

Abstract
Background/Aims
Transforming growth factor-β1 (TGF-β1) induction of epithelial-mesenchymal transition (EMT) is one of the mechanisms by which colorectal cancer (CRC) cells acquire migratory and invasive capacities, and subsequently metastasize. Parthenolide (PT) expresses multiple anti-cancer and anti-inflammatory activities that inhibit nuclear factor κB by targeting the IκB kinase complex. In the present study, we aimed to investigate whether PT can inhibit TGF-β1-induced EMT in CRC cell lines.
Methods
HT-29 and SW480 cell lines were used in the experiment. Cell viability was detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and sub-G1 analysis was measured by flow cytometry. The induction of EMT by TGF-β1 and inhibition of the process by PT was analyzed by phase contrast microscopy, wounding healing, cellular migration and invasion assays, and Western blotting.
Results
TGF-β1 inhibits HT-29 cell proliferation, but has no effect on SW480 cell proliferation; different concentrations of TGF-β1 did not induce apoptosis in HT-29 and SW480 cells. PT attenuates TGF-β1-induced elongated, fibroblast-like shape changing in cells. PT inhibits TGF-β1-induced cell migration and cell invasion. In addition, other EMT markers such as β-catenin, Vimentin, Snail, and Slug were suppressed by PT, while E-cadherin was increased by PT.
Conclusions
Our findings show that PT inhibits TGF-β1-induced EMT by suppressing the expression of the mesenchymal protein and increasing expression of the epithelial protein. These findings suggest a novel approach for CRC treatment by suppression of TGF-β1-induced EMT.
Keywords: Transforming growth factor beta 1, Epithelial-mesenchymal transition, Parthenolide, Colorectal neoplasms
INTRODUCTION
Colorectal cancer (CRC) is the third dominant cancer by incidence and mortality worldwide in both sexes [1]. Patients show distant metastases to the liver most frequently. The liver, lung, and peritoneum are common locations for metastasis [2,3]. The estimated 5-year survival rate of metastatic CRC remains low [4]. Therefore, more knowledge of the molecular mechanisms involved in metastases is vital to the development of more effective therapies.
Transforming growth factor β (TGF-β) is a 25-kDa cytokine that plays a central role in proliferation, differentiation, apoptosis, and extracellular matrix formation in many biologic processes. Three isoforms of TGF-β1, 2, and 3 are expressed in the normal colonic epithelium. TGF-β perform tumor suppress function in the normal colon by inhibiting proliferation and induction of apoptosis [5,6]. TGF-β is a multi-functional cytokine expressed by both cancer and stromal cells [7]. This cytokine is overexpressed in CRC and functions as a tumor promoter in the late stages of carcinogenesis [8]. TGF-β has a dual role in the progression and metastasis of cancer. During the early stage of carcinogenesis, TGF-β acts a tumor suppressor, but paradoxically, it serves as tumor promoter by inducing metastasis during late stages of carcinogenesis [9,10].
Epithelial-mesenchymal transition (EMT) is a process by which cells lose their epithelial characteristics, including their cell polarity and epithelial markers, along with increased expression of mesenchymal markers and gain of migratory and invasive capacities [11]. In cancer cells, TGF-β leads to EMT and cancer stem cell-like traits. Cells that undergo EMT demonstrate a migratory ability that favors metastasis. The cells become spindle-shaped and suffer cytoskeletal changes that make cells more prone to disseminate and colonize other organs [12,13]. EMT is driven by an interactive network of transcriptional factors including Snail1 and Snail2 (also known as Slug and Twist). In mammary epithelial cells, TGF-β-activated Smad proteins directly motivate the expression of Snail1 and Twist1 (Twist-related protein 1) [14].
Parthenolide (PT), a sesquiterpene lactone extract, has been showed to have anti-inflammatory and anticancer properties and PT inhibits the function of nuclear factor κB (NF-κB) through targeting the IκB kinase complex. PT has been predominately investigated as an inducer of apoptosis in human cancer cells [15]. A number of studies have reported sesquiterpene lactones have an effect on cell migration/invasion and the EMT process in human cancer cells [16]. Antrocin showed inhibitory activity about the growth, migration, and invasion of human bladder cancer cells. One mechanism for motility inhibition is through inactivation of focal adhesion kinase-paxillin (FAK-paxillin) and extracellular signal-regulated kinases-c-Fos-matrix metalloproteinases (ERK-c-Fos-MMP2) pathways [17]. PT has been reported to repress migration and invasion in pancreatic cancer cells and also inhibit proliferation [18]. Codonolactone also inhibits TGF-β1-mediated EMT and motility of breast cancer cells by inhibiting TGF-β signaling and Runx2 phosphorylation [16]. We previously demonstrated that PT inhibits the migration and invasion of SW620 cells by wound healing, migration, and invasion test and also downregulated EMT regulated markers [19]. Therefore, we hypothesized that PT may attenuate TGF-β1-induced EMT progression in CRC cell lines.
METHODS
1. Chemicals and Reagents
PT was attained from Calbiochem (San Diego, CA, USA) and dissolved in dimethylsulfoxide (DMSO; Sigma, St. Louis, MO, USA) to obtain a concentration of 100 μM and then saved at –20°C in the dark. Growth factor-reduced Matrigel was purchased from BD Biosciences (San Diego, CA, USA). Recombinant human TGF-β1 protein was acquired from R&D Systems (Minneapolis, MN, USA) and was dissolved in Ultra-Pure Bovine Serum Albumin (GenDEPOT, Hanam, Korea) to obtain a concentration of 20 μg/mL and then stored at –20°C. Anti-E-cadherin, anti-β-catenin, and anti-Vimentin were from Cell Signaling Technology (Danvers, MA, USA). Anti-Slug and anti- Snail were from Abcam (Cambridge, UK). Anti-Actin was obtained from Sigma-Aldrich (St. Louis, MO, USA).
2. Cell Culture, Treatments, and Observation of Morphological Changes
HT-29 and SW480 cells (American Type Culture Collection, Rockville, MD, USA) were representative human CRC cells cultured in RPMI-1640 medium (GIBCO-BRL; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; GIBCO-BRL), 100 μg/mL penicillin (GIBCO-BRL), and 100 μg/mL streptomycin (GIBCO-BRL). HT-29 and SW480 cells belong to microsatellite instability (MSI) cell lines. Those 2 cell lines can be induced to EMT by TGF-β1 cause no mutant TGF-β receptor II (TGFBR2) exists in 2 cell lines. The cell passage number from 2 to 5 was used in the experiment. PT was diluted through FBS-free media to attain the indicated concentrations. The morphological alterations in the HT-29 and SW480 cells were observed using an inverted microscope. Images were captured using an inverted microscope (Olympus IX71, Melville, NY, USA).
3. Cell Viability Assay
HT-29 and SW480 cells were seeded at a density of 2×104 cells per well in 24-well plates and treated by TGF-β1 and PT for 48 hours. Thereafter, the media were discarded from each well and substituted with 200 μL of fresh media and 15 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; 2.5 mg dissolved in 50 μL of DMSO). After 3 hours of incubation at 37°C, the culture medium containing MTT was discarded and 250 μL of DMSO was added. Plates were located on a plate shaker till the crystals were dissolved. Cell viability was detected by measuring the absorbance at 570 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
4. Cell Cycle and Sub-G1 Analysis
Distribution of cells in different phases of the cell cycle including sub-G1 was determined by staining DNA with propidium iodide (Thermo Fisher, Eugene, OR, USA). Briefly, 1×106 cells were incubated with various concentrations of TGF-β1 and PT for 48 hours. Thereafter, cells were washed with cold 1× phosphate-buffered saline (PBS) and fixed in 70% ethanol overnight. The cells were washed again with 1× PBS, incubated with propidium iodide (10 μg/mL), and combinedly treated with RNase at 37°C for 1 hour. The percentages of cells in the distinct phases of the cell cycle were measured using a BD LSR flow cytometer and analyzed by the CellQuest software (Becton Dickinson, NY, USA). The experiments were repeated independently at least three times.
5. Wound Healing Assay
HT-29 and SW480 cells were grown-up to ~80% confluence in 6-well plates. A straight scratch was made slowly across the center of the well by a P200 pipette tip. Detached cells were washed away with PBS and added serum-free medium with various concentrations of TGF-β1 and PT. Cell migration to scratch was monitored at the indicated time periods in the same area. The capability of the cells to close the wound, and cell motility, were evaluated by determining the healed area. The images were taken at ×100 magnification (Olympus IX71). Wounded areas were measured twice time in the images by AxioVision SE64 Release 4.9.1 SP1. The measured times were 0 hours and 48 hours after treated by TGF-β1 and PT. The experiments were repeated three times.
6. Transwell Migration and Matrigel Invasion Assays
Migration was analyzed via a modified Transwell chamber assay using cell culture inserts with a polycarbonate filter (24-wells, 8-μm pore size with polycarbonate membrane; SPL Life Sciences, Pocheon, Korea). Cells were pretreated with TGF-β1 or PT for 48 hours. Invasion assays were completed via application a 24-well Transwell chamber (24-wells, 8-μm pore size with a polycarbonate membrane; SPL Life Sciences) with growth factor-reduced Matrigel. Briefly, after treatment 1×104 cells per well were planted into the upper chamber contained serum-free media. Especially in Matrigel invasion assays, the 100 μL Matrigel was coated on each well overnight before seeding the cells. The low chamber was occupied with 800 μL medium comprising 10% FBS as a chemoattractant and permitted to invade for 24 hours. Cells remaining above the insert membrane were removed by suction. Then, the cells on the lower border of the membrane were fixed in 3.8% formaldehyde for 20 minutes and stained with 0.1% crystal violet solution. The inserts were rinsed three times in ×1 PBS and air-dried. The numbers of invaded cells in 5 randomly selected fields were counted by an inverted microscope (Olympus IX71) at ×10 magnification and analyzed statistically. Five areas were counted per filter in each group. The number of invaded cells for each sample expressed the average of triplicate wells over 3 experiments.
7. Western Blotting
The protein concentration in cell lysates was determined by a Protein Quantification kit (Bio-Rad, Dallas-Fort Worth, TX, USA). Protein (50 μg) was loaded onto an SDS-polyacrylamide gel. The polyvinylidene difluoride (PVDF) membrane was probed with specific antibodies after transferring and blocking. The bindings of antibodies were identified by enhanced ECL prime (GE Healthcare, Piscataway, NJ, USA), captured and examined through the Las-3000 luminescent Image Analyzer (Fuji Film, Tokyo, Japan). The relative intensities of the protein bands were scanned and quantified by Image J. The marker protein bands were quantified adjusted with loading control (Actin).
8. Statistical Analysis
All data were collected into the Microsoft Excel 5.0 (Microsoft Corp., Redmond, WA, USA), and GraphPad Prism 5.0 (S GraphPad Software, San Diego, CA, USA) was used to perform the Student t-tests for the differences between 2 groups or the analysis of the variance, where appropriate. The data are expressed as the mean±SD of at least 3 independent experiments. P-values of <0.05 were evaluated statistically significant.
RESULTS
1. Effect of TGF-β1 on Cell Proliferation and Apoptosis in CRC Cell Lines
We treated the 2 CRC cell lines (HT-29 and SW480) with different concentrations of TGF-β1. There are no significant differences in growth rate were observed between untreated and treated SW480 cells. A statistically significant 10% reduction in cell viability was observed in HT-29 cells treated with TGF-β1 after 48 hours (Fig. 1A). Therefore, we treated the cells with 5 ng/mL TGF-β1 and different concentrations of PT. There was significant cell growth inhibition when cells were treated with both 5 ng/mL TGF-β1 and 15 μM PT in the 2 cell lines compared to cells treated by 5 ng/mL TGF-β1 (Fig. 1B). Cell growth was inhibited when the 2 cell lines were treated by both 5 ng/mL TGF-β1 and 15 μM PT compared to cells treated with 5 ng/mL TGF-β1 (Fig. 1A). A previous study demonstrated that PT promotes apoptotic cell death and inhibits cell growth in SW620 cancer cells [19]. To determine whether TGF-β and PT induce cell apoptosis in cell lines, we used FACS to detect the fraction of cells in sub-G1. No differences in apoptosis were detected between untreated and TGF-β1-treated HT-29 and SW480 cells (Fig. 1C). Additionally, as shown by flow cytometry, treatment by 5 ng/mL TGF-β1 and 5 μM PT for 48 hours had a minimal effect on cell cycle distribution and did not induce apoptosis in either of the 2 cell lines (Fig. 1D). Thus, 5 ng/mL TGF-β1 and 5 μM PT were used in subsequent experiments.
Fig. 1.

Effect of transforming growth factor β1 (TGF-β1) on cell proliferation and apoptosis in colorectal cancer cell lines. HT-29 and SW480 cells were treated with different concentrations of TGF-β1 or with 5 ng/mL TGF-β1 combined with different concentrations of parthenolide (PT) for 48 hours. Subsequently, the cell viability was detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (A, B) and cell apoptosis (sub-G1 fraction) was detected by flow cytometry (C, D). Data are shown as the mean±SD and based on 3 independent experiments. aP<0.01.
2. PT Attenuates the TGF-β1-Induced Elongation into Fibroblast-Like Shape in CRC Cell Lines
Trans-differentiation of CRC cells from an epithelial to a fibroblast-like morphology was induced by treatment by TGF-β1 for 48 hours. After 48 hours, treated cells showed a significant change towards a more lengthened fibroblastic-like shape compared to untreated cells. PT could reverse morphological changes in both cells (Fig. 2), demonstrating that PT can reverse TGF-β1-induced morphological changes in CRC cells such that they became elongated with a fibroblast-like shape.
Fig. 2.
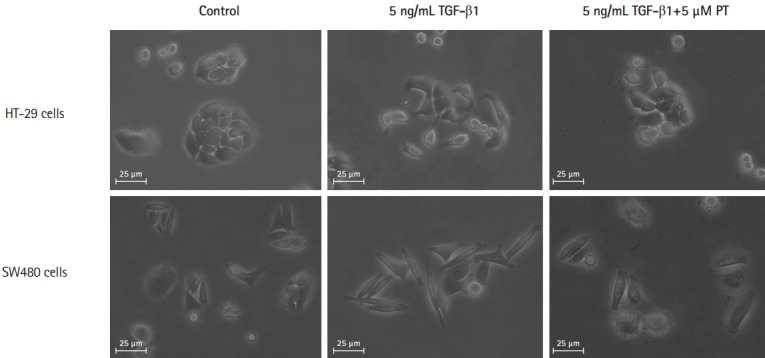
Parthenolide (PT) attenuates transforming growth factor β1 (TGF-β1)-induced elongated, fibroblast-like shape changing in colon cancer cell lines. Phase-contrast photomicrographs of control cells, cells treated with 5 ng/mL TGF-β1, cells treated with 5 ng/mL TGF-β1+5 μM PT for 48 hours.
3. PT Inhibits TGF-β1-Induced Cell Migration and Invasion in CRC Cell Lines
It has been established that down-regulation of E-cadherin and loss of intercellular adhesion is related to improved motility and invasive ability [20]. To verify that PT inhibits TGF-β1-induced EMT, cell migration and invasion were evaluated in wound healing, using migration and invasion assays with a Matrigel-coated Transwell chamber. In the wound healing assays, wounded areas were reduced after 48 hours with control and 5 ng/mL TGF-β1, and 5 ng/mL TGF-β1+5 μM PT conditions. Among the 3 conditions, 5 ng/mL TGF-β1 statistically reduced the wounded area more than control or 5 ng/mL TGF-β1+5 μM PT in both cell lines (Fig. 3A). In migration and invasion assays, TGF-β1 statistically increased the number of migrated cells and PT reversed this situation (Fig. 3B). These results mean PT inhibits TGF-β1-mediated cell migration and invasion.
Fig. 3.

Parthenolide (PT) represses transforming growth factor β1 (TGF-β1)-induced cell migration and invasion in colon cancer cell lines. HT-29 and SW480 cells were treated with 5 ng/mL TGF-β1 or 5 ng/mL TGF-β1+5 μM PT for 48 hours, and subsequently, the changes in migratory capacity were measured by the (A) wound healing assay and (B) Transwell migration assay. Scratch closure was monitored for 48 hours; microscopic images were taken at 0- and 48-hours post-scratching is shown. The percentage of wound area is shown in the histogram (A). The result is presented as the mean±SD, and the graph bar represent the mean±SD of 3 independent experiments. aP<0.05.
4. PT Suppresses TGF-β1-Induced EMT Pathway
To determine whether TGF-β1 induces a mesenchymal phenotype transition associated with EMT in colon cancer cells, we chose SW480 and HT-29 cell lines. It was observed that a decrease in E-cadherin (epithelial marker) and induction of β-catenin and Vimentin (mesenchymal marker) in TGF-β1-treated SW480 and HT-29 cell lines (Fig. 4). PT reversed the reduction in E-cadherin and increase of Vimentin in TGF-β1-treated SW480 and HT-29 cell lines. Suppression of E-cadherin in epithelial cells can be mediated by numerous transcription factors, including Snail and Slug [21]. The expression of Snail and Slug was dramatically enhanced by TGF-β1 in SW480 and HT-29 cell lines when compared to untreated controls. PT reversed the TGF-β1-induced expression of Snail and Slug in SW480 and HT-29 cell lines. These results indicate that PT inhibits EMT progression through the upregulation of epithelial markers and the downregulation of mesenchymal markers.
Fig. 4.
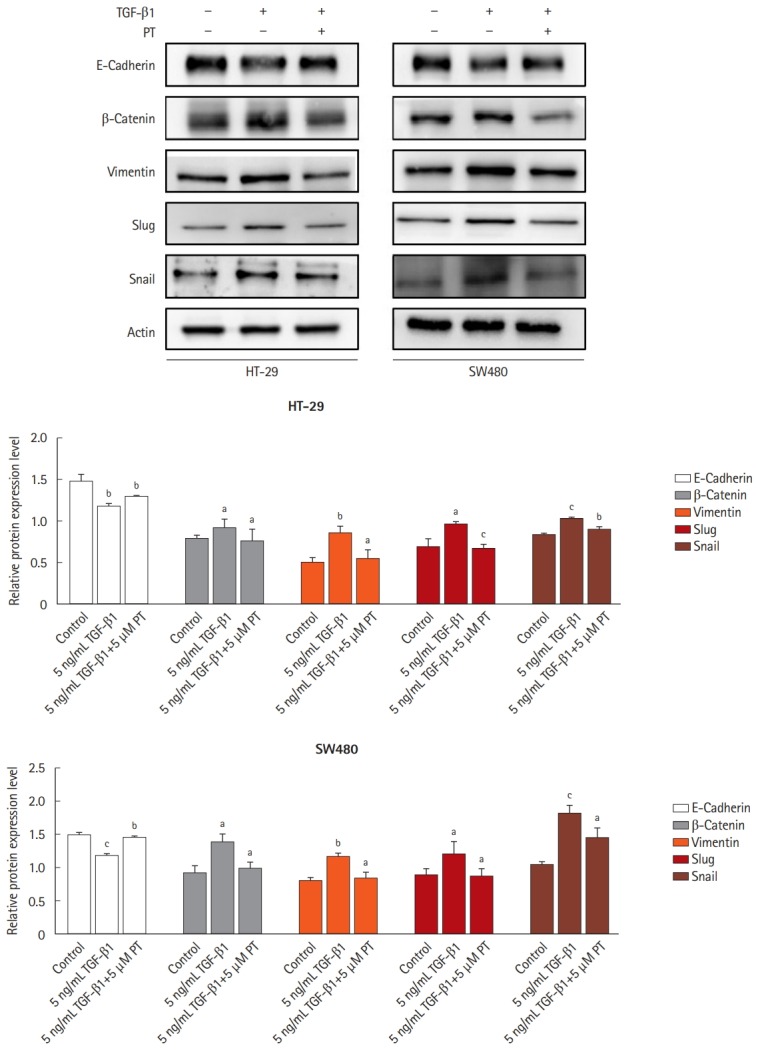
Parthenolide (PT) inhibits transforming growth factor β1 (TGF-β1) induces epithelial-mesenchymal transition pathway in colon cancer cell lines. Western blotting analysis of all protein expression in total lysates of untreated cells and cells treated with 5 ng/mL TGF-β1 or 5 ng/mL TGF-β1+5 μM PT for 48 hours by the indicated primary antibodies. Actin was used as a loading control. The results represent the mean of their independent experiments. Values represent mean±SD. Significant difference versus control group. aP<0.05, bP<0.01, and cP<0.001.
DISCUSSION
The findings from this study suggest that PT inhibits TGF-β1-induced EMT progression in CRC cell lines. TGF-β1 is a multifunctional growth factor that regulates various cell behaviors, such as cell proliferation and apoptosis. TGF-β demonstrates contextual pleiotropy in function; in normal and premalignant cells, TGF-β maintains the homeostasis and has tumor-suppressive effects (cytostatic, differentiation, and apoptosis). In late stage cancer progression, once a mutation occurs that causes cancer cells to lose TGF-β tumor-suppressive responses, TGF-β can promote tumorigenesis [22].
There is an established signaling pathway in which Smad4 protein forms heteromeric complexes with the receptor-phosphorylated Smad2 and Smad3 and then translocates from the cytoplasm to the nucleus [22]. Loss of Smad4, a tumor suppressor gene, may led to loss of transcription of genes critical to cell-cycle control [23]. Pathological forms of TGF-β signaling that are created by the tumor stroma or cancer cells themselves can lead to EMT, and ultimately cause invasiveness and dissemination [24]. It is well documented that TGF-β inhibits the cell cycle in phase G1 through mobilization of cyclin-dependent kinase inhibitors and inhibition of c-Myc dependent Smad3/4 complexes in epithelial cells. Abrogation of Smad4 translation may switch the pathway and cause loss of cell-cycle inhibition.
Null Smad4 cancer cells may escape from TGF-β-induced growth control and apoptosis. Two colon cancer cell lines, SW480 and HT-29, have loss of Smad4 expression [23], resulting in abolition of TGF-β tumor suppression and cell cycle arrest without inhibiting EMT activation by the Ras/Erk pathway [25]. We have observed that TGF-β1 does not induced apoptosis in HT-29 and SW480 cells. TGF-β1 can inhibit cell proliferation in HT-29 cells after 48 hours, but TGF-β1 did not inhibit cell proliferation in SW480 cells. At this point, it is not clear whether both of the null-Smad4 cell lines, HT-29 and SW480, showed a different rate of growth after treatment by TGF-β1. HT-29 cell and SW480 cell lines both belong to MSI-cell lines that loss at Smad4 but HT-29 loss at Smad4 in 2/3 markers and SW480 shows completely loss of Smad4. SW480 cell line has KRAS mutation, BRAF wild type, and PIK3CA wild type, while HT-29 cell line has KRAS wild type, BRAF mutation, PIK3CA mutation. Those kinds of different mutations maybe leads HT-29 and SW480 showed a different rate of growth after treatment with TGF-β1. In the previous study, we observed that PT inhibited the rate of growth in SW620 cells and PT promoted apoptosis in SW620 cells [19]. Similar to the previous study, we also have observed that 15 μM PT combined with 5 ng/mL TGF-β1 can inhibit the growth rate of cancer cells.
It is widely believed that TGF-β signaling evolved to enforce homeostasis and suppression of tumor progression in normal and premalignant cells can result in the tumorigenic effects in late stage cancer. One of the major genetic alterations associated with colorectal tumorigenesis is mutation of Smad4 or TGFBR2 that emerges during the adenoma to carcinoma transition [26]. Less Smad4 protein is involved with poor prognosis in CRC and a high occurrence of Smad4 gene mutation is involved in CRC distant metastasis [27,28]. Missing of Smad4 in CRC cells leads to a switch in the role of TGF-β from tumor suppressor to a tumor promoter [29]. In a previous study, TGF-β1 induced Smad2/3 transfer into the nucleus from the cytoplasm in HT-29 and SW480 cells [30]. Snail2 (Slug) is a transcription factor that induces EMT and is characterized by an increase in cell spreading and cell-cell separation. Induction of Slug is Smad4-independent and might be a Smad3-dependent process. The complexes Smad2/3 without Smad4 means loss of growth arrest but some target genes induced by EMT still exist, such as c-JUN and Slug, which both induce EMT. Smad4 inactivation selectively takes out only the tumor suppressive gene, allowing TGF-β activate pro-oncogenic genes [25]. TGF-β induced Smad-independent pathway, MEK/ERK is vital to EMT and plays a role during colorectal tumor invasion and metastasis [25,30].
Along the process of EMT, these factors are important like EMT effectors and EMT core regulators-transcription factors. EMT effectors are subcellular structure proteins like E-cadherin and Vimentin. Snail and Slug are regulators-transcription factors and over expression of them is involved with invasiveness, metastasis, and poor prognosis of CRC. Upregulation of Snail is a primary way of promoting EMT primarily through inhibition of E-cadherin [31]. Loss of E-cadherin promotes to release of β-catenin and accelerates EMT. Without TGF-β1, both contact β-catenin and E-cadherin are immediately degraded after contact disassembly. In contrast, TGF-β1 induces β-catenin separation from the epithelial contacts and stabilizes β-catenin in the cytoplasm, transferring it to the nucleus [32]. Snail and Slug and β-catenin, Vimentin are epithelial markers. E-cadherin is a typical marker for mesenchymal. Consistent with previous study, PT suppresses hypoxia-induced EMT in HT-29 cells through reverse epithelial and mesenchymal makers [33]. The figures of morphology were acquired by phase contrast microscopy. When β-catenin transfer into the nucleus and loss of E-cadherin expression happened, cell exposure to TGF-β1 changed the cells from invasive phenotype as mesenchymal shape [34]. PT reversed the elongated, fibroblast-like shape cells back to the epithelial shape.
No previous studies have investigated the ability of PT to inhibit TGF-β1-induced EMT in CRC cells. From the study of cell function and expression of protein level, we observed that PT suppressed the TGF-β1-induced EMT in CRC cells, but we still need to determine the mechanism by which PT inhibits the pathway. NF-κB is vital for EMT and metastasis in breast cancer cells [35]. Repression of NF-κB activity can reverse EMT in mesenchymal cells [35]. However, no activation of NF-κB is observed in null-Smad4 SW480 cells induced by TGF-β1 [36]. It well demonstrated that inhibition of TGF-β receptors and ERK pathway can suppress TGF-β1-induced EMT in human cancer cells [37,38].
In conclusion, TGF-β1 does not induce apoptosis in HT-29 and SW480 cell lines but can decrease HT-29 cell viability. TGF-β1 can induce EMT in HT-29 and SW480 cell lines. On the other hand, PT can reverse the TGF-β1-induced EMT in HT-29 and SW480 cell lines through inhibition of mesenchymal maker proteins, which also reduces cell migration and invasion. These findings suggest a novel approach for CRC treatment by inhibition of TGF-β1-induced EMT. Further studies will focus on metastatic animal models to determine how PT inhibits metastasis of CRC.
Footnotes
FINANCIAL SUPPORT
The authors received no financial support for the research, authorship, and/or publication of this article.
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION
Study design, data collection, data analysis, figures, and writing: Zhu SM, Kim SW. Data analysis, data interpretation: Park YR, Seo SY, Kim IH, Lee ST. Approval of final manuscript: all authors.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Raval M, Bande D, Pillai AK, et al. Yttrium-90 radioembolization of hepatic metastases from colorectal cancer. Front Oncol. 2014;4:120. doi: 10.3389/fonc.2014.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villalba M, Evans SR, Vidal-Vanaclocha F, Calvo A. Role of TGF-beta in metastatic colon cancer: it is finally time for targeted therapy. Cell Tissue Res. 2017;370:29–39. doi: 10.1007/s00441-017-2633-9. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Zubeldia I, Dotor J, Redrado M, et al. Co-migration of colon cancer cells and CAFs induced by TGFbeta1 enhances liver metastasis. Cell Tissue Res. 2015;359:829–839. doi: 10.1007/s00441-014-2075-6. [DOI] [PubMed] [Google Scholar]
- 5.Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 6.Lampropoulos P, Zizi-Sermpetzoglou A, Rizos S, Kostakis A, Nikiteas N, Papavassiliou AG. TGF-beta signalling in colon carcinogenesis. Cancer Lett. 2012;314:1–7. doi: 10.1016/j.canlet.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 7.Bremnes RM, Dønnem T, Al-Saad S, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol. 2011;6:209–217. doi: 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- 8.Padua D, Massagué J. Roles of TGFbeta in metastasis. Cell Res. 2009;19:89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 9.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 10.Akhurst RJ, Derynck R. TGF-beta signaling in cancer: a double-edged sword. Trends Cell Biol. 2001;11:S44–S51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 11.Lim J, Thiery JP. Epithelial-mesenchymal transitions: insights from development. Development. 2012;139:3471–3486. doi: 10.1242/dev.071209. [DOI] [PubMed] [Google Scholar]
- 12.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22:396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peñuelas S, Anido J, Prieto-Sánchez RM, et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009;15:315–327. doi: 10.1016/j.ccr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Massagué J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jafari N, Nazeri S, Enferadi ST. Parthenolide reduces metastasis by inhibition of vimentin expression and induces apoptosis by suppression elongation factor alpha-1 expression. Phytomedicine. 2018;41:67–73. doi: 10.1016/j.phymed.2018.01.022. [DOI] [PubMed] [Google Scholar]
- 16.Fu J, Ke X, Tan S, et al. The natural compound codonolactone attenuates TGF-beta1-mediated epithelial-to-mesenchymal transition and motility of breast cancer cells. Oncol Rep. 2016;35:117–126. doi: 10.3892/or.2015.4394. [DOI] [PubMed] [Google Scholar]
- 17.Chiu KY, Wu CC, Chia CH, Hsu SL, Tzeng YM. Inhibition of growth, migration and invasion of human bladder cancer cells by antrocin, a sesquiterpene lactone isolated from Antrodia cinnamomea, and its molecular mechanisms. Cancer Lett. 2016;373:174–184. doi: 10.1016/j.canlet.2015.11.046. [DOI] [PubMed] [Google Scholar]
- 18.Liu JW, Cai MX, Xin Y, et al. Parthenolide induces proliferation inhibition and apoptosis of pancreatic cancer cells in vitro. J Exp Clin Cancer Res. 2010;29:108. doi: 10.1186/1756-9966-29-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu YC, Kim SL, Park YR, Lee ST, Kim SW. Parthenolide promotes apoptotic cell death and inhibits the migration and invasion of SW620 cells. Intest Res. 2017;15:174–181. doi: 10.5217/ir.2017.15.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol. 2005;17:559–564. doi: 10.1016/j.ceb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Natalwala A, Spychal R, Tselepis C. Epithelial-mesenchymal transition mediated tumourigenesis in the gastrointestinal tract. World J Gastroenterol. 2008;14:3792–3797. doi: 10.3748/wjg.14.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massagué J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodford-Richens KL, Rowan AJ, Gorman P, et al. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc Natl Acad Sci U S A. 2001;98:9719–9723. doi: 10.1073/pnas.171321498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung B, Staudacher JJ, Beauchamp D. Transforming growth factor beta superfamily signaling in development of colorectal cancer. Gastroenterology. 2017;152:36–52. doi: 10.1053/j.gastro.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. 2005;25:8108–8125. doi: 10.1128/MCB.25.18.8108-8125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones S, Chen WD, Parmigiani G, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alazzouzi H, Alhopuro P, Salovaara R, et al. SMAD4 as a prognostic marker in colorectal cancer. Clin Cancer Res. 2005;11:2606–2611. doi: 10.1158/1078-0432.CCR-04-1458. [DOI] [PubMed] [Google Scholar]
- 28.Miyaki M, Iijima T, Konishi M, et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene. 1999;18:3098–3103. doi: 10.1038/sj.onc.1202642. [DOI] [PubMed] [Google Scholar]
- 29.Zhang B, Halder SK, Kashikar ND, et al. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology. 2010;138:969–980. doi: 10.1053/j.gastro.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pino MS, Kikuchi H, Zeng M, et al. Epithelial to mesenchymal transition is impaired in colon cancer cells with microsatellite instability. Gastroenterology. 2010;138:1406–1417. doi: 10.1053/j.gastro.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vu T, Datta PK. Regulation of EMT in colorectal cancer: a culprit in metastasis. Cancers (Basel) 2017;9:E171. doi: 10.3390/cancers9120171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian X, Liu Z, Niu B, et al. E-cadherin/beta-catenin complex and the epithelial barrier. J Biomed Biotechnol. 2011;2011:567305. doi: 10.1155/2011/567305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SL, Park YR, Lee ST, Kim SW. Parthenolide suppresses hypoxia-inducible factor-1alpha signaling and hypoxia induced epithelial-mesenchymal transition in colorectal cancer. Int J Oncol. 2017;51:1809–1820. doi: 10.3892/ijo.2017.4166. [DOI] [PubMed] [Google Scholar]
- 34.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huber MA, Azoitei N, Baumann B, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grau AM, Datta PK, Zi J, Halder SK, Beauchamp RD. Role of Smad proteins in the regulation of NF-kappaB by TGF-beta in colon cancer cells. Cell Signal. 2006;18:1041–1050. doi: 10.1016/j.cellsig.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 37.Zhao L, Liu S, Che X, et al. Bufalin inhibits TGF-beta-induced epithelial-to-mesenchymal transition and migration in human lung cancer A549 cells by downregulating TGF-beta receptors. Int J Mol Med. 2015;36:645–652. doi: 10.3892/ijmm.2015.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y, Lou Z, Lee SH. Arctigenin represses TGF-beta-induced epithelial mesenchymal transition in human lung cancer cells. Biochem Biophys Res Commun. 2017;493:934–939. doi: 10.1016/j.bbrc.2017.09.117. [DOI] [PubMed] [Google Scholar]