

Abstract

Nucleoside analogues are among the most common medications given for the treatment of viral infections and cancers. The therapeutic effectiveness of nucleoside analogues can be dramatically improved by phosphorylation. The ProTide approach was developed using a phosphorylated nucleoside that is masked by esterification with an amino acid and phenol forming a chiral phosphorus center. The biological activity of the ProTides depends, in part, on the stereochemistry at phosphorus, and thus, it is imperative that efficient methods be developed for the chemical synthesis and isolation of diastereomerically pure ProTides. Chiral ProTides are often synthesized by direct displacement of a labile phenol (p-nitrophenol or pentafluorophenol) from a chiral phosphoramidate precursor with the appropriate nucleoside analogue. The ability to produce these chiral products is dictated by the synthesis of the chiral phosphoramidate precursors. The enzyme phosphotriesterase (PTE) from Pseudomonas diminuta is well-known for its high stereoselectivity and broad substrate profile. Screening PTE variants from enzyme evolution libraries enabled the identification of variants of PTE that can stereoselectively hydrolyze the chiral phosphoramidate precursors. The variant G60A-PTE exhibits a 165-fold preference for hydrolysis of the RP isomer, while the variant In1W-PTE has a 1400-fold preference for hydrolysis of the SP isomer. Using these mutants of PTE, the SP and RP isomers were isolated on a preparative scale with no detectable contamination of the opposite isomer. Combining the simplicity of the enzymatic resolution of the precursor with the latest synthetic strategy will facilitate the production of diastereometrically pure nucleotide phosphoramidate prodrugs.
With more than 20 clinically available examples, nucleoside analogues have become the standard of care for viral infections and cancer (reviewed in refs (1) and (2)). These compounds typically inhibit DNA and RNA polymerases or result in accumulated mutations and chain terminations, which further disrupt genomic replication. The specificity of these compounds, such as acyclovir, for viral polymerases over host polymerases has made it the standard of care for herpes simplex virus and varicella zoster virus.2 While the value of nucleoside analogues is well proven clinically, their use has been limited because of poor bioavailability, the need for active transport into cells, and the requirement for cellular phosphorylation to be therapeutically active (reviewed in refs (3−5)).
The most successful method for overcoming the challenges of nucleoside analogues has been the development of the nucleoside phosphoramidate prodrugs (ProTides, prodrug and nucleotides).1,2 For many nucleoside analogues, the limiting step in the cellular activation of these compounds is phosphorylation, resulting in the development of drug resistance by downregulation of the relevant kinases.3,5 The ProTides, such as Sofosbuvir and Tenofovir Alafenamide, overcome this deficiency by starting with a phosphorylated or phosphonylated prodrug (Scheme 1). The multiple negative charges on the phosphorylated nucleoside analogues, which would normally lead to poor bioavailability and cellular uptake, are masked in the prodrugs by amide and ester bond formation.6 This strategy of masking the phosphate moiety significantly increases the hydrophobic nature of the prodrugs, which enables cellular entry via passive diffusion across the membrane, and gives the added benefit of substantially increasing the oral bioavailability, as well as overcoming resistance due to downregulation of the transporter.7,8
Scheme 1. Structures of Selected ProTides.

The success of the ProTide approach has led to the Food and Drug Administration approval of Sofosbuvir and Tenofovir Alafenamide for viral infections, and several other compounds are in clinical trials to treat HIV, hepatitis C, and Ebola, as well as two compounds being tested as cancer treatments.1,2 Regardless of the condition being treated, all ProTides are delivered to the cell as prodrugs that must undergo intracellular activation to form the active compound. In the case of the nucleotide analogues, the mechanism of activation is fairly well understood.9−11 In the first activation step, the ester group of the amino acid is hydrolyzed by a peptidase or esterase. The enzymes responsible for the in vivo cleavage of this bond are known to be stereoselective, leading to widely differing activities depending on the stereochemistry of the prodrug.9,10,12,13 For example, the SP isomer of Sofosbuvir exhibits activty that is 18-fold better than that of the RP isomer against hepatitis C.14 Tenofovir Alafenamide shows a similar dependence, with the SP isomer being 12-fold more active against HIV than the RP isomer.15
For both Sofosbuvir and Tenofovir Alafenamide, the SP isomer is more active. However, MK-3682, developed by Merck, is the RP isomer and is activated approximately 20-fold faster than the SP isomer.16 MK-3682 was found to be highly effective in clinical trials, demonstrating that for some ProTides the SP isomer will be more active while with others the RP isomer is more desirable.17
The ProTide approach is not limited to nucleoside analogues. There are currently efforts to apply the ProTide technology to deliver bioactive compounds for the treatment of osteoarthritis, Parkinson’s disease, multiple sclerosis, African sleeping sickness, and tuberculosis, making apparent the need to develop robust methods for the synthesis of diastereomerically pure ProTides.18−22 The most common synthetic route for ProTides involves reaction of an O-aryl, N-amino acid phosphochloridate precursor with the desired nucleoside analogue (Scheme 2).2,23 The simple base-catalyzed reaction can couple the nucleoside to the 5′- or 3′-hydroxyl group, and the desired product is formed as a diastereomeric mixture.23,24 Various protection schemes have been employed to prevent side product formation, but the required deprotection is generally inefficient.2,25 Merck has reported improved stereoselective syntheses using small molecule catalysts to enhance the yield of the RP isomers.26 However, these methods are not universal for all nucleoside analogues and still face the difficulties of side product formation and the need to separate the two diastereomers. An alternative method was recently developed to generate a chiral phosphoramidate diester intermediate with a labile aromatic group (p-nitrophenol or pentafluorophenol), which allows for a crystallographic separation of the two isomers prior to base-catalyzed substitution of the labile leaving group resulting in diastereomerically pure ProTide products.27 This strategy was further advanced by the use of Lewis acids to catalyze the substitution reaction to control the regioselectivity without the need for protecting groups (Scheme 3).24 Using dimethylaluminum chloride as the Lewis acid, a wide variety of ProTides can be synthesized in good yields and with minimal side products.
Scheme 2. Base-Catalyzed Synthesis of a Nucleotide Phosphoramidate Prodrug.

Scheme 3. Lewis Acid-Catalyzed Synthesis of a Diastereomerically Pure Nucleotide Phosphoramidate Diester.

The current state of the art in synthetic methodology enables the production of various ProTides with high purities However, the ultimate obstacle to diastereomeric purity lies in the efficient diastereomeric separation of the chiral precursor. The chiral separation of these compounds generally requires differential crystallization or chiral chromatography, which are both difficult and inefficient. The most common precursors used in the synthesis of the ProTides are chiral phosphoramidate diesters such as compound 1. The enzyme phosphotriesterase from Pseudomonas diminuta (PTE) has a broad substrate specificity and excellent stereoselectivity, which has allowed its use in chiral resolution with multiple types of phosphorus-containing compounds.28−30 The adaptation of PTE for both detoxification and chemical synthesis methodologies has yielded many variants of the enzyme with enhanced, as well as reversed, stereoselectivity.28,31−35 To explore whether PTE could be utilized to prepare diastereomerically pure precursors for ProTide synthesis, variants from several enzyme libraries were screened for their ability to selectively hydrolyze a chiral p-nitrophenyl-containing ProTide precursor (1). The phosphotriesterase from Sphingobium sp. TCM1 (Sb-PTE) was also tested and found to hydrolyze both isomers of the precursor, but with the SP isomer, any one of the three bonds to phosphorus is cleaved. Variants of the P. diminuta PTE were identified with >100-fold chiral selectivity for either the SP or RP isomers. The use of the simple PTE variant G60A (G60A-PTE), as well as a previously uncharacterized variant of PTE with a 10-amino acid insertion in loop 7 (In1W-PTE), allowed the preparative isolation of pure SP and RP isomers of compound 1. The purified isomers were recovered by simple organic extraction, allowing facile isolation on a preparative scale, with no apparent contamination of the opposing isomer.
Materials and Methods
Chemicals and Enzymes
Laboratory chemicals were from Sigma-Aldrich, general laboratory supplies from VWR, and growth media from RPI Corp. Protein production and purification were as previously described.36,37 The purified enzymes were stored at −80 °C prior to use.
Chemical Synthesis of Compound 1
Compound 1 was synthesized using a modified method of Ross et al.27 Briefly, a stirred solution of phenyl dichlorophosphate (1.1 mL, 7.0 mmol, 95%) and p-nitrophenol (0.97 g, 7.0 mmol) in 100 mL of anhydrous ether was cooled to −15 °C. Triethylamine (0.98 mL, 7.0 mmol) was added dropwise, and the reaction mixture stirred for 4 h at 0 °C. Solid byproducts were filtered, washed with ether, and discarded. l-Alanine isopropyl ester hydrochloride (1.2 g, 7.0 mmol) was added to the liquid phase, followed by triethylamine (1.96 mL, 14.0 mmol), and the mixture stirred for 16 h at room temperature (23 °C). The reaction mixture was filtered, and after concentration, the residue was purified by silica gel column chromatography (3:1, 2:1, and 1:1 hexanes/ethyl acetate mixtures) yielding 0.72 g (25%) of the product as a mixture of diastereomers in a 93:100 ratio as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.25 (dd, J = 9.0 and 1.8 Hz, 2H), 7.44–7.33 (m, 4H), 7.28–7.18 (m, 3H), 5.08–4.98 (m, 1H), 4.17–4.06 (m, 1H), 3.92–3.85 (m, 1H), 1.44–1.40 (m, 3H), 1.27–1.23 (m, 6H); 31P NMR (160 MHz, CDCl3) δ −3.17 (s), −3.21 (s).
PTE Variants
The mutant A80V/K185R/I274N is a wild-type-like variant of PTE with high expression levels and will be called wild-type PTE hereafter.38,39 The variants G60A and I106G/F132G/H257Y were previously identified in prior investigations that focused on altering the stereoselectivity of PTE.29,30 The variant H257Y/L303T was identified as stereoselective for phosphonate substrates.35 PTE-In1W was originally found during the screening of a 254X/257X enzyme library for catalytic activity against DEVX.36 This variant has a 10-amino acid insertion in the sequence for loop 7, which originally arose as a cloning artifact. The genetic identity of PTE In1W is F132L/H254S/H257W/257_258insSAIGLDPIPN. Additional variants were screened on the basis of their observed stereoselectivity with V-agent analogues.28
Screening for Stereoselective Variants
PTE variants were screened for their ability to stereoselectively hydrolyze the two isomers of compound 1 by a total hydrolysis reaction.35 Reactions were conducted in a final volume of 1.0 mL with 60 μM compound 1, 100 μM CoCl2, 50 mM CHES (pH 9.0), and 1% methanol. Reactions were initiated by the addition of 10 μL of the enzyme (final concentrations of 0.5–2 μM) or 1 M KOH, and the reactions were followed at 400 nm (E400 = 17000 M–1 cm–1 for p-nitrophenol) with a Molecular Devices Spectramax 384 Plus spectrophotometer. Stock solutions of compound 1 were prepared in methanol.
Complementation Assays
Variants that demonstrated enhanced stereoselective hydrolysis of compound 1 were used in complementation assays to determine if these enzymes preferred the same or opposite stereochemistry at the phosphorus center. The relative concentrations of each enzyme were determined so that they hydrolyzed the faster isomer at roughly the same rate. Reaction mixtures had a total volume of 1.0 mL with 60 μM compound 1, 100 μM CoCl2, 50 mM CHES (pH 9.0), and 1% methanol. The reactions were initiated by the addition of the first enzyme. Once the first enzyme had consumed its preferred diastereomer, the second enzyme was added to the reaction mixture. If the two variants preferred the opposite diastereomer, the addition of the second enzyme enabled the hydrolysis of the other remaining isomer. If the two variants preferred the same diastereomer, no additional hydrolysis was observed.
31P Nuclear Magnetic Resonance (NMR) Analysis of Enzymatic Reactions
To enable the identification of the specific diastereomer being hydrolyzed by wild-type PTE, G60A-PTE, In1W-PTE, and Sb-PTE, the reaction products were interrogated by 31P NMR. Reactions were conducted in a reaction volume of 4.0 mL with 2.0 mM compound 1, 30% methanol, and 50 mM HEPES (pH 8.0). The progress of the reaction was monitored by removing small aliquots and measurement of the absorbance at 400 nm. At various time points, 1.0 mL samples were removed from the reaction mixture, and the enzyme was removed by ultrafiltration using a Vivaspin 500 (GE Healthcare) centrifugal filtration device. The sample was brought to 10 mM EDTA in 20% D2O, and the 31P NMR spectrum recorded using a sample volume of 750 μL.
Isolation of Individual Diastereomers
The RP diastereomer of compound 1 was isolated by selective hydrolysis of the SP diastereomer using In1W-PTE. Compound 1 (50 mg) was dissolved in methanol and added to a 50 mL reaction mixture, containing 30% methanol, 50 mM HEPES (pH 8.0), and 500 nM In1W-PTE. The progress of the reaction was monitored by removing 10 μL aliquots, which were then diluted to 1.0 mL, and the absorbance at 400 nM was recorded. Once the reaction had exceeded 50% completion, the methanol was removed by rotary evaporation and the remaining RP diastereomer was extracted using dichloromethane (3 × 30 mL). The organic phase was washed with copious amounts of 50 mM HEPES (pH 8.0) to remove contaminating p-nitrophenol, dried over Na2SO4, and the solvent removed by rotary evaporation. A total of 20 mg of pure RP-1 was recovered. The SP diastereomer of compound 1 was isolated by selective hydrolysis of the RP diastereomer from the racemic mixture using G60A-PTE. Conditions and purification were performed as described above except 5.0 μM G60A-PTE was used and the reaction was allowed to proceed to ∼65% completion. A total of 14 mg of the pure SP isomer was recovered.
Steady State Kinetics
The steady state kinetic constants with racemic compound 1 and the isolated SP and RP diastereomers were determined for WT-PTE, G60A-PTE, In1W-PTE, and Sb-PTE. Reactions were conducted in a volume of 250 μL with 10% methanol, 50 mM CHES (pH 9.0), and 10–250 μM compound 1 at 30 °C. Reactions with WT-PTE, G60A-PTE, and In1W-PTE were supplemented with 0.1 mM CoCl2. The reactions with Sb-PTE were supplemented with 1.0 mM MnCl2. Reactions were initiated by the addition of 10 μL of the appropriately diluted enzyme and followed using a Molecular Devices Spectramax 384 Plus spectrophotometer in a 96-well plate format. None of the tested enzymes demonstrated saturation by the substrate at 250 μM, so the data were fit to a linear equation to enable the calculation of kcat/Km.
Results and Discussion
Screening of PTE Variants
Variants of PTE with differing stereochemical preferences were screened for their ability to hydrolyze the two isomers of compound 1. Of the 57 variants screened, 52 exhibited no or weak stereoselectivity with compound 1. Of the five variants that did show selectivity, four showed a preference for one diastereomer, and only G60A-PTE preferred the other diastereomer. The preliminary screening results are summarized in Table S1.
Wild-type PTE completely hydrolyzed racemic compound 1 with an apparent single-exponential phase (Figure 1a). At relatively low concentrations, the reactions appear to stop at approximately 50% of the expected value with G60A-PTE and In1W-PTE, suggesting that these variants preferentially hydrolyzed one diastereomer. At a 20-fold higher concentration, G60A-PTE clearly hydrolyzes the second diastereomer of compound 1, but at a much slower rate (Figure 1b). In1W-PTE did not exhibit significant hydrolysis of the second diastereomer even at a 20-fold higher enzyme concentration, suggesting a very strong preference for one of the two diastereomers. To determine if the identified variants preferred the same diastereomer or the opposite diastereomer, a complementation assay was utilized. As demonstrated in Figure 1c, the hydrolysis reaction is first initiated by one enzyme and once the first diastereomer is consumed the second enzyme is added. If the two variants prefer the same diastereomer, the rate will not increase. However, if the second variant prefers the other diastereomer, the rate will dramatically increase as observed in the case of In1W-PTE and G60A-PTE. Sb-PTE was found to apparently hydrolyze ∼60% of the sample in the screen; however, neither G60A-PTE nor In1W-PTE could complement this catalytic activity.
Figure 1.
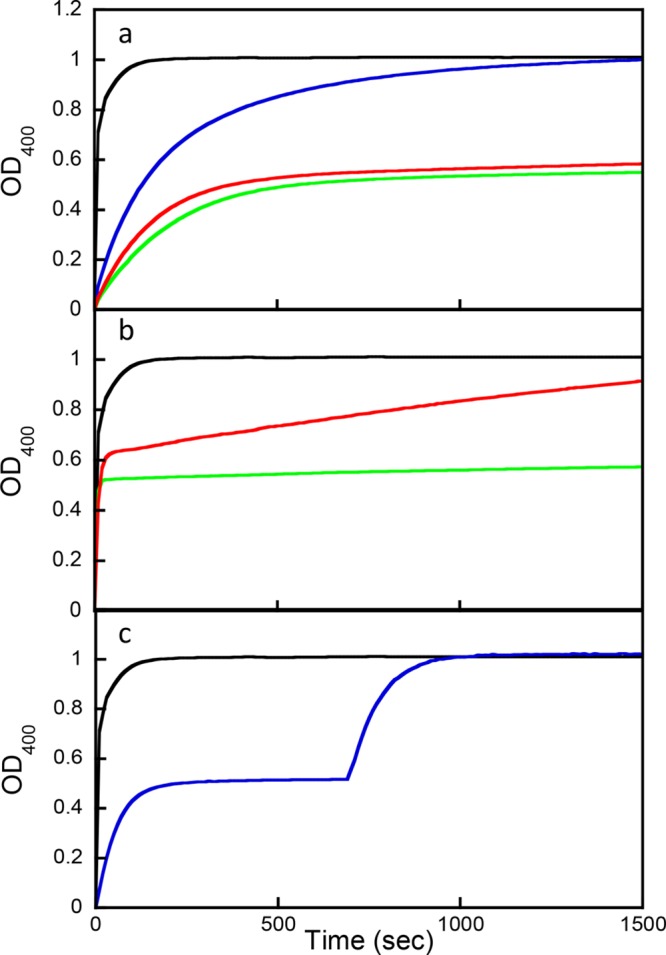
Stereoselective hydrolysis of 60 μM compound 1. (a) Chemical hydrolysis by 1 M KOH (black) and enzymatic hydrolysis by 100 nM wild-type PTE (blue), 700 nM G60A-PTE (red), and 3.6 nM In1W-PTE (green). (b) Chemical hydrolysis by 1 M KOH (black) and enzymatic hydrolysis by 14 μM G60A-PTE (red) or 72 nM In1W-PTE (green). (c) Complementation assay with In1W-PTE and G60A. Chemical hydrolysis by 1 M KOH (black). Enzymatic hydrolysis (blue) was initiated by addition of 7.2 nM In1W-PTE, and then 1.0 μM G60A-PTE was added at 700 s.
Identification of the Preferred Isomers
The 31P NMR chemical shifts of the two diastereomers of compound 1 have been previously identified with the resonance for the SP isomer being downfield of the RP isomer in dimethyl sulfoxide.27 While the chemical shift changes significantly, a similar relationship was found using either methanol or water as the solvent with the SP isomer always being downfield of the RP isomer (Figure S1). When wild-type PTE is used to hydrolyze compound 1, there is a small, but observable, preference for hydrolysis of the RP isomer (Figure 2). The chemical synthesis of 1 results in a small excess of the RP diastereomer.
Figure 2.

31P NMR spectra of compound 1 and hydrolysis product in water. (a) Two diastereomers of compound 1. The chemical shift of the SP isomer is at −1.43 ppm, while the RP isomer is at −1.54 ppm. (b) Hydrolysis of compound 1 by wild-type PTE at ∼60% completion. The single phosphorus-containing hydrolysis product is observed at 2.05 ppm. (c) Complete hydrolysis of compound 1 by wild-type PTE.
The hydrolysis of 1 by G60A-PTE results in a much more dramatic stereoselective effect. With G60A, the RP isomer is depleted much faster than the SP isomer, and thus when the reaction reaches ∼30% completion, the ratio of the SP isomer to the RP isomer is approximately 2:1 (Figure 3b). When the reaction is ∼60% complete, the SP:RP ratio has increased to 4.6:1 (Figure 3c). Among the variants that preferred the opposite isomer to G60A-PTE, the In1W-PTE mutant had the highest activity. As observed during the initial screening, In1W-PTE also has a stronger stereoselectivity than G60A-PTE. In the reaction catalyzed by In1W at ∼40% completion, the SP:RP ratio is 1:3.5 (Figure 3d). When the reaction is just more than 50% complete, there is no detectable resonance for the SP isomer.
Figure 3.

31P NMR spectra showing the differential hydrolysis of 1 by variants of PTE. (a) 31P NMR spectrum of compound 1 (2 mM). SP-1 resonates at −1.43 ppm, while RP-1 resonates at −1.54 ppm. (b) Hydrolysis of 1 by 3 μM PTE-G60A. The reaction is ∼28% complete. The single phosphorus-containing product resonates at 2.05 ppm. (c) Reaction from spectrum b at ∼60% completion. (d) Hydrolysis of 1 by 0.1 μM PTE-In1W. The reaction is ∼38% complete. (e) Reaction from spectrum d at ∼50% completion.
Preparative Isolation of Isomers
The strong stereoselectivity obtained with G60A-PTE and In1W-PTE suggested that these variants could be used to isolate pure diastereomers of compound 1. Large-scale preparative reactions were initiated to resolve 50 mg of the diastereomeric mixture. To ensure complete removal of the RP isomer, the reaction catalyzed by 5.0 μM G60A-PTE was allowed to proceed to approximately 65% completion (∼4 h). The recovery of the remaining unreacted isomer of 1 resulted in the isolation of 14 mg of the pure SP isomer (Figure 4b). The higher selectivity of In1W enabled the reaction to be stopped at ∼50% completion (∼1 h), and 20 mg of the pure RP isomer was recovered (Figure 4c). In both cases, there was no detectable contamination from the opposite isomer (≥98% enantiomeric excess).
Figure 4.

31P NMR spectra of isolated RP and SP isomers of 1 in methanol. (a) NMR spectrum of the initial mixture of the RP and SP isomers of 1. The SP isomer resonates at −1.52 ppm, and the RP isomer resonates at −1.69 ppm. (b) Isolated SP isomer obtained by selective hydrolysis using PTE-G60A. (c) Isolated RP isomer obtained by selective hydrolysis using PTE-In1W. (d) Mixture of samples from spectra b and c.
Stereochemical Preferences of Sb-PTE
When tested in the initial screen, Sb-PTE did not appear to completely hydrolyze the mixture of diastereomers of compound 1. To determine the stereoselectivity of Sb-PTE for the hydrolysis of 1, the purified SP and RP isomers of 1 were hydrolyzed separately and the products analyzed by 1H and 31P NMR spectroscopy (Figure 5 and Figure S4). With the SP isomer, ∼27% of the product formed was due to the hydrolysis of the bond to p-nitrophenol. Phenol cleavage accounted for 65% of the product formed, and ∼7% was due to hydrolysis of the phosphorus–nitrogen bond. The aromatic region of the 1H NMR spectrum is in good agreement with this product distribution with two clear resonances present for the p-nitrophenyl substituent attached to phosphorus and as p-nitrophenol in solution (Figure S4). The hydrolysis of the RP isomer resulted in the nearly exclusive hydrolysis of the p-nitrophenol substituent with a minor product (4%), which may be due to hydrolysis of the isopropyl ester bond.
Figure 5.

Hydrolysis of 1 by Sb-PTE. (a) Hydrolysis of the isolated SP isomer by Sb-PTE. The resonance at 2.31 ppm is the product of hydrolysis of p-nitrophenol. The resonance at 1.61 ppm is due to the product from hydrolysis of phenol, and the resonance at −10.45 ppm is due to the product of hydrolysis of the phosphoramidate bond. (b) Hydrolysis of the isolated RP isomer by Sb-PTE. (c) Hydrolysis of the isolated RP isomer by wild-type PTE.
Steady State Kinetics
To determine the magnitude of the stereochemical preferences, the isolated isomers were used to measure steady state kinetic constants of the various enzymes used in this investigation (Table 1). Due to the limited solubility of compound 1 in water, the highest concentration possible was ∼250 μM. None of the variants tested displayed substrate saturation at this concentration, and thus, only the kcat/Km values were obtained from linear fits to the data. WT-PTE demonstrated reasonable catalytic activity with compound 1 with a kcat/Km of 4.7 × 104 M–1 s–1 for the faster isomer and a preference for the RP isomer relative to the SP isomer of 6:1. G60A-PTE was considerably slower with a kcat/Km of 2.8 × 103 M–1 s–1 for the faster RP isomer and a preference of 170, relative to the SP isomer. The mutant In1W-PTE showed the best activity of any variant tested with a kcat/KM of 2.3 × 105 M–1 s–1 for the preferred S -isomer and a catalytic preference of 1400 relative to the RP isomer. Sb-PTE exhibited essentially no stereoselectivity for compound 1. However, as has been seen with other compounds, the major hydrolysis product from the SP isomer is due to the cleavage of phenol rather than p-nitrophenol.40
Table 1. Kinetic Constants for the Enzymatic Hydrolysis of Compound 1.
| substrate | enzyme | kcat/Km (M–1 s–1) |
|---|---|---|
| RP/SP-1 | WT-PTE | (4.2 ± 0.1) × 104 |
| G60A-PTE | (2.7 ± 0.1) × 103 | |
| In1W-PTE | (1.9 ± 0.4) × 105 | |
| Sb-PTE | (1.92 ± 0.03) × 103 | |
| SP-1 | WT-PTE | (7.4 ± 0.1) × 103 |
| G60A-PTE | (1.69 ± 0.03) × 10 | |
| In1W-PTE | (2.25 ± 0.08) × 105 | |
| Sb-PTE (p-NP) | (6.6 ± 0.2) × 102 | |
| Sb-PTE (phenol)a | (2.3 ± 0.1) × 103 | |
| RP-1 | WT-PTE | (4.7 ± 0.1) × 104 |
| G60A-PTE | (2.83 ± 0.04) × 103 | |
| In1W-PTE | (1.6 ± 0.2) × 102 | |
| Sb-PTE | (3.0 ± 0.1) × 103 |
Calculated from product ratios.
Conclusion
The ProTides are chiral phosphoramidates, which require activation by intracellular enzymes to achieve their active forms. The first enzymatic activation step is stereoselective, leading to widely different biological activity based on the stereochemistry at the phosphorus center. The need to selectively synthesize pure diastereomers of the ProTides has led to much progress in the synthesis, but the ability to make pure diastereomers depends on the isolation of diastereomerically pure precursors such as compound 1. Previous efforts have utilized differential crystallization or chiral column chromatography to prepare these chiral precursors. While these efforts have proven to be successful, they are difficult and time-consuming. As an alternative approach, diastereometrically pure precursors can be obtained by enzymatic resolution. In this approach, the undesired isomer is preferentially hydrolyzed by a stereoselective enzyme. The PTE mutant G60A-PTE displays an ∼165-fold preference for the RP isomer of compound 1, allowing the SP isomer to be easily isolated. The In1W-PTE variant exhibits even higher selectivity with a 1400-fold preference for the SP isomer, making the isolation of the RP isomer relatively straightforward. The hydrophobic nature of compound 1 allows for the facile recovery of the desired isomer via simple organic extraction on a preparative scale. Combining this enzymatic methodology with the regioselective synthesis methods developed by others will greatly simplify the preparation of diasteromerically pure ProTides of either stereochemistry.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00530.
Additional data, tables, and figures (PDF)
Accession Codes
Pd-PTE, UniProt entry P0A434; Sb-PTE, UniProt entry A0A077JBW9.
This work was supported in part by National Institutes of Health Grant GM 116894.
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
Supplementary Material
References
- Mehellou Y.; Rattan H. S.; Balzarini J. (2018) The ProTide Prodrug Technology: From the Concept to the Clinic. J. Med. Chem. 61, 2211–2226. 10.1021/acs.jmedchem.7b00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarczyk M.; Serpi M.; Pertusati F. (2018) Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antiviral Chem. Chemother. 26, 204020661877524. 10.1177/2040206618775243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander P.; Kucera G.; Pardee T. S. (2016) Improving nucleoside analogs via lipid conjugation: Is fatter any better?. Crit. Rev. Oncol. Hematol. 100, 46–56. 10.1016/j.critrevonc.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey J. R.; Baldwin S. A.; Young J. D.; Cass C. E. (1998) Nucleoside transport and its significance for anticancer drug resistance. Drug Resist. Updates 1, 310–324. 10.1016/S1368-7646(98)80047-2. [DOI] [PubMed] [Google Scholar]
- Stein D. S.; Moore K. H. (2001) Phosphorylation of nucleoside analog antiretrovirals: a review for clinicians. Pharmacotherapy 21, 11–34. 10.1592/phco.21.1.11.34439. [DOI] [PubMed] [Google Scholar]
- Mehellou Y.; Balzarini J.; McGuigan C. (2009) Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 4, 1779–1791. 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- Slusarczyk M.; Lopez M. H.; Balzarini J.; Mason M.; Jiang W. G.; Blagden S.; Thompson E.; Ghazaly E.; McGuigan C. (2014) Application of ProTide technology to gemcitabine: a successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 57, 1531–1542. 10.1021/jm401853a. [DOI] [PubMed] [Google Scholar]
- Murakami E.; Wang T.; Park Y.; Hao J.; Lepist E. I.; Babusis D.; Ray A. S. (2015) Implications of efficient hepatic delivery by tenofovir alafenamide (GS-7340) for hepatitis B virus therapy. Antimicrob. Agents Chemother. 59, 3563–3569. 10.1128/AAC.00128-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkus G.; Wang R.; Liu X. H.; Kutty N.; MacArthur H.; Cihlar T.; Gibbs C.; Swaminathan S.; Lee W.; McDermott M. (2007) Cathepsin A is the major hydrolase catalyzing the intracellular hydrolysis of the antiretroviral nucleotide phosphonoamidate prodrugs GS-7340 and GS-9131. Antimicrob. Agents Chemother. 51, 543–550. 10.1128/AAC.00968-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami E.; Tolstykh T.; Bao H.; Niu C.; Steuer H. M.; Bao D.; Chang W.; Espiritu C.; Bansal S.; Lam A. M.; Otto M. J.; Sofia M. J.; Furman P. A. (2010) Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 285, 34337–34347. 10.1074/jbc.M110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazkova E.; Navratil R.; Janeba Z.; Roithova J.; Baszczynski O. (2019) Reactive cyclic intermediates in the ProTide prodrugs activation: trapping the elusive pentavalent phosphorane. Org. Biomol. Chem. 17, 315–320. 10.1039/C8OB02870B. [DOI] [PubMed] [Google Scholar]
- Derudas M.; Carta D.; Brancale A.; Vanpouille C.; Lisco A.; Margolis L.; Balzarini J.; McGuigan C. (2009) The application of phosphoramidate protide technology to acyclovir confers anti-HIV inhibition. J. Med. Chem. 52, 5520–5530. 10.1021/jm9007856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Fleming C. D.; Nishi K.; Redinbo M. R.; Hammock B. D. (2005) Stereoselective hydrolysis of pyrethroid-like fluorescent substrates by human and other mammalian liver carboxylesterases. Chem. Res. Toxicol. 18, 1371–1377. 10.1021/tx050072+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia M. J.; Bao D.; Chang W.; Du J.; Nagarathnam D.; Rachakonda S.; Reddy P. G.; Ross B. S.; Wang P.; Zhang H. R.; Bansal S.; Espiritu C.; Keilman M.; Lam A. M.; Steuer H. M.; Niu C.; Otto M. J.; Furman P. A. (2010) Discovery of a beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53, 7202–7218. 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- Lee W. A.; He G. X.; Eisenberg E.; Cihlar T.; Swaminathan S.; Mulato A.; Cundy K. C. (2005) Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob. Agents Chemother. 49, 1898–1906. 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandre F. R.; Badaroux E.; Bilello J. P.; Bot S.; Bouisset T.; Brandt G.; Cappelle S.; Chapron C.; Chaves D.; Convard T.; Counor C.; Da Costa D.; Dukhan D.; Gay M.; Gosselin G.; Griffon J. F.; Gupta K.; Hernandez-Santiago B.; La Colla M.; Lioure M. P.; Milhau J.; Paparin J. L.; Peyronnet J.; Parsy C.; Pierra Rouviere C.; Rahali H.; Rahali R.; Salanson A.; Seifer M.; Serra I.; Standring D.; Surleraux D.; Dousson C. B. (2017) The discovery of IDX21437: Design, synthesis and antiviral evaluation of 2′-alpha-chloro-2′-beta-C-methyl branched uridine pronucleotides as potent liver-targeted HCV polymerase inhibitors. Bioorg. Med. Chem. Lett. 27, 4323–4330. 10.1016/j.bmcl.2017.08.029. [DOI] [PubMed] [Google Scholar]
- Wyles D.; Wedemeyer H.; Ben-Ari Z.; Gane E. J.; Hansen J. B.; Jacobson I. M.; Laursen A. L.; Luetkemeyer A.; Nahass R.; Pianko S.; Zeuzem S.; Jumes P.; Huang H.-C.; Butterton J.; Robertson M.; Wahl J.; Barr E.; Joeng H.-K.; Martin E.; Serfaty L. (2017) Grazoprevir, ruzasvir, and uprifosbuvir for hepatitis C virus after NS5A treatment failure. Hepatology 66, 1794–1804. 10.1002/hep.29358. [DOI] [PubMed] [Google Scholar]
- James E.; Pertusati F.; Brancale A.; McGuigan C. (2017) Kinase-independent phosphoramidate S1P1 receptor agonist benzyl ether derivatives. Bioorg. Med. Chem. Lett. 27, 1371–1378. 10.1016/j.bmcl.2017.02.011. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Derudas M.; Gonczy B.; Hinsinger K.; Kandil S.; Pertusati F.; Serpi M.; Snoeck R.; Andrei G.; Balzarini J.; McHugh T. D.; Maitra A.; Akorli E.; Evangelopoulos D.; Bhakta S. (2014) ProTides of N-(3-(5-(2′-deoxyuridine))prop-2-ynyl)octanamide as potential anti-tubercular and anti-viral agents. Bioorg. Med. Chem. 22, 2816–2824. 10.1016/j.bmc.2014.02.056. [DOI] [PubMed] [Google Scholar]
- Osgerby L.; Lai Y. C.; Thornton P. J.; Amalfitano J.; Le Duff C. S.; Jabeen I.; Kadri H.; Miccoli A.; Tucker J. H. R.; Muqit M. M. K.; Mehellou Y. (2017) Kinetin Riboside and Its ProTides Activate the Parkinson’s Disease Associated PTEN-Induced Putative Kinase 1 (PINK1) Independent of Mitochondrial Depolarization. J. Med. Chem. 60, 3518–3524. 10.1021/acs.jmedchem.6b01897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruda G. F.; Alibu V. P.; Mitsos C.; Bidet O.; Kaiser M.; Brun R.; Barrett M. P.; Gilbert I. H. (2007) Synthesis and biological evaluation of phosphate prodrugs of 4-phospho-D-erythronohydroxamic acid, an inhibitor of 6-phosphogluconate dehydrogenase. ChemMedChem 2, 1169–1180. 10.1002/cmdc.200700040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpi M.; Bibbo R.; Rat S.; Roberts H.; Hughes C.; Caterson B.; Alcaraz M. J.; Gibert A. T.; Verson C. R.; McGuigan C. (2012) Novel phosphoramidate prodrugs of N-acetyl-(D)-glucosamine with antidegenerative activity on bovine and human cartilage explants. J. Med. Chem. 55, 4629–4639. 10.1021/jm300074y. [DOI] [PubMed] [Google Scholar]
- Vanboom J. H.; Burgers P. M. J.; Crea R.; Luyten W. C. M. M.; Vink A. B. J.; Reese C. B. (1975) Phosphorylation of Nucleoside Derivatives with Aryl Phosphoramidochloridates. Tetrahedron 31, 2953–2959. 10.1016/0040-4020(75)80318-8. [DOI] [Google Scholar]
- Simmons B.; Liu Z.; Klapars A.; Bellomo A.; Silverman S. M. (2017) Mechanism-Based Solution to the ProTide Synthesis Problem: Selective Access to Sofosbuvir, Acelarin, and INX-08189. Org. Lett. 19, 2218–2221. 10.1021/acs.orglett.7b00469. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Gilles A.; Madela K.; Aljarah M.; Holl S.; Jones S.; Vernachio J.; Hutchins J.; Ames B.; Bryant K. D.; Gorovits E.; Ganguly B.; Hunley D.; Hall A.; Kolykhalov A.; Liu Y.; Muhammad J.; Raja N.; Walters R.; Wang J.; Chamberlain S.; Henson G. (2010) Phosphoramidate ProTides of 2′-C-methylguanosine as highly potent inhibitors of hepatitis C virus. Study of their in vitro and in vivo properties. J. Med. Chem. 53, 4949–4957. 10.1021/jm1003792. [DOI] [PubMed] [Google Scholar]
- DiRocco D. A.; Ji Y.; Sherer E. C.; Klapars A.; Reibarkh M.; Dropinski J.; Mathew R.; Maligres P.; Hyde A. M.; Limanto J.; Brunskill A.; Ruck R. T.; Campeau L. C.; Davies I. W. (2017) A multifunctional catalyst that stereoselectively assembles prodrugs. Science 356, 426–430. 10.1126/science.aam7936. [DOI] [PubMed] [Google Scholar]
- Ross B. S.; Ganapati Reddy P.; Zhang H.-R.; Rachakonda S.; Sofia M. J. (2011) Synthesis of diastereomerically pure nucleotide phosphoramidates. J. Org. Chem. 76, 8311–8319. 10.1021/jo201492m. [DOI] [PubMed] [Google Scholar]
- Bigley A. N.; Desormeaux E.; Xiang D. F.; Bae S. Y.; Harvey S. P.; Raushel F. M. (2019) Overcoming the Challenges of Enzyme Evolution To Adapt Phosphotriesterase for V-Agent Decontamination. Biochemistry 58, 2039–2053. 10.1021/acs.biochem.9b00097. [DOI] [PubMed] [Google Scholar]
- Chen-Goodspeed M.; Sogorb M. A.; Wu F.; Hong S. B.; Raushel F. M. (2001) Structural determinants of the substrate and stereochemical specificity of phosphotriesterase. Biochemistry 40, 1325–1331. 10.1021/bi001548l. [DOI] [PubMed] [Google Scholar]
- Chen-Goodspeed M.; Sogorb M. A.; Wu F.; Raushel F. M. (2001) Enhancement, relaxation, and reversal of the stereoselectivity for phosphotriesterase by rational evolution of active site residues. Biochemistry 40, 1332–1339. 10.1021/bi001549d. [DOI] [PubMed] [Google Scholar]
- Li W. S.; Li Y.; Hill C. M.; Lum K. T.; Raushel F. M. (2002) Enzymatic synthesis of chiral organophosphothioates from prochiral precursors. J. Am. Chem. Soc. 124, 3498–3499. 10.1021/ja017840d. [DOI] [PubMed] [Google Scholar]
- Li Y.; Aubert S. D.; Maes E. G.; Raushel F. M. (2004) Enzymatic resolution of chiral phosphinate esters. J. Am. Chem. Soc. 126, 8888–8889. 10.1021/ja048457m. [DOI] [PubMed] [Google Scholar]
- Li Y.; Aubert S. D.; Raushel F. M. (2003) Operational control of stereoselectivity during the enzymatic hydrolysis of racemic organophosphorus compounds. J. Am. Chem. Soc. 125, 7526–7527. 10.1021/ja035625m. [DOI] [PubMed] [Google Scholar]
- Nowlan C.; Li Y.; Hermann J. C.; Evans T.; Carpenter J.; Ghanem E.; Shoichet B. K.; Raushel F. M. (2006) Resolution of chiral phosphate, phosphonate, and phosphinate esters by an enantioselective enzyme library. J. Am. Chem. Soc. 128, 15892–15902. 10.1021/ja0658618. [DOI] [PubMed] [Google Scholar]
- Tsai P. C.; Bigley A.; Li Y.; Ghanem E.; Cadieux C. L.; Kasten S. A.; Reeves T. E.; Cerasoli D. M.; Raushel F. M. (2010) Stereoselective hydrolysis of organophosphate nerve agents by the bacterial phosphotriesterase. Biochemistry 49, 7978–7987. 10.1021/bi101056m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigley A. N.; Xu C.; Henderson T. J.; Harvey S. P.; Raushel F. M. (2013) Enzymatic neutralization of the chemical warfare agent VX: evolution of phosphotriesterase for phosphorothiolate hydrolysis. J. Am. Chem. Soc. 135, 10426–10432. 10.1021/ja402832z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang D. F.; Bigley A. N.; Ren Z.; Xue H.; Hull K. G.; Romo D.; Raushel F. M. (2015) Interrogation of the Substrate Profile and Catalytic Properties of the Phosphotriesterase from Sphingobium sp. Strain TCM1: An Enzyme Capable of Hydrolyzing Organophosphate Flame Retardants and Plasticizers. Biochemistry 54, 7539–7549. 10.1021/acs.biochem.5b01144. [DOI] [PubMed] [Google Scholar]
- Mee-Hie Cho C.; Mulchandani A.; Chen W. (2006) Functional analysis of organophosphorus hydrolase variants with high degradation activity towards organophosphate pesticides. Protein Eng., Des. Sel. 19, 99–105. 10.1093/protein/gzj007. [DOI] [PubMed] [Google Scholar]
- Roodveldt C.; Tawfik D. S. (2005) Directed evolution of phosphotriesterase from Pseudomonas diminuta for heterologous expression in Escherichia coli results in stabilization of the metal-free state. Protein Eng., Des. Sel. 18, 51–58. 10.1093/protein/gzi005. [DOI] [PubMed] [Google Scholar]
- Bigley A. N.; Narindoshvili T.; Xiang D. F.; Raushel F. M. (2018) Multiple Reaction Products from the Hydrolysis of Chiral and Prochiral Organophosphate Substrates by the Phosphotriesterase from Sphingobium sp. TCM1. Biochemistry 57, 1842–1846. 10.1021/acs.biochem.8b00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.