

Abstract
Fragile X syndrome (FXS) is the leading cause of inherited intellectual disability and a significant genetic contributor of Autism spectrum disorder. In addition to autistic-like phenotypes, individuals with FXS are subject to developing numerous comorbidities, one of the most prevalent being seizures. In the present study, we investigated how a single early-life seizure superimposed on a genetic condition impacts the autistic-like behavioral phenotype of the mouse. We induced status epilepticus (SE) on postnatal day (PD) 10 in Fmr1 wildtype (WT) and knockout (KO) mice. We then tested the mice in a battery of behavioral tests during adulthood (PD90) to examine the long-term impact of an early-life seizure. Our findings replicated prior work that reported a single instance of SE results in behavioral deficits, including increases in repetitive behavior, enhanced hippocampal-dependent learning, and reduced sociability and prepulse inhibition (p < 0.05). We also observed genotypic differences characteristic of the FXS phenotype in Fmr1 KO mice, such as enhanced prepulse inhibition and repetitive behavior, hyperactivity, and reduced startle responses (p < 0.05). Superimposing a seizure on deletion of Fmr1 significantly impacted repetitive behavior in a nose poke task. Specifically, a single early-life seizure increased consecutive nose poking behavior in the task in WT mice (p < 0.05), yet seizures did not exacerbate the elevated stereotypy observed in Fmr1 KO mice (p < 0.05). Overall, these findings help to elucidate how seizures in a critical period of development can impact long-term behavioral manifestations caused by underlying gene mutations in Fmr1. Utilizing double-hit models, such as superimposing seizures on the Fmr1 mutation, can help to enhance our understanding of comorbidities in disease models.
Keywords: Fragile X syndrome, seizures, autism, double-hit, repetitive behavior, prepulse inhibition
1. Introduction
Fragile X syndrome (FXS) is a neurodevelopmental disorder and the most common inherited cause of intellectual disability (Hagerman et al., 2008). FXS results from an expanded trinucleotide mutation in the Fmr1 gene, leading to an absence of the RNA binding protein, Fragile X mental retardation protein (FMRP). Mutations in Fmr1 are the largest genetic contributor to Autism spectrum disorder (ASD), with a comorbidity rate of approximately 2-6% (Hagerman, 2006). As a result, FXS and ASD have similar behavioral phenotypes, including hyperactivity, altered social interactions and communication, and repetitive or stereotypical behaviors (King et al., 2014; Niu et al., 2017). In addition to behavioral impairments, individuals with FXS are susceptible to developing a number of other medical conditions, including gastrointestinal problems, recurrent otitis media, and seizures (Berry-Kravis et al., 2010; Kidd et al., 2014).
Approximately 10-20% of individuals with FXS experience seizures, as well as many have subclinical seizure activity and abnormal electroencephalograph (EEG) recordings without overt seizures (Berry-Kravis, 2002; Berry-Kravis et al., 2010; Hagerman and Stafstrom, 2009; Musumeci et al., 1999). Abnormal EEGs in FXS individuals typically display patterns of paroxysmal discharges, often characteristic of centrotemporal spikes in certain childhood epilepsies, such as benign childhood epilepsy with centrotemporal spikes (BCECTS) (Berry-Kravis et al., 2010; Musumeci et al., 1988; Musumeci et al., 1999). EEG endophenotypes, specifically individuals with an epileptiform phenotype, have indicated a trend demonstrating that the incidence and degree of epileptic EEG is correlated with worsened behavioral symptoms in children with FXS and seizures, specifically with impairments in attention (Cowley et al., 2016). The most common type of seizures observed in FXS are complex focal seizures, and less frequently observed generalized seizures (Berry-Kravis, 2002; Berry-Kravis et al., 2010; Cowley et al., 2016; Musumeci et al., 1999).
The increased frequency of seizures in FXS may be due to elevated neuronal excitability, stemming from reduced translational inhibition typically provided by FMRP and enhanced responses to metabotropic glutamate receptor (mGluR) stimulation (Bear et al., 2004; Bianchi et al., 2009; Contractor et al., 2015; Gross et al., 2011). The neuronal hyperexcitability present in individuals with FXS is paralleled by many similar phenotypes in the Fmr1 knockout (KO) mouse model of the disorder, a commonly used genetic model to study ASD neurobiology (Budimirovic and Kaufmann, 2011; Gibson et al., 2008). For instance, Fmr1 KO mice have an excess of immature dendritic spines, enhanced group 1 mGluR activation, and numerous deficits in neuronal wiring that together results in synaptic abnormalities and hyperexcitability (Berry-Kravis et al., 2010; Brooks-Kayal, 2011; Chuang et al., 2005; Huber et al., 2002). In addition, Fmr1 KO mice have an increased frequency and enhanced susceptibility to experiencing audiogenic seizures (Musumeci et al., 2000; Musumeci et al., 2007). Knockout mice also display EEG phenotypes similar to those observed in humans with FXS, including enhanced EEG gamma (30-80 Hz) power at the resting state and reduced evoked gamma oscillations (Lovelace et al., 2018; Wang et al., 2017; Wen et al., 2019).
Epileptic phenotypes in FXS individuals are most common in childhood, and the presence of spontaneous seizures usually resolve by adolescence (Berry-Kravis et al., 2010). However, the impact of seizures during early developmental periods on the existing FXS behavioral impairments is largely unknown. One study examining FXS populations found that FXS males with comorbid seizures were more likely to develop a co-diagnosis of autism, as well as have increased aggressive behavior and poor overall health compared to FXS males without seizures (Berry-Kravis et al., 2010). Additionally, several studies have found that neonatal seizures in rodent models can lead to behavioral deficits in adulthood, including impairments in sociability, learning and memory, and changes in repetitive behavior (Bernard and Benke, 2015; Bernard et al., 2015; Lugo et al., 2014). There is evidence in rodent models suggesting a link between early-life seizures and the development of autistic-like behavior, however, how mutations in Fmr1 may effect this relationship requires further investigation (Stafstrom and Benke, 2015).
In the present study, we used Fmr1 KO and wild type mice to examine how a single seizure on postnatal day (PD) 10 effects the FXS behavioral phenotype in adulthood at PD90. Specifically, we examine the effects of a kainic acid seizure during the early postnatal period on activity and anxiety levels, sociability, learning and memory, repetitive behavior, and prepulse inhibition. This double-hit model allows us to investigate how seizures in early developmental periods can influence the genetic predisposition of FXS, and potentially exacerbate behavioral impairments observed in the disorder. Seizures can have profound effects on the developing brain that persist, or potentially manifest, in adulthood (Holmes, 2016). Understanding the long-term behavioral impact of early-life seizures in FXS is critical to elucidate the underlying mechanism of this comorbid relationship and could provide novel insights to potential therapeutic interventions for FXS.
2. Methods
2.1. Animals
Subject mice included male Fmr1 knockout (KO) and wild type (WT) mice on a FVB/NJ background strain (Jackson Labs Stock No: 004624). A total of 69 mice were utilized in the study. Mice were bred and group housed at Baylor University in standard laboratory conditions with an ambient temperature of 22°C and a 12-hour light, 12-hour dark diurnal cycle. Food and water were provided to mice ad libitum. Breeding pairs consisted of WT males and Fmr1 heterozygous dams to produce Fmr1 KO and WT male pups. On postnatal day (PD) 10, mice received either seizure or control treatment, followed by having their toes clipped for identification purposes and to be sent to Mouse Genotype for genotyping information (Escondido, CA, USA). All mice were weaned at PD21, and behavioral testing was conducted once mice reached adulthood at PD90. Testing was performed in order from least invasive to most invasive to minimize any test order effects: open field, elevated plus maze, nosepoke assay, social partition, delay fear conditioning, prepulse inhibition (McIlwain et al., 2001) (Fig. 1). Procedures were conducted in compliance with the Baylor University Institutional Animal Care and Use Committee and the National Institute of Health Guidelines for the Care and Use of Laboratory Animals.
Figure 1. Behavioral testing timeline.
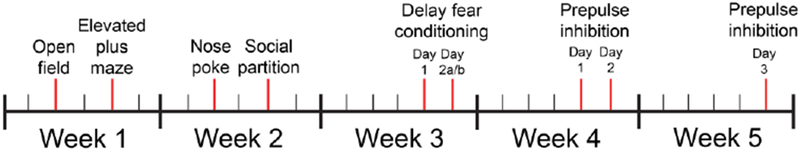
All mice went through the same schedule of behavioral testing across a 5 week time period. Each individual test was conducted at least 48 hours apart to minimize interference between tests. Delay fear conditioning consists of 3 trials, with Day 2a conducted 24 h following Day 1, and Day 2b 2 h following Day 2a. Prepulse inhibition also consisted of 3 days, with habituation on Day 1, prepulse inhibition the following day (Day 2), and startle response a week later (Day 3).
2.2. Seizure induction
On PD10, Fmr1 KO and WT pups were randomly assigned to either the treatment group or control group. Treatment pups were administered intraperitoneal (i.p.) injections of 0.5% kainate (2.0 mg/kg) and control pups were given an equivalent dose of 0.9% physiological saline. Pups from both conditions were then placed into individual containers with clean bedding and warmed on a heating pad to a temperature of ~35°C for the duration of the seizure induction period. Seizure pups entered status epilepticus (SE) within approximately 30-40 min following kainate administration, which was characterized by continuous tonic-clonic seizure activity lasting 1-2 h. The seizure pups were monitored until natural cessation of all seizure activity, along with control pups simultaneously being monitored for an equivalent time. Following recovery from SE, both seizure and control pups were returned to their home cages, weaned at PD21, and housed with mixed genotype littermates until behavioral testing in adulthood at PD90.
2.3. Nosepoke assay
The nosepoke assay was used to evaluate repetitive and exploratory behavior. The testing apparatus consisted of a clear, acrylic chamber (40 cm x 40 cm x 30 cm), with a flat board inserted comprised of 16 equidistant holes of 1” diameter and 0.75” depth. During the testing period, an experimenter live scored the number and placement of nose pokes made for 10 min. A nose poke was considered anytime the nose of a mouse extended into the hole to the level of their eyes. The experimenter was blinded to experimental condition. Following testing, mice were returned to a separate cage until all mice in the home cage were tested, and then placed back into the original home cage with littermates. The apparatus was cleaned with 30% isopropyl alcohol in between testing sessions. An increase in total nose poking behavior or in consecutive nose pokes in the same hole and decreased latency to the first nose poke is indicative of repetitive behavior, a component of the autistic-like phenotype (Moy et al., 2008; Silverman et al., 2010).
2.4. Prepulse inhibition
Sensorimotor gating abilities were evaluated in mice with a prepulse inhibition (PPI) paradigm. The testing chamber consisted of an acrylic hollow constraint tube mounted on a sensor platform, able to detect and transduce startle response amplitude via the SR-Lab System (San Diego Instruments, San Diego, CA, USA). The PPI paradigm consisted of 3 testing days, with background noise levels maintained at 68 dB. On day 1, mice habituated to the testing apparatus for 5 min, followed by the presentation of 80 startle stimuli every 15 s. The startle stimulus was a 40 ms, 120 dB noise, with a rise/fall time < 1 ms. The following day (testing day 2), PPI was examined. Mice habituated to the testing apparatus for 5 min, and then were presented with a 90-trial prepulse phase. The 1st three trial types were weak, 20 ms prepulse stimuli (rise/fall time < 1 ms) at intensities of 70, 75, and 80 dB. The 2nd three trial types consisted of the same three prepulse stimuli, paired with the original startle stimulus (40 ms, 120 dB) 100 ms after the prepulse. The 90 trials were randomly organized, and all spaced by a 15 s inter-trial interval. A week after PPI testing, the startle threshold of mice was examined (testing day 3). Mice habituated to the testing apparatus for 5 min, followed by the presentation of 99 trials, consisting of 11 trial types (no stimulus, 75 dB, 80 dB, 85 dB, 90 dB, 95 dB, 100 dB, 105 dB, 110 dB, 115 dB, 120 dB) 10 startle stimuli ranging from 75 – 120 dB at 5 dB intervals). The startle stimuli were 40 ms noise bursts (rise/fall time < 1 ms) and were spaced by a 15 s inter-trial interval. The 11 trial types were presented in a pseudorandom order, with each type presented once within a block of 11 trials. Startle threshold is defined as the minimal intensity in which the response is significantly greater than the response during no stimulus trials (Frankland et al., 2004).
2.5. Social partition
The social partition test was used to evaluate the social behavior of mice, specifically their preference for interacting with a familiar versus an unfamiliar mouse. The mice were housed overnight for 24 h in a cage separated into two chambers by a clear partition with 0.6 cm diameter holes. On the opposite side of the partition, a conspecific matched for age and weight remained in the cage overnight with the test mouse. The following day, an experimenter lived scored the frequency of visits to the partition and the duration of time spent at the partition. Mice were tested in the same cage they were housed in overnight. Each test session consisted of 3 conditions, each 5 min in duration. In the first condition, the “familiar” mouse that the test mouse was housed with overnight was in the opposite chamber, the 2nd condition consisted of an “unfamiliar” mouse in the opposite chamber, and the 3rd condition was the same “familiar” mouse that the test mouse was housed with the previous night. Mice remained in their testing cages until all mice were tested, and then placed back into their original home cages with littermates. Fewer visits to the partition and reduced time spent at the partition is indicative of social deficits, a component of the autistic-like phenotype (Silverman et al., 2010).
2.6. Delay fear conditioning
Mice were tested in delay fear conditioning to examine learning and memory abilities in the mice. The testing apparatus consisted of an operant conditioning chamber (26 cm x 22 cm x 18 cm), comprised of two clear acrylic sides and two metal sides, with metal grid flooring able to deliver a mild shock. The operant conditioning chamber was placed inside an additional sound attenuating chamber. For all trials, freezing behavior was measured with FreezeFrame 3 automated detection software (Coulbourn, Ohio). Testing consisted of 3 trials, across 2 separate days. During the 1st trial on day 1, mice received two pairings of a 30 s, 80 decibel (dB) white noise (conditioned stimulus [CS]) followed immediately by a 2 s 0.7 mA shock stimulus (unconditioned stimulus [US]). There was a 20 s interval in between the tone-shock pairings, with the entire 1st trial lasting 334 s. On day 2 of testing there were 2 separate trials. On the 1st trial of day 2, mice were placed back into the original context and freezing was measured while the mice freely explored for 300 s. Two hours following the original context trial, mice were placed back in the apparatus for the 2nd trial of day 2 in a novel context. The apparatus was altered by changing the shape and flooring of the chamber, placing a vanilla-scented odor under the flooring, changing transfer cage bedding to shredded paper towels, and cleaning the apparatus with 70% ethanol instead of 30% isopropanol in between testing sessions. Mice were placed in the novel context for 360 s, with the mouse acclimating to the novel environment in the first 180 s, and during the 2nd 180 s, mice were presented the CS (80 dB tone) continuously while freezing behavior was measured. Following testing, mice were returned to a separate cage until all mice in the home cage were tested, and then placed back into the original home cage with littermates. Increased freezing in response to the context or cue is an innate fear response and indicative of the learned association between the CS and US (Wehner and Radcliffe, 2004).
2.7. Open field
The open field test was conducted to evaluate changes in activity and anxiety levels, as well as repetitive behavior. The testing apparatus consisted of an acrylic chamber (40 cm x 40 cm x 30 cm) in an isolated room controlled for light levels, temperature, and background noise. During the 30 min testing period, mice were individually placed into the apparatus and locomotor activity was measured with automatic optical animal detection software (Fusion by Omnitech Electronics, Inc., Columbus, OH). In addition to locomotion or total distance moved, clockwise revolutions, a measure of repetitive behavior, was quantified. Anxiety behaviors were also examined by computing the distance and time spent in the center of the apparatus (inner 50%, 20 cm by 20 cm) compared to the surround region. Following testing, mice were returned to a separate cage until all mice in the home cage were tested, and then placed back into the original home cage with littermates. The apparatus was cleaned with 30% isopropyl alcohol in between testing sessions.
2.8. Elevated plus maze
Mice were tested in the elevated plus maze to examine changes in baseline anxiety levels. The plus maze apparatus consisted of 4 arms (all 30 cm x 5 cm) positioned 40 cm above the floor, with a center platform (5 cm x 5 cm), and 2 arms enclosed with acrylic walls. The plus maze was in an isolated room controlled for light levels, temperature, and background noise. The testing period was 10 min, in which Ethovision XT video tracking software (Noldus, Netherlands) quantified the frequency of entries and duration of time spent in each of the arms. Following testing, mice were returned to a separate cage until all mice in the home cage were tested, and then placed back into the original home cage with littermates. The apparatus was cleaned with 30% isopropyl alcohol in between testing sessions. Increased time spent in the closed arms of the maze compared to the open arms is indicative of increased anxiety (Walf and Frye, 2007).
2.9. Statistical analysis
Data were analyzed using either GraphPad Prism 7 software (San Diego, CA, USA) or SPSS 21.0 (IBM, USA). Two-way analysis of variance (ANOVA) (genotype [wild type, knockout] x treatment [seizure, control]) were utilized to evaluate results of each behavioral task. The assumptions for an ANOVA were met for all statistical analyses, including independent cases, normality, and homogeneity of the variances. Specifics on each analysis can be found in the respective result section. Significant interactions were followed by separate group analyses, with mice split into four unique groups: WT/control, KO/control, WT/seizure, KO/seizure. Any interactions for group analyses were examined with Fisher’s LSD post-hoc multiple comparison tests. A statistical interaction between genotype (wild type, knockout) and treatment (seizure, control) would support our hypothesis, in that the double-hit of an early-life seizure significantly impacted the behavioral phenotype caused by gene mutations in Fmr1. The level of significance was set at p < 0.05 for all comparisons. Data are expressed as the mean ± standard error of the mean (SEM).
3. Results
3.1. Seizure induction
A total of 69 mice were utilized for seizure induction and went through behavioral testing. Of the 20 Fmr1 wild type (WT) mice injected with kainic acid, 5 mice (25%) did not go into status epilepticus (SE) and were removed from all analyses. Of the 20 Fmr1 knockout (KO) mice injected with kainic acid, 7 (35%) did not go into SE and were removed from analyses. A Pearson Chi-Square revealed no significant difference in the proportion of mice from each genotype that underwent SE (X2[1, N = 40] = 0.49, p = 0.73). Only mice that entered full SE were included in behavioral analyses to ensure mice were in the same category of seizure severity. Of the mice that underwent SE, there was no significant difference in latency to SE between WT (40.16 ± 3.00 min) and Fmr1 KO mice (31.75 ± 3.80 min), t(26) = 1.76, p = 0.09. The final sample sizes for all behavioral tests unless noted otherwise were as follows: Fmr1 WT/control – 21, Fmr1 KO/control – 8, Fmr1 WT/seizure – 15, Fmr1 KO/seizure – 13 (total of 57 mice in analyses).
3.2. Nosepoke assay
The nosepoke assay was used to evaluate changes in repetitive behavior. For latency to first nose poke, a two-way ANOVA detected no main effect of treatment, F(1, 53) = 0.26, p = 0.61, or genotype, F(1, 53) = 0.23, p = 0.64. However, there was an interaction of treatment and genotype, F(1, 53) = 8.64, p < 0.05. Given the significant interaction, animals were subdivided into four groups for post-hoc analysis: Fmr1 WT/Control, Fmr1 KO/Control, Fmr1 WT/Seizure, Fmr1 KO/Seizure. Fisher’s LSD multiple comparisons revealed that early-life seizures reduced the latency to first nose poke in Fmr1 WT mice (p < 0.05), but had the opposite effect in Fmr1 KO mice and a seizure increased the latency to first nose poke in KO mice (p < 0.05). In addition, Fmr1 KO/Control mice had decreased latency to first nose poke compared to Fmr1 WT/Control mice (p < 0.05) (Fig. 2A).
Figure 2. Nosepoke assay.
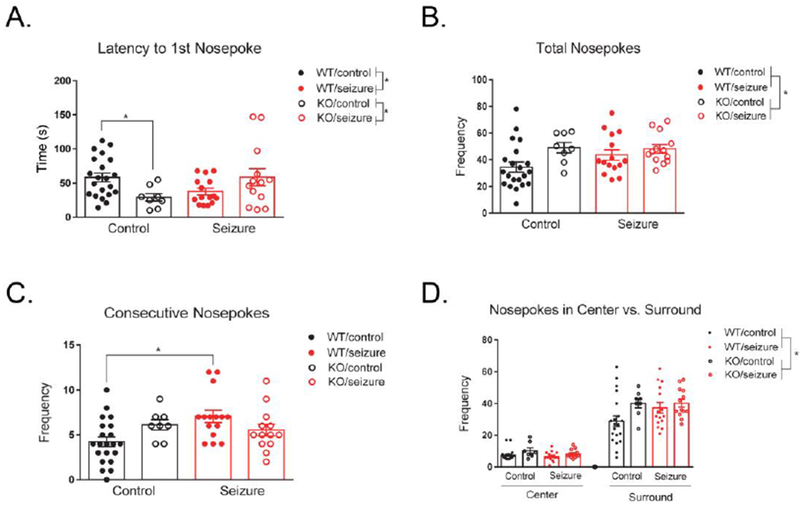
There was an interaction between treatment and genotype for the latency to first nosepoke. In wild type (WT) mice, a seizure significantly reduced the latency to first nose poke, while in Fmr1 knockout (KO) mice a seizure increased the latency. The latency to first nose poke was also significantly decreased in Fmr1 KO mice compared to WT mice (A). Fmr1 KO mice exhibited an increased number of total nosepokes compared to WT mice, with no effect of treatment (B). A seizure in WT mice significantly increased their tendency to return to the same hole consecutively, with no effect in Fmr1 KO mice (C). When comparing nosepokes in the center versus surround regions of the chamber, there was no effect of treatment or genotype (D). Fmr1 KO mice had an overall increased number of nosepokes compared to WT mice, not dependent on the chamber region (D). Data are expressed as mean ± standard error of the mean (SEM), * p < .05.
We next analyzed the total number of nose pokes exhibited by each group. A two-way ANOVA revealed no significant effect of treatment, F(1, 53) = 0.97, p = 0.33. There was a significant effect of genotype, F(1, 53) = 5.34, p < 0.05, with Fmr1 KO mice exhibiting more nose poking behavior than Fmr1 WT mice, indicative of increased repetitive behavior (Fig. 2B). There was no interaction of treatment and genotype for total number of nosepokes, F(1, 53) = 1.46, p = 0.23. We also examined the number of times mice returned to the same hole consecutively. No main effect was detected for treatment, F(1, 53) = 2.71, p = 0.12, or genotype, F(1, 53) = 0.07, p = 0.79. However, there was an interaction between treatment and genotype for consecutive nose pokes, F(1, 53) = 6.29, p < 0.05. Given the significant interaction, animals were subdivided into four groups for post-hoc analysis: Fmr1 WT/Control, Fmr1 KO/Control, Fmr1 WT/Seizure, Fmr1 KO/Seizure. Fisher’s LSD multiple comparisons showed that seizures in WT mice increased their tendency to return to the same hole consecutively (p < 0.01), as well as there was a trending increase in Fmr1 KO/Control mice compared to Fmr1 WT/Control mice (p = 0.07) (Fig. 2C).
In addition to repetitive behavior, we measured anxiety in the nosepoke assay by examining nose poking behavior in the center versus surround regions of the testing apparatus. A two-way repeated measures ANOVA with a within-subjects variable of “location” revealed a significant effect of location F(1, 53) = 276.21, p < 0.001. Location did not interact with treatment, F(1, 53) = 2.70, p = 0.11, or genotype, F(1, 53) = 1.99, p = 0.16. There was also no three-way interaction between treatment, genotype, and location, F(1, 53) = 1.05, p = 0.31. Between-subjects effects indicated a main effect of genotype, F(1, 53) = 5.83, p< 0.05, with Fmr1 KO mice poking more holes than Fmr1 WT mice (Fig. 2D). However, there was no between-subjects effect for treatment, F(1, 53) = 0.57, p = 0.45, or an interaction between treatment and genotype, F(1, 53) = 1.57, p = 0.22.
3.3. Prepulse inhibition
Habituation to startle stimuli was examined on Day 1 of testing. A two-way repeated measures ANOVA, with a within-subjects variable of “time” (80 startle stimuli condensed into 8 time bins with 10 trials per bin), revealed no significant effect of time on treatment F(7, 371) = 0.59, p = 0.77, or genotype, F(7, 371) = 1.17, p = 0.32. There was also no interaction between time, treatment, and genotype, F(7, 371) = 0.92, p = 0.49. However, between-subjects effects revealed a significant effect of genotype, F(1, 53) = 25.96, p < 0.001, with Fmr1 KO mice showing reduced responding to the startle stimulus compared to WT mice. There was no between-subjects effects of treatment F(1, 53) = 2.27, p = 0.14, or an interaction between genotype and treatment, F(1, 53) = 0.19, p = 0.66 (Fig. 3A).
Figure 3. Prepulse inhibition.
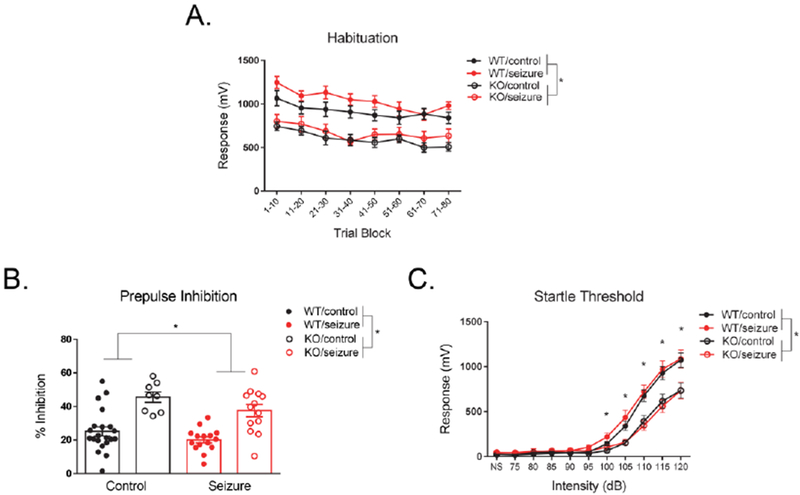
Fmr1 knockout (KO) mice had significantly enhanced prepulse inhibition compared to wild type (WT) mice, as well as seizure mice had significantly reduced inhibition compared to control mice (A). Fmr1 KO mice had significantly reduced startle responses across all decibel (dB) levels compared to WT mice, as well as specifically at higher dB levels (100, 105, 110, 115, 120) (B). Data are expressed as mean ± standard error of the mean (SEM), * p < .05.
A two-way ANOVA was utilized to examine changes in total prepulse inhibition on the 2nd day of testing. There was a main effect of genotype, F(1, 53) = 36.37, p < 0.001, with Fmr1 KO mice showing exaggerated levels of prepulse inhibition (Fig. 3B). There was also a main effect of treatment, F(1, 53) = 4.22, p < 0.05, with seizure mice showing reduced prepulse inhibition compared to control mice (Fig. 3B). The interaction between treatment and genotype was not significant, F(1, 53) = 0.24, p = 0.63.
One week following PPI testing, startle threshold was assessed. A two-way repeated measures ANOVA with a within-subjects factor of “dB” (11 dB levels) was utilized. There was a significant interaction between the startling stimulus and genotype, F(10, 530) = 12.43, p < 0.001. However, decibel level did not interact with treatment, F(10, 530) = 0.19, p = 1.00, nor was there a three-way interaction between decibel, treatment, and genotype, F(10, 530) = 0,20, p = 1.00. When further analyzing the significant interaction between decibel and genotype, an ANOVA revealed that Fmr1 KO mice had reduced startle responses at higher stimulus levels compared to WT mice (dB levels: 100, 105, 110, 115, 120) (p < 0.05) (Fig. 3C). Between-subjects effects indicated a main effect of genotype, F(1, 53) = 19.08, p < 0.001, with Fmr1 KO mice demonstrating overall reduced startle responding (Fig. 3C). There was no main effect of treatment, F(1, 53) = 0.60, p = 0.44, or an interaction between treatment and genotype, F(1, 53) = 0.37, p = 0.55.
3.4. Social partition
To measure sociability in the social partition task, a two-way repeated measures ANOVA was utilized. A within-subjects variable of “trial” was used to examine the frequency of visits to the partition across the three trials of the task (1: familiar mouse, 2: unfamiliar mouse, 3: familiar mouse). There was a significant within-subjects effect of trial, F(2, 106) = 25.74, p < 0.001. Trial did not interact with treatment, F(2, 106) = 2.50, p = 0.09, or genotype, F(2, 106) = 1.09, p = 0.34, nor was there an interaction between treatment, genotype, and trial, F(2, 106) = 0.71, p = 0.49. Between-subjects effects indicated a main effect of treatment, F(1, 53) = 7.62, p < 0.05, with seizure mice visiting the partition a significantly decreased number of times compared to control mice (Fig. 4A). No between-subjects effects were detected for genotype, F(1, 53) = 1.24, p = 0.27, or an interaction between treatment and genotype, F(1, 53) = 0.08, p = 0.78.
Figure 4. Social partition task.
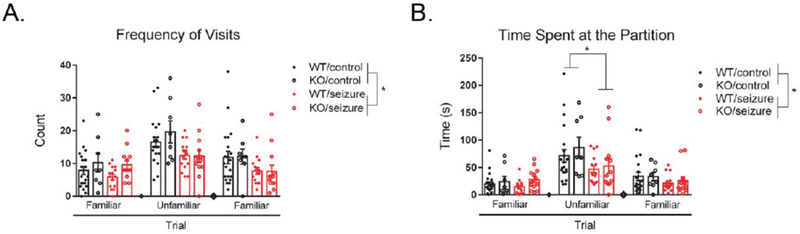
Seizure mice visited the partition significantly less than control mice across all trials, with no effect of genotype (A). Seizure mice also spent significantly less time interacting at the partition compared to control mice across all trials, as well as specifically during trial 2 in which the unfamiliar mouse was present (B). Data are expressed as mean ± standard error of the mean (SEM), * p < .05.
We also examined the duration of time mice spent at the partition for each of the three trials. Similarly, we used a two-way repeated measures ANOVA with a within factor of “trial.” There was a significant within-subjects effect of trial, F(2, 106) = 35.15, p < 0.001. We found a significant interaction between treatment and trial, F(2, 106) = 3.48, p < 0.05. However, there was no interaction between genotype and trial, F(2, 106) = 0.33, p = 0.72, or a three-way interaction between treatment, genotype, and trial, F(2, 106) = 0.38, p = 0.68. Further analyses to investigate the interaction between treatment and trial revealed that seizure mice spent significantly less time interacting at the partition only during the 2nd trial (unfamiliar mouse) compared to control mice (p < 0.05). Between-subjects effects revealed a main effect for treatment, F(1, 53) = 3.91, p < 0.05, with seizure mice spending significantly less time interacting at the partition (Fig. 4B). There was no significant effect of genotype, F(1, 53) = 1.16, p = 0.29, or an interaction between treatment and genotype, F(1, 53) = 0.02, p = 0.88.
3.5. Delay fear conditioning
During the training phase (Day 1) of delay fear conditioning, mice were presented with two pairings of a conditioned stimulus (CS) and unconditioned stimulus (US). Results were analyzed with a two-way repeated measures ANOVA, with the within-subjects variable defined as “time” (baseline, tone 1, intertrial interval 1, tone 2 and intertrial interval 2). There was a significant interaction between time and treatment, F(4, 212) = 4.60, p < 0.01, as well as a significant interaction between time and genotype, F(4, 212) = 2.60, p < 0.05. The three-way interaction between treatment, genotype, and time was not significant, F(4, 212) = 0.94, p = 0.44. An ANOVA was used to further analyze the significant interaction of time and treatment, and detected that seizure mice froze significantly more than control mice during the first intertrial interval (p < 0.05) and during the presentation of the 2nd tone (p < 0.01) (Fig. 5A). When further analyzing the interaction of time and genotype, no discernable patterns were revealed. Between-subjects effects indicated a main effect of treatment, F(1, 53) = 6.91, p < 0.05, with seizure mice freezing significantly more than control mice (Fig. 5A). There was no significant between-subjects effects for genotype, F(1, 53) = 1.38, p = 0.25, or an interaction between treatment and genotype, F(1, 53) = 0.07, p = 0.80.
Figure 5. Delay fear conditioning.
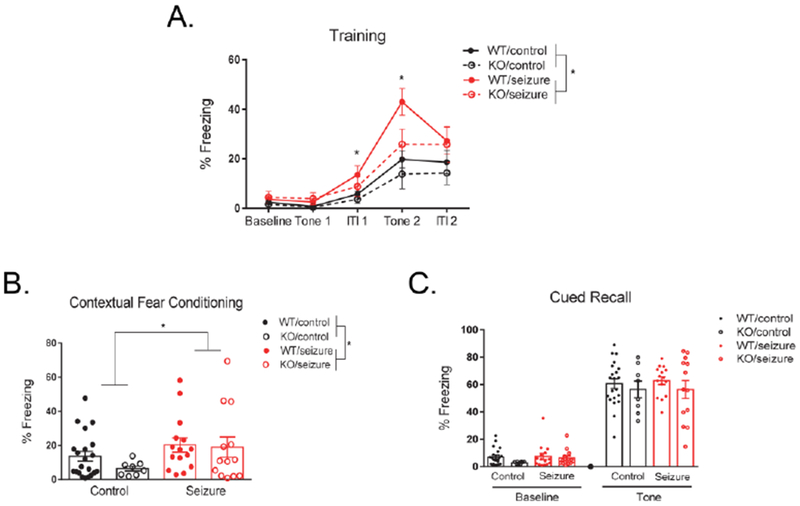
During training, seizure mice froze significantly more than control mice across the entire period, as well as specifically during the first intertrial interval (ITI1) and during the 2nd presentation of the tone (Tone 2) (A). On part 1 of testing on day 2 (contextual fear conditioning), seizure mice froze significantly more than control mice, with no effect of genotype (B). There were no effects of treatment or genotype on the 2nd part of testing on day 2 (cued recall) (C). Data are expressed as mean ± standard error of the mean (SEM), * p < .05.
For the first part of testing on Day 2 (contextual fear conditioning), mice were placed back into the original context from the previous day and freezing behavior was evaluated for 5 minutes. A two-way ANOVA detected a significant effect of treatment, F(1, 53) = 4.84, p < 0.05, with seizure mice freezing significantly more than control mice (Fig. 5B). No significant main effect was detected for genotype, F(1, 53) = 0.66, p = 0.42, or an interaction between treatment and genotype for total amount of time spent freezing, F(1, 53) = 0.61, p = 0.42.
Two hours later during the second part of testing on Day 2 (cued recall), mice were placed in a novel context and freezing behavior in response to the CS (tone) and at baseline was evaluated. A two-way repeated measures ANOVA was used with a within-subjects variables of “time,” consisting of two levels examining percent freezing during baseline and during the tone presentation. Time did not significantly interact with treatment, F(1, 53) = 0.04, p = 0.84, or genotype, F(1, 53) = 0.31, p = 0.58. There was no significant interaction between treatment, genotype, and time, F(1, 53) = 0.33, p = 0.57. Between-subjects effects also indicated no significant effects for treatment, F(1, 53) = 0.29, p = 0.59, genotype, F(1, 53) = 2.08, p = 0.16, or an interaction between treatment and genotype, F(1, 53) = 0.01, p = 0.94 (Fig. 5C).
3.6. Open field
The open field test was utilized to examine changes in activity and exploratory behavior. A two-way ANOVA revealed no significant main effect of treatment in activity levels, F(1, 53) = 0.31, p = 0.58. However, there was a significant effect of genotype, F(1, 53) = 6.26, p < 0.05, with Fmr1 KO mice showing increased total distance moved (Fig. 6A). There was no interaction of treatment and genotype, F(1, 53) = 0.05, p = 0.83. We also measured clockwise revolutions as a parameter of repetitive behavior in the open field test and found that seizures did not affect the number of revolutions, F(1, 53) = 0.25, p = 0.62. However, there was a significant genotype effect, F(1, 53) = 5.38, p < 0.05, with Fmr1 KO mice exhibiting an increased number of revolutions (Fig. 6B). There was no interaction between treatment and genotype for clockwise revolutions, F(1, 53) = 0.11, p = 0.74.
Figure 6. Open field.
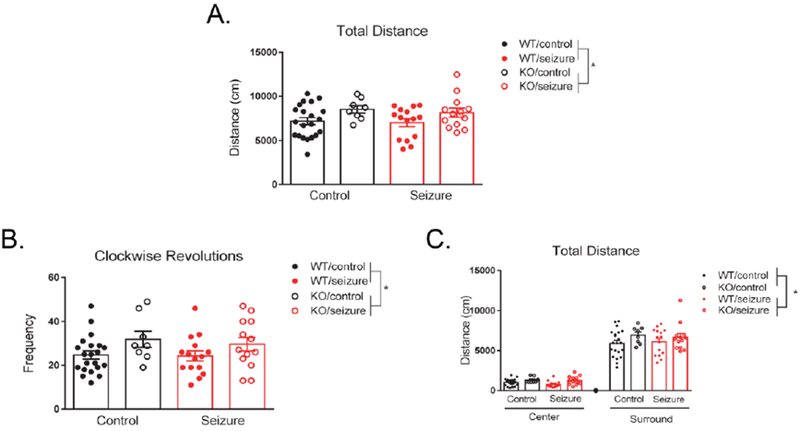
Fmr1 knockout (KO) mice exhibit hyperactivity in the open field compared to wild type (WT) mice, with no effect of seizures on activity levels (A). Fmr1 KO mice displayed increased clockwise revolutions compared to WT mice, with no effect of treatment (B). There was no effect of treatment or genotype in whether mice preferred the center versus surround area of the testing chamber, indicating no differences in anxiety levels in the open field (C). Fmr1 KO mice had increased activity levels compared to WT mice, not dependent on the region of the chamber (C). Data are expressed as mean ± standard error of the mean (SEM), * p < .05.
We next examined anxiety in the open field by measuring total distance moved in the center versus surround region of the testing apparatus. Using a within-subjects variable of “location” to measure activity in the center versus surround regions, there was a significant effect of location, F(1, 53) = 575.43, p < 0.001. However, location did not interact with treatment, F(1, 53) = 0.06, p = 0.81, or genotype, F(1, 53) = 0.79, p = 0.38. The three-way interaction between location, treatment, and genotype, F(1, 53) = 0.62, p = 0.44, was also not significant. Between-subjects effects indicated a main effect of genotype, F(1, 53) = 5.80, p < 0.05, with Fmr1 KO mice having significantly increased total distance moved (Fig. 6C). There was no between-subjects main effect for treatment, F(1, 53) = 0.11, p = 0.68, or an interaction between treatment and genotype, F(1, 53) = 0.07, p = 0.79.
3.7. Elevated plus maze
Anxiety behavior was examined using the elevated plus maze. One Fmr1 WT/control mouse was removed from analyses due to falling off the maze during testing. To analyze total time spent in the arms of the maze, we used a two-way repeated measures ANOVA with a within-subjects variable of “location” to designate open versus closed arms. For total time spent in the arms, there was a significant within-subjects effect of location, F(1, 52) = 99.29, p < 0.001. Location did not interact with treatment, F(1, 52) = 0.05, p = 0.83, or genotype, F(1, 52) = 1.57, p = 0.22. The three-way interaction between location, treatment, and genotype, F(1, 52) = 1.05, p = 0.31, was also not significant. Between-subjects effects indicated no effect of treatment, F(1, 52) = 0.01, p = 0.93, genotype, F(1, 52) = 0.28, p = 0.60, or an interaction between treatment and genotype, F(1, 52) = 0.75, p = 0.39 (Fig. 7A).
Figure 7. Elevated plus maze.
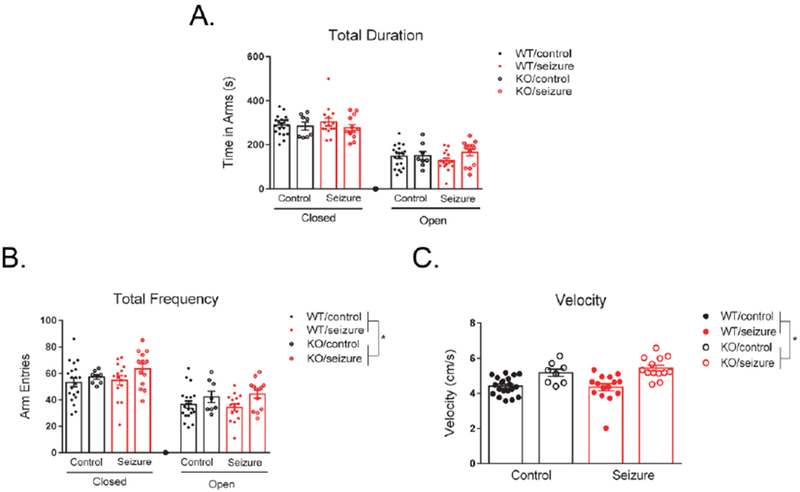
There was no effect of treatment or genotype in total time spent in the closed versus open arms of the maze, indicating no differences in anxiety levels (A). Fmr1 KO mice had an overall increased number of arm entries in the maze compared to WT mice, not dependent on the arms being closed or open (B). Fmr1 knockout (KO) mice had increased velocity in the elevated plus maze compared to wild type (WT) mice, with no effect of treatment on velocity (C). Data are expressed as mean ± standard error of the mean (SEM), * p < .05.
We also measured the total number of visits to the open versus closed arms in the maze. Using the same within-subjects variable of “location,” we found there to be a significant within-subjects location effect, F(1, 52) = 41.51, p < 0.001. However, location did not interact with treatment, F(1, 52) = 0.51, p = 0.48, or genotype, F(1, 52) = 0.07, p = 0.79, for frequency of visits. There was also no three-way interaction between location, treatment, and genotype, F(1, 52) = 0.001, p = 0.97. Between-subjects effects indicated a significant main effect of genotype, F(1, 52) = 11.37, p < 0.01, with Fmr1 KO mice visiting the arms an increased number of times compared to WT mice (Fig. 7B). There was no significant between-subjects main effect for treatment, F(1, 52) = 0.86, p = 0.36, or an interaction between treatment and genotype for frequency of visits, F(1, 52) = 1.12, p = 0.29.
We also measured velocity in EPM, with a two-way ANOVA revealing a significant main effect for genotype, F(1, 52) = 27.06, p < 0.001, with Fmr1 KO mice exhibiting increased velocity in the maze (Fig. 7C). There was no significant main effect of treatment, F(1, 52) = 0.33, p = 0.57, or an interaction between treatment and genotype, F(1, 52) = 0.93, p = 0.34, for velocity.
4. Discussion
There is considerable evidence for how early-life seizures, and separately mutations in Fmr1, can result in aberrant behavioral phenotypes (Bernard and Benke, 2015; Gross et al., 2015; Lugo et al., 2014). However, how seizures in early developmental periods may exacerbate or impact the behavioral manifestations of an Fmr1 mutation and impact FXS pathophysiology is less understood. Utilizing the Fmr1 knockout (KO) mouse model, we observed several long-term behavioral changes following a single kainic acid seizure on postnatal day (PD) 10. For example, seizures had differential effects in Fmr1 KO and wild type (WT) mice, with seizures inducing repetitive behavior in WT mice, but not enhancing the stereotypy observed in Fmr1 KO mice. In addition, early-life seizures themselves resulted in long-term changes, such as a reduction in social interaction with an unfamiliar mouse and enhanced freezing in a contextual fear conditioning task. Many alterations commonly associated with the FXS phenotype were also observed in the Fmr1 KO mouse, such as hyperactivity, increased repetitive or stereotypical behavior, enhanced prepulse inhibition (PPI), and reduced startle responses (Kazdoba et al., 2014; Spencer et al., 2011).
Stereotypical or repetitive behavior is a core component of the autistic-like phenotype and has been observed in both individuals with FXS and the Fmr1 KO mouse (DeFilippis and Wagner, 2016; Kazdoba et al., 2014; Oakes et al., 2016). In humans, repetitive behavior in FXS is characterized by repetition and rigidity of unvarying movements, ritualistic behaviors, restricted interests, and perseverative speech (Niu et al., 2017; Oakes et al., 2016). Our findings from the nosepoke assay are in line with other rodent studies, as Fmr1 KO mice poked significantly more holes across multiple measures when compared to WT mice (Bhattacharya et al., 2012; Dolan et al., 2013). Interestingly, a single PD10 seizure in WT mice induced similar stereotypical behavior in the task, at levels comparable to Fmr1 KO mice. A single seizure not only reduced the latency to the first nose poke, but significantly enhanced the tendency of mice to return to the same hole during the task. This decrease in latency exhibited by WT seizure mice is not due to hyperactivity, as there was no effect of treatment on activity levels.
Evidence has shown that seizures early in life can result in long-term alterations in cognition and behavior, as well as impairments characteristic of the autistic-like phenotype (Bernard and Benke, 2015; Holmes, 2016; Nickels et al., 2016; Sayin et al., 2004). However, there are inconsistent findings in regard to whether stereotyped behaviors are elevated following early-life seizures in rodent models. For example, pilocarpine seizures in PD9 rats resulted in increased self-grooming in an anxiogenic environment in adulthood, a measure indicative of increased stereotypy (Castelhano et al., 2013). In contrast, rats given a single kainic acid seizure on PD7 exhibited reduced marble burying, suggestive of restricted interests, as well as reduced grooming in adult seizure rats (Bernard et al., 2015). In addition, early-life flurothyl seizures from PD7-11 in C57BL/6 mice resulted in no change in nosepoke or marble burying in adulthood (Lugo et al., 2014). These variable results could be due to the differences in the animal strain and seizure model, as well as the developmental timeline of seizure induction. Future studies should explore the association between early-life seizures and stereotypical behavior to further elucidate whether this could be a long-term behavioral phenotype resultant from early-life seizures.
Another consistent phenotype of the Fmr1 KO mouse that was observed in the present study was enhanced PPI and reduced startle responses (Frankland et al., 2004; Spencer et al., 2011; Thomas et al., 2011; Veeraragavan et al., 2012). Prepulse inhibition is a measure of sensorimotor gating which examines the attenuation of a subjects startle reflex when a startle stimulus (the pulse) is preceded by a lower-intensity sensory stimulus (the prepulse) (Aguilar et al., 2018; Matsuo et al., 2018). While enhanced PPI has been found in the Fmr1 KO mouse, the opposite is often exhibited by humans with FXS, who display reductions in PPI (Chen and Toth, 2001; Frankland et al., 2004; Hessl et al., 2009; Yuhas et al., 2011). Interestingly, the reduction in PPI in FXS individuals parallels what we found following seizures in our rodent model. While seizures did not impact the phenotype in Fmr1 KO mice, a single early-life seizure resulted in reduced PPI in adulthood in WT animals. The reduction in PPI following seizures supports prior evidence, and has been suggested to be indicative of the development of schizophrenia-like phenotypes in animal models (Aguilar et al., 2018; Labbate et al., 2014; Wolf et al., 2016). Early-life seizures have not only been associated with reduced PPI in adulthood, but evidence has shown as the number of spontaneous seizures in rats increases, PPI efficacy declines (Labbate et al., 2014; Wolf et al., 2016). Specifically, 22 days following pilocarpine-induced status epilepticus in adult rats, the total number of spontaneous recurrent seizures in the chronic phase of epilepsy was negatively correlated with PPI performance (Wolf et al., 2016). In humans, temporal lobe seizures are capable of inducing psychosis with schizophrenic-like symptoms, including deficits in PPI which is often a biomarker of schizophrenia (Howland et al., 2007; Ma and Leung, 2016; McKenna et al., 1985; Mena et al., 2016; Slater and Beard, 1963; Takeda et al., 2001). Individuals with non-epileptic seizures have also been shown to have reduced PPI, with deficits being greater in unmedicated individuals (Pouretemad et al., 1998).
Long-term alterations in PPI efficacy following an early-life seizure have been associated with a bilateral decrease in dorsal hippocampal volume in seizure rats (Labbate et al., 2014). It is possible some of the behavioral impairments observed in the present study are also due to structural changes in the hippocampus. The chemoconsulvant pilocarpine utilized in Labbate et al., 2014 has similar functions as kainic acid used in the present study, both modeling features of temporal lobe epilepsy in humans (Kandratavicius et al., 2014). While seizures did not impact the effect Fmr1 deletion had on PPI at the behavioral level, future studies should investigate whether seizures early in development could impact FXS pathology on a molecular level in adulthood. Specifically, whether seizures superimposed on a Fmr1 deletion could impact hippocampal dendritic and synaptic abnormalities commonly associated with Fragile X syndrome (Kazdoba et al., 2014; Lai et al., 2016). Furthermore, utilizing the same seizure model as in the present study, our lab has previously shown that a single seizure in 129 SvEvTac mice resulted in acute changes in PI3K/Akt/mTOR signaling in the hippocampus in males on PD12. Specifically, seizure mice had upregulations in % total phosphorylated S6(s235/236) expression and decreased % total phosphorylated FMRP(s499) compared to control male mice (Reynolds et al., 2016). Future studies could expand on these findings to determine how deletions in Fmr1 effect these changes in the hippocampus, and if these alterations persist long-term and could underlie behavioral impairments found in this study. Other signaling mechanisms, such as dysregulated metabotropic glutamate receptor (mGluR) signaling could also underlie the deficits observed in PPI (Brody et al., 2003; Brody et al., 2004). Altered glutamatergic signaling is implicated in seizures, FXS, and more recently in schizophrenia, which all have been associated with abnormalities in sensorimotor gating and PPI (Barker-Haliski and White, 2015; Frankland et al., 2004; Ma and Leung, 2016; Maksymetz et al., 2017; Mena et al., 2016; Muddashetty et al., 2007).
We also found early-life seizures to significantly enhance freezing in delay fear conditioning, with deletions in Fmr1 not impacting learning and memory. In both the training phase of the task, as well as during contextual fear conditioning, seizure mice froze significantly more than control mice. Increased freezing could be indicative of enhanced learning in the task, as an early-life seizure had no long-term impact on activity levels in the mice which could be another potential reason for the enhanced freezing. No differences were detected in the novel context cued recall phase between seizure and control mice, suggesting that this aberrant phenotype is hippocampal-dependent. Other studies have also provided evidence for seizure-induced learning impairments to be localized to the hippocampus (Holley et al., 2018; Lugo et al., 2014; Pearson et al., 2014; Zhou et al., 2007). Early-life seizures can enhance dendritic complexity in the hippocampus, specifically increasing the number of large mossy fiber terminals, which could potentially strengthen the learned association between the context and aversive stimulus pairing (Raijmakers et al., 2016; Tao et al., 2016). Enhanced learning following seizures has also been shown in clinical populations, with febrile convulsions in early childhood leading to significantly better mnemonic capacity, memory consolidation, and sequential memory retrieval in comparison to control subjects (Chang et al., 2001).
Our lab has previously found that a single seizure in adult neuron subset-specific Pten (NS-Pten) heterozygous mice also enhanced contextual fear learning (Smith et al., 2016). Deletions in Pten are a monogenic cause of Autism spectrum disorder (ASD) similar to Fmr1, and when PTEN is absent, it results in hyperactive PI3K/Akt/mTOR signaling and alterations in Fragile X mental retardation protein, the protein product of Fmr1 (Hulbert and Jiang, 2016; Lugo et al., 2013). However, unlike Smith et al. (2016), we did not find that superimposing a seizure on top of a genetic cause of ASD altered memory and learning in the Fmr1 KO mouse. This could potentially be due to the method and timeline of seizure induction, as seizures in the NS-Pten heterozygous mice were administered in adulthood, as well as could be due to the differential functions that deletion of Pten and Fmr1, and a lack of their respective proteins, have in the brain.
Utilizing double-hit models, such as superimposing seizures on the Fmr1 mutation, can enhance our understanding of comorbidities in disease models. Double-hit models have demonstrated the impact of early-life seizures on heightened seizure susceptibility later in life, as well as other models have examined how environmental perturbations, such as immune insults, can exacerbate seizure-induced behavioral deficits and brain damage in adulthood (Chrzaszcz et al., 2010; Galic et al., 2008; Koh et al., 2004; Riazi et al., 2010; Yin et al., 2013). However, few studies have examined how seizures impact underlying gene mutations, especially in relation to monogenic causes of ASD. Recently, a study found that repeated seizures at the age of disease onset in a Scnla mouse model (Scn1aRHI+), transformed the milder phenotype into a more severe phenotype, characteristic of Dravet syndrome (severe myoclonic epilepsy of infancy) (Salgueiro-Pereira et al., 2019). Mutations in Scn1a are a genetic cause of Dravet syndrome, genetic epilepsy with febrile seizures plus (GEFS+), as well as are implicated in ASDs (Escayg and Goldin, 2010; Han et al., 2012). While findings from Salgueiro-Pereira et al. (2019) and our study provide evidence for how seizures impact Sc1a and Fmr1 mutations, how seizures superimposed on genetic mutations can affect ASD phenotypes remains largely unknown. Future studies should expand on these findings by examining other monogenic causes of ASD, as double-hit models can be advantageous in understanding how comorbidities exacerbate the molecular and behavioral basis of neurodevelopmental disorders.
5. Conclusions
Findings from the present study provide insight into how seizures may impact the FXS phenotype. A single early-life seizure increased repetitive behavior in adulthood, a core component of the ASD phenotype, to levels comparable to those observed in the Fmr1 KO mouse, yet seizures did not exacerbate stereotypy in the KO. This study also contributes to evidence of how early-life seizures can impact long-term behavioral outcomes in control mice, exhibited by reduced sociability, enhanced learning and memory, and a reduction in prepulse inhibition in seizure mice. Separately, several commonly found behavioral impairments were also observed in the Fmr1 KO mouse, such as hyperactivity, and enhanced prepulse inhibition and repetitive behavior. Apart from changes in repetitive behavior, inducing a single kainic acid seizure on postnatal day 10 did not drastically exacerbate the behavioral manifestations caused by mutations in Fmr1. However, it is critical to continue investigating how seizures may impact the behavioral phenotype of ASDs, as other seizure types and early-life injuries at other developmental time points could have diverse effects on behavioral outcomes.
Highlights.
Early-life seizures increased repetitive behavior in adult wild type (WT) mice
Repetitive behavior in Fmr1 knockout mice was not exacerbated following seizures
Deletion of Fmr1 and seizures had opposite effects on prepulse inhibition
Early-life seizures resulted in reduced sociability in adult WT mice
An early-life seizure enhanced hippocampal-dependent learning in adult WT mice
Acknowledgements
We would like to acknowledge the Baylor University Molecular Biosciences Core for the use of equipment for this study. We would also like to thank Paige Womble for her critical review of the paper.
Funding
This work was supported by the National Institutes of Health (NIH) to JNL [Grant Number: NS088776].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The authors have no competing interests to declare.
Data statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon request.
References
- Aguilar BL, Malkova L, N’Gouemo P, Forcelli PA, 2018. Genetically Epilepsy-Prone Rats Display Anxiety-Like Behaviors and Neuropsychiatric Comorbidities of Epilepsy. Front Neurol 9, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker-Haliski M, White HS, 2015. Glutamatergic Mechanisms Associated with Seizures and Epilepsy. Cold Spring Harb Perspect Med 5, a022863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST, 2004. The mGluR theory of fragile X mental retardation. Trends Neurosci 27, 370–377. [DOI] [PubMed] [Google Scholar]
- Bernard PB, Benke TA, 2015. Early life seizures: Evidence for chronic deficits linked to autism and intellectual disability across species and models. Exp Neurol 263, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard PB, Castano AM, Beitzel CS, Carlson VB, Benke TA, 2015. Behavioral changes following a single episode of early-life seizures support the latent development of an autistic phenotype. Epilepsy Behav 44, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, 2002. Epilepsy in fragile X syndrome. Dev Med Child Neurol 44, 724–728. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Raspa M, Loggin-Hester L, Bishop E, Holiday D, Bailey DB, 2010. Seizures in fragile X syndrome: characteristics and comorbid diagnoses. Am J Intellect Dev Disabil 115, 461–472. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E, 2012. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76, 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi R, Chuang SC, Zhao W, Young SR, Wong RK, 2009. Cellular plasticity for group I mGluR-mediated epileptogenesis. J Neurosci 29, 3497–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody SA, Conquet F, Geyer MA, 2003. Disruption of prepulse inhibition in mice lacking mGluR1. Eur J Neurosci 18, 3361–3366. [DOI] [PubMed] [Google Scholar]
- Brody SA, Dulawa SC, Conquet F, Geyer MA, 2004. Assessment of a prepulse inhibition deficit in a mutant mouse lacking mGlu5 receptors. Mol Psychiatry 9, 35–41. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal A, 2011. Molecular mechanisms of cognitive and behavioral comorbidities of epilepsy in children. Epilepsia 52 Suppl 1, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budimirovic DB, Kaufmann WE, 2011. What can we learn about autism from studying fragile X syndrome? Dev Neurosci 33, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelhano AS, Cassane Gdos S, Scorza FA, Cysneiros RM, 2013. Altered anxiety-related and abnormal social behaviors in rats exposed to early life seizures. Front Behav Neurosci 7, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Guo NW, Wang ST, Huang CC, Tsai JJ, 2001. Working memory of school-aged children with a history of febrile convulsions: a population study. Neurology 57, 37–42. [DOI] [PubMed] [Google Scholar]
- Chen L, Toth M, 2001. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103, 1043–1050. [DOI] [PubMed] [Google Scholar]
- Chrzaszcz M, Venkatesan C, Dragisic T, Watterson DM, Wainwright MS, 2010. Minozac treatment prevents increased seizure susceptibility in a mouse “two-hit” model of closed skull traumatic brain injury and electroconvulsive shock-induced seizures. J Neurotrauma 27, 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK, 2005. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci 25, 8048–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, Portera-Cailliau C, 2015. Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron 87, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley B, Kirjanen S, Partanen J, Castren ML, 2016. Epileptic Electroencephalography Profile Associates with Attention Problems in Children with Fragile X Syndrome: Review and Case Series. Front Hum Neurosci 10, 353–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis M, Wagner KD, 2016. Treatment of Autism Spectrum Disorder in Children and Adolescents. Psychopharmacol Bull 46, 18–41. [PMC free article] [PubMed] [Google Scholar]
- Dolan BM, Duron SG, Campbell DA, Vollrath B, Shankaranarayana Rao BS, Ko HY, Lin GG, Govindarajan A, Choi S-Y, Tonegawa S, 2013. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc Natl Acad Sci 110, 5671–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, Goldin AL, 2010. Sodium channel SCN1A and epilepsy: Mutations and mechanisms. Epilepsia 51, 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ, 2004. Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry 9, 417–425. [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ, 2008. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 28, 6904–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, Huber KM, 2008. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol 100, 2615–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Hoffmann A, Bassell GJ, Berry-Kravis EM, 2015. Therapeutic Strategies in Fragile X Syndrome: From Bench to Bedside and Back. Neurotherapeutics 12, 584–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Yao X, Pong DL, Jeromin A, Bassell GJ, 2011. Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci 31, 5693–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman PJ, Stafstrom CE, 2009. Origins of epilepsy in fragile X syndrome. Epilepsy Curr 9, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, 2006. Lessons from fragile X regarding neurobiology, autism, and neurodegeneration. J Dev Behav Pediatr 27, 63–74. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ, Rivera SM, Hagerman PJ, 2008. The fragile X family of disorders: a model for autism and targeted treatments. Curr Pediatr Rev 4, 40–52. [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA, 2012. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessl D, Berry-Kravis E, Cordeiro L, Yuhas J, Ornitz EM, Campbell A, Chruscinski E, Hervey C, Long JM, Hagerman RJ, 2009. Prepulse inhibition in fragile X syndrome: feasibility, reliability, and implications for treatment. Am J Med Genet B Neuropsychiatr Genet 150B, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley AJ, Hodges SL, Nolan SO, Binder M, Okoh JT, Ackerman K, Tomac LA, Lugo JN, 2018. A single seizure selectively impairs hippocampal-dependent memory and is associated with alterations in PI3K/Akt/mTOR and FMRP signaling. Epilepsia Open 3, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GL, 2016. Effect of Seizures on the Developing Brain and Cognition. Semin Pediatr Neurol 23, 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland JG, Hannesson DK, Barnes SJ, Phillips AG, 2007. Kindling of basolateral amygdala but not ventral hippocampus or perirhinal cortex disrupts sensorimotor gating in rats. Behav Brain Res 177, 30–36. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF, 2002. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99, 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulbert SW, Jiang YH, 2016. Monogenic mouse models of autism spectrum disorders: Common mechanisms and missing links. Neuroscience 321, 3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandratavicius L, Balista PA, Lopes-Aguiar C, Ruggiero RN, Umeoka EH, Garcia-Cairasco N, Bueno-Junior LS, Leite JP, 2014. Animal models of epilepsy: use and limitations. Neuropsychiatr Dis Treat 10, 1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazdoba TM, Leach PT, Silverman JL, Crawley JN, 2014. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis Res 3, 118–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd SA, Lachiewicz A, Barbouth D, Blitz RK, Delahunty C, McBrien D, Visootsak J, Berry-Kravis E, 2014. Fragile X syndrome: a review of associated medical problems. Pediatrics 134, 995–1005. [DOI] [PubMed] [Google Scholar]
- King BH, Navot N, Bernier R, Webb SJ, 2014. Update on diagnostic classification in autism. Curr Opin Psychiatry 27, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S, Tibayan FD, Simpson JN, Jensen FE, 2004. NBQX or topiramate treatment after perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia 45, 569–575. [DOI] [PubMed] [Google Scholar]
- Labbate GP, da Silva AV, Barbosa-Silva RC, 2014. Effect of severe neonatal seizures on prepulse inhibition and hippocampal volume of rats tested in early adulthood. Neurosci Lett 568, 62–66. [DOI] [PubMed] [Google Scholar]
- Lai JK, Lerch JP, Doering LC, Foster JA, Ellegood J, 2016. Regional brain volumes changes in adult male FMR1-KO mouse on the FVB strain. Neuroscience 318, 12–21. [DOI] [PubMed] [Google Scholar]
- Lovelace JW, Ethell IM, Binder DK, Razak KA, 2018. Translation-relevant EEG phenotypes in a mouse model of Fragile X Syndrome. Neurobiol Dis 115, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo JN, Smith GD, Morrison JB, White J, 2013. Deletion of PTEN produces deficits in conditioned fear and increases fragile X mental retardation protein. Learn Mem 20, 670–673. [DOI] [PubMed] [Google Scholar]
- Lugo JN, Swann JW, Anderson AE, 2014. Early-life seizures result in deficits in social behavior and learning. Exp Neurol 256, 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Leung LS, 2016. Dual Effects of Limbic Seizures on Psychosis-Relevant Behaviors Shown by Nucleus Accumbens Kindling in Rats. Brain Stimul 9, 762–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksymetz J, Moran SP, Conn PJ, 2017. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Molecular brain 10, 15–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo J, Ota M, Hidese S, Teraishi T, Hori H, Ishida I, Hiraishi M, Kunugi H, 2018. Sensorimotor Gating in Depressed and Euthymic Patients with Bipolar Disorder: Analysis on Prepulse Inhibition of Acoustic Startle Response Stratified by Gender and State. Front Psychiatry 9, 123–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain KL, Merriweather MY, Yuva-Paylor LA, Paylor R, 2001. The use of behavioral test batteries: effects of training history. Physiol Behav 73, 705–717. [DOI] [PubMed] [Google Scholar]
- McKenna PJ, Kane JM, Parrish K, 1985. Psychotic syndromes in epilepsy. Am J Psychiatry 142, 895–904. [DOI] [PubMed] [Google Scholar]
- Mena A, Ruiz-Salas JC, Puentes A, Dorado I, Ruiz-Veguilla M, De la Casa LG, 2016. Reduced Prepulse Inhibition as a Biomarker of Schizophrenia. Front Behav Neurosci 10, 202–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Poe MD, Nonneman RJ, Young NB, Koller BH, Crawley JN, Duncan GE, Bodfish JW, 2008. Development of a mouse test for repetitive, restricted behaviors: relevance to autism. Behav Brain Res 188, 178–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Kelic S, Gross C, Xu M, Bassell GJ, 2007. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci 27, 5338–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M, Ferri R, Oostra BA, 2000. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 41, 19–23. [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Calabrese G, Bonaccorso CM, D’Antoni S, Brouwer JR, Bakker CE, Elia M, Ferri R, Nelson DL, Oostra BA, Catania MV, 2007. Audiogenic seizure susceptibility is reduced in fragile X knockout mice after introduction of FMR1 transgenes. Exp Neurol 203, 233–240. [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Colognola RM, Ferri R, Gigli GL, Petrella MA, Sanfilippo S, Bergonzi P, Tassinari CA, 1988. Fragile-X syndrome: a particular epileptogenic EEG pattern. Epilepsia 29, 41–47. [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Hagerman RJ, Ferri R, Bosco P, Dalla Bernardina B, Tassinari CA, De Sarro GB, Elia M, 1999. Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 40, 1092–1099. [DOI] [PubMed] [Google Scholar]
- Nickels KC, Zaccariello MJ, Hamiwka LD, Wirrell EC, 2016. Cognitive and neurodevelopmental comorbidities in paediatric epilepsy. Nat Rev Neurol 12, 465–476. [DOI] [PubMed] [Google Scholar]
- Niu M, Han Y, Dy ABC, Du J, Jin H, Qin J, Zhang J, Li Q, Hagerman RJ, 2017. Autism symptoms in fragile X syndrome. J Child Neurol 32, 903–909. [DOI] [PubMed] [Google Scholar]
- Oakes A, Thurman AJ, McDuffie A, Bullard LM, Hagerman RJ, Abbeduto L, 2016. Characterising repetitive behaviours in young boys with fragile X syndrome. J Intellect Disabil Res 60, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JN, Schulz KM, Patel M, 2014. Specific alterations in the performance of learning and memory tasks in models of chemoconvulsant-induced status epilepticus. Epilepsy Res 108, 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouretemad HR, Thompson PJ, Fenwick PB, 1998. Impaired sensorimotor gating in patients with non-epileptic seizures. Epilepsy Res 31, 1–12. [DOI] [PubMed] [Google Scholar]
- Raijmakers M, Clynen E, Smisdom N, Nelissen S, Brone B, Rigo JM, Hoogland G, Swijsen A, 2016. Experimental febrile seizures increase dendritic complexity of newborn dentate granule cells. Epilepsia 57, 717–726. [DOI] [PubMed] [Google Scholar]
- Reynolds CD, Smith G, Jefferson T, Lugo JN, 2016. The effect of early life status epilepticus on ultrasonic vocalizations in mice. Epilepsia 57, 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazi K, Galic MA, Pittman QJ, 2010. Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res 89, 34–42. [DOI] [PubMed] [Google Scholar]
- Salgueiro-Pereira AR, Duprat F, Pousinha PA, Loucif A, Douchamps V, Regondi C, Ayrault M, Eugie M, Stunault MI, Escayg A, Goutagny R, Gnatkovsky V, Frassoni C, Marie FL, Bethus F, Mantegazza M, 2019. A two-hit story: Seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol Dis 125, 31–44. [DOI] [PubMed] [Google Scholar]
- Sayin U, Sutula TP, Stafstrom CE, 2004. Seizures in the developing brain cause adverse long-term effects on spatial learning and anxiety. Epilepsia 45, 1539–1548. [DOI] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN, 2010. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater E, Beard AW, 1963. The Schizophrenia-like Psychoses of Epilepsy: i. Psychiatric Aspects. Br J Psychiatry 109, 95–112. [DOI] [PubMed] [Google Scholar]
- Smith GD, White J, Lugo JN, 2016. Superimposing Status Epilepticus on Neuron Subset-Specific PTEN Haploinsufficient and Wild Type Mice Results in Long-term Changes in Behavior. Sci Rep 6, 36559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Alekseyenko O, Hamilton SM, Thomas AM, Serysheva E, Yuva-Paylor LA, Paylor R, 2011. Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res 4, 40–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE, Benke TA, 2015. Autism and Epilepsy: Exploring the Relationship Using Experimental Models. Epilepsy Curr 15, 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y, Inoue Y, Tottori T, Mihara T, 2001. Acute psychosis during intracranial EEG monitoring: close relationship between psychotic symptoms and discharges in amygdala. Epilepsia 42, 719–724. [DOI] [PubMed] [Google Scholar]
- Tao K, Ichikawa J, Matsuki N, Ikegaya Y, Koyama R, 2016. Experimental febrile seizures induce age-dependent structural plasticity and improve memory in mice. Neuroscience 318, 34–44. [DOI] [PubMed] [Google Scholar]
- Thomas AM, Bui N, Graham D, Perkins JR, Yuva-Paylor LA, Paylor R, 2011. Genetic reduction of group 1 metabotropic glutamate receptors alters select behaviors in a mouse model for fragile X syndrome. Behav Brain Res 223, 310–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraragavan S, Graham D, Bui N, Yuva-Paylor LA, Wess J, Paylor R, 2012. Genetic reduction of muscarinic M4 receptor modulates analgesic response and acoustic startle response in a mouse model of fragile X syndrome (FXS). Behav Brain Res 228, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walf AA, Frye CA, 2007. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc 2, 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ethridge LE, Mosconi MW, White SP, Binder DK, Pedapati EV, Erickson CA, Byerly MJ, Sweeney JA, 2017. A resting EEG study of neocortical hyperexcitability and altered functional connectivity in fragile X syndrome. J Neurodev Disord 9, 11–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner JM, Radcliffe RA, 2004. Cued and Contextual Fear Conditioning in Mice. Curr Protoc Neurosci 27, 8.5C.1–8.5C.14. [DOI] [PubMed] [Google Scholar]
- Wen TH, Lovelace JW, Ethell IM, Binder DK, Razak KA, 2019. Developmental Changes in EEG Phenotypes in a Mouse Model of Fragile X Syndrome. Neuroscience 398, 126–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DC, Bueno-Junior LS, Lopes-Aguiar C, Do Val Da Silva RA, Kandratavicius L, Leite JP, 2016. The frequency of spontaneous seizures in rats correlates with alterations in sensorimotor gating, spatial working memory, and parvalbumin expression throughout limbic regions. Neuroscience 312, 86–98. [DOI] [PubMed] [Google Scholar]
- Yin P, Liu J, Li Z, Wang YY, Qiao NN, Huang SY, Li BM, Sun RP, 2013. Prenatal immune challenge in rats increases susceptibility to seizure-induced brain injury in adulthood. Brain Res 1519, 78–86. [DOI] [PubMed] [Google Scholar]
- Yuhas J, Cordeiro L, Tassone F, Ballinger E, Schneider A, Long JM, Ornitz EM, Hessl D, 2011. Brief Report: Sensorimotor Gating in Idiopathic Autism and Autism Associated with Fragile X Syndrome. J Autism Dev Disord 41, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JL, Shatskikh TN, Liu X, Holmes GL, 2007. Impaired single cell firing and long-term potentiation parallels memory impairment following recurrent seizures. Eur J Neurosci 25, 3667–3677. [DOI] [PubMed] [Google Scholar]