

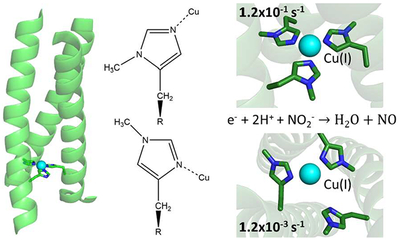
Abstract
Copper proteins have the capacity to serve as both redox active catalysts and purely electron transfer centers. A longstanding question in this field is how the function of histidine ligated Cu centers are modulated by δ vs ε-nitrogen ligation of the imidazole. Evaluating the impact of these coordination modes on structure and function by comparative analysis of deposited crystal structures is confounded by factors such as differing protein folds and disparate secondary coordination spheres that make direct comparison of these isomers difficult. Here, we present a series of de novo designed proteins using the noncanonical amino acids 1-methyl-histidine and 3-methyl-histidine to create Cu nitrite reductases where δ- or ε-nitrogen ligation is enforced by the opposite nitrogen’s methylation as a means of directly comparing these two ligation states in the same protein fold. We find that ε-nitrogen ligation allows for a better nitrite reduction catalyst, displaying 2 orders of magnitude higher activity than the δ-nitrogen ligated construct. Methylation of the δ nitrogen, combined with a secondary sphere mutation we have previously published, has produced a new record for efficiency within a homogeneous aqueous system, improving by 1 order of magnitude the previously published most efficient construct. Furthermore, we have measured Michaelis–Menten kinetics on these highly active constructs, revealing that the remaining barriers to matching the catalytic efficiency (kcat/KM) of native Cu nitrite reductase involve both substrate binding (KM) and catalysis (kcat).
Graphical Abstract
INTRODUCTION
Copper is an essential metal for life whose relatively easily accessed redox chemistry fulfills roles ranging from electron transfer (ET), oxygen binding and activation, and nitrite or nitrous oxide reduction.1–3 This wide range of functionality also comes with a wide range of coordination environments, resulting in three predominant classifications of copper proteins. Type 1 copper proteins are typified by blue copper proteins such as plastocyanin with high intensity low energy ligand-to-metal charge-transfer (LMCT) bands and compressed hyperfine coupling constant due to a uniquely covalent Cu–S bond from Cys ligation.4–10 Type 1 (T1) copper proteins largely function as electron transfer centers though some research indicates their involvement in nitric oxide sensing.11–13 Type 3 (T3) copper proteins contain dinuclear coupled copper centers, each ligated by three His residues and commonly function as oxygen binders or activators.1,14 Type 2 (T2) copper proteins, the focus of this work, were first grouped due to their lack of unique spectroscopic features, having “normal” copper EPR spectra and lacking intense electronic absorption features.1,15 T2 copper centers are often His3 coordinated but include all mononuclear Cu sites lacking a ligated sulfur. Perhaps due to this variety in ligation, T2 copper centers have been found to encompass a large variety of functions.3,14,15
Our lab is particularly interested in investigating the structural component of differing functions within T2 copper centers. These extremes of function within T2 Cu sites are well exemplified by comparing Cu only nitrite reductase (CuNiR) and peptidylglycine alpha-hydroxylating monooxygenase (PHM; Figure 1).16–18 While both enzymes contain T2 Cu sites with His3 coordination, their function differs drastically. CuNiR’s T2 site is capable of reducing nitrite via electron transfer from a T1 Cu site,19,20 while PHM’s His3 T2 site (CuH) acts to transfer electrons to the CuM active site.21 Examination of CuNiR and PHM crystal structures through databases such as MetalPDB indicates that the differences between these two constructs arise from variations in histidine ligation, namely that the His residues of PHM bind through the Nδ nitrogen while those of CuNiR bind through Nε.22 A previous study comparing crystal structures of metalloproteins deposited in the PDB led to the hypothesis that metal ligation through Nδ, which is more often seen in electron transfer sites such as T1 copper proteins, may increase the stability of the coordination sphere and decrease reorganization energy, while Nε ligation, found largely in metalloprotein active sites, allows for greater flexibility and improved catalytic efficiency.23 It is also possible that more direct differences such as the greater basicity of the Nε position may be the origin for this disparity in functionality.24
Figure 1.

Pymol representations of (left) Cu nitrite reductase (PDB: 4YSE) compared to those of (right) peptidylglycine alpha-hydroxylating monooxygenase (PHM; PDB: 1PHM).
Directly comparing the native proteins in question is difficult or even impossible due to large structural differences between CuNiR and PHM (Figure 1). Previously, our lab has shown that a Cu(His)3 active site can be designed within the TRI system of three stranded coiled coil (3SCC) peptides.25 Using this construct and the noncanonical amino acids δmHis and εmHis (histidine methylated at the δ- or ε-nitrogen respectively, depicted in Figure 2) we have explored the effect of Nδ or Nε ligation within variations of this de novo protein system to circumvent the complications that come with making such comparisons within native systems. This is the first systematic study of the effect of tris-Nδ vs tris-Nε ligation on the structural and electronic properties of both Cu(I) and Cu(II) within a single protein scaffold. Through a variety of spectroscopic techniques we determine that enforcing Nδ or Nε ligation affects the coordination number of the complex as well as the NiR activity increases 100-fold in δNmetHis compared to εNmetHis. It is difficult to disentangle the electronic and structural changes of Nδ or Nε ligation. One would expect not only electronic differences between Nδ or Nε ligation, but steric differences that lead to the observed change in coordination geometry. However, previous studies indicate that coordination number of the Cu(I) complex of our constructs is unlikely to cause an activity change and we are thus confident that this activity enhancement is due to Nδ or Nε electronic differences.
Figure 2.

Chemical structures of (left) histidine, (middle) δmHis (N(pros)-methyl-l-histidine), or 3-methyl-l-histidine, and (right) εmHis (N(tau)-methyl-l-histidine or 1-methyl-l-histidine).
While methylated histidine-like Cu ligands have been utilized extensively in small molecule studies, their use in protein design has been limited to studying the incorporation of δ or ε-methylated histidine in amyloid-β to investigate whether bridging His ligands between two Cu(II) ions were a factor in fibril formation.26–28 While lower neurotoxicity was observed for both delta and epsilon methylated Histidine, it was also found that methylated His amyloid-β peptides have higher efficiency in hydrogen peroxide generation than standard amyloid-β. This was explained by the bridging His of amyloid-β acting as an inhibitor to H2O2 production.
The only example of native mHis bound to Cu can be found in the polysaccharide monooxygenase (PMO) family of enzymes secreted by several varieties of fungi.29–32 The unique copper binding site of this protein family contains not only a mHis but also a “histidine brace” motif due to binding of both the N-terminal mHis residue’s amine and side chain. The purpose of including a mHis residue in the binding site is currently unknown; however, based on the results of the amyloid-β study referenced above, this may serve to increase the efficiency of the enzyme as compared to a His derivative.
We have previously employed a variety of strategies to maximize the catalytic efficiency of nitrite reduction in a series of functional de novo CuNiR catalysts. Toward this end, our approach has included mutations to outer sphere residues with little effect on NiR activity and second coordination sphere mutations which successfully increased our system’s efficiency to the highest for any aqueous model of CuNiR.33,34 The latter study used mutations of the hydrophobic layers above or below the CuHis3 binding site to Asp or Ala to enhance NiR efficiency up to 75 times. In the present work we demonstrate that Cu(I)(TRIW-δmH)3 displays an increased efficiency of 300 times that of Cu(I)(TRIW-H)3 (Table 1). TRIδmHis L19A was created to test if these two strategies could be combined and whether their effects on activity were additive. Finally, Michaelis Menten kinetic studies were performed for Cu(I)(TRIW-δmH)3 and Cu(I)(TRIW-δmH L19A)3 to explore the activity enhancement of these constructs relative to Cu(I)(TRIW-H L19A)3. These studies not only further our understanding of how one could improve de novo CuNiR catalysts but have produced the most efficient nitrite reductase model within an aqueous medium with a pseudo first order rate constant of 0.32 s−1, an enhancement from our first model of over 700-fold.
Table 1.
TRI Family Peptide Sequences Used in This Studya
| peptide | 1 | 2 | 9 | 16 | 23 | 30 |
|---|---|---|---|---|---|---|
| TRIW-H | Ac-G | WKALEEK | LKALEEK | LKALEEK | HKALEEK | G-NH2 |
| TRIW-δmH | Ac-G | WKALEEK | LKALEEK | LKALEEK | δmHKALEEK | G-NH2 |
| TRIW-εmH | Ac-G | WKALEEK | LKALEEK | LKALEEK | εmHKALEEK | G-NH2 |
| TRIW-δmH L19A | Ac-G | WKALEEK | LKALEEK | LKAAEEK | δmHKALEEK | G-NH2 |
The C- and N-termini of the peptides are amidated and acetylated, respectively. The first residue within each heptad is labeled with its overall position within the peptide. δmH and εmH represent the delta and epsilon nitrogen methylated forms of histidine, respectively.
METHODS
Peptide Synthesis.
Fmoc protected amino acids and MBHA rink amide resin were purchased from Novabiochem except Fmoc-1-methyl-l-histidine and Fmoc-3-methyl-l-histidine which were purchased from Millipore. Hydroxybenzotriazole (HOBt) and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from Anaspec while diisopropylethylamine (DIEA), acetic anhydride, and pyridine were purchased from Aldrich and piperadine was bought from Sigma. N,N-Dimethylformamide (DMF) was obtained from Fisher Scientific. All peptides in this study were synthesized on a Biotage Initiator + Alstra microwave peptide synthesizer using standard protocols and purified by reverse-phase HPLC on a C18 column at a flow rate of 20 mL/min using a linear gradient varying from 0.1% trifluoroacetic acid (TFA) in water to 0.1% TFA in 9:1 CH3CN/H2O as previously reported.35 Peptides were characterized by electrospray mass spectrometry. Peptide concentrations were determined based on the tryptophan absorbance at 280 nm (ε = 5500 M−1 cm−1). Amino acids sequences of all peptides used within this study are detailed in Table 1.
UV–vis and EPR Spectroscopy.
Electronic absorption data were collected on a Cary 100 UV–vis Spectrometer. Electron paramagnetic resonance (EPR) spectra were collected using a Bruker EMXE 200 EPR cooled to 100 K.
X-ray Absorption Spectroscopy.
Cu(I) samples were prepared as follows. Tetrakis(acetonitrile)Cu(I)hexafluorophosphate (1 mM) was added to a 4.5 mM peptide (1.5 mM 3SCC trimer) in 50 mM MES buffer at pH 5.9 with 50% glycerol (added as a glassing agent) in the glovebox maintained in anaerobic conditions. Sample solutions were then loaded into a sample cell and frozen in liquid nitrogen. 0.5 mM excess apo peptide trimer was included to ensure the free Cu(I) concentration was minimal (<0.01%).
Cu(II) samples were prepared with 1 mM Cu(II)Cl2 and 4.5 mM peptide (1.5 mM 3SCC trimer) in 50 mM MES buffer at pH 5.9 in aerobic conditions. Samples were then lyophilized before transferring to sample cells. During collection the Cu edge energy and 1s → 4p transition of Cu(I)peptide were monitored. We estimate that no more than 10% of the sample was photoreduced by the final scan included in fitting.
Measurements were carried out at Stanford Synchrotron Radiation Lightsource (SSRL) beamlines 7–3 and 9–3. A Si(220) double-crystal monochromator was employed to select the incident energy, and a flat Rh-coated mirror was used for harmonic rejection. Samples were maintained below 10 K with an Oxford Instruments liquid helium cryostat. Fluorescence detected measurements were performed using either a 30- or 100-element Canberra Ge detector array (for beamlines 7–3 or 9–3 respectively) normalized to incident intensity measured with a N2 filled ion chamber. Data were measured using steps of 0.25 eV in the XANES region (1 s integration time) and 0.05 Å−1 in the EXAFS region to k = 13.5 Å−1 (1–20 s integration, k3 weighted). Energies were calibrated by assigning the lowest energy inflection point of a copper metal foil to 8980.3 eV. The threshold energy, E0, was defined as 8990 eV based on fits of model compounds; this was used to convert data to k-space, and the background was removed using a 3-region cubic spline. XANES data were normalized using MBACK.36 For analysis of the 1s → 3d transitions (Figure 4), the data were fitted with an arctan background + a pseudo-Voigt peak to model the rising edge and the 1s → 3d peak, and this fitted background was then subtracted from the data.
Figure 4.

Cu(I) XANES at pH 5.8 of all constructs reported compared to that of TRIW-H.
Single- and multiple-scattering path fitting of EXAFS data were performed using EXAFSPAK37 with ab initio amplitude and phase parameters calculated using FEFF 9.0.38 An initial model of Cu-imidazole coordination was built based on the average Cu–N bond distances determined by single-scattering fitting of EXAFS data, with the imidazole bond-lengths and angles taken as the average of all Cu-imidazole structures contained in the Cambridge Structural Database. All significant non-H paths, defined as those having an amplitude greater than 4% of the Cu–N amplitude, from this model were then loaded into EXAFSPAK and modeled as a rigid ligand. Initial estimates of the Debye–Waller factors for each Cu-imidazole shell were taken from calculations by Dimakis and Bunker.39 The Cu–N distance and Debye–Waller factor were allowed to vary, with the distance and Debye–Waller factor for the other paths calculated based on the Cu–N values. Thus, the long distance scatterering from the Cu-Imid was modeled while only varying two independent variables.
Kd Determinations.
Cu(I) binding constants were determined by UV–visible titration of the peptide with Cu(I) in the presence of disodiumcuproinedisulfonate (Na2BCS) as a competitive chelator in 50 mM MES buffer pH 5.9 using a logβ2 of 19.8 for the formation of Cu(I)(BCS)2.40 Cu(II) binding constants were determined by direct titration of the peptide with Cu(II) while monitoring Trp fluorescence quenching in 50 mM MES pH 5.9 buffer. Redox potentials for TRIW-δmH and TRIW-εmH were calculated based on the Nernst equation and the Cu(I)/(II) binding constants for those peptides at pH 5.9.
NiR Activity.
NiR activity was measured by UV–visible spectroscopy using previously published procedures monitoring ascorbate oxidation.25 Pseudo first order rate constants were determined in solutions containing ~30 mM nitrite, ~1.2 mM ascorbate and varying metallopeptide catalyst. For cases in which the initial rates were sufficiently high, the initial rate of the metallopeptide-catalyzed reduction of nitrite by ascorbate was determined at varying nitrite concentrations and the data were fit to the Michaelis–Menten equation to obtain Vmax, kcat, and KM.34
RESULTS
Cu(II) Absorption and EPR Spectroscopy.
Absorption spectra were obtained at pH 5.8 for a series of solutions containing 300 μM Cu(II)Cl2 and 300 μM (TRIW-δmH)3 or (TRIW-εmH)3 (Table 2). Electron paramagnetic resonance spectra were obtained for Cu(II)(TRIW-H)3, Cu(II)-(TRIW-δmH)3 and Cu(II)(TRIW-εmH)3 at pH 5.8 and fit using the program Spincount.41 The A‖ measured for the new constructs matches closely with our previously reported TRIW-H, and is comparable to other His3 coordinated Cu(II) systems. The fit g-tensors indicate axial symmetry around the copper center.
Table 2.
Cu(II) Spectroscopic Parameters at pH 5.8
| construct | λmax (nm) | ελ (M−1 cm−1) | gx, gy, gz | A‖ |
|---|---|---|---|---|
| TRIW-H | 643 | 135 | 2.064, 2.028, 2.271 | 167 |
| TRIW-δmH | 670 | 67 | 2.045, 2.047, 2.282 | 167 |
| TRIW-εmH | 700 | 78 | 2.046, 2.034, 2.296 | 151 |
Cu(II) X-ray Absorption Spectroscopy.
X-ray absorption spectra (XAS) were collected for Cu(II)(TRIW-H)3, Cu(II)-(TRIW-δmH)3, and Cu(II)(TRIW-εmH)3 to compare the electronic and geometric structures of these three constructs (Table 3). In almost every case (including the Cu(I) spectra discussed below) the fitted Debye–Waller factors are somewhat larger than seen in small model compounds. This is typical of our findings for de novo designed peptides and may reflect a small degree of disorder in the metal sites. The average nearest-neighbor bond-length for the Cu(II) is 1.93–1.95 Å. This distance, which is well-defined and does not depend on the details of the fitting, is consistent with ligation by four oxygen or nitrogen-containing ligands. However, all three constructs exhibit long distance scatterers indicative of His ligation, and in all cases at least two imidazole ligands were required in order to account for the outer shell scattering. Unfortunately, it is difficult to definitively distinguish between two and three imidazoles. For TRIW-H and TRIW-δmH, fits using Cu(imid)3O were somewhat better, while for TRIW-εmH a model using Cu(imid)2O2 gave somewhat better fits. This is consistent with the observation that the Fourier transform of the TRIW-εmH EXAFS has noticeably weaker outer-shell scattering than is seen for the other samples (Figure S2). Fitting parameters for all models attempted are included in the Supporting Information.
Table 3.
Cu(II) EXAFS Fitting Parameters
| Cu–O |
Cu–N |
|||||
|---|---|---|---|---|---|---|
| construct | model | R (Å) | σ2 × 10−3 (Å2) | R (Å) | σ2 × 10−3 (Å2) | Avg. bond length (Å) |
| TRIW-H | CuHis3O | 2.023 | 1.0 | 1.910 | 7.9 | 1.938 |
| TRIW-δmH | CuHis3O | 1.980 | 1.3 | 1.913 | 10.5 | 1.930 |
| TRIW-εmH | CuHis2O2 | 1.974 | 4.7 | 1.929 | 11.3 | 1.952 |
The 1s → 3d transition of Cu(II), presenting as a small local maximum at 8979 eV, was analyzed to investigate geometric differences between constructs as determined by the relative intensity of this peak which arises from 3d-4p mixing.42,43 Comparing Cu(II)(TRIW-H)3, Cu(II)(TRIW-δmH)3, and Cu(II)(TRIW-εmH)3 we find that TRIW-H and TRIW-δmH have very similar peak intensities while TRIW-εmH has a higher intensity (Figures 3 and S3).
Figure 3.

Isolated 1s → 3d region of the Cu(II) XANES at pH 5.8 of the mHis constructs reported compared to that of TRIW-H.
Cu(I) X-ray Absorption Spectroscopy.
XAS data were collected on Cu(I)(TRIW-δmH)3, Cu(I)(TRIW-εmH)3, and Cu(I)(TRIW-δmH L19A)3 and compared to our previously published measurements of Cu(I)(TRIW-H)3. EXAFS data for the three new constructs all exhibited long distance scatterers indicative of His ligation and all could be reasonably fit using either Cu(imid)2 or Cu(imid)3 models. For TRIW-εmH and TRIW-δmH L19A the fit quality was nearly identical for 2 or 3 coordinate models, while for TRIW- δmH the 2-coordinate model gave a noticeably poorer fit. Although EXAFS fit quality alone cannot unambiguously differentiate between these models, the invariance of the Cu(imid) distance regardless of model employed leaves us confident in the determined Cu–N distances (Table 4); the smaller average Cu–N distance for TRIW-εmH and TRIW-δmH L19A is consistent with a lower average coordination number for these sites. Fitting parameters for all models attempted are included in Table S1 of the Supporting Information.
Table 4.
Cu(I) EXAFS Fitting Parameters
| Cu–N |
|||
|---|---|---|---|
| construct | model | R (Å) | σ2 × 10−3 (Å2) |
| TRIW-H25 | 3 Cu-His | 1.93 | 9 |
| TRIW-δmH | 2 Cu-His | 1.93 | 5.5 |
| 3 Cu-His | 1.94 | 7.2 | |
| TRIW-δmH L19A | 2 Cu-His | 1.89 | 8.1 |
| 3 Cu-His | 1.89 | 10.4 | |
| TRIW-εmH | 2 Cu-His | 1.895 | 9.3 |
| 3 Cu-His | 1.895 | 11.8 | |
The 1s → 4p transition of Cu(I), presenting as a local maximum at 8982–8985 eV, was analyzed to investigate coordination number differences between constructs as determined by the intensity of this peak with higher peak intensities being indicative of lower coordination number.44 Figure 4 shows a comparison of Cu(I)(TRIW-H)3, Cu(I)-(TRIW-δmH)3, Cu(I)(TRIW-εmH)3, and Cu(I)(TRIW-δmH L19A)3. Upon closer examination we find that the four constructs can be divided into two groups with TRIW-H and TRIW-δmH having almost identical peak intensity, while TRIW-εmH and TRIW-δmH L19A are similarly matching. Interestingly, while the L19A mutation of TRIW-δmH shifts the 1s → 4p transition to higher energy relative to TRIW-H (in a similar way as reported for L19A) the intensity of the peak does not increase to the same degree.34 This suggests that there is less of a shift to a two coordinate species when His23 is methylated.44
Cu(I/II) Binding Constants and Redox Potential.
Cu(I) and Cu(II) Kd’s were determined for (TRIW-δmH)3 and (TRIW-εmH)3 and compared to the original (TRIW-H)3 model construct (Table 5). The Cu(I) binding constants were surprisingly insensitive both to nitrogen methylation and mode of His coordination. Cu(II) binding constants were more sensitive, with binding affinities decreasing by up to 2 orders of magnitude going from (TRIW-H)3 and (TRIW-εmH)3 to (TRIW-δmH)3. This decrease in the Cu(II) KD for (TRIδmH)3 leads to a concomitant increase in the calculated redox potential while the redox potential of Cu(TRIW-εmH)3 is identical to that of Cu(TRIW-H)3.
Table 5.
Equilibrium Cu Binding Constants at pH 5.9
| construct | Cu(I) Kd (pM) | Cu(II) Kd (μM) | redox potential (mV vs NHE) |
|---|---|---|---|
| (TRIW-H)3 | 3.1 ± 0.7 | 0.040 ± 0.008 | 400 ± 10 |
| (TRIW-δmH)3 | 1.7 ± 0.2 | 3.3 ± 1.0 | 530 ± 10 |
| (TRIW-εmH)3 | 5.8 ± 3.2 | 0.04 ± 0.02 | 390 ± 30 |
Kinetic Analysis of NiR Activity.
Previously published protocols were used to determine the pseudo first order rate constants for Cu(I)(TRIW-εmH)3−, Cu(I)(TRIW-δmH)3−, and Cu(I)(TRIW-δmH L19A)3-catalyzed reduction of nitrite by ascorbate under a standard set of conditions.25,34 Cu(I)-(TRIW-δmH)3 and Cu(I)(TRIW-δmH L19A)3 performed efficiently enough that Michaelis—Menten kinetic analyses were pursued. Parameters from these kinetic analyses and those of some previously published TRIW-H based constructs are compared to other model constructs and native CuNiR enzymes below (Table 6).
Table 6.
Kinetic Parameters for Nitrite Reductase Activity at pH 5.8 Compared to Previously Published Results and Other Model Complexes or the Native Enzymea
| construct | rate (s−1) | Vmax (M s−1) | KM (M) | kcat (s−1) | kcat/KM (s−1 M−1) |
|---|---|---|---|---|---|
| TRIW-H25 | 4.6 × 10−4 | N/A | N/A | N/A | N/A |
| TRIW-H L19A34 | 3.5 × 10−2 | 2.3 ± 0.3 × 10−6 | 0.24 ± 0.05 | 0.23 ± 0.03 | 1.0 ± 0.3 |
| TRIW-δmH | 0.12 | 1.5 ± 0.1 × 105 | 0.18 ± 0.02 | 1.5 ± 0.1 | 8.2 ± 0.1 |
| TRIW-δmH L19A | 0.30 | 1.5 ± 0.1x 105 | 0.13 ± 0.01 | 1.5 ± 0.1 | 11.3 ± 0.1 |
| TRIW-εmH | 1.2 × 10−3 | N/A | N/A | N/A | N/A |
| [CuMe2bpa(H2O)(ClO4)]+ on electrode pH 5.545 | N/A | N/A | 1.1 × 103 | 0.063 | 57.3 |
| [CuMe2bpa(H2O)(ClO4)]+ in solution pH 5.545 | N/A | N/A | 2.5 × 103 | 5.3 × 105 | 0.02 |
| AfCuNiR pH 6.546 | N/A | N/A | 1.5 × 104 | 620 | 4.1 × 106 |
| AxCuNiR pH 7.0, 4 °C47 | N/A | N/A | 2.7 × 103 | 89 | 3.3 × 105 |
The rate value above refers to the pseudo-first order rate constant for metallopeptide-catalyzed reduction of nitrite by ascorbate in solutions containing 30 mM nitrite and 1.2 mM ascorbate.25
DISCUSSION
We have previously reported on efficient nitrite reductase activity in de novo designed 3SCC scaffolds by incorporating copper into the (TRI-L2WL23H)3 [hereafter designated (TRIW-H)3]. Our initial studies demonstrated that this copper protein was a remarkably robust catalytic model of CuNiR, completing over 1000 turnovers without a measurable decrease in activity.25 We have since proceeded to systematically modify this system to enhance activity. We began by changing the surface charge of the coiled-coils outside the direct first and second coordination sphere of the copper center. These studies successfully tuned the redox potential of the bound copper by up to 100 mV; however, they led only to modest changes in reaction rate (a range of a factor of 4) and none led to enhanced activity compared to the original construct.33 Our next study investigated the influence of steric constraints in the metal’s second coordination sphere, leading to further modifications which increased the NiR first order rate constant up to 75-fold compared to the original (TRIW-H)3 construct.34 We have now moved our attention to the primary coordination sphere of the metal with the objective of testing the effects of histidine δ- or ε-nitrogen coordination (enforced by methylation of ε- or δ-nitrogen atoms) on the electronic and geometric structure, redox potential, and catalytic activity of the copper center. These changes in coordination, developed within a well-defined scaffold, influence the chemistry of the Cu site through both electronic effects (e.g., inductive effects) and by steric. In doing so, we have directly interrogated the influence of copper–histidine coordination isomers on the physical and enzymatic properties of a Cu active site in a well understood protein environment.
As a ligand, N-methyl histidine provides numerous alterations to the physical properties of the imidazole side chain compared to standard l-histidine. The obvious first change is that imidazole has three protonation forms: imidazolium, imidazole, and imidazolate. The first and last species occur at very low and very high pH, respectively, but can be emulated in a protein environment through hydrogen bond donors and acceptors in the metal’s second coordination sphere. Upon alkylation, the imidazolate form can no longer exist, nor can local H-bonding residues perturb the protonation state of the bound ligand. The second obvious change is that a methyl group is much more electron donating than a simple proton. Therefore, one can consider εmH and δmH substitution leading to a more electron rich ligand. However, the consequence of this additional electron density is not straightforward to interpret. This is because the two nitrogen atom lone pairs of the imidazole have different degrees of σ versus π character. As an example, one may consider the pKas for imidazole and N-methylated imidazole, which leads to a −0.21 pKa shift upon addition of the alkyl group. At the same time, the observed pKas for histidine, εmH, and δmH are 6.03, 5.91, and 6.52, respectively.24 Thus, alkylation can either increase or decrease the acidity of the imidazole ring relative to the parent histidine. When the pKa values for the histidine are corrected to give microscopic constants specifically reflecting the acidities of the εHHistidine and δHHistidine species (which measure the dissociation of protons at either the δ or ε positions) it was found that the pKas were 6.12 and 6.73, respectively.24 This means that the σ donor capacity of the two nitrogen atoms of imidazole itself are markedly different. It is for this reason that the two histidine imidazole nitrogen atoms have been referred to as pyrrole (δ) and pyridine (ε) like nitrogens. These differences in ligating character and basicity should lead to different positions within the spectrochemical series for each coordination mode, altered binding affinities for Cu(I)/Cu(II), and modulation of the metal reduction potential.
Besides electronic effects, N-methylation imposes steric constraints within the protein interior. The first consideration is how the protein scaffold accommodates the more bulky methyl group in the active site. The second issue is that the δ- and ε-nitrogen positions correspond to the 1 and 3 positions of a 5 membered ring (which is attached to the backbone at the 5 position). Thus, the length and angle of coordination of these two types of nitrogen ligands with Cu are considerably different. Finally, as the methyl group cannot hydrogen bond, stabilizing interactions with second coordination sphere ligands that may orient the imidazole within the active site are largely removed. Therefore, one must consider a myriad of both electronic and steric perturbations to interpret fully changes observed through side chain methylation. It is our proposition that understanding the impact of these changes is best completed within a minimal, well-defined scaffold as is provided by de novo design.
The N-methyl histidine modified compounds εmH and δmH have been used in enzyme design previously as a proximal ligand to heme in order to create de novo heme models of deoxymyoglobin or to increase the peroxidase activity of myoglobin.48,49 Examples also exist where N-methylated derivatives serve as a substitute for the His ligands within the zinc-containing metalloenzyme mannose-6-phosphate isomerase (ManA) to create organisms which were dependent on supplementation due to this noncanonical amino acid substitution.50 In addition, a few examples of natural proteins, which contain methylated His ligated to Cu, are deposited to the PDB.29–31 While the amyloid-β peptide study discussed in the Introduction did compare εmH and δmH substitution, this was done within the context of fibril formation and not as an investigation of the electronic and structural differences that result from these changes. Thus, an exploration of the structural and electronic effects of either Nδ or Nε nitrogen methylation on copper coordination as it relates to the tautomeric preferences of native copper enzymes, and the impact of these modifications on a catalytic reaction is warranted. In this study we have used TRI-L2WL23εmH (TRIW-εmH) and TRI-L2WL23δmH (TRIW-δmH) derivatives to address these issues.
Structural and Spectroscopic Perturbations Resulting from Histidine N-Methylation.
Cu(II) Geometry.
Combining the results from electronic absorption, EPR, and XAS spectroscopies we can evaluate the geometric and electronic changes that occur at the metal center upon histidine methylation. Furthermore, we can assess whether Cu(II)-(TRIW-δmH)3 impacts these properties differently than Cu(II)(TRIW-εmH)3. The energy of the d–d transition of Cu(II) complexes has been shown to be dependent on the identity of the ligands around the Cu(II) and we have previously used this as evidence that Cu(II)(TRIW-H)3 binds Cu(II) using all three available His residues.25,51 If we follow the spectrochemical series as imidazole > pyridine > pyrrole, then one would expect TRIW-H to have the highest energy d—d transitions if the imidazoles in TRIW-H bind to Cu via the ε-nitrogen, while TRIW-δmH would have the highest energy if the binding in TRIW-H was via the δ-nitrogen. In either case, TRIW-εmH is expected to have the lowest d–d transition energy. It is therefore not surprising that this series shows a progressive shift to lower energy moving from Cu(II)(TRIW-H)3 (643 nm, 15 550 cm−1, ε = 135 M−1 cm−1) to Cu(II)(TRIW-δmH)3 (670 nm, 14925 cm−1, ε = 67 M−1 cm−1) to Cu(II)(TRIW-εmH)3 (700 nm, 14 286 cm−1, ε = 78 M−1 cm−1) (Table 2). This trend follows the estimated basicity of the δ and ε nitrogen atoms of the three employed ligands εNHistidine, 6.73; δmHistidine, 6.52; and εmHistidine, 5.91) possibly suggesting that σ basicity is dominant and supporting a model in which the TRIW-H peptide coordinates to Cu via the ε-nitrogen. Ligation of Cu by the ε-nitrogen is consistent with Zn(II) ligation seen in the crystal structure (Hg(II)S[Zn-(II)N(H2O)](CSL9CL23H)3+).52 Our lab has also been unsuccessful in attempts to model 3-coordinate δ-nitrogen ligation of a metal by a His3 site within a 3SCC. These data, however, are the first empirical evidence for this assignment with Cu(II).
The explanation provided in the previous paragraph is potentially complicated by two other factors. First, the δmH and εmH substitutions remove hydrogen bonding that could alter the unsubstituted imidazole ligand field. Second, if Cu(II)(TRIW-εmH)3 does indeed have a CuN2O2 coordination environment, as suggested by the EXAFS (Table 3), the replacement of histidine with a weaker-field water ligand could account for part of the red-shift seen for the TRIW-εmH d–d transition. With these caveats in mind we still tentatively conclude from the data that the Cu(II)(TRIW-δmH)3 more closely mimics the original Cu(II)(TRIW-H)3 than Cu(II)-(TRIW-εmH)3 does. This conclusion simplifies analyzing other comparative parameters such as Cu(II) binding constants and NiR activity.52 Taken together, Figure 5 provides our best interpretation of the Cu(II) structure in these systems.
Figure 5.

Models of the metal binding sites of (A) Cu(II)-(TRIW-δmH)3 and (B) Cu(I)(TRIW-εmH)3. Models were made using the program pymol and based on the Zn(II)(His)3 site of Hg(II)S[Zn(II)N(H2O)](CSL9CL23H)3+ [PDB 3PBJ] as well as spectroscopic analysis of Cu(II)(TRIW-δmH)3 or Cu(II)-(TRIW-εmH)3.
Further clues as to the geometric differences between these three constructs can be elucidated from interpretation of EPR and XANES spectroscopies. These data again show that Cu(II)(TRIW-εmH)3 differs from Cu(II)(TRIW-H)3 and Cu(II)(TRIW-δmH)3, with a more compressed hyperfine coupling constant (Table 2) and an increased 1s → 3d intensity (Figure 3). Both observations are consistent with Cu(II)(TRIW-εmH)3 having a geometry of somewhat more tetrahedral nature than the two other constructs. The fits to the EXAFS data (Table 3) are also consistent with a change from N3O to N2O2 for Cu(II)(TRIW-εmH)3. This could occur as a consequence of steric effects: either three methyl groups on the Nε nitrogen being unable to fit at the active site or copper being unable to complex three Nδ nitrogen atoms within a 3SCC.
Cu(I) Geometry.
Similar to the previous comparison of the structures of the Cu(II) peptides, XAS shows that the geometry of Cu(I)(TRIW-δmH)3 is indistinguishable from that of Cu(I)(TRIW-H)3 while Cu(I)(TRIW-εmH)3 differs from both (Table 4). As predicted by a previous comparative study on deposited crystal structures,23 coordination of Cu through the delta nitrogen of His is more prone to be two coordinate than with the epsilon nitrogen. The shorter bond length, as determined by EXAFS (1.89 Å compared to 1.94 Å), and increased 1s → 4p transition intensity within the XANES region of Cu(I)(TRIW-εmH)3 are consistent with an equilibrium that favors bis-coordinate for this species to a greater extent than Cu(I)(TRIW-δmH)3 or Cu(I)(TRIW-H)3 (Figure 4).44 The Debye–Waller factors of these three constructs show that Cu(I)(TRIW-εmH)3 is in a more disordered state than Cu(I)(TRIW-δmH)3 or Cu(I)(TRIW-H)3. This is likely because the δ nitrogen derivative is a mixture of 2 and 3 coordinate structures while the other two constructs seem to fit well within the expected values of 3 coordinate Cu(I)His3 complexes. That Cu(I)(TRIW-εmH)3 exists in an equilibrium between two and three coordinate rather than purely two coordinate may be a consequence of our choice of a 3SCC as our scaffold. Metalated TRI family peptides have had several examples in which higher coordination numbers are enforced such as the case of HgS3.35,53 Recently, we reported that the mutation of L19A above the His layer in Cu(I)-(TRIW-H)3 relieved the steric constraints imposed on histidine movement at the active site, allowing for a two coordinate construct. It appears that Cu(I) prefers the two coordinate structure in all of these designed peptides when sufficient space is available for the third imidazole to remain unbound.34,35,53 Thus, one could argue that Cu(I)-(TRIW-εmH)3 would be fully two coordinate if not for the preferences inherent in the scaffold. The resulting mixed coordination would then increase the disorder of this coordination, which is consistent with our current results. Finally, the observation, from both EXAFS and XANES, that the Cu(I)(TRIW-εmH)3 site is different from the Cu(I)-(TRIW-H)3 and Cu(I)(TRIW-δmH)3 sites is consistent with the hypothesis that it is not possible to form a Cu(His)3 site when histidine is coordinated via Nδ within a 3SCC, as was also seen with Cu(II) coordination. These data thus suggest that Cu(I)(TRIW-H)3 is likely bound to Cu(I) via the Nε nitrogen of His. Our model structures for the Cu(I) forms are shown in Figure 6.
Figure 6.

Models of the metal binding sites of (A) Cu(I)-(TRIW-δmH)3 and (B) Cu(I)(TRIW-εmH)3. Models were made using the program pymol and based off the Zn(II)(His)3 site of Hg(II)S[Zn(II)N(H2O)](CSL9CL23H)3+ [PDB 3PBJ] as well as spectroscopic analysis of Cu(II)(TRIW-δmH)3 or Cu(II)-(TRIW-εmH)3.
Given that the Cu(I)(TRIW-H L19A)3 converts to a partially 2-coordinate Cu(I), we can next compare the changes seen between Cu(I)(TRIW-δmH)3 and Cu(I)(TRIW-δmH L19A)3. Interestingly, although the type of changes seen are similar in both cases, with a slightly shorter Cu–N distance and the exact same shift in 1s → 4p transition energy, the intensity of this transition for the Cu(I)(TRIW-δmH L19A)3 construct is much smaller than Cu(I)(TRIW-H L19A)3, suggesting that the 3 coordinate–2 coordinate equilibrium does not shift to the same degree as it does in the case of unmodified histidine (Figure 7). This is likely due to the steric bulk of the methyl group on the δ nitrogen inhibiting the movement of one histidine away from the copper atom. That L19A mutation of TRIW-δmH increases NiR efficiency, though to a lesser degree than in the case of TRIW-H, while increasing the 1s → 4p transition energy serves as further evidence of the link between these two observables.
Figure 7.

Cu(I) XANES at pH 5.8 of Cu(I)(TRIWδmH)3 and Cu(I)(TRIWδmH L19A)3 compared to previously published data for Cu(I)(TRIW-H)3 and Cu(I)(TRIW-H L19A)3 from refs 25 and 34, respectively.
Cu(I/II) Thermodynamics: Binding Equilibria and Reduction Potentials.
Given that spectroscopic evidence suggests (TRIW-H)3 binds copper in both oxidation states via the Nε nitrogen (vida supra), we can use a comparison of binding constants for Cu(I) and Cu(II) to (TRIH)3, (TRIδmH)3, and (TRIεmH)3 to investigate the effects of histidine methylation, and therefore Cu–Nδ or Cu–Nε ligation, on the stabilities of these systems. The data in Table 5 show that while the Cu(I) Kds for all three constructs are relatively similar, in the range 2–6 pM, the Cu(II) Kd for (TRIδmH)3 differs by 2 orders of magnitude from the other 3SCCs. These results are surprising in at least two ways. First, two of the three Cu(I) compounds are three coordinate, whereas the third is in an equilibrium biased toward two coordinate, yet all binding constants are within a factor of 3 and essentially within the errors of the determinations. So, the cuprous binding affinities are invariant to modifications such as the number of ligands, the basicity of the ligands, the presence or absence of methyl groups, the bonding of Nε or Nδ nitrogen atoms or the polyhedral preferences (alternatively, differences due to these factors may fortuitously cancel). Second, the 3.3 μM value for Cu(II)-(TRIδmH)3 compares to 40 nM for the other two Cu(II) constructs. At first examination this appears odd since, as previously mentioned, spectroscopic evidence indicates that Cu(II)(TRIW-H)3 binds to the same Nε nitrogen as Cu(II)(TRIδmH)3. Thus, the coordination environment and relative ligand basicity for these two peptides are expected to be very similar, whereas the Cu(I)(TRIW-εmH)3 has imidazoles that are the least basic of the three and may, in addition, have one fewer bound histidine. We can only conclude that the difference between Cu(II)(TRIW-H)3 and Cu(II)(TRIδmH)3 is due to methylation of the His rather than the type of nitrogen coordination or geometry. The simplest model is that while a tris imidazole structure can still form, the methyl groups of Cu(II)(TRIδmH)3 are disposed to cause severe steric clashes that diminish the copper affinity. It may also be that H-bonding, unavailable in the methylated derivative, may stabilize Cu(II)(TRIW-H)3. Previous studies support this possibility as mutations of surface carboxylates suggested that hydrogen bonding influenced the reduction potentials significantly.33
It is interesting that the amyloid-β study of Tickler et al. did not observe any difference between EPR spectra of Cu(II)-amyloid-β(δmH) compared to Cu(II)-amyloid-β(εmH).28 This discrepancy likely arises from the geometric differences we hypothesize for Cu(II)(TRIW-δmH)3 versus Cu(II)-(TRIW-εmH)3. The more confined space within the 3SCC environment limits whether the same geometry can be adopted by all methylated derivatives. We also find an opposite trend for the KD’s determined for methylated His forms of amyloid-β for Cu(II). In that system, tighter Cu(II) binding was observed for amyloid-β and amyloid-β(δmH) than amyloid-β(εmH), while we find that methylation itself leads to a looser KD for Cu(II) by 2 orders of magnitude and (TRIW-εmH)3 exhibits tighter binding than (TRIW-δmH)3 by 2 orders of magnitude. Again, this may be a consequence of the differing secondary structures adopted by TRI peptide and amyloid-β.
The consequence of invariant cuprous affinities and divergent cupric affinities is that the calculated reduction potential for Cu(II)(TRIδmH)3 is significantly more positive than for the other nitrite reductase mimics. Table 5 shows that the reduction potential for Cu(II)(TRIWδmH)3 is 130 mV more positive than observed for the other two peptides. This directly reflects the weaker Cu(II) affinity and suggests this protein is the most likely to be in the reduced state. Not surprisingly, it is also the construct with the highest Cu(I) affinity, even though the two-coordinate form seen in Cu(I)(TRIW-εmH)3 might have been predicted to be the preferred coordination geometry. Below we discuss the implications of these thermodynamic parameters on the nitrite reductase activity.
Effect of N-Methylation on Nitrite Reductase Catalysis.
Based on either first order rate constants or kcat, the TRI family nitrite reductases are the most active artificial complexes for the conversion of nitrite to NO in homogeneous aqueous solution. Figure 8 illustrates the over 1000 fold range of first order rate constants seen across these proteins. Clearly, modification of the surface residues, shown in blue, had minor influence on activities, despite spreading the reduction potentials over a range greater than 130 mV. In contrast, second coordination sphere modifications, shown in green, led to significant rate enhancements, with k values nearly 100-fold higher than the original TRIW-H construct. In the present studies, we see that modification of the properties of the first coordination sphere histidine by methylation leads to the most significant increases in activities. Most interesting, combining one of the best second coordination sphere mutations (TRIW-L19A, 75-fold enhancement) with the best alkylated derivative (TRIWδmH, 260-fold), only led to a modest increase in rate (TRIWL19AδmH 640-fold or ~2.5-fold better than TRIWδmH).
Figure 8.

Pseudo first order rate constants of the original TRIW-H construct (red) reported in ref 25 compared to the current manuscripts results (magenta) and previously published mutation series of either helical interface residues (blue) reported in ref 33 or interior residues (green) reported in ref 34
Probably the most interesting observation from this comparison of rate data is that methylation of the delta nitrogen leads to much greater rate enhancements (260-fold) as compared to methylation of the epsilon nitrogen (2.6-fold). Since both TRIW-H and TRIW-δmH bind to Cu in both oxidation states through the Nε, this comparison provides a direct reflection of methylation on catalytic activity. An inductive-like effect has been previously hypothesized to account for the difference in activity of pyrazole containing small molecule models of CuNiR and those containing more electron-rich imidazole ligands.54,55 This was hypothesized to be linked to the Cu(II)(NO2) HOMO having increased energy with more electron-rich ligands. It is possible that similar inductive effects could be relevant here between Cu(I)(TRIH)3 and Cu(I)(TRIδmH)3.
Because we cannot evaluate directly the TRIW-H Nδ isomer, we do not know whether we can apply the above 260-fold factor to assess TRIW-H Nδ isomer rates; however, it is rather likely that Cu–Nδ coordination in general leads to far diminished catalysis. This is emphasized by the 2 orders of magnitude difference in rates between the two N-methylated isomers probed here; consistent with hypotheses on the differing roles of Nδ or Nε nitrogen ligation to Cu, but this is the first time such a difference has been quantified.23 This comparison is not perfect, however, due to the differing coordination numbers of Cu(I)(TRIWδmH)3 (3 coordinate) and Cu(I)(TRIW-εmH)3 (mix of 2 and 3 coordinate) which complicates the comparison. Previous results with secondary sphere mutations to Cu(I)(TRIW-H)3 above or below the His plane have shown that several Cu(I)(TRIW-H)3 constructs with coordination numbers of two or three have similar activities.34 Further, comparing the activities of Cu(I)-(TRIW-εmH)3 with Cu(I)(TRIWδmH L19A)3, which have the same coordination number according to XANES analysis (Figure 4), shows a similar enhancement of Cu–Nε ligation compared to Cu–Nδ as was observed between Cu(I)-(TRIWδmH)3 and Cu(I)(TRIW-εmH)3. These results leave us confident that the differing coordination number does not account for the 2 orders of magnitude enhancement in NiR activity between Cu(I)(TRIWδmH)3 and Cu(I)(TRIW-εmH)3.
We previously have shown, using surface modifications of TRIW-H, that nitrite reductase activity was not dependent on the reduction potential of the copper center in these 3SCCs.33 With the present derivatives, the Cu(II)(TRIWδmH)3 has a 130 mV more positive reduction potential than either Cu(II)(TRIWH)3 or Cu(II)(TRIWεmH)3. This 2 orders of magnitude difference (based on the Cu(II) Kd) is close to what is observed for the 2 orders of magnitude increase in rate; however, given that other derivatives, such as TRIW-EH, have reduction potentials even more positive (+587 mV) with rates nearly 500 times slower than Cu(I)(TRIWδmH)3, it is unlikely that the catalytic enhancement is due to this thermodynamic difference. Furthermore, making the reduction potential more positive should slow the catalytic reaction, not enhance it. Therefore, we conclude that electron transfer is not rate limiting in any of the 3SCC derivatives. This conclusion fits with the rapid reduction of Cu(II) by ascorbate, but now also addresses the Cu(I) reoxidation reaction as well.25
Finally, with the improved nitrite reductase activity of Cu(I)(TRIWδmH)3 and Cu(I)(TRIWδmH L19A)3, Michaelis–Menten kinetics were investigated to probe further the cause of the improved catalytic efficiencies of these constructs. Having quantitative values for kcat, KM, and kcat/KM allows us to separate binding effects from catalytic effects incurred by each modification of the 3SCC. Having these Michaelis values also allows us to compare activities more easily to other reported catalysts for NiR activity (Table 6). It is instructive to first compare these new systems to native enzymes.
For homogeneous aqueous soluble systems, even using Michaelis Menten kinetics criteria for maximal rate (kcat) or catalytic efficiency (kcat/KM), the TRI peptides remain the most efficient synthetic copper catalysts for nitrite reduction. That said, they are extremely humble as compared to either of the reported native enzymes. Based on kcat/KM, the best TRI peptide is nearly 400 000-fold less efficient than the best Cu nitrite reductase. The present kinetic analysis reveals that the lower efficiency is due both to factors that influence the maximal rate of catalysis and factors controlling substrate access and binding. The ratio of kcat values between the AfCuNiR and Cu(TRIWL19δmH)3 is approximately 400, whereas the ratio of KM values is just shy of 3 orders of magnitude (~900). Considering that no effort has yet been made to include the critical Asp98 acid base catalyst at the active site which is required for proper proton transfer, the similarity in maximal rates between the native enzyme and the designed systems is actually quite respectable. We previously sought to incorporate an Asp in a second coordination sphere position, but the enforced symmetry of the TRI system means that three Asp residues were simultaneously above the CuHis3 layer.34 All evidence points to these carboxylic acid side chains interacting with each other rather than interacting with nitrite. Thus, to achieve this goal may require development of scaffolds that allow asymmetric modification of the 3SCC. Work to transplant the TRIW-H binding site into such a scaffold is underway.
The best small inorganic molecule untethered to an electrode has a kcat value nearly 30 000-fold lower than our optimized construct and more than 10 000 000-fold lower than the enzyme.45 Where the present synthetic protein catalysts fall short is in the KM values, suggesting that substrate recognition and access is the primary limiting feature of the 3SCC scaffold. Previous mutation studies on CuNiR from Alcaligenes xylosoxidans found that mutation of the secondary sphere amino acids Asp98 or His255 both decreased the kcat and increased the KM by as much as 2 orders of magnitude.56 Thus, the most logical step forward to improve CuNiR catalytic efficiency further within the TRI 3SCC system is to incorporate similar residues to interact with the bound substrate and improve both KM and kcat at the same time. However, the data in Table 6 indicate that optimization of maximal rate versus substrate recognition with the TRI peptides may be to some extent uncoupled. For example, the entirety of the increase in catalytic efficiency between Cu(TRIWδmH)3 and Cu(TRIWL19AδmH)3 can be assigned to a lower KM for the second peptide. In contrast, the 11-fold enhancement in catalytic efficiency between Cu-(TRIWL19AδmH)3 and Cu(TRIWH L19A)3 is predominantly due to a kcat effect. This suggests that future modifications of these systems to optimize catalytic efficiency may be able to fine-tune one parameter independently of the other.
CONCLUSION
The present studies have used methylation of a coordinated histidine to make significant advances in the understanding of designed redox active metalloenzymes. In addition to isolating an enzyme with a 10-fold enhancement in catalytic efficiency from what had previously been reported, we have dissected the basis for these changes through kinetic analysis. Furthermore, by selective methylation of Nε or Nδ nitrogen atoms, we have been able to assign the binding mode of copper in the unmethylated histidine protein and to compare the differences in physical properties associated with the coordination of pyrrole versus pyridine like imidazole nitrogens. We have found that in these systems, the Nε binding mode appears to be preferred and this complexation leads to the most active catalysts. For the first time, one can assess directly the rate for an enzymatic reaction where the scaffold remains unperturbed but the Cu is bound to Nε or Nδ nitrogen atoms. In this case, the Nε form is 100-fold faster than the Nδ protein.
Using the peptides Cu(TRIWL19AδmH)3 and Cu(TRIWH L19A)3, we observed that methylation of imidazole leads to a 10-fold increase in catalytic efficiency. This appears to be a consequence primarily of electronic features and not due to steric constraints associated with the more bulky methyl group. In contrast, by comparing Cu(TRIWL19AδmH)3 and Cu-(TRIWδmH)3, more space in the second coordination sphere of the peptides affects the KM for the reaction without impacting kcat. We also can conclude that the rate limiting processes here are not dependent on the reduction potentials of the systems.
Directing copper coordination modes using N-methyl histidines can impact the spectroscopy, binding affinities, and reduction potential for the system. We have demonstrated how it is possible to exploit these perturbations to enhance catalysis and also probe structure and mechanism. Finally, these results suggest that other noncoded amino acids could be used to alter the chemistry of these systems.
Supplementary Material
ACKNOWLEDGMENTS
V.L.P. thanks the National Institutes of Health for financial support of this research (ES012236). T.B.J.P. thanks the Natural Sciences and Engineering Research Council of Canada for their support in the form of a postdoctoral fellowship. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b00196.
XAS, EPR, and UV–visible spectra example Cu(I) or Cu(II) titrations for Kd determination; example kinetic analysis figures (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Malkin R; Malmström BG The State and Function of Copper in Biological Systems In Adv. Enzymol. Relat. Areas Mol. Biol; Nord FF, Ed.; John Wiley & Sons, Inc.: New York, 2006; Vol. 33, pp 178–244. [DOI] [PubMed] [Google Scholar]
- (2).Kaim W; Rall J Copper—A “Modern” Bioelement. Angew. Chem., Int. Ed. Engl 1996, 35 (1), 43–60. [Google Scholar]
- (3).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114 (7), 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Solomon EI; Hare JW; Gray HB Spectroscopic studies and a structural model for blue copper centers in proteins. Proc. Natl. Acad. Sci. U. S. A 1976, 73 (5), 1389–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Solomon EI; Hare JW; Dooley DM; Dawson JH; Stephens PJ; Gray HB Spectroscopic studies of stellacyanin, plastocyanin, and azurin. Electronic structure of the blue copper sites. J. Am. Chem. Soc 1980, 102 (1), 168–178. [Google Scholar]
- (6).Gray HB Centenary Lecture. Long-range electron-transfer in blue copper proteins. Chem. Soc. Rev 1986, 15 (1), 17–30. [Google Scholar]
- (7).Gray HB; Malmström BG; Williams RJP Copper coordination in blue proteins. JBIC, J. Biol. Inorg. Chem 2000, 5 (5), 551–559. [DOI] [PubMed] [Google Scholar]
- (8).Penfield KW; Gay RR; Himmelwright RS; Eickman NC; Norris VA; Freeman HC; Solomon EI Spectroscopic studies on plastocyanin single crystals: a detailed electronic structure determination of the blue copper active site. J. Am. Chem. Soc 1981, 103 (15), 4382–4388. [Google Scholar]
- (9).Penfield KW; Gewirth AA; Solomon EI Electronic structure and bonding of the blue copper site in plastocyanin. J. Am. Chem. Soc 1985, 107 (15), 4519–4529. [Google Scholar]
- (10).Colman PM; Freeman HC; Guss JM; Murata M; Norris VA; Ramshaw JAM; Venkatappa MP X-ray crystal structure analysis of plastocyanin at 2.7 [angst] resolution. Nature 1978, 272 (5651), 319–324. [Google Scholar]
- (11).Zhang S; Melzer MM; Sen SN; Çelebi-Ölçüm N; Warren TH A motif for reversible nitric oxide interactions in metalloenzymes. Nat. Chem 2016, 8, 663. [DOI] [PubMed] [Google Scholar]
- (12).Tian S; Liu J; Cowley RE; Hosseinzadeh P; Marshall NM; Yu Y; Robinson H; Nilges MJ; Blackburn NJ; Solomon EI; Lu Y Reversible S-nitrosylation in an engineered azurin. Nat. Chem 2016, 8, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Samanta S; Lehnert N A switch for blue copper proteins? Nat. Chem 2016, 8, 639. [DOI] [PubMed] [Google Scholar]
- (14).Solomon EI; Chen P; Metz M; Lee S-K; Palmer AE Oxygen Binding, Activation, and Reduction to Water by Copper Proteins. Angew. Chem. Int. Ed 2001, 40 (24), 4570–4590. [DOI] [PubMed] [Google Scholar]
- (15).MacPherson IS; Murphy MEP Type-2 copper-containing enzymes. Cell. Mol. Life Sci 2007, 64 (22), 2887–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Iwasaki H; Noji S; Shidara S Achromobacter cycloclastes Nitrite Reductase The Function of Copper, Amino Acid Composition, and ESR Spectra1. J. Biochem 1975, 78 (2), 355–361. [DOI] [PubMed] [Google Scholar]
- (17).Klinman JP Mechanisms Whereby Mononuclear Copper Proteins Functionalize Organic Substrates. Chem. Rev 1996, 96 (7), 2541–2562. [DOI] [PubMed] [Google Scholar]
- (18).Prigge ST; Kolhekar AS; Eipper BA; Mains RE; Amzel LM Amidation of Bioactive Peptides: The Structure of Peptidylglycine α-Hydroxylating Monooxygenase. Science 1997, 278 (5341), 1300–1305. [DOI] [PubMed] [Google Scholar]
- (19).Libby E; Averill BA Evidence that the Type 2 copper centers are the site of nitrite reduction by Achromobacter cycloclastes nitrite reductase. Biochem. Biophys. Res. Commun 1992, 187 (3), 1529–1535. [DOI] [PubMed] [Google Scholar]
- (20).Kukimoto M; Nishiyama M; Murphy MEP; Turley S; Adman ET; Horinouchi S; Beppu T X-ray Structure and Site-Directed Mutagenesis of a Nitrite Reductase from Alcaligenes Faecalis S-6: Roles of Two Copper Atoms in Nitrite Reduction. Biochemistry 1994, 33 (17), 5246–5252. [DOI] [PubMed] [Google Scholar]
- (21).Chufán EE; Prigge ST; Siebert X; Eipper BA; Mains RE; Amzel LM Differential Reactivity between Two Copper Sites in Peptidylglycine α-Hydroxylating Monooxygenase. J. Am. Chem. Soc 2010, 132 (44), 15565–15572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Andreini C; Cavallaro G; Lorenzini S; Rosato A MetalPDB: a database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2012, 41 (D1), D312–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Karlin S; Zhu Z-Y; Karlin KD The extended environment of mononuclear metal centers in protein structures. Proc. Natl. Acad. Sci. U. S. A 1997, 94 (26), 14225–14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tanokura M 1H-NMR study on the tautomerism of the imidazole ring of histidine residues: I. Microscopic pK values and molar ratios of tautomers in histidine-containing peptides. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1983, 742 (3), 576–585. [DOI] [PubMed] [Google Scholar]
- (25).Tegoni M; Yu F; Bersellini M; Penner-Hahn JE; Pecoraro VL Designing a functional type 2 copper center that has nitrite reductase activity within α-helical coiled coils. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (52), 21234–21239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Himes RA; Park GY; Barry AN; Blackburn NJ; Karlin KD Synthesis and X-ray Absorption Spectroscopy Structural Studies of Cu(I) Complexes of HistidylHistidine Peptides: The Predominance of Linear 2-Coordinate Geometry. J.Am. Chem. Soc 2007, 129 (17), 5352–5353. [DOI] [PubMed] [Google Scholar]
- (27).Park GY; Lee JY; Himes RA; Thomas GS; Blackburn NJ; Karlin KD Copper–Peptide Complex Structure and Reactivity When Found in Conserved His-Xaa-His Sequences. J. Am. Chem. Soc 2014, 136 (36), 12532–12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tickler AK; Smith DG; Ciccotosto GD; Tew DJ; Curtain CC; Carrington D; Masters CL; Bush AI; Cherny RA; Cappai R; Wade JD; Barnham KJ Methylation of the Imidazole Side Chains of the Alzheimer Disease Amyloid-β Peptide Results in Abolition of Superoxide Dismutase-like Structures and Inhibition of Neurotoxicity. J. Biol. Chem 2005, 280 (14), 13355–13363. [DOI] [PubMed] [Google Scholar]
- (29).Quinlan RJ; Sweeney MD; Lo Leggio L; Otten H; Poulsen J-CN; Johansen KS; Krogh KBRM; Jørgensen CI; Tovborg M; Anthonsen A; Tryfona T; Walter CP; Dupree P; Xu F; Davies GJ; Walton PH Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (37), 15079–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li X; Beeson WTIV; Phillips CM; Marletta MA; Cate JHD Structural Basis for Substrate Targeting and Catalysis by Fungal Polysaccharide Monooxygenases. Structure 2012, 20 (6), 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hansson H; Karkehabadi S; Mikkelsen N; Douglas NR; Kim S; Lam A; Kaper T; Kelemen B; Meier KK; Jones SM; Solomon EI; Sandgren M High-resolution structure of a lytic polysaccharide monooxygenase from Hypocrea jecorina reveals a predicted linker as an integral part of the catalytic domain. J. Biol. Chem 2017, 292 (46), 19099–19109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Beeson WT; Vu VV; Span EA; Phillips CM; Marletta MA Cellulose Degradation by Polysaccharide Monooxygenases. Annu. Rev. Biochem 2015, 84 (1), 923–946. [DOI] [PubMed] [Google Scholar]
- (33).Yu F; Penner-Hahn JE; Pecoraro VL De Novo-Designed Metallopeptides with Type 2 Copper Centers: Modulation of Reduction Potentials and Nitrite Reductase Activities. J. Am. Chem. Soc 2013, 135 (48), 18096–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Koebke KJ; Yu F; Salerno E; Stappen CV; Tebo AG; Penner-Hahn JE; Pecoraro VL Modifying the Steric Properties in the Second Coordination Sphere of Designed Peptides Leads to Enhancement of Nitrite Reductase Activity. Angew. Chem., Int. Ed 2018, 57 (15), 3954–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Farrer BT; Harris NP; Balchus KE; Pecoraro VL Thermodynamic Model for the Stabilization of Trigonal Thiolato Mercury(II) in Designed Three-Stranded Coiled Coils. Biochemistry 2001, 40 (48), 14696–14705. [DOI] [PubMed] [Google Scholar]
- (36).Weng T-C; Waldo GS; Penner-Hahn JE A method for normalization of X-ray absorption spectra. J. Synchrotron Radiat 2005, 12 (4), 506–510. [DOI] [PubMed] [Google Scholar]
- (37).George GN; Pickering IJ EXAFSPAK. http://ssrl.slac.stanford.edu/~george/exafspak/exafs.htm
- (38).Ankudinov AL; Rehr JJ Relativistic calculations of spin-dependent x-ray-absorption spectra. Phys. Rev. B: Condens. Matter Mater. Phys 1997, 56 (4), R1712–R1716. [Google Scholar]
- (39).Dimakis N; Bunker G XAFS Debye-Waller factors for Zn metalloproteins. Phys. Rev. B: Condens. Matter Mater. Phys 2004, 70 (19), 195114. [Google Scholar]
- (40).Xiao Z; Loughlin F; George GN; Howlett GJ; Wedd AG C-Terminal Domain of the Membrane Copper Transporter Ctr1 from Saccharomyces cerevisiae Binds Four Cu(I) Ions as a Cuprous-Thiolate Polynuclear Cluster: Sub-femtomolar Cu(I) Affinity of Three Proteins Involved in Copper Trafficking. J. Am. Chem. Soc 2004, 126 (10), 3081–3090. [DOI] [PubMed] [Google Scholar]
- (41).Hendrich M SpinCount; Carnegie Mellon University: Pittsburgh, PA, 2018. [Google Scholar]
- (42).Hahn JE; Scott RA; Hodgson KO; Doniach S; Desjardins SR; Solomon EI Observation of an electric quadrupole transition in the X-ray absorption spectrum of a Cu(II) complex. Chem. Phys. Lett 1982, 88 (6), 595–598. [Google Scholar]
- (43).de Groot F; Vanko V; Glatzel P The 1s x-ray absorption preedge structures in transition metal oxides. J. Phys.: Condens. Matter 2009, 21 (10), 104207. [DOI] [PubMed] [Google Scholar]
- (44).Kau LS; Spira-Solomon DJ; Penner-Hahn JE; Hodgson KO; Solomon EI X-ray absorption edge determination of the oxidation state and coordination number of copper. Application to the type 3 site in Rhus vernicifera laccase and its reaction with oxygen. J. Am. Chem. Soc 1987, 109 (21), 6433–6442. [Google Scholar]
- (45).Isoda N; Yokoyama H; Nojiri M; Suzuki S; Yamaguchi K Electroreduction of nitrite to nitrogen oxide by a copper-containing nitrite reductase model complex incorporated into collagen film. Bioelectrochemistry 2010, 77 (2), 82–88. [DOI] [PubMed] [Google Scholar]
- (46).Tocheva EI; Eltis LD; Murphy MEP Conserved Active Site Residues Limit Inhibition of a Copper-Containing Nitrite Reductase by Small Molecules. Biochemistry 2008, 47 (15), 4452–4460. [DOI] [PubMed] [Google Scholar]
- (47).Leferink NGH; Han C; Antonyuk SV; Heyes DJ; Rigby SEJ; Hough MA; Eady RR; Scrutton NS; Hasnain SS Proton-Coupled Electron Transfer in the Catalytic Cycle of Alcaligenes xylosoxidans Copper-Dependent Nitrite Reductase. Biochemistry 2011, 50 (19), 4121–4131. [DOI] [PubMed] [Google Scholar]
- (48).Zhuang J; Amoroso JH; Kinloch R; Dawson JH; Baldwin MJ; Gibney BR Design of a Five-Coordinate Heme Protein Maquette: A Spectroscopic Model of Deoxymyoglobin. Inorg. Chem 2004, 43 (26), 8218–8220. [DOI] [PubMed] [Google Scholar]
- (49).Pott M; Hayashi T; Mori T; Mittl PRE; Green AP; Hilvert D A Noncanonical Proximal Heme Ligand Affords an Efficient Peroxidase in a Globin Fold. J.Am. Chem. Soc 2018, 140 (4), 1535–1543. [DOI] [PubMed] [Google Scholar]
- (50).Gan F; Liu R; Wang F; Schultz PG Functional Replacement of Histidine in Proteins To Generate Noncanonical Amino Acid Dependent Organisms. J. Am. Chem. Soc 2018, 140 (11), 3829–3832. [DOI] [PubMed] [Google Scholar]
- (51).Prenesti E; Daniele PG; Berto S; Toso S Spectrum–structure correlation for visible absorption spectra of copper(II) complexes showing axial co-ordination in aqueous solution. Polyhedron 2006, 25 (15), 2815–2823. [Google Scholar]
- (52).Zastrow ML; Peacock AFA; Stuckey JA; Pecoraro VL Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat. Chem 2012, 4 (2), 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Dieckmann GR; McRorie DK; Tierney DL; Utschig LM; Singer CP; O’Halloran TV; Penner-Hahn JE; DeGrado WF; Pecoraro VL De Novo Design of Mercury-Binding Two- and Three-Helical Bundles. J.Am. Chem. Soc 1997, 119 (26), 6195–6196. [Google Scholar]
- (54).Kujime M; Izumi C; Tomura M; Hada M; Fujii H Effect of a Tridentate Ligand on the Structure, Electronic Structure, and Reactivity of the Copper(I) Nitrite Complex: Role of the Conserved Three-Histidine Ligand Environment of the Type-2 Copper Site in Copper-Containing Nitrite Reductases. J. Am. Chem. Soc 2008, 130 (19), 6088–6098. [DOI] [PubMed] [Google Scholar]
- (55).Johnson BJ; Mankad NP Model Compounds of Copper-Containing Enzymes Involved in Bacterial Denitrification In Metalloenzymes in Denitrification: Applications and Environmental Impacts; Moura I, Moura JJG, Pauleta SR, Maia LB, Eds.; The Royal Society of Chemistry: London, 2017; pp 225–251. [Google Scholar]
- (56).Boulanger MJ; Kukimoto M; Nishiyama M; Horinouchi S; Murphy MEP Catalytic Roles for Two Water Bridged Residues (Asp-98 and His-255) in the Active Site of Copper-containing Nitrite Reductase. J. Biol. Chem 2000, 275 (31), 23957–23964. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.