

Abstract
Two methods are reported for the 1,2- and 1,1-arylboration of α-methyl vinyl arenes. In the case of 1,2-arylboration, the formation of a quaternary center occurred through a rare cross-coupling reaction of a tertiary organometallic complex. 1,1-Arylboration was enabled by catalyst optimization and occurred through a β-hydride elimination/reinsertion cascade. Enantioselective variants of both processes are presented as well as mechanistic investigations.
Keywords: carboboration, copper, cross-coupling, homogeneous catalysis, palladium
In recent years, carboboration of alkenes has emerged as a valuable strategy for chemical synthesis due to the rapid buildup of complexity from simple components.[1] While several approaches have been described, Cu/Pd-catalyzed arylboration has been demonstrated on a variety of activated alkenes (e.g., alkenyl arenes, 1,3-dienes, vinyl silanes, strained alkenes).[2–4] These reactions operate by the general catalytic cycles illustrated in Scheme 1. Upon generation of LnCuBpin (II), syn addition to the alkene occurs to generate C(sp3)—Cu complex III. Transmetalation with Pd complex VI ensues to provide IV, which upon reductive elimination generates the product and Pd0 complex V. As well as the difunctionalization of an alkene, the stereo-selective cross-coupling of a stereo-defined secondary C(sp3) nucleophile is achieved, which is a significant challenge in organic chemical synthesis.[5]
Scheme 1.

Approaches to arylboration by Cu/Pd cooperative catalysis.
In all reported examples of Cu/Pd-catalyzed arylboration, terminal and 1,2-disubstituted alkenes are used, which leads to the formation of tertiary stereogenic centers via secondary C(sp3)—Cu complexes.[2] Reactions to form quaternary centers through palladium-catalyzed cross-coupling of tertiary C-(sp3)—Cu complexes have not been previously demonstrated.[6] In general, palladium-catalyzed cross-coupling reactions with tertiary C(sp3) nucleophiles is challenging owing to slow transmetalation as a result of steric hindrance and a high propensity towards β-hydride elimination in the alkyl–palladium intermediate.[7] Thus, only one study has demonstrated palladium-catalyzed cross-coupling of fully substituted alkyl metals.[7,8] Herein, we describe a method for the arylboration of α-alkyl alkenyl arenes that allows for the synthesis of quaternary carbon centers, with control of enantioselectivity. These studies also led to the development of a 1,1-arylboration reaction through tuning of the Pd catalyst. With respect to the latter reaction, the only known example of 1,1-arylboration was reported by Toste and co-workers and operates on monosubstituted alkenes.[9,10] The process described herein operates on α-alkyl alkenyl arenes and leads to the controlled formation of two stereocenters.
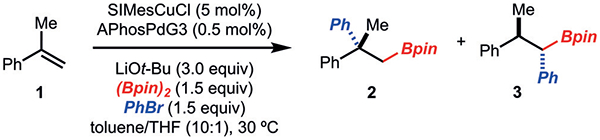
We initiated our investigations with the arylboration of α-methylstyrene (1; Scheme 2). Under conditions previously disclosed by our research group for the arylboration of alkenyl arenes,[2b] the formation of product 2 with a quaternary center was observed along with 1,1-arylboration adduct 3 (> 20:1 dr). Product 2 most likely arises from reductive elimination of alkyl–palladium complex 5. On the basis of the formation of product 3, we propose that a β-hydride elimination/reinsertion sequence occurs to generate 6, which then undergoes reductive elimination.[9]
Scheme 2.

Initial investigations. SIMes= 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene, XPhosPdG3= (2-dicyclohexylphosphanyl-2’,4’,6’-triisopropyl-1,1’-biphenyl)[2-(2’-amino-1,1’-biphenyl)]palladium-(II) methanesulfonate.
The process generating 2 was optimized to the conditions shown in the reaction scheme above Table 1. Key to optimization was the identification of APhosPdG3[11,12] as a suitable catalyst that suppressed the formation of 3. Other ligands, such as t-Bu2PhP, gave rise to preferential generation of 2, albeit in lower yield (Table 1, entry 6). Interestingly, the use of t-Bu3PPdG3 and Cy3PPdG3 led to preferential formation of the 1,1-arylboration adduct (Table 1, entries 4 and 7).
Table 1:
Investigation of the reaction conditions.[a]
 | |||
|---|---|---|---|
| Entry | Change from standard conditions | 2 [%] | 3 [%] |
| 1 | none | 75 | 12 |
| 2 | 1 mol% APhosPdG3 instead of 0.5 mol% | 77 | 11 |
| 3 | XPhosPdG3 instead of APhosPdG3 | 22 | 20 |
| 4 | t-Bu3PPdG3 instead of APhosPdG3 | 29 | 54 |
| 5 | t-Bu2CyPPdG3 instead of APhosPdG3 | 52 | 40 |
| 6 | t-Bu2PhPPdG3 instead of APhosPdG3 | 58 | 8 |
| 7 | Cy3PPdG3 instead of APhosPdG3 | 11 | 27 |
| 8 | 1.5 equiv LiOt-Bu instead of 3.0 equiv | 61 | 3 |
| 9 | toluene instead oftoluene/THF (10:1) | 29 | < 1 |
Yields were determined by 1H NMR analysis of the crude reaction mixture with an internal standard. Cy=cyclohexyl.
With an optimized set of conditions in hand, the scope of the method was explored (Scheme 3). The method was found to tolerate aryl bromides that bear electron-donating (product 7) and electron-withdrawing groups (product 8). In the case of electron-rich aryl bromides, increased quantities of the 1,1-arylboration product formed, most likely due to a slower reductive elimination of Pd complexes analogous to 5, thus allowing for β-hydride elimination to compete. Sterically demanding aryl bromides were not tolerated, presumably due to a slow transmetalation (product 10). Whereas a Boc-protected indole was tolerated (product 11), other hetero-cycles with more basic motifs, such as pyridines, suppressed the reaction. With respect to the alkene component, electron-donating (products 7 from 4-methoxy-α-methylstyrene and 13) and electron-withdrawing groups (product 14) did not impede the reaction. However, α-ethylstyrene did not function well in this reaction due to incomplete conversion and poor selectivity (product 16). The yield and selectivity could be improved if the substituent was constrained within a ring (product 17). Finally, an alkenyl boronic ester could serve as the alkene component to provide access to 15.
Scheme 3.

Scope of quaternary center formation. Yields are for the isolated purified alcohol product (following the oxidation step), unless noted otherwise. Yields in parentheses were determined by 1H NMR analysis of the crude reaction mixture with an internal standard. [a] Isolated as the Bpin product. [b] The 1,1-arylboration product was also observed (20% yield as determined by NMR analysis). Boc= tert-butoxycarbonyl.
Enantioselective variants of the reaction were achieved through the use of chiral NHC catalyst 18 to generate products 9, 12, and 14 (Scheme 4).[13] This Cu catalyst was previously used by our research group to promote the enantioselective arylboration of cis-β-alkyl alkenyl arenes.[2e,6a] Although the yields are moderate, these reactions represent a rare example of a stereoselective palladium-catalyzed cross-coupling of a tertiary alkyl metal (e.g., 20).[7] Based on the absolute configuration of product 13, the mechanistic pathway most likely involves an enantioselective borylcupration via 19, followed by stereoretentive transmetalation (Scheme 4).[14]
Scheme 4.

Enantioselective formation of quaternary centers. Yields are for the isolated purified alcohol product (following the oxidation step), unless noted otherwise. Yields in parentheses were determined by 1H NMR analysis of the crude reaction mixture with an internal standard.
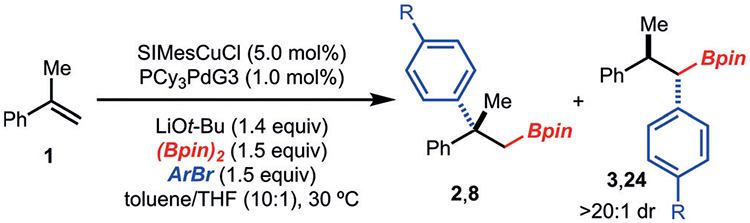
On the basis of the observation that the use of PdPCy3G3 led to 1,1-arylboration (Table 1, entry 7), we directed our efforts towards optimizing the formation of this product (Table 2, entry 1). It was found that using less LiOt-Bu and increased Pd-catalyst loading improved yields. Evaluation of other related ligands did not enhance yields (Table 2, entries 2 and 3). An unexpected finding is that the preformed (PCy3)2Pd complex was not competent in the reaction (Table 2, entry 4). Upon addition of PCy3 (1.0 mol%) to PdPCy3G3, product formation was not observed, which further substantiated this result (Table 2, entry 5). Furthermore, phosphine-free conditions with solely PdG3 dimer functioned well (Table 2, entry 6). Based on this result, other Pd0 precatalysts were evaluated, and it was found that the reaction proceeded in comparable yields with Pd2dba3 to those observed with PdPCy3G3 (Table 2, entry 7). However, initial evaluation of aryl bromides bearing electron-with-drawing groups revealed Pd2dba3 to be less general (Table 2, entry 9). Furthermore, it appears that dba may have a slight inhibitory effect, as the use of PdG3 dimer led to slightly improved yields as compared to Pd2dba3 (Table 2, entry 10). Therefore, since PdPCy3G3 demonstrated the highest yield and broadest initial aryl bromide scope it was used to evaluate the scope of the method.
Table 2:
Investigation of reaction conditions for the preferential formation of the 1,1-arylboration product.[a]
 | ||||
|---|---|---|---|---|
| Entry | Change from standard conditions | R | 2,8 [%] | 3,24 [%] |
| 1 | no change | H (2/3) | 3 | 63 |
| 2 | Pi-Bu3PdG3 instead of PCy3PdG3 | H (2/3) | 2 | 39 |
| 3 | Pt-Bu3PdG3 instead of PCy3PdG3 | H (2/3) | 21 | 33 |
| 4 | (PCy3)2Pd instead of PCy3PdG3 | H (2/3) | < 2 | <2 |
| 5 | with 1.0 mol% PCy3 | H (2/3) | < 2 | <2 |
| 6[b] | PdG3 dimer instead of PCy3PdG3 | H (2/3) | 3 | 50 |
| 7[b] | Pd2dba3 instead of PCy3PdG3 | H (2/3) | 3 | 65 |
| 8 | no change | CF3 (8/24) | 4 | 64 |
| 9 | Pd2dba3 instead of PCy3PdG3 | CF3 (8/24) | 2 | 24 |
| 10[b] | PdG3 dimer instead of PCy3PdG3 | CF3 (8/24) | 5 | 42 |
Yields were determined by 1H NMR analysis of the crude reaction mixture with an internal standard.
Pd catalyst loading: 0.5 mol%.
Under the optimized conditions, a range of aryl bromides were investigated (Scheme 5). Aryl bromides that bear electron-donating (products 25 and 27), electron-withdrawing (products 24 and 26), and sterically demanding groups (product 27) functioned well. Electronic modification of the aryl alkene was tolerated (products 28–30); however, a substrate with extended conjugation resulted in poor selectivity for the 1,1-arylboration product 31, most likely owing to formation of a stabilized π-naphthyl complex. Sterically modified α-substituted alkenyl arenes were tolerated in these conditions to provide 32–34.
Scheme 5.

1,1-Diarylation scope. Yields are for the isolated purified alcohol product (following the oxidation step), unless noted otherwise. Yields in parentheses were determined by 1H NMR analysis of the crude reaction mixture with an internal standard. [a] Isolated as the Bpin product. [b] The reaction was carried out with PhBr (3 equiv).
Enantioselective variants of the reaction were also probed, and whereas the reaction to generate (1S,2R)-3 proceeded with good enantioselectivity and diastereoselectivity, the yield was moderate (Scheme 5B). Based on the absolute and relative configuration of the product, we propose the following reaction pathway: The alkyl–palladium complex 35 was most likely generated through an analogous pathway to that shown in Scheme 4. At this stage, β-hydride elimination ensued to selectively generate E-alkene Pd complex 36. Since the enantioselectivity in the formation of (1S,2R)-3 is similar to that in the generation of (R)-9, (R)-12, and (S)-14 shown in Scheme 4, it is likely that the alkene does not readily dissociate.[15] Crossover experiments have also been carried out that corroborate the stability of Pd complex 36.[16] Therefore, hydropalladation occurs selectively on the Si face of the bound alkene to provide 37 and subsequently (1S,2R)-3 upon reductive elimination.
With respect to the catalytic cycles of the reaction, the primary avenue of inquiry regards the divergence in reactivity with different Pd catalysts. To probe this question, we investigated reactions with preformed alkyl–copper complex 39, which was prepared from α-methyl styrene and SIMes-CuBpin (Scheme 6A). It has been shown that the reaction of (APhos)2Pd or PdAPhosG4/LiOt-Bu with PhBr gives rise to the monoligated complex 38.[17] The reaction of Pd complex 38 with Cu complex 39 led to formation of the expected product 2 containing a quaternary center in 45% yield. It is proposed that the large APhos ligand disfavors the β-agostic interaction necessary for β-hydride elimination and promotes reductive elimination by relief of steric congestion.
Scheme 6.

Mechanistic investigations. Yields were determined by 1H NMR analysis of the crude reaction mixture with an internal standard.
Regarding the formation of the 1,1-arylboration product, since alkyl–copper complex 39 is stable and does not undergo β-hydride elimination even when heated to 50°C,[18] the divergence in reactivity appears to occur after transmetalation with ArPdL(Br). In contrast to APhos–Pd complexes, the reaction of (PCy3)2Pd with PhBr resulted in the formation of bis-ligated complex 41, and the reaction of PdPCy3-G4/LiOt-Bu with PhBr resulted in the generation of a mixture of Pd complexes (Scheme 6B).[19] It has been confirmed that the bis-ligated complex 41 was not catalytically competent (Table 2, entry 4); thus, L2 versus L1 Pd complexes cannot be the justification for the divergence in reactivity. It is suspected that one (or more) of the complexes generated from reaction of PdPCy3-G4/LiOt-Bu with PhBr is responsible for catalysis. Since phosphine-free Pd catalysts also function in the reaction (see Table 2, entries 6 and 7), it is proposed that PdPh(Br)(PCy3)n (42) complexes undergo ligand exchange to form PdPh(Br) (43) and PdPh(Br)-(PCy3)n+1 (44), and that PdPh(Br) (43) is the catalytically competent species. Transmetalation of Cu complex 39 with PdPh(Br) (43) results in the formation of 45. The tertiary alkyl–palladium complex 45, having vacant coordination sites (or at least coordination sites occupied with weakly coordinating ligands, such as solvent), can readily undergo β-hydride elimination to ultimately form the 1,1-arylboration product.
In summary, through the tuning of Pd catalysts a regiodivergent process for the 1,2- and 1,1-selective arylboration of α-alkyl alkenyl arenes was developed. Importantly, this method represents a rare cross-coupling of a tertiary alkyl metal. These studies also serve to showcase the subtleties of ligand–metal dynamics and their impact on reaction rates and selectivity.
Supplementary Material
Acknowledgements
We thank Indiana University and the NIH (R01GM114443) for generous financial support. This project was partially funded by the Vice Provost for Research through the Research Equipment Fund and the NSF (CHE1726633).
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201812533.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].For reviews, see:; a) Shimizu Y, Kanai M, Tetrahedron Lett. 2014, 55, 3727; [Google Scholar]; b) Semba K, Fujihara T, Terao J, Tsuji Y, Tetrahedron 2015, 71, 2183; [Google Scholar]; c) Lazreg F, Nahra F, Cazin CSJ, Coord. Chem. Rev 2015, 293–294, 48; [Google Scholar]; d) Neeve EC, Geier SJ, Mkhalid IAI, Westcott SA, Marder TB, Chem. Rev 2016, 116, 9091; [DOI] [PubMed] [Google Scholar]; e) Hemming D, Fritzemeier R, Westcott SA, Santos WL, Steel PG, Chem. Soc. Rev 2018, 47, 7477–7494. [DOI] [PubMed] [Google Scholar]
- [2].For Cu/Pd-catalyzed arylboration of alkenes, see:; a) Semba K, Nakao Y, J. Am. Chem. Soc 2014, 136, 7567; [DOI] [PubMed] [Google Scholar]; b) Smith KB, Logan KM, You W, Brown MK, Chem. Eur. J 2014, 20, 12032; [DOI] [PubMed] [Google Scholar]; c) Logan KM, Smith KB, Brown MK, Angew. Chem. Int. Ed 2015, 54, 5228; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 5317; [Google Scholar]; d) Semba K, Ohtagaki Y, Nakao Y, Org. Lett 2016, 18, 3956; [DOI] [PubMed] [Google Scholar]; e) Logan KM, Brown MK, Angew. Chem. Int. Ed 2017, 56, 851; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 869; [Google Scholar]; f) Chen B, Cao P, Yin X, Liao Y, Jiang L, Ye J, Wang M, Liao J, ACS Catal. 2017, 7, 2425; [Google Scholar]; g) Smith KB, Brown MK, J. Am. Chem. Soc 2017, 139, 7721; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Sardini SR, Brown MK, J. Am. Chem. Soc 2017, 139, 9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].For palladium-catalyzed arylboration, see:; a) Yang K, Song Q, J. Org. Chem 2016, 81, 1000; [DOI] [PubMed] [Google Scholar]; b) Yang K, Song Q, Org. Lett 2016, 18, 5460. [DOI] [PubMed] [Google Scholar]
- [4].For nickel-catalyzed arylboration, see:; Logan KM, Sardini SR, White SD, Brown MK, J. Am. Chem. Soc 2018, 140, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].For reviews, see:; a) Jana R, Pathak TP, Sigman MS, Chem. Rev 2011, 111, 1417; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Swift EC, Jarvo ER, Tetrahedron 2013, 69, 5799; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang C-Y, Derosa J, Biscoe MR, Chem. Sci 2015, 6, 5105; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cherney AH, Kadunce NT, Reisman SE, Chem. Rev 2015, 115, 9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Carboboration reactions of α-alkyl vinyl arenes are known; see:; a) Gong T-J, Yu S-H, Li K, Su W, Lu X, Xiao B, Fu Y, Chem. Asian J. 2017, 12, 2884; [DOI] [PubMed] [Google Scholar]; b) Cheng F, Lu W, Huang W, Wen L, Li M, Meng F, Chem. Sci 2018, 9, 4992; [DOI] [PMC free article] [PubMed] [Google Scholar]; for protoboration, see:; c) Corberán R, Mszar NW, Hoveyda AH, Angew. Chem. Int. Ed 2011, 50, 7079–7082; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2011, 123, 7217–7220. [Google Scholar]
- [7].For the stereoselective cross-coupling of an oxygen-substituted tertiary alkyl metal, see:; Zhang H, Buchwald SL, J. Am. Chem. Soc 2017, 139, 11590–11594. [DOI] [PubMed] [Google Scholar]
- [8].For representative examples of the cross-coupling of tertiary alkyl metals under the catalysis of other transition metals, see: Ni catalysis:; a) Joshi-Pangu A, Wang C-Y, Biscoe MR, J. Am. Chem. Soc 2011, 133, 8478; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lohre C, Droge T, Wang C, Glorius F, Chem. Eur. J 2011, 17, 6052; [DOI] [PubMed] [Google Scholar]; c) Ando S, Mawatari M, Matsunaga H, Ishizuka T, Tetrahedron Lett. 2016, 57, 3287; [Google Scholar]; d) Primer DN, Molander GA, J. Am. Chem. Soc 2017, 139, 9847; [DOI] [PMC free article] [PubMed] [Google Scholar]; Cu catalysis:; e) Ren P, Stern L-A, Hu X, Angew. Chem. Int. Ed 2012, 51, 9110; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2012, 124, 9244; [Google Scholar]; f) Iwasaki T, Imanishi R, Shimizu R, Kuniyasu H, Terao J, Kambe N, J. Org. Chem 2014, 79, 8522; [DOI] [PubMed] [Google Scholar]; g) Dai W, Shi H, Zhao X, Cao S, Org. Lett 2016, 18, 4284; [DOI] [PubMed] [Google Scholar]; h) Shi H, Dai W, Wang B, Cao S, Organometallics 2018, 37, 459; [Google Scholar]; i) Dai W, Lin Y, Wan Y, Cao S, Org. Chem. Front 2018, 5, 55; [Google Scholar]; Fe catalysis:; j) Cahiez G, Avedissian H, Synthesis 1998, 8, 1199. [Google Scholar]; Co-catalysis:; k) Iwasaki T, Takagawa H, Singh SP, Kuniyasu H, Kambe N, J. Am. Chem. Soc 2013, 135, 9604. [DOI] [PubMed] [Google Scholar]
- [9].For a palladium-catalyzed 1,1-arylboration, see:; Nelson HM, Williams BD, Miró J, Toste FD, J. Am. Chem. Soc 2015, 137, 3213–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].For 1,1-diarylation reactions, see:; a) Urkalan KB, Sigman MS, Angew. Chem. Int. Ed 2009, 48, 3146–3149; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2009, 121, 3192–3195; [Google Scholar]; b) Werner EW, Urkalan KB, Sigman MS, Org. Lett 2010, 12, 2848–2851; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Saini V, Sigman MS, J. Am. Chem. Soc 2012, 134, 11372–11375; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Saini V, Liao L, Wang Q, Jana R, Sigman MS, Org. Lett 2013, 15, 5008–5011; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Orlandi M, Hilton MJ, Yamamoto E, Toste FD, Sigman MS, J. Am. Chem. Soc 2017, 139, 12688–12695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guram AS, King AO, Allen JG, Wang X, Schenkel LB, Chan J, Bunel EE, Faul MM, Larsen RD, Martinelli MJ, Reider PJ, Org. Lett 2006, 8, 1787–1789. [DOI] [PubMed] [Google Scholar]
- [12].Bruno NC, Tudge MT, Buchwald SL, Chem. Sci 2013, 4, 916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Park JK, Lackey HH, Rexford MD, Kovnir K, Shatruk M, McQuade DT, Org. Lett 2010, 12, 5008; [DOI] [PubMed] [Google Scholar]; b) Park JK, McQuade DT, Angew. Chem. Int. Ed 2012, 51, 2717; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2012, 124, 2771; [Google Scholar]; c) Park J, McQuade D, Synthesis 2012, 44, 1485; [Google Scholar]; d) Delvos L, Hensel A, Oestreich M, Synthesis 2014, 46, 2957. [Google Scholar]
- [14].Casares JA, Espinet P, Salas G, Chem. Eur. J 2002, 8, 4843–4853. [DOI] [PubMed] [Google Scholar]
- [15].Mei T-S, Patel HH, Sigman MS, Nature 2014, 508, 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].See the Supporting Information for details.
- [17].Kosaka K, Uchida T, Mikami K, Ohta Y, Yokozawa T, Macromolecules 2018, 51, 364. [Google Scholar]
- [18].a) Laitar DS, Tsui EY, Sadighi JP, Organometallics 2006, 25, 2405; [Google Scholar]; b) Lee J, Radomkit S, Torker S, del Pozo J, Hoveyda AH, Nat. Chem 2017, 10, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Stambuli JP, Incarvito CD, Buhl M, Hartwig JF, J. Am. Chem. Soc 2004, 126, 1184; [DOI] [PubMed] [Google Scholar]; b) Barrios-Landeros F, Carrow BP, Hartwig JF, J. Am. Chem. Soc 2009, 131, 8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.