHM-CT-2006-037593). The Cardiogenics Consortium provided transcriptomic data. Author contributions: Designing research studies: T.N.D., H.L.W., A.H.G., D.J.R., C.C.S., S.E.F., and E.K.-T. Conducting experiments and acquiring data: J.M.J., A.A., R.C.B., K.B., T.N.D., M.T., and S.E.F. Analyzing data: J.M.J., R.C.B., S.H., S.K.S., K.B., Z.H., T.N.D., H.L.W., A.H.G., C.C.S., S.E.F., and E.K.-T. Writing the manuscript: J.M.J., H.L.W., A.H.G., S.E.F., C.C.S., and E.K.-T. Competing interests: The authors declared that they have no conflict of interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax9183/DC1
Fig. S1. Expected and observed numbers of 8-week-old offspring with specified Trib1 genotypes.
Fig. S2. Trib1mKO and Trib1mTg mice have normal tissue anatomy and F4/80+ macrophage numbers.
Fig. S3. Plasma lipid levels of chimera and Pcsk9 mice.
Fig. S4. Atherosclerotic burden in mTrib1➔ApoE−/− mice, clinical grading of lesions, and presence of foam cells.
Fig. S5. Reciprocal regulation of OLR1 and SCARB1 RNA levels in polarized MDMs.
Table S1. Fold changes and P values of genes differentially expressed in both MDMs and monocytes.
Table S2. Top-ranking biological processes enriched in differentially expressed gene lists of (1) Human TRIB1High versus TRIB1Low monocytes and (2) between TRIB1High versus TRIB1Low MDMs.
Table S3. The most significantly altered pathways in TRIB1High versus TRIB1Low macrophages.
Table S4. Linoleic (LiA), oleic (OA), and lauric acid (LA) in vitro polarized human MDMs recapitulate the Olr1High/LplHigh/Scarb1Low/CD36WT RNA profile of Trib1mTg BMDM.
Table S5. Primer sequences.
REFERENCES AND NOTES
- 1.Rocha V. Z., Libby P., Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol. 6, 399–409 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Uchida Y., Recent advances in fluorescent angioscopy for molecular imaging of human atherosclerotic coronary plaque. J. Atheroscler. Thromb. 24, 539–551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore K. J., Sheedy F. J., Fisher E. A., Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 13, 709–721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skålén K., Gustafsson M., Rydberg E. K., Hultén L. M., Wiklund O., Innerarity T. L., Borén J., Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 417, 750–754 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Schrijvers D. M., De Meyer G. R., Kockx M. M., Herman A. G., Martinet W., Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 25, 1256–1261 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Tabas I., Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 10, 36–46 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gustafsson M., Levin M., Skålén K., Perman J., Fridén V., Jirholt P., Olofsson S. O., Fazio S., Linton M. F., Semenkovich C. F., Olivecrona G., Borén J., Retention of low-density lipoprotein in atherosclerotic lesions of the mouse: Evidence for a role of lipoprotein lipase. Circ. Res. 101, 777–783 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Takahashi M., Yagyu H., Tazoe F., Nagashima S., Ohshiro T., Okada K., Osuga J.-I., Goldberg I. J., Ishibashi S., Macrophage lipoprotein lipase modulates the development of atherosclerosis but not adiposity. J. Lipid Res. 54, 1124–1134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann A., Brunssen C., Morawietz H., Contribution of lectin-like oxidized low-density lipoprotein receptor-1 and LOX-1 modulating compounds to vascular diseases. Vascul. Pharmacol. 107, 1–11 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Hundal R. S., Gómez-Muñoz A., Kong J. Y., Salh B. S., Marotta A., Duronio V., Steinbrecher U. P., Oxidized low density lipoprotein inhibits macrophage apoptosis by blocking ceramide generation, thereby maintaining protein kinase B activation and Bcl-XL levels. J. Biol. Chem. 278, 24399–24408 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Getz G. S., Reardon C. A., Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 50, S156–S161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baitsch D., Bock H. H., Engel T., Telgmann R., Müller-Tidow C., Varga G., Bot M., Herz J., Robenek H., von Eckardstein A., Nofer J.-R., Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler. Thromb. Vasc. Biol. 31, 1160–1168 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sung H. Y., Francis S. E., Arnold N. D., Holland K., Ernst V., Angyal A., Kiss-Toth E., Enhanced macrophage tribbles-1 expression in murine experimental atherosclerosis. Biology 1, 43–57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varbo A., Benn M., Tybjӕrg-Hansen A., Grande P., Nordestgaard B. G., TRIB1 and GCKR polymorphisms, lipid levels, and risk of ischemic heart disease in the general population. Arterioscler. Thromb. Vasc. Biol. 31, 451–457 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Bauer R. C., Yenilmez B. O., Rader D. J., Tribbles-1: A novel regulator of hepatic lipid metabolism in humans. Biochem. Soc. Trans. 43, 1079–1084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satoh T., Kidoya H., Naito H., Yamamoto M., Takemura N., Nakagawa K., Yoshioka Y., Morii E., Takakura N., Takeuchi O., Akira S., Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 495, 524–528 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Tao H., Yancey P. G., Babaev V. R., Blakemore J. L., Zhang Y., Ding L., Fazio S., Linton M. F., Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J. Lipid Res. 56, 1449–1460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clausen B. E., Burkhardt C., Reith W., Renkawitz R., Förster I., Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Drechsler M., Megens R. T., van Zandvoort M., Weber C., Soehnlein O., Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 122, 1837–1845 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Hasty A. H., Linton M. F., Brandt S. J., Babaev V. R., Gleaves L. A., Fazio S., Retroviral gene therapy in ApoE-deficient mice: ApoE expression in the artery wall reduces early foam cell lesion formation. Circulation 99, 2571–2576 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Bjørklund M. M., Hollensen A. K., Hagensen M. K., Dagnæs-Hansen F., Christoffersen C., Mikkelsen J. G., Bentzon J. F., Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ. Res. 114, 1684–1689 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Norata G. D., Tavori H., Pirillo A., Fazio S., Catapano A. L., Biology of proprotein convertase subtilisin kexin 9: Beyond low-density lipoprotein cholesterol lowering. Cardiovasc. Res. 112, 429–442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arndt L., Dokas J., Gericke M., Kutzner C. E., Müller S., Jeromin F., Thiery J., Burkhardt R., Tribbles homolog 1 deficiency modulates function and polarization of murine bone marrow-derived macrophages. J. Biol. Chem. 293, 11527–11536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schunkert H., König I. R., Kathiresan S., Reilly M. P., Assimes T. L., Holm H., Preuss M., Stewart A. F., Barbalic M., Gieger C., Absher D., Aherrahrou Z., Allayee H., Altshuler D., Anand S. S., Andersen K., Anderson J. L., Ardissino D., Ball S. G., Balmforth A. J., Barnes T. A., Becker D. M., Becker L. C., Berger K., Bis J. C., Boekholdt S. M., Boerwinkle E., Braund P. S., Brown M. J., Burnett M. S., Buysschaert I.; Cardiogenics, Carlquist J. F., Chen L., Cichon S., Codd V., Davies R. W., Dedoussis G., Dehghan A., Demissie S., Devaney J. M., Diemert P., Do R., Doering A., Eifert S., Mokhtari N. E., Ellis S. G., Elosua R., Engert J. C., Epstein S. E., de Faire U., Fischer M., Folsom A. R., Freyer J., Gigante B., Girelli D., Gretarsdottir S., Gudnason V., Gulcher J. R., Halperin E., Hammond N., Hazen S. L., Hofman A., Horne B. D., Illig T., Iribarren C., Jones G. T., Jukema J. W., Kaiser M. A., Kaplan L. M., Kastelein J. J., Khaw K. T., Knowles J. W., Kolovou G., Kong A., Laaksonen R., Lambrechts D., Leander K., Lettre G., Li M., Lieb W., Loley C., Lotery A. J., Mannucci P. M., Maouche S., Martinelli N., McKeown P., Meisinger C., Meitinger T., Melander O., Merlini P. A., Mooser V., Morgan T., Mühleisen T. W., Muhlestein J. B., Münzel T., Musunuru K., Nahrstaedt J., Nelson C. P., Nöthen M. M., Olivieri O., Patel R. S., Patterson C. C., Peters A., Peyvandi F., Qu L., Quyyumi A. A., Rader D. J., Rallidis L. S., Rice C., Rosendaal F. R., Rubin D., Salomaa V., Sampietro M. L., Sandhu M. S., Schadt E., Schäfer A., Schillert A., Schreiber S., Schrezenmeir J., Schwartz S. M., Siscovick D. S., Sivananthan M., Sivapalaratnam S., Smith A., Smith T. B., Snoep J. D., Soranzo N., Spertus J. A., Stark K., Stirrups K., Stoll M., Tang W. H., Tennstedt S., Thorgeirsson G., Thorleifsson G., Tomaszewski M., Uitterlinden A. G., van Rij A., Voight B. F., Wareham N. J., Wells G. A., Wichmann H. E., Wild P. S., Willenborg C., Witteman J. C., Wright B. J., Ye S., Zeller T., Ziegler A., Cambien F., Goodall A. H., Cupples L. A., Quertermous T., März W., Hengstenberg C., Blankenberg S., Ouwehand W. H., Hall A. S., Deloukas P., Thompson J. R., Stefansson K., Roberts R., Thorsteinsdottir U., O’Donnell C. J., McPherson R., Erdmann J.; CARDIoGRAM Consortium, Samani N. J., Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 43, 333–338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramji D. P., Davies T. S., Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 26, 673–685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inoue K., Arai Y., Kurihara H., Kita T., Sawamura T., Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E-null mice. Circ. Res. 97, 176–184 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Mehta J. L., Sanada N., Hu C. P., Chen J., Dandapat A., Sugawara F., Satoh H., Inoue K., Kawase Y., Jishage K. I., Suzuki H., Takeya M., Schnackenberg L., Beger R., Hermonat P. L., Thomas M., Sawamura T., Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ. Res. 100, 1634–1642 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Schaeffer D. F., Riazy M., Parhar K. S., Chen J. H., Duronio V., Sawamura T., Steinbrecher U. P., LOX-1 augments oxLDL uptake by lysoPC-stimulated murine macrophages but is not required for oxLDL clearance from plasma. J. Lipid Res. 50, 1676–1684 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ference B. A., Ginsberg H. N., Graham I., Ray K. K., Packard C. J., Bruckert E., Hegele R. A., Krauss R. M., Raal F. J., Schunkert H., Watts G. F., Borén J., Fazio S., Horton J. D., Masana L., Nicholls S. J., Nordestgaard B. G., van de Sluis B., Taskinen M. R., Tokgözoğlu L., Landmesser U., Laufs U., Wiklund O., Stock J. K., Chapman M. J., Catapano A. L., Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 38, 2459–2472 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chui P. C., Guan H.-P., Lehrke M., Lazar M. A., PPARγ regulates adipocyte cholesterol metabolism via oxidized LDL receptor 1. J. Clin. Invest. 115, 2244–2256 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guan H., Shuaib A., Leon D. D., Angyal A., Salazar M., Velasco G., Holcombe M., Dower S. K., Kiss-Toth E., Competition between members of the tribbles pseudokinase protein family shapes their interactions with mitogen activated protein kinase pathways. Sci. Rep. 6, 32667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hunerdosse D. M., Morris P. J., Miyamoto D. K., Fisher K. J., Bateman L. A., Ghazaleh J. R., Zhong S., Nomura D. K., Chemical genetics screening reveals KIAA1363 as a cytokine-lowering target. ACS Chem. Biol. 9, 2905–2913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahman K., Vengrenyuk Y., Ramsey S. A., Vila N. R., Girgis N. M., Liu J., Gusarova V., Gromada J., Weinstock A., Moore K. J., Loke P., Fisher E. A., Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Invest. 127, 2904–2915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peled M., Nishi H., Weinstock A., Barrett T. J., Zhou F., Quezada A., Fisher E. A., A wild-type mouse-based model for the regression of inflammation in atherosclerosis. PLOS ONE 12, e0173975 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki Y., Derudder E., Hobeika E., Pelanda R., Reth M., Rajewsky K., Schmidt-Supprian M., Canonical NF-κB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity 24, 729–739 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Houthuys E., Movahedi K., De Baetselier P., Van Ginderachter J. A., Brouckaert P., A method for the isolation and purification of mouse peripheral blood monocytes. J. Immunol. Methods 359, 1–10 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Ghosn E. E., Cassado A. A., Govoni G. R., Fukuhara T., Yang Y., Monack D. M., Bortoluci K. R., Almeida S. R., Herzenberg L. A., Herzenberg L. A., Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc. Natl. Acad. Sci. U.S.A. 107, 2568–2573 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stary H. C., Chandler A. B., Dinsmore R. E., Fuster V., Glagov S., Insull W. Jr., Rosenfeld M. E., Schwartz C. J., Wagner W. D., Wissler R. W., A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. Vasc. Biol. 15, 1512–1531 (1995). [DOI] [PubMed] [Google Scholar]
- 39.Park Y. M., CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 46, e99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y., Wang X., Ben J., Yue S., Bai H., Guan X., Bai X., Jiang L., Ji Y., Fan L., Chen Q., The di-leucine motif contributes to class a scavenger receptor-mediated internalization of acetylated lipoproteins. Arterioscler. Thromb. Vasc. Biol. 26, 1317–1322 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax9183/DC1
Fig. S1. Expected and observed numbers of 8-week-old offspring with specified Trib1 genotypes.
Fig. S2. Trib1mKO and Trib1mTg mice have normal tissue anatomy and F4/80+ macrophage numbers.
Fig. S3. Plasma lipid levels of chimera and Pcsk9 mice.
Fig. S4. Atherosclerotic burden in mTrib1➔ApoE−/− mice, clinical grading of lesions, and presence of foam cells.
Fig. S5. Reciprocal regulation of OLR1 and SCARB1 RNA levels in polarized MDMs.
Table S1. Fold changes and P values of genes differentially expressed in both MDMs and monocytes.
Table S2. Top-ranking biological processes enriched in differentially expressed gene lists of (1) Human TRIB1High versus TRIB1Low monocytes and (2) between TRIB1High versus TRIB1Low MDMs.
Table S3. The most significantly altered pathways in TRIB1High versus TRIB1Low macrophages.
Table S4. Linoleic (LiA), oleic (OA), and lauric acid (LA) in vitro polarized human MDMs recapitulate the Olr1High/LplHigh/Scarb1Low/CD36WT RNA profile of Trib1mTg BMDM.
Table S5. Primer sequences.