

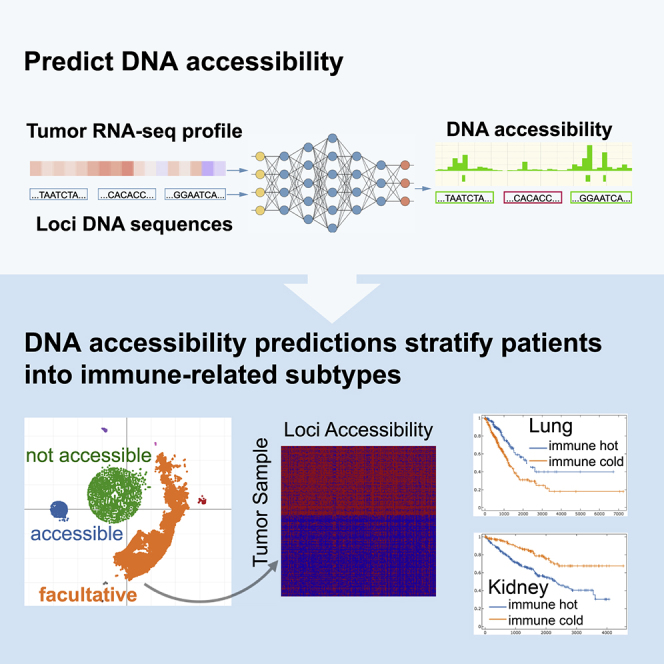
Summary
DNA accessibility is a key dynamic feature of chromatin regulation that can potentiate transcriptional events and tumor progression. To gain insight into chromatin state across existing tumor data, we improved neural network models for predicting accessibility from DNA sequence and extended them to incorporate a global set of RNA sequencing gene expression inputs. Our expression-informed model expanded the application domain beyond specific tissue types to tissues not present in training and achieved consistently high accuracy in predicting DNA accessibility at promoter and promoter flank regions. We then leveraged our new tool by analyzing the DNA accessibility landscape of promoters across The Cancer Genome Atlas. We show that in lung adenocarcinoma the accessibility perspective uniquely highlights immune pathways inversely correlated with a more open chromatin state and that accessibility patterns learned from even a single tumor type can discriminate immune inflammation across many cancers, often with direct relation to patient prognosis.
Subject Areas: Bioinformatics, Neural Networks, Cancer
Graphical Abstract
Highlights
-
•
Tissue-specific aspects of DNA can be predicted in new tissue types if given RNA-seq
-
•
DNA accessibility prediction is most reliable at promoter and promoter flank regions
-
•
Clustering global chromatin state highlights immune pathway activity in tumors
-
•
Accessibility patterns discriminate immune active tumors that can differ in prognosis
Bioinformatics; Neural Networks; Cancer
Introduction
DNA accessibility plays a key role in the regulatory machinery of DNA transcription. Locations where DNA is not tightly bound in nucleosomes, detectable as DNase I hypersensitivity sites (DHSs), render the sequence accessible to other DNA-binding proteins, including a wide range of transcription factors (TFs). DHSs are cell specific and play a crucial role in determining transcriptional events that differentiate cells.
Furthermore, genome-wide association studies (GWAS) have revealed that the vast majority of genetic variants significantly associated with many diseases and traits are located in non-coding regions (Deplancke et al., 2016) and well over half non-coding single nucleotide polymorphisms (SNPs) affect DHSs (Maurano et al., 2012). Thus variable access to DNA regulatory elements plays a key role not only in normal cell development but also in altered expression profiles associated with disease states (Deplancke et al., 2016, Xu et al., 2015), including cancer.
In an effort to go beyond association studies and gain deeper insight into how changes in DNA sequence affect transcriptional regulation, some groups have developed predictive models for a multitude of genomic phenomena. Several works have recently made significant advances in accuracy of such DNA-sequence-based prediction tasks by applying neural network models to problems such as TF binding (Alipanahi et al., 2015, Zhou and Troyanskaya, 2015, Quang and Xie, 2016, Lanchantin et al., 2016), promoter-enhancer interactions (Singh et al., 2016), DNA accessibility (Hoffman et al., 2018, Kelley et al., 2016, Zhou and Troyanskaya, 2015), and DNA methylation states (Angermueller et al., 2017, Hoffman et al., 2018).
One common issue that limits the broad applicability of these models is the cell-type-specific nature of many of the underlying biological mechanisms, such as DHSs. All the above examples approached their prediction problems by learning to estimate the conditional probability, , where is the accessibility (or other attributes) of a segment of DNA sequence, , and is a discrete label of tissue type. In practice, this meant either training a separate model for each cell or tissue type or having a single model output multiple tissue-specific (multi-task) predictions. This made it difficult to apply the models to new data and limits them from being integrated into broader scope pathway models (Vaske et al., 2010).
Conveniently, a number of studies have demonstrated that gene expression levels from RNA sequencing (RNA-seq) can be used to discriminate cell types (Sudmant et al., 2015, Breschi et al., 2016, Conesa et al., 2016), providing evidence that can be learned (where is a vector of RNA-seq gene expression measurements). In addition, DNase sequencing (DNase-seq) and microarray-based gene expression levels from matched samples were found to cluster similarly according to biological relationships, with many DHSs found to significantly correlate with gene expressions (Sheffield et al., 2013).
Our work focuses on overcoming the barrier to broad applicability due to cell-type-specific phenomena by putting the burden on a deep neural network classifier to handle the complex relationship between expression and DNA sequence accessibility without intermediate discrete tissue labels. Our model directly estimates , and thus implicitly handles the space of possible tissue types and states, , because . This allows the model to exploit similarity information in the space of tissue types and make predictions for previously unseen tissues whose gene expressions were similar but unique from samples in the training data.
We build on the Basset neural network architecture of Kelley et al., which recently demonstrated state-of-the-art results on DNA accessibility prediction (Kelley et al., 2016). They factored the cell-specific DHS issue into their work by first creating a binary matrix of sample tissues and their respective accessibility for a list of genomic sites. The universal list of (potentially accessible) sites was found by agglomerative clustering of all overlapping DNase-seq peaks across all samples before training. These potentially accessible sites defined the 600-base pair DNA segments used as inputs. Second, they set up the model's final layer as a multitask output, with a distinct prediction unit for each tissue type.
We began by showing that neural network performance on the original task of Kelley et al., predicting accessibility at held-out sites across 164 tissue types, could be improved by strategic factorization of convolutional layers. Then, based on our hypothesis that a neural network should be capable of learning to modulate appropriately its prediction of the DNase-seq signal if informed with a global RNA-seq state, we extended the network to handle a vector of gene expression values as input. To train this model, we constructed a new dataset of samples from the ENCODE project (ENCODE Project Consortium, 2012) consisting only of RNA-seq and DNase-seq measurements whose correspondence could be determined. Our new model achieved compelling results for held-out tissue types and proved to be very reliable at predicting accessibility at promoter and promoter flank genomic regions.
We then applied our accessibility prediction model to whole genomes across six cancer cohorts from The Cancer Genome Atlas (TCGA) (TCGA, 2018), as summarized in Figure 1A, and highlighted that accessibility complements RNA-seq. For example, clustering lung adenocarcinoma (LUAD) samples based on predicted accessibility was distinct from any RNA-seq-based cluster assignment and revealed a group of patients showing enrichment for pathways involved in immune response. Furthermore, splitting the same cohort by immune cell composition revealed a difference in survival and enabled training of a classifier to detect an immune-inflamed tumor state in LUAD, based only on accessibility predictions for a small set of sites. This classifier allowed us to discriminate immune-active patient groups with significant differences in survival in several distinct cancers, often aligning with findings from other cancer immunology studies. To the extent of our knowledge, this was the first time that a prediction of DNA accessibility had been applied to whole genomes in TCGA cancer cohorts to infer the chromatin landscape across cancers. In parallel with our work, accessibility was measured using assay for transposase-accessible chromatin using sequencing (ATAC-seq) for select samples across several TCGA cohorts (Corces et al., 2018), and we have shown that these new empirical results validate our predictions (Figure 7).
Figure 1.

Overview of Our Pipeline from Training to Application
(A) DHS, hg19 DNA, and RNA-seq information are all used to train the neural network. With tumor RNA-seq and DNA-seq data input the DNA accessibility model can then be used to predict chromatin state in tumors.
(B–D) (B) The neural network architectures for the tissue-specific baseline model, (C) the tissue-specific factorized convolutions model, and (D) the expression-informed model are shown. Depth (d) is provided for all fully connected (fc) layers. Convolution (conv) layers also list their width (w). Max pooling (mp) is indicated where present between layers and is always applied with equal size and stride (s).
See also Figures S1, S3, and S4.
Figure 7.

Validation of Our Promoter and Promoter Flank DNA Accessibility Predictions in TCGA with Empirical ATAC-Seq Measurements
(A) The top violin plots show the distributions of per ATAC-seq peak means of normalized counts in lung and kidney cohorts, for sites we labeled as constitutively (const.) accessible, facultative, or const. not accessible (based on our analysis shown in Figure 3D). Peak count values along all y axes were log transformed and quantile normalized as provided by the authors of the empirical study.
(B and C) Distributions of ATAC-seq peak normalized counts for all prediction sites across all available samples were further broken down per cohort by classification decision (accessible, , and not accessible, ) in addition to site category. Site categories were either facultative (facult.) or constitutive (const.), the latter including both const. accessible as well as const. not accessible. The number of TCGA samples that contributed to each plot is shown (N = ). (B and C) Only TCGA samples for which we had made predictions and were also empirically measured were used, but (A) utilized all available measured samples. The distribution plots were informed by N * 61,342 data points in (B and C), whereas for (A), where we considered the mean value for each site, there were only 61,342 data points total within each cohort.
We anticipate that our expression-informed model not only may provide detailed information regarding DNA accessibility across tissues and enable discrimination of immune-inflamed tumors but also might be used to predict individual patient response to various immune-based therapies. We also expect our approach to be a useful tool in understanding other conditions where chromatin state is suspected to play an important role, including aging (Moskowitz et al., 2017), neurodegenerative disease (Berson et al., 2018), autoimmune diseases (Farh et al., 2014), as well as autism (Zhao et al., 2018). Finally, we stress that our approach to implicitly handling tissue type and state can be used in any DNA-based prediction task.
Results
Convolutional Layer Factorization Improves Accuracy
As a baseline we first implemented our own version of the Basset architecture (Figure 1B), with minor changes not related to network structure. Compared with an receiver operating characteristic area under the curve (ROC AUC) = 0.895 reported by Kelley et al. on their benchmark test set (Kelley et al., 2016), and confirmed by applying the pre-trained model provided by the authors, our baseline implementation achieved a mean ROC AUC = 0.903 (Table 1).
Table 1.
Tissue-Specific Model Results on Basset Benchmark Dataset
| Tissue-Specific Model | Mean ROC AUC | Mean PR AUC |
|---|---|---|
| Basset (pre-trained) | 0.895 | 0.561 |
| Our baseline | 0.903 | 0.582 |
| Our factorized convolutions | 0.910 | 0.605 |
Following the success of many works demonstrating that deeper hierarchies of small convolutional kernels tend to improve neural network performance (Simonyan and Zisserman, 2014, Szegedy et al., 2016, He et al., 2016), we experimented with factorization of large convolutional layers in the baseline model. We found that factorization of layers closest to the data was the most significant for improving accuracy (Figure S1). When only the second convolutional layer was factorized, the speed of learning improved during the early epochs of training, but final accuracy was not noticeably affected compared with our baseline implementation. An overall improvement in both rate of learning and final accuracy was achieved when both the first and second convolutional layers were factorized (Figure 1C), leading to a mean ROC AUC = 0.910 (Table 1).
Furthermore, despite following the same training procedure and taking no additional steps to account for class imbalance, our final tissue-specific model's mean precision-recall area under the curve (PR AUC) = 0.605 compared favorably with the mean PR AUC = 0.561 reported as the best result obtained by the Basset model.
ENCODE DNase-Seq and RNA-Seq Dataset
To train a model for predicting accessibility that is informed implicitly about tissue state through gene expression it was necessary to build a new dataset where both DNase-seq and RNA-seq were available for a large and diverse collection of different tissue types. We collected all human samples from the ENCODE project (ENCODE Project Consortium, 2012) for which correspondence between RNA-seq and DNase-seq measurements could be determined. After errors were filtered out, the final dataset consisted of 74 unique tissue types, with 220 DNase-seq files and 304 RNA-seq files. A validation set of 5% randomly held-out samples was split from the data so that tissue types were diverse but still independent measurements of tissues also appeared in training.
Two sets of paired test and training partitions were created (Figure S2). The first partition pair (tissue overlap in Table 2) was constructed in the same way as the validation data by randomly holding out test samples and allowing for tissue type overlap with training. The second partition pair (held-out tissue in Table 2) was constructed such that the test set was composed only of samples from tissue types that were not present in either training or validation partitions. The latter was meant to more accurately simulate the intended application scenario and was thus the main focus throughout our analysis.
Table 2.
File- and Tissue-Type Distribution per Dataset Partition
| Partition | Unique Tissues | DNase-Seq Files | RNA-Seq Files |
|---|---|---|---|
| Validation | 10 | 11 | 12 |
| Tissue overlap train | 73 | 195 | 277 |
| Tissue overlap test | 12 | 14 | 15 |
| Held-out tissue train | 66 | 198 | 281 |
| Held-out tissue test | 8 | 11 | 11 |
The data partitions were revised once from their first iteration when several erroneous samples were discovered and revoked by the ENCODE consortium. Once revoked samples were removed, we saw a significant decrease in spurious DHSs. Table 2 shows the distributions of the final dataset.
Expression-Informed Model Can Predict Accessibility in Held-Out Tissues
We explored several alternative versions of our expression-informed neural network along with a range of different hyperparameters. Based on validation performance, we found that concatenating the global gene set expression vector directly with output from the convolutional layers, using a large batch size with appropriately matched learning rate, and weight initialization from a model trained using more noisy training data made the most impact. Changing the fraction of positive samples per training batch from 0.5 to 0.25 also led to a minor improvement.
Table S2 shows that final model (Figure 1D) performance on the validation set, both overall and by tissue type, was consistent across each of the two training partitions with respect to both ROC AUC as well as PR AUC. Tables 3 and 4 summarize the results of applying our model across whole genomes, at all potential DHSs. For tissue types with more than a single file pair in the test set, each sample's results are listed.
Table 3.
Tissue Overlap Whole-Genome Test Results, with Scores Computed across All Tissues in Bold
| Sample Tissue Type | ROC AUC | PR AUC |
|---|---|---|
| A172 | 0.959 | 0.721 |
| Left renal pelvis | 0.967 | 0.838 |
| Small intestine | 0.951, 0.926 | 0.737, 0.571 |
| Muscle of arm | 0.968 | 0.843 |
| Forelimb muscle | 0.968 | 0.808 |
| Keratinocyte | 0.939 | 0.644 |
| Skin fibroblast | 0.948, 0.947 | 0.770, 0.763 |
| Large intestine | 0.964 | 0.727 |
| Muscle of back | 0.967, 0.954 | 0.853, 0.854 |
| Adrenal gland | 0.957 | 0.743 |
| SK-N-DZ | 0.898 | 0.652 |
| Fibroblast of lung | 0.942 | 0.840 |
| Mean tissue type AUC | 0.950 | 0.758 |
| Overall AUC | 0.947 | 0.748 |
Table 4.
Held-Out Tissue Whole-Genome Test Results, with Scores Computed across All Tissues in Bold
| Sample Tissue Type | ROC AUC | PR AUC |
|---|---|---|
| Left kidney | 0.965 | 0.778 |
| OCI-LY7 | 0.899, 0.899, 0.886, 0.886 | 0.654, 0.654, 0.655, 0.654 |
| Prostate gland | 0.865 | 0.516 |
| Hindlimb muscle | 0.943 | 0.824 |
| Spleen | 0.913 | 0.582 |
| Astrocyte | 0.919, 0.944 | 0.787, 0.613 |
| Fibroblast of skin of abdomen | 0.964 | 0.826 |
| G401 | 0.739, 0.846 | 0.459, 0.516 |
| Mean tissue type AUC | 0.898 | 0.655 |
| Overall AUC | 0.897 | 0.621 |
As expected, overall the model was less accurate on completely new tissue types; however, even in the more challenging scenario, the overall PR AUC was higher than the best tissue-specific models evaluated on known tissue types. Note that several of the results in Table 4 were within similar ranges as predictions whose sample types overlapped with training.
Expression-Informed Model Predictions Are Highly Reliable at Promoter and Promoter Flank Genomic Sites
To better understand the performance characteristics and limitations of our model, we broke down our ENCODE validation and test results by genomic site type. Table 5 details the distribution of annotations applied to the 1.71 million sites considered in the held-out tissue training set, the percentage of all positive samples that fall within each annotation, and the percentage of samples per each annotation type that are positive. Note that a single site may overlap with more than one annotation.
Table 5.
Distribution of Potentially Accessible Sites by Annotation
| Site Type | % of All Sites | % of All Positive Examples | % per Site Type that Are Positive |
|---|---|---|---|
| Exon | 3.47 | 5.08 | 9.74 |
| Intragenic | 49.94 | 45.25 | 6.04 |
| Intergenic | 39.89 | 34.11 | 5.70 |
| Promoter and flank | 6.37 | 30.33 | 31.75 |
| Enhancer | 1.08 | 3.81 | 23.47 |
| Other | 5.39 | 5.20 | 6.43 |
We found that even for samples in which the model performed poorly overall (Figure 2C), predictions within promoter and promoter flank regions consistently attained a high level of accuracy (Figures 2A and 2D, Table 6), achieving a PR AUC = 0.839 over all held-out tissue types and a PR AUC = 0.911 over randomly held-out samples (validation set).
Figure 2.

Promoter and Promoter Flank Accessibility Is Highly Predictable, but Enhancers Show Variability
(A) Promoter flank (pf) accessibility is highly predictable (PR AUC = 0.839), as shown by the genomic site performance breakdown over all samples in the held-out tissues test set. The orange line indicates overall PR AUC computed across all test samples and all sites.
(B) No clear performance difference was observed when genomic sites across the held-out tissue test set were split into those that did (in L1000) and did not (non-L1000) overlap the L1000 RNA-seq input gene set. Note that not all sites overlapped with known gene regions, so the union of the L1000 and non-L1000 subsets did not always make up the complete set of sites of a certain type.
(C) Overall metrics separated by tissue type show that some held-out tissues in the test set were more challenging as reflected by lower AUCs.
(D) Predictions at enhancers were highly variable between samples, even with good PR AUC, and performance on pf regions remained consistently high, even for tissues where overall results were lowest.
Table 6.
Held-Out Tissue Test Results Restricted to Promoter and Promoter Flank Sites, with Scores Computed across All Tissues in Bold
| Sample Tissue Type | ROC AUC | PR AUC |
|---|---|---|
| Left kidney | 0.949 | 0.905 |
| OCI-LY7 | 0.869, 0.868, 0.864, 0.864 | 0.842, 0.842, 0.859, 0.859 |
| Prostate gland | 0.897 | 0.826 |
| Hindlimb muscle | 0.935 | 0.938 |
| Spleen | 0.867 | 0.782 |
| Astrocyte | 0.925, 0.914 | 0.946, 0.838 |
| Fibroblast of skin of abdomen | 0.951 | 0.929 |
| G401 | 0.798, 0.828 | 0.807, 0.757 |
| Overall AUC | 0.876 | 0.839 |
We also confirmed that the accuracy of these predictions was independent of whether the promoter and promoter flank sites overlapped with the regions of genes used in our RNA-seq input gene set (Figure 2B). Selecting a threshold for classification of only promoter and promoter flank sites such that precision is 80% (20% false discovery rate) on the held-out tissue test set, our trained model recalls 65.3% of accessible promoter regions, with a false-positive rate of 10%. Applying this same threshold to the validation set where tissues are allowed to overlap with the training set, the model achieves a precision of 93.4%, recalling 62.6% of accessible promoter regions, and has a false-positive rate of only 3.5%.
We also investigated the accuracy at enhancer sites, finding a PR AUC = 0.732 over held-out tissues and PR AUC = 0.889 over randomly held-out samples (validation set). Differently from promoter and promoter flank regions, however, enhancer prediction accuracies showed a high variance between test samples (Figure 2D, Table S3). Thus, more investigation is necessary before relying on accessibility predictions at enhancers in further analysis.
To quantify the effect of similarity to training data on prediction performance we looked at correlation between PR AUC (computed independently for all predictions in each whole genome sample) and distance of each test and validation sample to its closest sample in the training set (Table 7). As might be expected from Figure 2D, we confirm that prediction performance is less correlated with test sample similarity to training data at promoter and promoter flank sites than when PR AUC is evaluated over all potentially accessible sites.
Table 7.
Correlation of PR AUC with Test Sample Distance to the Nearest Training Sample
| PR AUC Evaluation Domain | Pearson Correlation | Pearson p Value | Spearman ρ | Spearman p Value |
|---|---|---|---|---|
| Overall | −0.7472 | 1.77 × 10−5 | −0.7080 | 7.52 × 10−5 |
| Promoter and flank | −0.6795 | 1.87 × 10−4 | −0.5417 | 5.16 × 10−3 |
Promoter Accessibility Patterns across Cohorts from The Cancer Genome Atlas
We applied our trained model to promoter and promoter flank sites in TCGA samples from six cohorts (LUAD, LUSC, KICH, KIRC, KIRP, and BRCA). Across all samples in these cohorts for which whole-genome sequencing (WGS) was available, 3,172 interest regions had one SNP, 78 had two SNPs, and only 9 regions had between three and five SNPs. A total of 465 sites included insertion or deletions (INDELs), and only 7 sites featured both an INDEL and an SNP. Lung cancers exhibited the highest average number of mutated sites per patient from our selected cohorts (Figure 3A).
Figure 3.

SNP and INDEL Mutations and Predicted Accessibility Landscape in Tumors
(A) The average number of SNP and insertion or deletion (INDEL) mutations that overlap prediction sites per patient across six TCGA cohorts is shown.
(B) When predictions at sites with mutations were compared with and without applying mutations to the input DNA sequence, the change in predicted accessibility exhibited a higher variance for INDELs than SNPs.
(C) In addition, a larger fraction of sites with INDELs were responsible for a change in the classification decision (flipped prediction) than the fraction of sites with SNPs.
(D) Using t-SNE (perplexity = 50) to visualize the predicted accessibility of individual promoter flank (pf) sites across our selected TCGA samples, we identified which sites were facultative (orange), constitutively accessible (blue), and constitutively not accessible (green).
(E and F) (E) Finally, t-SNE applied to patient samples exhibited different relationships (such as a clear split in BRCA samples) when based on RNA-seq gene expression of the L1000 gene set, than (F) when based on predicted accessibility at all pf sites within each sample (in which case lung and breast cancers appeared to share some common characteristics).
To observe the effect of region changes on accessibility, we compared predictions with and without SNPs and INDELs present. INDELs had the greatest impact on predicted accessibility, exhibiting a higher variance than SNPs (Figure 3B) and leading to a change in accessibility classification in 5.46% cases (at the previously defined accessibility threshold that achieved 80% precision) (Figure 3C). As there were generally few somatic mutations impacting accessibility prediction sites, and the percentage of those that actually impacted classifier decisions was even lower, we can conclude that any patterns we observed at the landscape scale of chromatin state predictions will be dictated more by gene expression levels providing global context of tissue state rather than somatic mutations.
To get a landscape view of how promoter and promoter flank sites behave, we embedded their binary accessibility decisions across all our selected TCGA samples in two dimensions with t-distributed stochastic neighbor embedding (t-SNE) (van der Maaten and Hintion, 2008). This clearly separated sites into constitutively accessible, constitutively not accessible, and facultative (Figure 3D). A few very small clusters of several hundred sites were also formed, which were groups of typically constitutive sites that acted uniquely in one or two individual patients.
Second, we stacked all predictions into a single vector per patient to form accessibility profiles for all samples in our six TCGA cohorts and again applied t-SNE to visualize relationships (Figure 3F). This qualitatively showed that looking at cancers from the viewpoint of DNA accessibility highlights different relationships than analysis of RNA-seq alone. For example, in the RNA-seq t-SNE space (Figure 3E) a clear separation emerges among breast cancers (BRCA), which correspond to basal-like versus luminal A/B and HER2-enriched clusters. In contrast, in accessibility t-SNE space (Figure 3F), the lung (LUAD, LUSC) and breast (BRCA) cancer samples appear to share some common characteristics. There also appears to be a slight partition into left and right groups in how lung cancer samples arrange in the embedding space, with LUAD forming a distinct subset away from the LUSC/BRCA modality within the lung/breast supercluster. Some similar clustering trends were observed in empirical ATAC-seq measurements performed concurrent to our work (Corces et al., 2018).
One of many potential biological factors that may contribute to overlap in the accessibility space is the impact of hormone activity on both breast and lung tumors; this activity in turn is epigenetically regulated (Zhang and Ho, 2011), thus some chromatin state patterns could be shared. Cell type composition is another factor that is likely to play a role in determining our observations of both expression and accessibility states of tissues; however, at this time we cannot isolate which biological confounders that determine tissue state contribute in what quantities to accessibility predictions.
Accessibility Is Linked to Immune Activity in Lung Adenocarcinoma
We subsequently explored the biological associations of our model's accessibility predictions by examining transcriptomic data from LUAD samples, as this tumor type has been shown to be of particular interest in chromatin accessibility studies due to the impact on progression (Polak et al., 2014, Kim and Kim, 2016). Upon clustering LUAD samples for which WGS was available according to their predicted accessibility, clear bifurcation into low- (C0, 21 samples) and high-accessibility (C1, 20 samples) samples was observed (Figure 4A). Cluster assignment based on predicted accessibility was distinct from any cluster assignments using the same methodology on RNA-seq directly (Figure 4B).
Figure 4.

Promoter and Promoter Flank Accessibility and Checkpoint Gene Expression in LUAD WGS Samples Only and Augmented with Non-WGS Samples
(A) The heatmap and patient sample cluster assignment based on the top 5% most variable promoter and promoter flank (pf) accessibility sites across LUAD samples with WGS available are shown. Cluster 0 (C0) has lower overall accessibility (blue = not accessible), and cluster 1 (C1) exhibits generally higher accessibility (red = accessible).
(B) Adjusted mutual information (AMI) (1) between label assignments based on different data shows higher values (red) between different RNA-seq cluster assignments and low values (blue) between accessibility (Access.) and clusters based on any other data type.
(C) Distribution of key checkpoint gene expression levels (with x axis sorted by significance of two-sided t test between C0 and C1) shows that the low-accessibility group tends to have higher checkpoint levels.
(D) Applying the same procedure to the full LUAD cohort, which also includes predictions for all non-WGS samples, we see a similar split into low- (C0) and high (C1)-accessibility groups.
(E) The same trend in checkpoint expression is observed, with FOXP3 again appearing as the most significant difference (two-sided t test with Benjamini-Hochberg adjusted p = 4.53 × 10−19).
(F) Plotting promoter and flank accessibility with respect to its first three principal components (PC1–3) and coloring points by total number of accessible sites in a sample reveals a smoothly varying relationship, motivating a correlation-based approach to exploring the relationship between overall accessibility and gene expression levels.
Differential KEGG pathway expression analysis with Enrichr (Kuleshov et al., 2016) showed the Chemokine Signaling Pathway (hsa04062) to be upregulated in the low DNA accessibility (C0) patient group. This association held true whether using TOIL RNA-seq data (Vivian et al., 2017; TOIL RNA-seq Recompute, 2016) (Enrichr adjusted p value [adj. p] = 1.191 × 10−6) or HiSeqV2 RNA-seq data (TCGA Genome Characterization Center UNC, 2017) (Enrichr adj. p = 0.0145) (see methods and Figure S5 for details). Chemokines are involved in multiple key processes in tumor growth and immune response (Nagarsheth et al., 2017, Rivas-Fuentes et al., 2015, Sarvaiya et al., 2013), and their regulation by epigenetic mechanisms has also previously been reported (Flavahan et al., 2017, Yasmin et al., 2015).
No difference in tumor mutation burden was found between the two clusters (two-sided t test: t = −0.696, p = 0.491), but interestingly the C0 group exhibited higher expression of immune checkpoint genes (Figure 4C). Cell type enrichment analysis (Aran et al., 2017) of lymphoids and myeloids also revealed a higher level of class-switched memory B cells (two-sided t test: t = 4.040, p = 0.000385, Benjamini-Hochberg [BH] adj. p = 0.0131) in C0, although estimated levels were generally low in both clusters. Other immune cell estimates exhibited no differences with adj. p < 0.1, which was largely limited by small sample set size.
Total Number of Accessible Sites Correlates with Activity in Immune Pathways
To enhance the scope of our findings, we extended our analysis to all LUAD patient samples for which WGS was not available by predicting accessibility using just the reference genome (hg19/GRCh37) and gene expression data. Although no mutation information was included for these additional samples, only 37 of 5,449 sites (6.79%) used to cluster all WGS data included any instances of mutations. With the additional consideration that only a small percentage of all mutations actually flip binary class predictions (Figure 3C), it is unlikely that cluster assignment of new non-WGS samples was significantly affected by this missing information. As before, the expanded set of patient samples was clustered into two groups according to accessibility.
The group with generally lower accessibility (C0) again exhibited generally higher checkpoint levels (Figures 4D and 4E). However, visualizing the first three principal components and coloring points by total number of accessible promoter and promoter flank sites (Figure 4F), we did find a smooth change in value along a continuous manifold of samples, primarily along the first principal component (Spearman correlation = 0.989, p = 0.0).
Therefore, instead of differential analysis, all protein-coding genes were filtered by correlation with the total number of accessible sites and evaluated for KEGG pathway enrichment. We found that all genes satisfying the threshold (correlation absolute value >0.4) had negative correlation values. As some relationships may not be linear but still monotonic, we focus on the Spearman measure (Table S4), although the Pearson measure yielded similar top pathways (Table S5). Osteoclast Differentiation (hsa04380, adj. p = 7.45 × 10−15) was the most significantly correlated pathway.
Interestingly, the process of osteoclast differentiation is controlled by two essential cytokines (Kim and Kim, 2016): macrophage colony stimulating factor and the receptor activator of nuclear factor (NF)-κB ligand. Tumor Necrosis Factor (TNF) Signaling Pathway (hsa04668, adj. p = 1.30 × 10−8) also appeared among the top three results. TNF has a pro-inflammatory effect and has been noted to play a critical role in the control of apoptosis, angiogenesis, proliferation, invasion, and metastasis (Yasmin et al., 2015).
When pathways were sorted by significance, the Chemokine Signaling Pathway, observed in WGS-only cluster analysis, appeared 11th (adj. p = 4.01 × 10−5). Other notable pathways appearing in the top 10 included Pathways in Cancer (hsa05200, adj. p = 6.50 × 10−7), Regulation of Actin Cytoskeleton (hsa04810, adj. p = 6.50 × 10−7), NF-κB Signaling Pathway (hsa04064, adj. p = 2.68 × 10−7), and Epstein-Barr Virus Infection (hsa05169, adj. p = 2.83 × 10−5). Focal Adhesion (hsa04510, adj. p value = 2.29 × 10−4) appeared 20th but is worth noting as it resurfaces in later analysis.
Majority of Genes with Differential Accessibility Exhibit Consistent Differential Expression in Immune Cell-Driven Clusters
To investigate accessibility patterns specifically in the context of different tumor immune environments, all LUAD samples were clustered into two groups according to lymphoid and myeloid levels based on xCell cell type enrichment analysis (Aran et al., 2017). Lymphoid and myeloid cells were selected for their roles in the adaptive and innate immune system, respectively. A thin margin was introduced between clusters to exclude samples with near-ambiguous label assignment (Figure S6).
Patients in X0 (141 samples) were enriched for many immune cells (Figure 5A), as well as checkpoint gene expression (Figure 5C), and tended to have narrower distributions of both number of accessible promoter and flank sites (generally lower than X1, two-sided t test p = 1.07 × 10−3) and total overall methylation (generally higher than X1, two-sided t test p = 1.29 × 10−7) (Figure 5B). These samples also reflected significantly favorable survival (Figure 5G). We therefore interpreted X0 as the group of “immune-hot” patients in LUAD and X1 as “immune-cold” patients.
Figure 5.

Enrichment in LUAD xCell-Derived Clusters (after Adding a Small Margin) by Cell Type, Checkpoint Expression, Methylation, Accessibility, and Survival
(A) Cell type enrichment distributions sorted by significance of two-sided t test for the two clusters (X0, X1), based on xCell lymphoid and myeloid cells, with Benjamini-Hochberg adjusted p value < 1.0 × 10−5 are shown.
(B) Total number of accessible promoter and promoter flank sites in each sample by cluster (two-sided t test p = 1.07 × 10−3) along with total methylation (two-sided t test p = 1.29 × 10−7).
(C) Checkpoint expression distributions, likewise sorted by significance, also point to a general difference in immune landscape between the two groups.
(D and E) (D) All sites with differences in accessibility based on a two-sided t test with Benjamini-Hochberg adjusted p values < 0.01 and (E) < 1.0 × 10−5 are illustrated on the t-SNE plot of promoter and promoter flank facultative sites. Sites with a difference satisfying the thresholds were assigned to the cluster in which they were more accessible.
(F) Accessibility differences are further broken down by how they align with direction of upregulation of corresponding nearby genes (ns gene, no significant difference in matching gene; consistent, direction of significant accessibility and gene expression differences are consistent; inconsistent, direction of significant accessibility and gene expression are inconsistent).
(G) Kaplan-Meier plots demonstrate better survival among X0 (immune hot) patients, shown with log rank test p value and hazard ratio (HR) based on a Cox proportional hazards (CoxPH) model regression using class assignment as the only explanatory variable.
See also Figures S6 and S7, and Tables S4–S7.
After eliminating sites that exhibited low standard deviation, we selected all significantly differentiated accessibility sites between the two clusters and mapped them to their nearest gene. Qualitatively, we observed that several groups of sites act together in different ways and that those different clusters of chromatin state behavior are stable across significance thresholds (Figures 5D and 5E). We found that when a majority of sites corresponding to a single gene were accessible more frequently in one cluster, that gene exhibited upregulated expression in the same cluster most of the time (64.7% for genes more accessible in X0 and 64.2% in X1).
Genes whose expression was consistent with increased accessibility in X0 showed near-significant levels of enrichment for some pathways that had previously surfaced in our correlation results such as Focal Adhesion (hsa04510, adj. p = 0.0355) and Osteoclast Differentiation (hsa04380, adj. p = 0.0936) (Table S6). No significant pathways were found for genes consistent with increased accessibility in X1, or those inconsistent with more accessibility in X0. The strongest significance in pathway enrichment existed in the set of genes inconsistent with increased accessibility in X1 (up in X0 despite accessibility predictions voting for upregulation in X1) (Table S7). The most prominent of the enriched pathways in this group were Platelet Activation (hsa04611, adj. p = 4.38 × 10−4), Inflammatory Mediator Regulation of TRP Channels (hsa0475, adj. p = 0.0109), and several with adj. p = 0.0235: Chemokine Signaling Pathway (hsa04062), Focal Adhesion (hsa04510), cGMP-PKG Signaling Pathway (hsa04022), Intestinal Immune Network for IgA Production (hsa04672), and Vascular Smooth Muscle Contraction (hsa04270).
These findings suggest that in LUAD tumors, partial regulation of immune- and cytokine-controlled pathways may be exerted via an activator mechanism at promoters. Furthermore, a more significant component of chemokine signaling and platelet activation that distinguishes immune-active patients may be subject to repressor regulation at promoter sites.
Patterns of Promoter Accessibility Predict Immune-Hot Tumors with Impact on Patient Survival across Several Cancers
To further explore the link between DNA accessibility, immune activity, and clinical outcomes, we trained an ensemble of three classifiers to detect an immune-hot tumor state in LUAD based only on a small subset of accessibility predictions. Applying the ensemble to all of LUAD (Figure 6A) led to a cleaner and more significant partition of patients (compared to Figures 5F and S6F) into immune-hot and immune-cold tumors. Further applying the classifier ensemble to accessibility predictions across 11 other cancers in TCGA revealed cases wherein the immune-hot state learned from LUAD was beneficial to patient survival (Figures 6A–6E), detrimental to survival (Figures 6J and 6K), or had little impact (Figures 6F–6I and 6L), with varying degrees of significance.
Figure 6.

Application of the Three SVM Ensembles for Classification of Immune-Hot Tumors (Trained on Subsets of LUAD) with the Only Input Being a Vector of 484 Promoter and Flank Predicted Accessibility Decisions
All Kaplan-Meier plots show group size (N) for patients of both predicted immune activity classes (hot/cold) that satisfy a confidence threshold (see Transparent Methods). Also provided are log rank test p values and hazard ratio (HR) based on a Cox proportional hazards (CoxPH) model regression using class assignment as the only explanatory variable. Note that the time axis range on subplots varies by cohort and that the immune-hot state learned based on LUAD is not always beneficial for patient survival in other tumor types. Tumor types included (A) LUAD, lung adenocarcinoma; (B) SKCM, skin cutaneous melanoma; (C) SARC, sarcoma; (D) BRCA, breast invasive carcinoma; (E) CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; (F) HNSC, head and neck squamous cell carcinoma; (G) LUSC, lung squamous cell carcinoma; (H) BLCA, bladder urothelial carcinoma; (I) GBM, glioblastoma multiforme; (J) KIRC, kidney renal clear cell carcinoma; (K) LGG, brain lower grade glioma; and (L) STAD, stomach adenocarcinoma.
See also Figure S8.
Our findings aligned very well with a comprehensive analysis of immune subtypes across TCGA (Thorsson et al., 2018), which characterized the influence of immune activations on survival. Despite very different methodologies, their plots also indicated that in cohorts such as LUAD, SKCM, and CESC activation of immune subtypes was associated with better outcomes; that the opposite was true in KIRC, LGG, and STAD; and that little impact was visible in LUSC. They did not, however, discuss accessibility as a potential additional biomarker for immune activity.
Significant negative impact of immune activity on survival in KIRC was also shown in a separate cohort of clinical data from Oulu University Hospital (Mella et al., 2015), confirming that this trend is not unique to TCGA. Interestingly, the study used CD8+ T cell count cutoffs to stratify patients with renal cell carcinoma into two groups. Based on xCell estimates this was the second most significantly enriched immune cell type (two-sided t test BH adj. p = 3.57 × 10−26) in KIRC immune-hot patients identified by our classifier, after activated dendritic cells (two-sided t test BH adj. p = 1.07 × 10−26) (Figure S8). Although the training cohort (LUAD) did express some difference in CD8+ T cell enrichment scores between hot and cold tumors (two-sided t test BH adj. p = 9.65 × 10−15), it was not in the top 10 most significantly different immune cells (Figure 5A). From this we see that our classifiers operating on accessibility predictions learned a more complex decision boundary than simply focusing on direct correlates with the most differentiated immune cell compositions in the training set.
An additional curiosity specific to the KIRC partition from our classifier ensemble is that little difference in CD274 (also called PD-L1) and PDCD1LG2 (also called PD-L2) expression is visible between the two predicted classes; however, the strong difference in PDCD1 (also called PD-1) expression levels that exists in other immune-hot versus immune-cold partitions does exist. This unique state of checkpoint-related gene expression may be linked to the low response rate in patients with renal cell carcinoma to anti-PD-L1 therapies, compared with more favorable responses found for anti-PD-1 drugs, in early-phase clinical trials (Weinstock and McDermott, 2015). Interestingly, an empirical study of ATAC-seq peaks across cancers (Corces et al., 2018) also found a link between four regulatory regions that exhibited distinct chromatin accessibility patterns across cancers and expression of CD274.
ATAC-Seq Measurements Validate Predictions in TCGA Samples
In parallel to our analysis, the chromatin state of select samples across several TCGA cohorts was empirically measured using ATAC-seq (Corces et al., 2018). At a minimum overlap threshold of 70% we found that 10.9% (61,342 of 562,709) of all pan-cancer peaks identified in the study corresponded directly with 56.3% of our (108,970) promoter and promoter flank sites at which we applied our model. At lower peak overlap thresholds this percentage increased significantly; for example, at 10% minimum overlap 83.6% of our promoter and promoter flank sites had corresponding pan-cancer ATAC-seq peaks. For correspondences defined by the 70% overlap threshold we first showed that the means of normalized count values for individual ATAC-seq peaks across lung and kidney cohorts had clearly distinct distributions (Figure 7A) between constitutive and facultative site categories (identified based on clustering our TCGA predictions, Figure 3D). Constitutive sites had consistently high mean peak counts in the accessible category and consistently low mean peak counts in the not accessible category, whereas facultative sites corresponded to ATAC-seq peaks with a broader distribution of mean normalized count values centered between the previous two categories (Figure 7A).
We then explored the distributions of ATAC-seq peak counts as stratified directly by our accessibility classifier predictions in addition to the above site categories (Figures 7B and 7C). In this case no means were computed across samples; every prediction site in every TCGA sample was considered as one data point. Normalized counts for peaks corresponding to sites predicted as accessible were generally distributed at higher values than peaks corresponding to sites predicted as not accessible, and the difference between predicted accessibility distributions was significantly more striking at constitutive sites than at facultative sites. This observation held true for all cohorts used in our immune classification experiments (Figure 6) for which there existed TCGA samples with both ATAC-seq measurements and our accessibility predictions. This result suggests that chromatin sites whose accessibility changes dynamically within a tissue type tend to remain closer to an accessibility threshold compared with less dynamic sites. The distributions did differ some in shape between cohorts, which may partly be explained by the fact that our constitutive and facultative site category labels used for this experiment were derived only based on predictions in lung and kidney cohorts; thus in different tissues wherein the dynamics of chromatin slightly differ those particular labels may not be as representative.
Discussion
We have demonstrated that predictive models operating on DNA sequence data, additionally conditioned on a global set of RNA-seq gene expression inputs, can predict DHSs in unseen tissue types in a way that allows application to new samples without re-training. We showed that these models were capable of achieving consistently high performance for predictions at promoter and promoter flank regions of the genome. Leveraging this new tool for analysis of tumor genomes across different cell and tissue types, we provided a unique perspective on the DNA accessibility landscape across TCGA data. Complementary to the exploration of the full range of variable accessibility sites across cancers made possible by empirical measurements (Corces et al., 2018), our analysis of sites at reliably predictable genomic regions explored a more limited and thus more subtle set of chromatin dynamics, which proved to still be very information rich. Despite the more limited view of accessibility sites in our case, both studies found some similar clustering trends and both concluded that chromatin state plays a significant role in cancer immune response.
DNA accessibility is one of many factors that determine expression, which makes inversion of the relationship not trivial; knowing expression levels does not uniquely define the pattern of DHSs. Our expression-informed model (Figure 1D) learns a most likely mechanism by which the DNA sequence immediately surrounding a potential DHS determines its accessibility, conditioned also on an observed global expression state. Therefore, accessibility prediction applied across the whole genome is an approach to approximately invert gene expression to obtain most likely DHSs.
Our results showed that viewing tumors by promoter accessibility highlights immune pathways that would otherwise be harder to detect from completely unsupervised analysis of RNA-seq data alone. For example, we found several pathways inversely correlated with an overall more open chromatin state. Through identification of facultative accessibility sites linked with differential gene expression in immune-inflamed LUAD tumors and training of a classifier ensemble, we showed that patterns of predicted chromatin state at a small subset of genomic regions are predictive of immune activity across many tumor types, with direct implications for patient prognosis. We see such predictive models playing a significant future role in matching patients to appropriate immunotherapy treatment regimens, as well as in analysis of other conditions wherein epigenetic state may play a significant role, such as autoimmune disease, autism, aging, and neurodegenerative disease.
It may also be interesting to pursue a deeper functional investigation of genes linked with accessibility. Genes with consistent behavior to accessibility are candidates that may be regulated via an activator mechanism at promoters, whereas genes with inconsistent behavior may be subject to alternative gene repression mechanisms, e.g., silencer elements or suppression via microRNAs.
In a few TCGA cohorts, our ensemble classification approach only identified a very small number of immune-hot tumor samples, making survival analysis impossible. The generalizability of our immune-related chromatin state across cancers was undoubtedly limited by only having trained the support vector machines (SVMs) on a single cohort, because immune cell composition and definition of an immune-active state varies across cancers (Thorsson et al., 2018); going forward, we will integrate accessibility signatures from multiple cohorts to train a more comprehensive subtyping of immune state.
Ideally, WGS for each of the samples in our training dataset should have been used to learn the most faithful representation of the true biology, as using only reference genome data introduces non-random noise in the input space. Unfortunately, such individual whole-genome data were not available for this project. Nonetheless, our work and that of others demonstrates that useful predictors can be learned despite this noise. Unlike models with multitask outputs, our architecture can easily support such individualized training without any changes, and when possible, it will be instituted in the future.
We saw high variance for enhancer sites, but these sites are also interesting with respect to chromatin state and immunotherapy, because they have been linked with T cell dysfunction with potential for therapeutic reprogrammability in mice (Philip et al., 2017). At this time, it needs to be determined whether the large variance in performance is due to limitations in the model, noise in the data, or lack of necessary information in the available inputs. To this end, we look forward to future exploration of a more complete set of genes instead of a manually curated set, such as the LINCS L1000. Many alternatives exist to learn gene embeddings as part of model training, and we believe that ultimately an approach that efficiently incorporates all genes as input will be most effective.
Furthermore, there are a multitude of alternative model architectures such as residual connections (He et al., 2016), densely connected convolutional networks (Huang et al., 2016), and recurrent neural networks (Hochreiter and Schmidhuber, 1997) with additions such as attention (Bahdanau et al., 2014, Xu et al., 2015), which we believe are likely to improve performance of our model. These have been left for future evaluation, such as one rigorous study that has independently verified and built on our architectural innovations (Nair et al., 2019). The key contribution of this work was movement beyond the cell-type-specific limitations of DNA sequence classifiers, demonstration of the application of our expression-informed model to predict accessibility, and the ability of these predictions to distinguish prognostically alternative immune states across human cancers.
Limitations of the Study
By design, convolutional neural networks only capture the influence of a small local neighborhood of DNA sequence (600 base pairs in our experiments) on predicted outputs, so impacts from more distal mutations on potential DNA accessibility sites can be captured implicitly only if they happen to influence expression levels of input of the global RNA-seq gene set. True biological function of DNA sequence may not be fully captured due to reliance on reference genome as a proxy for WGS in all training samples. At genomic regions where predictions demonstrate consistently good accuracy, we suspect that a fair amount of noise due to this approximation has averaged out over training data. Regions at which prediction accuracies have high variance across samples, such as enhancers, may be hard to predict for this reason, or other limitations with the training data or model assumptions. Both DNase-seq and RNA-seq measurements are taken from tissue samples, which feature heterogeneous cell type populations of varying proportions. The addition of RNA-seq data has enabled models to implicitly handle some degree of this type of noise; however, due to the massive possible permutations in how such variations can manifest, trained models may not perform well in test cases where tissue types, compositions, or the RNA-seq expression quantification pipeline are drastically different than samples seen in training.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Patricia Spilman for editing support and Hermes Garban for discussions regarding the biology of chromatin and regulation at promoters.
Author Contributions
K.W. conceived the project, curated the data, developed the model, performed analysis, and wrote the manuscript; J.S. contributed to data processing software; K.B.G. conceived and performed initial clustering analysis in LUAD and contributed to writing and editing; P.S.-S. secured funding; S.R. suggested RNA-seq as a suitable tissue-type signature; S.R., C.S., and C.V. provided cancer bioinformatics expertise and feedback and contributed to editing; C.V. and C.S. provided TCGA mutation calls.
Declaration of Interests
This work was funded by NantWorks affiliates (ImmunityBio, NantOmics, NantHealth) and performed by its employees; there are no other conflicts of interest.
Published: October 25, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.09.018.
Supplemental Information
References
- Alipanahi B., Delong A., Weirauch M.T., Frey B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015;33:831–838. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- Angermueller C., Lee H.J., Reik W., Stegle O. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017;18:67. doi: 10.1186/s13059-017-1189-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D., Hu Z., Butte A.J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahdanau D., Cho K., Bengio Y. Neural machine translation by jointly learning to align and translate. arxiv.org. 2014 arXiv:1409.0473. [Google Scholar]
- Berson A., Nativio R., Berger S.L., Bonini N.M. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 2018;41:587–598. doi: 10.1016/j.tins.2018.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breschi A., Djebali S., Gillis J., Pervouchine D.D., Dobin A., Davis C.A., Gingeras T.R., Guigo R. Gene-specific patterns of expression variation across organs and species. Genome Biol. 2016;17:151. doi: 10.1186/s13059-016-1008-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A., Madrigal P., Tarazona S., Gomez-Cabrero D., Cervera A., Mcpherson A., Szczesniak M.W., Gaffney D.J., Elo L.L., Zhang X., Mortazavi A. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13. doi: 10.1186/s13059-016-0881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R., Granja J.M., Shams S., Louie B.H., Seoane J.A., Zhou W., Silva T.C., Groeneveld C., Wong C.K., Cho S.W. The chromatin accessibility landscape of primary human cancers. Science. 2018;362 doi: 10.1126/science.aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplancke B., Alpern D., Gardeux V. The genetics of transcription factor DNA binding variation. Cell. 2016;166:538–554. doi: 10.1016/j.cell.2016.07.012. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh K.K.-H., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R.J.H., Shishkin A.A. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2014;518:337. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, K., Zhang, X., Ren, S. & Sun, J. 2016. Deep residual learning for image recognition. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, 770–778.
- Flavahan W.A., Gaskell E., Bernstein B.E. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357 doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter S., Schmidhuber J. Long short-term memory. Neural Comput. 1997;9:1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- Hoffman G.E., Schadt E.E., Roussos P. Functional interpretation of genetic variants using deep learning predicts impact of the epigenome. bioRxiv. 2018 doi: 10.1093/nar/gkz808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G., Liu Z., Weinberger K.Q., van der Mateen L. Densely Connected Convolutional Networks. arxiv.org. 2016 arXiv:160806993. [Google Scholar]
- Kelley D.R., Snoek J., Rinn J.L. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016;26:990–999. doi: 10.1101/gr.200535.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.H., Kim N. Signaling pathways in osteoclast differentiation. Chonnam Med. J. 2016;52:12–17. doi: 10.4068/cmj.2016.52.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanchantin J., Singh R., Lin Z., Qi Y. Deep motif: visualizing genomic sequence classifications. 2016. arXiv.org arXiv:1605.01133v2 [cs.LG], 1–5.
- van der Maaten L., Hintion G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mella M., Kauppila J.H., Karihtala P., Lehenkari P., Jukkola-Vuorinen A., Soini Y., Auvinen P., Vaarala M.H., Ronkainen H., Kauppila S. Tumor infiltrating CD8(+) T lymphocyte count is independent of tumor TLR9 status in treatment naïve triple negative breast cancer and renal cell carcinoma. Oncoimmunology. 2015;4:e1002726. doi: 10.1080/2162402X.2014.1002726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz D.M., Zhang D.W., Hu B., Le Saux S., Yanes R.E., Ye Z., Buenrostro J.D., Weyand C.M., Greenleaf W.J., Goronzy J.J. Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol. 2017;2:eaag0192. doi: 10.1126/sciimmunol.aag0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarsheth N., Wicha M.S., Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017;17:559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S., Kim D.S., Perricone J., Kundaje A. Integrating regulatory DNA sequence and gene expression to predict genome-wide chromatin accessibility across cellular contexts. bioRxiv. 2019;605717 doi: 10.1093/bioinformatics/btz352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M., Fairchild L., Sun L., Horste E.L., Camara S., Shakiba M., Scott A.C., Viale A., Laeur P., Merghoub T. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P., Lawrence M.S., Haugen E., Stoletzki N., Stojanov P., Thurman R.E., Garraway L.A., Mirkin S., Getz G., Stamatoyannopoulos J.A., Sunyaev S.R. Reduced local mutation density in regulatory DNA of cancer genomes is linked to DNA repair. Nat. Biotechnol. 2014;32:71–75. doi: 10.1038/nbt.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D., Xie X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016;44:e107. doi: 10.1093/nar/gkw226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas-Fuentes S., Salgado-Aguayo A., Pertuz Belloso S., Gorocica Rosete P., Alvarado-Vasquez N., Aquino-Jarquin G. Role of chemokines in non-small cell lung cancer: angiogenesis and inflammation. J. Cancer. 2015;6:938–952. doi: 10.7150/jca.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarvaiya P.J., Guo D., Ulasov I., Gabikian P., Lesniak M.S. Chemokines in tumor progression and metastasis. Oncotarget. 2013;4:2171–2185. doi: 10.18632/oncotarget.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield N.C., Thurman R.E., Song L., Safi A., Stamatoyannopoulos J.A., Lenhard B., Crawford G.E., Furey T.S. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res. 2013;23:777–788. doi: 10.1101/gr.152140.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonyan K., Zisserman A. Very deep convolutional networks for large-scale image recognition. 2014. ArXiv.org arXiv:1409.1556 [cs.CV]
- Singh S., Yang Y., Poczos B., Ma J. Predicting enhancer-promoter interaction from genomic sequence with deep neural networks. bioRxiv. 2016;85241 doi: 10.1007/s40484-019-0154-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant P.H., Alexis M.S., Burge C.B. Meta-analysis of RNA-seq expression data across species, tissues and studies. Genome Biol. 2015;16:287. doi: 10.1186/s13059-015-0853-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegedy, C., Vanhoucke, V., Ioffe, S., Shlens, J. & Wojna, Z. 2016. Rethinking the inception architecture for computer vision. 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), 2818–2826.
- TCGA Genome Characterization Center UNC . 2017. Dataset: gene expression RNAseq - IlluminaHiSeq - TCGA.LUAD.sampleMap/HiSeqV2. [Google Scholar]
- TCGA The Cancer Genome Atlas Program. 2018. https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga
- Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.-H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A. The immune landscape of cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toil RNA-seq Recompute, U. C. S. C. TCGA Pan-Cancer (PANCAN) - Gene Expression RNAseq - TOIL RSEM tpm. 2016. https://xenabrowser.net/datapages/?dataset=tcga_RSEM_gene_tpm&host=https://toil.xenahubs.net
- Vaske C.J., Benz S.C., Sanborn J.Z., Earl D., Szeto C., Zhu J., Haussler D., Stuart J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivian J., Rao A.A., Nothaft F.A., Ketchum C., Armstrong J., Novak A., Pfeil J., Narkizian J., Deran A.D., Musselman-Brown A. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017;35:314–316. doi: 10.1038/nbt.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock M., McDermott D. Targeting PD-1/PD-L1 in the treatment of metastatic renal cell carcinoma. Ther. Adv. Urol. 2015;7:365–377. doi: 10.1177/1756287215597647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K., Ba J., Kiros R., Cho K., Courville A., Salakhudinov R., Zemel R., Yoshua B. Show, attend and tell: neural image caption generation with visual attention. Int. Conf. Mach. Learn. 2015;37:2048–2057. [Google Scholar]
- Yasmin R., Siraj S., Hassan A., Khan A.R., Abbasi R., Ahmad N. Epigenetic regulation of inflammatory cytokines and associated genes in human malignancies. Mediators Inflamm. 2015;2015:201703. doi: 10.1155/2015/201703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Ho S.M. Epigenetics meets endocrinology. J. Mol. Endocrinol. 2011;46:R11–R32. doi: 10.1677/jme-10-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.T., Kwon D.Y., Johnson B.S., Fasolino M., Lamonica J.M., Kim Y.J., Zhao B.S., He C., Vahedi G., Kim T.H., Zhou Z. Long genes linked to autism spectrum disorders harbor broad enhancer-like chromatin domains. Genome Res. 2018;28:933–942. doi: 10.1101/gr.233775.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Troyanskaya O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015;12:931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.