

Abstract
Atrial fibrillation (AF) is the most common heart rhythm disorder and a major cause of stroke. Unfortunately, current therapies for AF are suboptimal, largely because the molecular mechanisms underlying AF are poorly understood. Since the autonomic nervous system is thought to increase vulnerability to AF, we used a rapid atrial pacing (RAP) canine model to investigate the anatomic and electrophysiological characteristics of autonomic remodeling in different regions of the left atrium. RAP led to marked hypertrophy of parent nerve bundles in the posterior left atrium (PLA), resulting in a global increase in parasympathetic and sympathetic innervation throughout the left atrium. Parasympathetic fibers were more heterogeneously distributed in the PLA when compared with other left atrial regions; this led to greater fractionation and disorganization of AF electrograms in the PLA. Computational modeling revealed that heterogeneously distributed parasympathetic activity exacerbates sympathetic substrate for wave break and reentry. We further discovered that levels of nerve growth factor (NGF) were greatest in the left atrial appendage (LAA), where AF was most organized. Preferential NGF release by the LAA — likely a direct function of frequency and regularity of atrial stimulation — may have important implications for creation of a vulnerable AF substrate.
Keywords: Cardiology
Keywords: Arrhythmias, Innervation
Region specific heterogeneity of parasympathetic nerve remodeling in the left atrium contributes to creation of a vulnerable substrate for persistence of atrial fibrillation.
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia in adults, affecting 2.7–6.1 million people in the United States and 33.5 million people worldwide. It is a cause of significant morbidity and mortality, being a leading risk factor for stroke (1). The diverse pathophysiology behind AF makes it a challenging therapeutic target. Recent years have seen the emergence of ablation as a major therapeutic advance in the treatment of AF. Unfortunately, even though ablation is at least moderately efficacious in patients with paroxysmal AF, its efficacy is suboptimal in patients with persistent AF (2). This is thought to be in good part because ablation is not targeted at the molecular mechanisms underlying AF (3). A better understanding of the molecular mechanisms underlying the genesis and maintenance of the AF disease state would allow for the development of newer, mechanism-guided therapies for this arrhythmia.
The autonomic nervous system is thought to be an important mechanism underlying the genesis and maintenance of AF. Previous studies have demonstrated sympathetic hyperinnervation in the fibrillating atrium (4), with evidence of increased sympathetic nerve firing in animal models, as well as in patients with AF (5–7). It is also known that increased parasympathetic signaling is a key contributor to refractory period shortening in the atrium. Heightened vagal tone was shown to precede the onset of AF paroxysms in patients (8, 9). In addition, the activity of ganglionated plexi (GP), which provides the majority of parasympathetic innervation to the atria, is increased both in animal models of AF (10) and before the onset of AF in patients undergoing open heart surgery. Recent studies also suggest that the parasympathetic nervous system may be the dominant limb in the creation of an arrhythmogenic substrate in the atrium, with the sympathetic nervous system playing a more modulatory role in creating conditions for triggered activity as well as reentry (11–15). In spite of these initial studies, the precise role of the autonomic nervous system — and the relative role of parasympathetic versus sympathetic signaling — in the creation of electrical remodeling in the fibrillating atrium is not known. It is also not known whether there are regional differences in autonomic remodeling within the left atrium in persistent AF and whether this contributes to the formation of a vulnerable AF substrate. A better understanding of regional differences in autonomic remodeling is especially important in view of mounting evidence that atrial regions outside the pulmonary veins (PVs) — such as the coronary sinus and the left atrial appendage (LAA) — are critical for the maintenance of persistent AF (16–18). Lastly, it is not known how neural remodeling affects the electrophysiological characteristics of the fibrillating atrium. Even though several reports suggest that pathophysiological substrate for AF may be reflected in the characteristics of AF electrograms (EGMs), the specific contribution of remodeled parasympathetic and sympathetic nerves to AF EGM characteristics is not known.
We therefore systematically assessed parasympathetic as well as sympathetic innervation in different regions of the normal canine left atrium — the posterior left atrium (PLA), left atrial free wall (LAFW), and LAA — and then determined autonomic nerve remodeling in these regions after rapid atrial pacing (RAP). We also evaluated the effect of RAP on autonomic postganglionic neuronal cell bodies in the atrial GPs and stellate ganglia. In order to understand the electrophysiological (functional) effects of these remodeled nerves, we also examined the relationship between autonomic innervation and the characteristics of overlying AF EGMs.
Since nerve growth factor (NGF) is a key cytokine known to underlie new nerve sprouting in the heart, we also assessed the relationship between autonomic remodeling and the pattern of NGF release in each atrial region. Our results suggest the presence of an intricate feedback loop between frequency and regularity of atrial myocyte stimulation and NGF release in the atrium, which leads to regional differences in NGF release within the left atrium, with the LAA demonstrating greater levels of NGF than other regions of the left atrium. Indeed, as retrograde transport of NGF from the myocardium by autonomic nerve fibers is a potent stimulus underlying hypertrophy of postganglionic neurons (19, 20), we believe that preferential NGF release from LAA myocytes may be a key mechanism underlying hypertrophy of parent parasympathetic nerve bundles in the fibrillating atrium, and thereby to parasympathetic hyperinnervation in the entire left atrium (with resulting creation of a vulnerable substrate for AF).
Results
Interregional differences in autonomic innervation in normal left atrium.
We first examined autonomic innervation in different regions of the left atrium in normal dogs (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.130532DS1). The PLA had significantly larger nerve bundles than the LAFW and the LAA, with nerve bundles in the LAA being the smallest (Supplemental Figure 1B). This was consistent with previous data that demonstrate the presence of “parent” nerve bundles in the PLA, in and around the atrial fat pads (12). Even though the size of the nerve bundles was smallest in the LAA, the number of nerve bundles (nerve bundle density) was greatest in the LAA (Supplemental Figure 1C). The number of parasympathetic nerve fibers within these bundles was significantly larger than the number of sympathetic fibers in each region (Supplemental Figure 1D). Due to larger bundle size in the PLA and LAFW as compared with the LAA, the number of parasympathetic fibers/bundle was significantly greater in the former 2 regions compared with the LAA (Supplemental Figure 1D).
To determine the contribution of these nerve trunks to the autonomic innervation of the myocardium, we next determined the density of individual parasympathetic and sympathetic nerve fibers in the myocardium of the PLA, LAFW, and LAA. Consistent with the predominance of parasympathetic nerve fibers in individual nerve trunks, each region exhibited a significantly higher concentration of parasympathetic as compared with sympathetic nerve fibers (Supplemental Figure 1E). There was no significant difference in nerve fiber density between the 3 regions. Taken together, the normal canine left atrium demonstrates a preponderance of parasympathetic nerve fibers compared with sympathetic nerve fibers. These fibers appear to largely originate in nerve bundles (trunks) situated in the PLA and, to a lesser extent, in the LAFW.
RAP-induced AF leads to marked nerve bundle hypertrophy in the PLA, resulting in a diffuse increase in parasympathetic and sympathetic innervation throughout the left atrium.
We then assessed autonomic innervation in left atria taken from dogs subjected to chronic RAP (with resulting persistent AF). There was no significant increase in nerve bundle density in any region in the AF dogs (Figure 1A). However, the combined median bundle size was significantly increased in the LAFW of dogs with AF, with a strong trend toward an increase in the PLA and LAA, as well (Figure 1B). Figure 1C shows examples of hypertrophied nerve bundles in the PLA of AF dogs (as compared with normal dogs). An interesting finding of our study was the presence of exceptionally large nerve bundles (>0.01 mm2) in both the PLA and LAFW of AF dogs (Figure 1D). Such large nerve trunks were rarely seen in normal dogs.
Figure 1. Persistent AF induced by rapid atrial pacing (RAP) causes a global increase in left atrial autonomic nervous fibers.

(A) Nerve bundle density in the posterior left atrium (PLA), left atrial free wall (LAFW), and left atrial appendage (LAA) of normal dogs and animals subjected to RAP is shown as median with interquartile range. n = 3, 4, 5, 5, 4, and 6. (B) Combined nerve bundle size in the PLA, LAFW, and LAA of normal dogs and animals subjected to RAP is shown as median with interquartile range. n = 53 and N = 4 for normal PLA, n = 69 and N = 6 for RAP PLA, n = 138 and N = 6 for normal LAFW, n = 125 and N = 6 for RAP LAFW, n = 112 and N = 5 for normal LAA, and n = 160 and N = 6 for RAP LAA. (C) Representative micrographs of nerve bundles in left atrial tissue of normal dogs (left) and after RAP (right) subjected to IHC for AChE (brown) and DBH (blue). (D) Examples of very large bundles seen in the PLA and LAFW after RAP. For C and D, scale bar: 500μm. (E) Sympathetic and (F) parasympathetic myocardial fiber density in the PLA, LAFW, and LAA of normal dogs or after RAP is shown mean ± SEM. For sympathetic fibers, n = 3, 4, 6, 4, 6, and 4. For parasympathetic fibers, n = 4, 6, 5, 6, 6, and 7. (G) Representative micrographs of IHC for AChE (brown) and DBH (blue) on left atrial tissue of normal dogs (left) and after RAP (right). Examples of parasympathetic (AChE+) fibers indicated with arrows. Examples of sympathetic (DBH+) fibers indicated with arrowheads. Scale bar: 250 μm. Two-way ANOVA significance indicated in graphs, after log transformation for nonparametric values. *P < 0.05; **P < 0.01; ***P < 0.001 for pairwise comparison with Holm-Sidak method.
Since nerve bundle size was increased globally, we then examined the relative contribution of parasympathetic and sympathetic nerve fibers to these hypertrophied nerve trunks. In the setting of persistent AF, both the number of sympathetic and parasympathetic nerve fibers per nerve bundle increased significantly in the PLA (Supplemental Figure 2, A and B), with sympathetic and parasympathetic fiber counts per bundle being significantly greater in the PLA than in the LAA. The preponderance of parasympathetic fibers as compared with sympathetic fiber per bundle was preserved in all 3 regions in the setting of AF. Taken together, these data indicate that RAP-induced AF leads to a marked hypertrophy of nerve bundles in the left atrium, with nerve hypertrophy being most pronounced in the PLA, and with a predominance of parasympathetic fibers.
In order to understand how these hypertrophied nerve trunks in persistent AF innervate the atrial myocardium, we then examined the density of individual nerve fibers in the myocardium in the setting of AF. Not unexpectedly, sympathetic as well as parasympathetic nerve fiber density was markedly increased in the myocardium of the fibrillating left atrium (Figure 1, E and F). Examples are shown in Figure 1G. This increase in the density of individual myocardial nerve fibers — both sympathetic and parasympathetic — was noted in all 3 regions (PLA, LAFW, and LAA) and was approximately equal across the 3 regions (Figure 1, E and F). As in the nerve trunks, parasympathetic nerve fibers predominated over sympathetic nerve fibers in the AF left atrium, with this preponderance being noted in each of the 3 regions. Taken together, these data demonstrate that, while the nerve bundle size increase is largely localized to the PLA, this leads to a global increase in both sympathetic and parasympathetic nerve fibers in the myocardium of the entire left atrium.
Although a systematic examination of right atrial innervation — and the corresponding electrophysiology of this region — was not the goal of this study, we did perform a qualitative assessment of autonomic nerve distribution in the right atrium. We examined both sympathetic and parasympathetic innervation in the posterior right atrium, right atrial free wall, and right atrial appendage (RAA) after RAP. As in the left atrium, we found evidence of large nerve trunks in the posterior right atrium (i.e., in the vicinity of the GP at the right atrium-inferior vena cava junction; Supplemental Figure 3). As in the left atrium, we found evidence of a large number of sympathetic and parasympathetic nerve fibers in the myocardium of the posterior right atrium, right atrial free wall, and RAA.
RAP-induced autonomic hyperinnervation of the left atrium is caused by postganglionic neuron hypertrophy and new nerve sprouting.
Given evidence of both nerve bundle hypertrophy and the increase in sympathetic and parasympathetic nerve fibers in the fibrillating atria, we examined the effect of RAP on postganglionic neurons. Representative examples of atrial GPs (providing parasympathetic innervation) and left stellate ganglia (LSG, providing sympathetic innervation) are shown in Figure 2, A and B. Importantly, RAP caused significant hypertrophy of both parasympathetic and sympathetic neurons, as determined by an increase in the area occupied by cell bodies when compared with unpaced animals. To further evaluate the mechanism of hyperinnervation in AF, we determined the expression of GAP43, a neuronal growth cone marker (19), in the atria of normal animals and after RAP. Similarly to previous reports (6), RAP caused a significant increase in GAP43 mRNA in the atria (Figure 2C). Consistent with our earlier findings of diffuse increase in parasympathetic and sympathetic fibers, the expression of GAP43 mRNA did not appear to vary between regions of the left atrium (Figure 2D). Taken together, these results suggest that the parasympathetic and sympathetic hyperinnervation seen in AF occurs secondary to postganglionic neuron hypertrophy in the GPs and LSG, respectively, which then facilitates new nerve sprouting in all regions of the left atrium via increased GAP43.
Figure 2. Persistent AF induced by rapid atrial pacing (RAP) causes hypertrophy of peripheral sympathetic and parasympathetic postganglionic neurons and nerve sprouting.

(A) Representative micrographs of IHC for AChE (brown) and DBH (blue) of left atrial ganglionated plexi (GP) and (B) left stellate ganglion (LSG) of normal animals (left) and after RAP (middle). The area occupied by postganglionic neurons (ratio of total cell area over ganglionic area) is shown on the right. For GP, n = 22 and N = 6 for normal, and n = 27 and N = 7 for RAP. For LSG, n = 18 and N = 3 in each group. (C) Relative GAP43 mRNA level by qPCR for normal atria and after RAP. N = 7 for normal and 6 for RAP. (D) NGF mRNA level in the posterior left atrium (PLA), left atrial free wall (LAFW), and left atrial appendage (LAA) after RAP. N = 8, 7, and 7, respectively. One-way ANOVA significance indicated in graph. ***P < 0.001 for pairwise comparison with t test.
Parasympathetic fibers in the RAP atrium are more heterogeneously distributed in the PLA than in the LAA.
Next, we examined the spatial distribution of autonomic nerve fibers in the rapidly paced left atrium. The coefficient of variation of sympathetic nerve fibers was approximately the same in the PLA, LAFW, and LAA (Figure 3A). However, coefficient of variation of parasympathetic nerve fibers was significantly greater in the PLA than in the LAA (Figure 3B), indicating more heterogeneous distribution of parasympathetic nerve fibers in the PLA. Figure 3C shows examples of increased heterogeneity of parasympathetic nerve distribution in the rapidly paced PLA as compared with the LAA.
Figure 3. Rapid atrial pacing (RAP) causes a heterogeneous parasympathetic nerve increase in the posterior left atrium (PLA).

(A) The coefficient of variation of sympathetic and (B) parasympathetic fiber density after RAP in the PLA, left atrial free wall (LAFW), and left atrial appendage (LAA) is shown as mean ± SEM. For sympathetic fibers, n = 4 in all groups. For parasympathetic fibers, n = 4, 5, and 6, respectively. ANOVA significance indicated in graph. **P < 0.01 for pairwise comparison with Holm-Sidak method. (C) Representative micrographs of IHC for AChE (brown) and DBH (blue) on tissue from the LAA (left) and PLA (right) after RAP. Examples of parasympathetic (AChE+) fibers indicated with arrows. Scale bar: 250 μm.
Effect of autonomic remodeling on AF characteristics in the intact atrium.
After examining the nature of neural remodeling of the rapidly paced left atrium, we examined the effects of this neural remodeling on the electrophysiology of the intact left atrium. Since these animals were in persistent AF, we examined the frequency characteristics and complexity of AF in the different regions of the left atrium at baseline and after administration of autonomic blockade.
At baseline, as shown in Figure 4A, organization index (OI) and fractional interval (FI) were significantly greater in the LAA than in the PLA and LAFW, indicating greater temporal organization/regularity and less fractionation of AF in the LAA than in the other 2 regions of the left atrium. Figure 4B shows examples of AF EGMs being more organized/regular and less fractionated in the LAA than in the PLA and LAFW. We also examined the spatial variation of AF parameters within each region, by determining the coefficient of variation of each AF parameter between the electrodes on the recording plaque. The coefficient of variation of dominant frequency (DF), OI, and recurrence cycle length (CLR) was found to be significantly greater in the PLA and LAFW than in the LAA (Figure 4C), indicating reduced spatial organization of AF EGMs in the former 2 regions as compared with the LAA.
Figure 4. Atrial fibrillation is less complex and more organized in the left atrial appendage.

(A) Dominant frequency (DF), organization index (OI), fractionation interval (FI), Shannon’s entropy (ShEn), and recurrence cycle length (CLR) in the posterior left atrium (PLA), left atrial free wall (LAFW), and left atrial appendage (LAA) is shown as mean ± SEM. n = 10 for PLA, n = 9 for LAFW, n = 11 for LAA except for CLR, where n = 8 for all groups. (B) Representative atrial electrograms in the PLA (top), LAFW (middle) and LAA (bottom). (C) Coefficient of variation of parameters shown in A. n = 11 for PLA, n = 8 for LAFW, n = 11 for LAA. ANOVA significance indicated in graphs. *P < 0.05; **P < 0.01; ***P < 0.001 for pairwise comparison with Holm-Sidak method.
Since the temporal and spatial heterogeneity of AF EGMs in each atrial region appears to correspond to the pattern of parasympathetic distribution in each region (with AF EGMs being most organized and regular in the LAA, where parasympathetic nerves are most homogeneously distributed), we postulated that spatial differences in distribution of parasympathetic nerve fibers at least partially underlie the observed differences in temporal and spatial organization of AF observed between the PLA, LAFW, and LAA.
To further investigate this possibility, we examined the effect of parasympathetic and dual autonomic blockade on AF EGMs. With atropine, there was a modest decrease in DF and a modest increase in OI in the PLA (Figure 5A). Similar trends were noted in the LAA, as well. However, when we compared the extent of change for each parameter with atropine, between all the electrodes on the mapping plaque, the change in DF, OI, fractionation interval (FI), Shannon’s entropy (ShEn), and CLR was markedly more heterogeneous in the PLA and LAFW than in the LAA (Figure 5, B and C). With administration of propranolol after atropine (double autonomic blockade), there was a further decrease in DF in all regions, and there was a further increase in FI and decrease in ShEn in the LAA (Figure 5A). However, there was no further increase in spatial heterogeneity of AF EGMs in response to double autonomic blockade in any atrial region (Figure 5, D and E). These findings further support our notion that spatial differences in AF EGM properties are at least partially related to the underlying distribution of parasympathetic nerve fibers.
Figure 5. Response to atropine is more homogeneous in the left atrial appendage (LAA), whereas propranolol does not induce a differential response.

(A) Effect of atropine or dual blockade on mean dominant frequency (DF), organization index (OI), fractionation interval (FI), Shannon’s entropy (ShEn), and recurrence cycle length (CLR) in the posterior left atrium (PLA) and left atrial appendage (LAA). n = 10 for baseline and atropine, n = 4 for dual blockade. Data is shown as scatterplot with mean ± SEM from mixed linear model. (B) Representative example of high-density electrode recordings of DF (left panels) and OI (right panels) in the PLA (top) and LAA (bottom) at baseline and after atropine. The change induced by atropine is shown as a Δ map. (C) SD of change induced by atropine in parameters shown in A for the PLA, left atrial free wall (LAFW), and LAA is shown as scatterplot with mean ± SEM. n = 10 in all groups. ANOVA significance indicated in graphs. (D) Representative examples of high-density electrode recordings of DF (right) and OI (left) in the PLA (top) and LAA (bottom) after atropine and dual blockade. The change induced by propranolol is shown as a Δ map. (E) SD of change induced by propranolol in parameters shown in A for the PLA and LAA is shown as scatterplot with mean ± SEM. n = 4 in all groups. ANOVA significance indicated in graphs. *P < 0.05; **P< 0.01; ***P < 0.001 for pairwise comparison with Holm-Sidak method.
Taken together, these data demonstrate that all 3 regions of the left atrium show significant responsiveness to autonomic blockade, with AF becoming slower, less fractionated, and more organized with autonomic blockade in each region. Furthermore, the spatial distribution of parasympathetic nerve fibers in the RAP left atrium appears to have a direct influence on the organizational characteristics of overlying AF EGMs, with EGMs being most organized in the LAA, where parasympathetic nerves are the most homogenously distributed.
Spatially heterogeneous distribution of parasympathetic nerve fibers contributes to disorganization of AF EGMs.
To further understand the relationship between distribution of parasympathetic and sympathetic nerves and frequency characteristics of AF EGMs, we first established the effect of sympathetic stimulation on atrial cardiomyocyte electrophysiology, before adding parasympathetic heterogeneity to the model. It is well established that sympathetic stimulation enhances a range of processes that control Ca2+ entry into the cell. We therefore increased the conductance of Ca2+ channels (ICa) to model the effect of sympathetic stimulation in order to explore how subcellular Ca2+ dynamics change with increased sympathetic tone. Figure 6, A–C, shows Ca2+ and voltage for 3 simulations where the conductance of ICa is increased systematically. For low ICa conductance (Figure 6A) we observed that the Ca2+ transient and action potential (AP) was essentially periodic from beat-to-beat, and that Ca2+ release occurred mainly at the cell boundary. As shown in our previous study (21), this response occurs because atrial myocytes have a poorly developed t-tubule system so that the bulk of Ca2+ signaling occurs at the cell boundary. After a 10% increase in ICa conductance (Figure 6B), the Ca2+ content in the cell is increased and Ca2+ waves begin to penetrate into the cell interior intermittently. When these Ca2+ waves occur, the global Ca2+ transient is increased due to the release of interior Ca2+. Consequently, we conclude that the AP duration (APD) on those beats is prolonged due to stimulation of sodium calcium exchanger (NCX) by the Ca2+ waves. After an additional 3% increase in ICa (Figure 6C), the frequency of subcellular Ca2+ waves increases substantially, leading to a highly aperiodic Ca2+ transient and APD. These results demonstrate that sympathetic stimulation promotes an increased frequency of subcellular Ca2+ waves that propagate inside the cell interior. These subcellular Ca2+ waves have been studied extensively by our group (21, 22) and have been shown to occur in normal atrial myocytes under rapid pacing conditions. In both cases, these waves originate under conditions of increased sarcoplasmic reticulum (SR) loading, where the interior of the atrial myocyte is sensitized and can support the propagation of Ca2+ waves.
Figure 6. Sympathetic stimulation enhances the frequency of subcellular Ca2+ waves and is modulated by parasympathetic tone in a computational model.

Spatially distributed atrial cell model is used to compute Ca2+ transient Ci, voltage VT, and a linescan through the center of the cell. The cell is paced for 50 beats at CL = 400 ms and only the last 20 beats are shown. (A) Reference model where ICa is adjusted to produce normal Ca2+ transients due to Ca2+ release at the cell boundary. (B) ICa conductance is increased by 10% above reference. (C) ICa conductance increased by 13% above reference. (D) Membrane voltage VT, and the Ca2+ transient Ci during pacing at CL = 300 ms with [ACh] = 1 × 10–4 μmol/l, and pb = 0.45, to simulate high sympathetic tone and low parasympathetic activity, or (E) with [ACh] = 5 × 10–3 μmol/l and pb = 0.45 to simulate high sympathetic and parasympathetic activity. Beats shown are at steady state.
Our 3-dimensional (3-D) computational model demonstrates that sympathetic stimulation is associated with an increased frequency of subcellular Ca2+ waves. However, this computational model, which simulates thousands of ion channels within a single atrial myocyte, cannot be incorporated into cardiac tissue simulations with several thousand electrically coupled cells. Thus, to investigate the interplay between parasympathetic and sympathetic stimulation, we have applied our phenomenological approach where only the population number of Ca2+ sparks is accounted for. Using this approach, we can explore the interactions between acetylcholine activated potassium channel (IKAch) and subcellular Ca2+ wave activity on the tissue scale.
To explore this interaction, we vary acetylcholine concentration ([ACh]) and the threshold for Ca2+ wave propagation from the atrial cell boundary to the interior (pb) to simulate different degrees of parasympathetic and sympathetic stimulation. In Figure 6D, we simulate high sympathetic tone and low parasympathetic activity. The threshold for Ca2+ wave propagation is lowered, causing aberrant Ca2+ release. The resulting activation of NCX leads to slowing of atrial AP repolarization and significant beat-to-beat variation of the maximum diastolic potential. In Figure 6E, we simulate a paced cardiac cell with high sympathetic and parasympathetic tone. The AP repolarization is accelerated due to increased IKACh, which opposes the effect of NCX. As a result, the voltage dynamics are periodic and lack the large variations in the maximum diastolic potential seen in Figure 6D. Also, since the APD is reduced, there is less Ca2+ entry into the cell. Thus, the effect of parasympathetic stimulation is to reduce fluctuations in the maximum diastolic potential that are caused by Ca2+ wave activity.
We next evaluated the effect of heterogeneous autonomic stimulation by exploring the stability of planar wave propagation in a 2-dimensional (2-D) sheet of 160 × 160 cells. We paced our tissue by periodically stimulating the left edge of the 2-D sheet. Figure 7A shows snapshots of the membrane voltage on our paced tissue under high sympathetic and parasympathetic stimulation. The AP wave front proceeds uniformly through the 2-D tissue. The bottom trace shows the voltage time course (VT) at the tissue center, which shows an approximately periodic response with minimal beat-to-beat fluctuations. To model a heterogeneous distribution of parasympathetic innervation, we used our light microscope data of fiber counts within tissue slices of size 294 × 220 μm. As these slices cut the tissue at an arbitrary angle, we estimated that the number of cells within each slice is roughly 50–150 cells. To approximate this data, we made the assumption that the local [ACh] concentration is proportional to the number of parasympathetic fibers. Thus, we divided our 160 × 160 tissue into squares of size 10 × 10 and then determined the local [ACh] concentration as [ACh] = (1 × 10–3 μmol⁄l) nfiber/nmax, where nfiber is the number of fibers and where nmax is the maximum number of fibers recorded from our samples. To determine nfiber for each tissue section, we picked the fiber count at random from a list of 68 distinct images. Figure 7B shows snapshots of the voltage from our paced 2-D tissue with heterogeneous parasympathetic activity. After several paced beats, the planar excitation emanating from the boundary begins to fractionate due to localized conduction block. The voltage time course for a cell at the tissue center shows a highly aperiodic time dependence due to the wave fractionation that is occurring in the tissue.
Figure 7. Spatially heterogeneous parasympathetic activity in the setting of high sympathetic activity leads to wave break and reentry in a computational model.

(A) Snapshots of the membrane voltage when the 2-D tissue is paced at the left edge at CL = 300 ms. Times shown are measured after the 21st paced beat. Model parameters are [ACh] = 1 × 10–2 μmol/l and pb = 0.45 to simulate high sympathetic and parasympathetic activity. Voltage trace shown is for a cell located at lx = 80 and ly = 80. (B) Same simulation with heterogeneous distribution of [ACh] proportional to the experimentally measured parasympathetic fiber counts. Times shown are measured after the 27th paced beat.
The main finding of these simulations is that, in the presence of enhanced sympathetic tone, a heterogeneous distribution of [ACh] yields a substrate for wave break and reentry. In the absence of parasympathetic stimulation, sympathetically induced Ca2+ waves cause AP fluctuations that slow repolarization. At rapid rates, this slowed repolarization leads to spatially heterogeneous conduction slowing since conduction velocity is highly sensitive to the maximum diastolic potential. This leads to localized wave break in regions of slowed repolarization. The presence of a heterogeneous distribution of [ACh] exacerbates this effect by amplifying these conduction heterogeneities. In particular, if [ACh] is reduced in a region of tissue, the elevated maximum diastolic potential will induce conduction block in that region during rapid pacing. Once this occurs, the fractionated wave propagates readily in regions of high [ACh] but not in regions of low [ACh]. Taken together, these results suggest that, in the presence of elevated sympathetic tone, the heterogeneous distribution of parasympathetic innervation, as modeled by heterogeneous [ACh], provides a substrate for wave break and reentry.
Frequency-dependent secretion of NGF by atrial myocytes — a likely mechanism underlying global autonomic remodeling in the fibrillating atrium.
NGF has been shown to be a key stimulus underlying sprouting of both sympathetic and parasympathetic nerves in the heart (19, 20, 23). Although NGF has been reported to increase in the rapidly paced atrium and also in the human AF atrium, the molecular mechanisms underlying this NGF release and the precise cellular source of this NGF have not been previously elucidated. Furthermore, it is not known whether there are regional differences in NGF secretion in the fibrillating atrium or whether these regional differences contribute to autonomic and electrophysiological remodeling in AF.
NGF expression was found to increase 6-fold in the rapidly paced atrial myocardium as compared with controls (Figure 8A). In comparison, NGF was only modestly increased in the atrial fat pads (containing GPs, Figure 8B), indicating that atrial myocytes may be the primary source of NGF release in the fibrillating atrium. We also compared myocardial NGF secretion between the PLA, LAFW, and LAA in both normal and RAP left atrium. In normal left atrium, there was no significant difference in NGF levels between these 3 regions (Figure 8C). In contrast, NGF levels were markedly higher in the rapidly paced LAA than in the other 2 regions of the left atrium (Figure 8D).
Figure 8. NGF secretion by atrial myocytes is dependent on frequency and regularity of activation.

(A) Relative NGF mRNA level by qPCR and NGF protein level by Western Blot for normal atria and after rapid atrial pacing (RAP). n = 8 for qPCR, n = 3 for Western blot. (B) Relative NGF mRNA level by qPCR from atrial fat pads (containing ganglionated plexi) of normal dogs and after RAP. n = 5. (C) NGF mRNA level in the posterior left atrium (PLA), left atrial free wall (LAFW), and left atrial appendage of normal dogs, and (D) after RAP. n = 3, 5, and 5 for normal regions, respectively. n = 7, 7, and 8 for RAP regions, respectively. (E) NGF mRNA level in HL-1 cells in control conditions or after pacing at 3 Hz or 6 Hz. n = 6, 4, and 6, respectively. (F) NGF mRNA level in HL-1 cells paced at 6 Hz regularly, with 25% or 50% variability. n = 6, 4, and 4, respectively. All data are shown as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 for pairwise comparison by t test or 1-way ANOVA with Holm-Sidak method when more than 2 groups were compared. Overall ANOVA significance indicated in graphs.
In view of the interregional differences in AF characteristics described earlier, we hypothesized that NGF is primarily secreted by atrial myocytes in a frequency-dependent fashion, with regional differences in NGF release being secondary to differences in frequency characteristics of AF between these regions. To investigate this hypothesis, we determined NGF secretion in an atrial myocyte cell line (HL-1 cells) at increasing frequency of pacing. As shown in Figure 8E, there was a progressive increase in NGF mRNA expression, with increasing frequency of tachypacing in HL-1 cells. We confirmed these findings in isolated canine atrial myocytes (Supplemental Figure 4). Furthermore, NGF secretion by atrial myocytes appeared to be a function of the regularity of atrial stimulation, with NGF release being significantly greater in the setting of regular as compared with irregular tachypacing (Figure 8F). Since AF is less fractionated, more regular, and more organized in the LAA, these data indicate that rapid and regular stimulation of atrial myocytes in the LAA may be a mechanism underlying preferential release of NGF in this region.
Since the concentration of parasympathetic and sympathetic nerve fibers in the RAP left atrium was approximately equal in the PLA, LAFW, and LAA, it also became apparent from our findings that the neural hyperinnervation observed in each atrial region was not directly related to local release of NGF. Local NGF release in the ventricular myocardium after a myocardial infarction has been shown to affect hyperinnervation at sites remote from the site of NGF release by virtue of a dynamic feedback between ventricular myocytes and neuronal cell bodies in the stellate ganglia (19). As represented in Figure 9, we therefore postulate that a similar feedback mechanism involving retrograde transport of NGF may underlie autonomic remodeling and perpetuation of electrical remodeling in the fibrillating atrium.
Figure 9. Schematic illustration of potential mechanism of sympathetic and parasympathetic hyperinnervation in atrial fibrillation (A–F).

Rapid activation leads to stimulation of atrial myocytes to secrete nerve growth factor (NGF), with organized activation in the left atrial appendage causing the most pronounced effect. Retrograde transport within autonomic axons leads to hypertrophy and stimulation of postganglionic cells in the stellate ganglia and the ganglionated plexi, and it induces global hyperinnervation of the left atrium. This, in turn, promotes persistence of atrial fibrillation.
Discussion
AF is the most common heart rhythm disorder and a major cause of stroke (1). Unfortunately, both pharmacological and ablative therapies for AF have suboptimal efficacy in patients with persistent AF. A better understanding of the fundamental, molecular mechanisms underlying AF is essential for the development of newer and potentially more efficacious therapies for this arrhythmia. The current study sheds new light on how the autonomic nervous system may be contributing to the creation and maintenance of electrical remodeling in AF. The study also highlights a potentially important role for the LAA — a chamber typically associated with thrombus formation in the fibrillating atrium but not considered a common site for the origin of AF triggers — for the creation of a vulnerable substrate for AF.
Interregional differences in parasympathetic remodeling in the fibrillating atrium may be a contributor to a vulnerable AF substrate.
Several studies have suggested that increased and/or aberrant autonomic innervation may be an important contributor to the creation of a vulnerable AF substrate (11, 14, 24–26). Stimulation of the parasympathetic nervous system activates IKAch, while activation of the sympathetic nervous system results in an increased Ca2+ transient (4, 14). When firing simultaneously, these 2 arms of the autonomic nervous system can abbreviate APD while increasing the Ca2+ transient amplitude and duration, thereby providing optimal conditions for the development of triggered activity and reentry, and therefore AF (14, 27, 28).
Recent studies indicate that the PVs have a unique pattern of autonomic innervation that may contribute to the emergence of focal AF triggers (29). It is also known that there is sympathetic hyperinnervation in the fibrillating atrium, with evidence of heightened sympathetic discharge from stellate ganglia in dogs with RAP-induced AF (4–7, 10). Although there is also evidence of intermittent parasympathetic discharge before episodes of atrial tachycardia in the RAP model, the precise nature of parasympathetic nerve remodeling in the fibrillating atrium is not known. A better understanding of regional differences in autonomic remodeling is especially important in view of recent evidence that atrial regions outside the PVs, such as the LAA, may be critical to the maintenance of persistent AF (16–18). Our results demonstrate that persistent AF, as modeled by chronic RAP, leads to marked hypertrophy of the nerve bundles located in and around the PVs, and of the parent postganglionic neuronal cell bodies innervating the heart (GPs and LSGs). This leads to a marked increase in markers of nerve sprouting and in the number of parasympathetic and sympathetic nerve elements in all regions of the atrial myocardium, but with a predominant increase in parasympathetic innervation. Furthermore, we noted significant interregional differences in the pattern of parasympathetic innervation, with parasympathetic nerve fibers being more heterogeneously distributed in the PLA and the LAFW as compared with the LAA.
We also discovered that AF is spatially and temporally more organized in the LAA — where parasympathetic innervation is most homogenous — than in the PLA or LAFW. Furthermore, atropine-induced changes in AF EGM characteristics appeared to closely parallel the heterogeneity of parasympathetic nerve fibers, indicating that nerve fiber distribution may be contributing to interregional differences in AF EGM characteristics.
Influence of sympatho-vagal interactions on formation of arrhythmogenic substrate and creation of disorganized AF EGMs.
The role of sympatho-vagal interactions in the genesis of ventricular arrhythmias has been well described, with vagal stimulation shown to attenuate sympathetic augmentation of arrhythmogenic substrate in the ventricle (30, 31). More recently, sympatho-vagal interactions have been invoked in the creation of a vulnerable substrate for AF. Combined firing of sympathetic and parasympathetic nerves has been demonstrated immediately prior to onset of atrial arrhythmias in animal models of AF (10, 32). Clinical studies also suggest altered sympatho-vagal balance — as assessed by heart rate variability — prior to onset of AF (33). However, the precise nature of sympatho-vagal interactions in the intact atrium — and how these interactions may lead to AF — are not known. We first modeled for heightened sympathetic tone by increasing Ca2+ load in rapidly stimulated atrial myocytes. This led to sarcoplasmic reticulum Ca2+ overload and the formation of Ca2+ waves; these Ca2+ waves, by altering resting membrane potential and by prolonging APD, create conditions for reentry. In the presence of spatially uniform parasympathetic signaling, the vulnerability of the atrium to sympathetic-induced reentry was attenuated. However, a nonuniform distribution of parasympathetic nerve fibers — as modeled based on our observations in the PLA and LAFW — appears to markedly exacerbate sympathetic-induced substrate for reentry and also facilitates increased fractionation of AF EGMs. These findings provide mechanistic insights into the unique interaction between heightened sympathetic tone and heterogeneous parasympathetic activity to create the electrophysiological substrate for AF.
Frequency-dependent release of NGF from atrial myocytes may underlie autonomic remodeling in the fibrillating atrium.
NGF is a key cytokine thought to promote autonomic nerve sprouting in the ventricle (19, 20) and atrium (4, 34, 35). NGF levels have been shown to increase in atrial tissue in an animal model of AF (36) and in the serum after ablation for AF; the latter is thought to contribute to hyperinnervation following AF ablation (37). In this study, we discovered that, despite the presence of an approximately equal number of parasympathetic and sympathetic nerve fibers in all regions of the left atrium, NGF levels were significantly higher in the LAA than in the PLA or LAFW. Importantly, previous studies have suggested that diffuse ventricular sympathetic hyperinnervation following myocardial infarction is secondary to local secretion of NGF in the peri-infarct region (19, 20). Retrograde transport of NGF by sympathetic nerve fibrils from the infarcted region to the stellate ganglia results in hypertrophy of postganglionic neurons in stellate ganglia. This, in turn, leads to diffuse sympathetic hyperinnervation of the left ventricle at regions remote from the infarct region, thereby promoting ventricular arrhythmias. Our findings in the left atrium — where neural hyperinnervation patterns do not directly correlate with local levels of NGF — suggest a similar mechanism: we postulate that NGF released from fibrillating atrial myocytes in the LAA is taken up by parasympathetic and sympathetic nerve fibrils in the atrial myocardium and is retrogradely transported to the atrial GPs and the stellate ganglia, respectively, thereby leading to hypertrophy of postganglionic neurons in these structures. This hypertrophy of “parent” ganglia then leads to diffuse parasympathetic and sympathetic hyperinnervation through all regions of the left atrium (as was seen in our study).
We also discovered that NGF release from atrial myocytes is a function of both the frequency and the regularity of atrial stimulation. It is therefore possible — even likely — that the preferential release of NGF in the LAA is at least partially due to AF being more organized and more regular in this region. Our findings may provide an important molecular mechanism by which the LAA may be contributing to the maintenance of electrical remodeling in persistent AF.
Translational and clinical implications.
A major clinical implication of our study is that the LAA may not be an “innocent bystander” in the creation of a vulnerable AF substrate but, instead, an important contributor to the genesis of autonomic remodeling in the fibrillating atrium (and therefore to the onset and persistence of electrical remodeling in AF). Indeed, a burgeoning amount of literature is beginning to implicate the LAA in the genesis and/or maintenance of electrical remodeling in persistent AF (16–18). In patients with persistent AF, addition of LAA ligation with the LARIAT device to conventional ablation appeared to improve the success rate of AF ablation (18, 38–40). Di Biase et al. demonstrated freedom from recurrence of atrial arrhythmias in patients who have undergone complete electrical isolation of the LAA, as compared with standard ablation for persistent AF alone (41). Similarly, surgical studies in which the LAA is removed at the time of open-heart surgery also demonstrate greater freedom from AF than patients that do not undergo appendage removal (42, 43). Adjunctive LAA isolation in patients undergoing PV isolation is therefore beginning to gain support as a treatment of persistent AF.
Study limitations.
Autonomic remodeling in the right atrium was not assessed in detail in this study. Furthermore, we did not investigate the mechanisms underlying the observed differences in homogeneity of parasympathetic innervation between the PLA, LAFW, and LAA. Future studies are also needed to more precisely establish the link between NGF release in the LAA and autonomic remodeling in the atria. For example, it would be important to determine whether LAA removal — or electrical isolation — will prevent or least attenuate parasympathetic and sympathetic remodeling in the RAP left atrium. Lastly, as this study was conducted in canines, the degree to which our conclusions can be extrapolated to human AF development is, as yet, undefined.
Methods
RAP model
Purpose-bred hounds weighing 25–35 kg over 1 year in age were used for this study. Eleven hounds underwent RAP for the AF EGM mapping with autonomic blockade. Sixteen normal and 18 rapid atrial paced hounds were used for the tissue nerve analysis. The RAP model for AF was performed similarly to previously published techniques. Sterile surgery for pacemaker implantation was performed for each dog. Endocardial pacing leads were placed into the RAA. The pacemakers were programmed to pace at 600 bpm at a minimum of 4 times the capture threshold. The dogs were paced for 3–6 weeks to induce sustained AF. Once persistent/sustained AF was induced, the animals were subjected to a terminal, open-chest electrophysiological mapping study. Before undergoing pacemaker implantation and electrophysiological mapping, all animals were premedicated with acepromazine (0.01–0.02 mg/kg, Vedco) and were induced with propofol (3–7 mg/kg, Zoetis). All experiments were performed under general anesthesia (inhaled) with isoflurane (1–3%; Patterson Veterinary). Adequacy of anesthesia was assessed by toe pinch and palpebral reflex.
In vivo electrophysiological mapping
AF EGM mapping.
At the terminal study, high-density epicardial activation mapping was performed using a UNEMAP mapping system (University of Auckland, Auckland, New Zealand). A triangular plaque containing 130 electrodes (interelectrode distance of 2.5 mm) was used to record 117 bipolar EGMs at a 1 kHz sampling rate. At least 4 successive recording were made on the LAA, PLA, and LAFW. In some animals, consecutive recordings were obtained using 2 rectangular plaques epicardially positioned on the LAFW and LAA with a GE Prucka Cardiolab system (GE Healthcare). Each plaque had 12 electrodes (3 × 4 electrodes; interelectrode distance, 5 mm). AF EGMs were recorded in order to determine the following EGM characteristics: (a) DF, (b) OI, (c) FI, (d) ShEn, and (e) CLR.
DF.
DF is a frequency domain measure of activation rate. Following bandpass filtering with cutoff frequencies of 40 and 250 Hz and rectification, the power spectrum of the EGM segment was computed using the fast Fourier transform. The frequency with the highest power in the power spectrum was considered the DF.
OI.
OI is a frequency domain measure of temporal organization or regularity (44, 45). It has been shown that AF episodes with recordings with high OI are more easily terminated with burst pacing and defibrillation. OI was calculated as the area under 1-Hz windows of the DF peak and the next 3 harmonic peaks divided by the total area of the spectrum from 3 Hz up to the fifth harmonic peak.
FI.
FI is the mean interval between deflections detected in the EGM segment (6). Deflections were detected if they meet the following conditions: (a) the peak-to-peak amplitude was greater than a user-determined noise level, (b) the positive peak was within 10 ms of the negative peak, and (c) the deflection was not within 50 ms of another deflection. The noise level was determined by selecting the amplitude level that would avoid detection of noise-related deflections in the iso-electric portions of the signal. FI is dependent on both the AF cycle length and the fractionation of the EGM.
ShEn.
ShEn is a statistical measure of complexity (46). The 4000 or 3908 (depending on the 1 kHz or 977 Hz sample rate) amplitude values of each EGM segment were binned into one of 29 bins with width of 0.125 SDs. ShEn was then calculated as:
(Equation 1)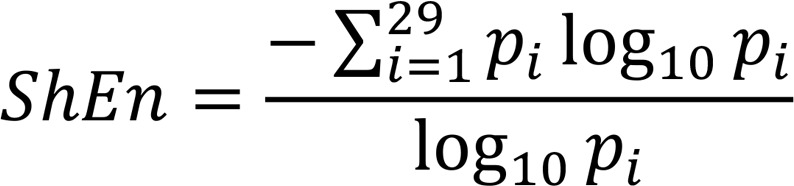
In this equation, pi is the probability of an amplitude value occurring in bin i.
CLR.
Cycle length (CL) of the most recurrent morphology was obtained by dividing the average CL for all EGMs by the recurrence percentage (47).
The above measures were assessed for each pixel/electrode on each plaque. There were a small number of electrodes (<10%) where signal (EGM) quality was inadequate (e.g., due to noise, poor contact) for assessment of the above measures. These pixels are shown as white in Figure 5.
Autonomic blockade.
Autonomic blockade consisted of (a) parasympathetic blockade with atropine (0.04 mg/kg; Med-Pharmex Inc.) and (b) double autonomic blockade with propranolol (0.2 mg/kg; Westward) and atropine (0.04 mg/kg). Atrial EGMs were recorded at baseline, after parasympathetic blockade and after dual blockade.
Data analysis.
Results are displayed as mean value averaged over the entire plaque. Spatial dispersion of atrial EGM characteristics was assessed by the coefficient of variation of values recorded in the entire plaque. We also determined at each electrode the change in an AF parameter (Δ) with parasympathetic blockade as compared with baseline. The SD of this change (Δ) across the entire plaque was determined as a measure of the spatial dispersion of parasympathetic responsiveness. As a Δ is not measured on a ratio scale, with both positive and negative values and with mean values frequently approaching zero, coefficient of variation could not be used for this purpose. Similarly, effect of sympathetic blockade was determined by the difference of measurements obtained after dual blockade (propranolol and atropine) and atropine alone.
Tissue analysis
RNA isolation and qPCR.
Total RNA from tissues or cells was isolated using the RNeasy kit (Qiagen). Total RNA was reverse transcribed into cDNA using Superscript III (Invitrogen), and quantitative PCR (qPCR) was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) on a 7500 Fast Real-Time PCR System (Applied Biosystems). Relative mRNA levels were calculated by the ΔΔCT method after normalization of each experimental sample to TATA-binding protein (TBP) mRNA level. The following primers were used: dog NGF forward 5′ - AAGCTTCAGCATTCCCTTGA - 3′, dog NGF reverse 5′ - TGCTCCTGTGAGTCCTGTTG - 3′, mouse NGF forward 5′ - CATGGGGGAGTTCTCAGTGT - 3′, mouse NGF reverse 5′ - GCACCCACTCTCAACAGGAT - 3′, dog GAP43 forward 5′ - GTGTGTGCAATGTTCCGTTC - 3′, dog GAP43 reverse 5′ - CTTAGAGCCGCAAGTTACGC - 3′, dog TBP forward 5′ - TGTATCTACAGTGAATCTTGGCTG - 3′, dog TBP reverse 5′ - GGTTCGGGGCTCTCTTATTCTC - 3′, mouse TBP forward 5′ - GTGTTACATTGCCCCTGCTT - 3′, mouse TBP reverse 5′ - GATGGGAATTCCAGGAGTCA - 3′.
Western blot analysis.
Snap-frozen samples were homogenized and total protein was extracted with the T-Per Tissue Protein Extraction Reagent (Thermo Fisher Scientific, 78510). Halt Protease & Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, 78446) were added to all buffers. Protein concentrations were determined using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23227). Proteins were fractionated by SDS-PAGE, transferred to polyvinyl difluoride (PVDF) membrane, blocked with 5% BSA, and blotted with polyclonal rabbit anti–nerve growth factor 2.5S (MilliporeSigma, N6655). Band densities were quantified using the ImageJ software (NIH) and normalized to HSP-90 rabbit monoclonal anti-HSP90 (Cell Signaling Technologies, 4877).
Immunohistochemical analysis of canine myocardium for nerve bundles, fibers, and ganglionic structures
Tissue sample preparation.
For both normal and RAP animals, immediately following the termination of the in vivo electrophysiology assays and after confirming a deep plane of anesthesia, the heart was promptly excised out of the chest and immersed in ice-cold cardioplegia solution containing (mmol/l) NaCl 128, KCl 15, HEPES 10, MgSO4 1.2, NaH2PO4 0.6, CaCl2 1.0, glucose 10, and heparin (0.0001 U/ml); pH 7.4. All solutions were equilibrated with 100% O2. The heart was cannulated via the aorta and perfused with ice cold cardioplegia solution containing protease inhibitors (MilliporeSigma, P8340) until vessels were clear of blood, and tissue was cold. The atria were separated from the associated ventricles, and tissue samples were taken from the PLA, LAFW, LAA, right atrial free wall, posterior right atrium, and RAA. In addition, the LSG was excised from the posterior chest wall. The samples were frozen in OCT tissue freezing medium (VWR) at approximately –50°C in 2-methyl butane cooled by dry ice and stored at –80°C until use. Alternatively, samples were fixed in 10% formalin and embedded in paraffin.
IHC.
IHC was performed to examine the size and distribution of the parasympathetic (AChE) and sympathetic (dopamine β-hydroxylase) nerves and ganglionic cells in the atria and LSG (48). Cryosections were air-dried and fixed in 75% acetone/25% ethanol for 10 minutes before being washed in Tris-buffered saline with 0.5% tween 20 (TBS-T). Paraffin sections were deparaffinized in xylene and rehydrated in series of ethanol, and they underwent antigen retrieval in citrate buffer in steamer for 20 minutes. Hydrogen peroxide (3%) was placed on the sections for 5 minutes, and the sections were washed in TBS-T. Protein block (Dako) was placed on the sections for 5 minutes. Anti-mouse antibody for acetylcholinesterase (AChE, MilliporeSigma, MAB303) was incubated overnight at 4°C. The next day, the sections were washed in TBS-T and incubated with HRP-conjugated anti-mouse secondary antibody (Dako, K4000) for 30 minutes at room temperature. The sections were washed in TBS-T and stained brown by incubation of 3,3′-diaminobenzidine (DAB). After reapplication of protein block, anti-rabbit antibody for dopamine β-hydroxylase (DBH; Chemicon, AB1538) was incubated for 1 hour at room temperature. The sections were washed in TBS-T and incubated with HRP-conjugated anti-rabbit secondary antibody (Dako, K4002) for 30 minutes at room temperature. The sections were washed in TBS-T and stained blue by incubation of 5-bromo-4-chloro-3-indolyl phosphate (BCIP) for 10 minutes. Cell nuclei were counter-stained in methyl green (Dako) for 10 minutes. Specimens were then dehydrated in alcohol, mounted in cytoseal mounting media (Thermo Fisher Scientific), and examined under light microscopy.
Quantification of sympathetic and parasympathetic fibers.
Quantification was performed manually with a light microscope. First, each slide was examined using a 4× objective. The total myocardial area was measured. Next, nerve bundle density and size were determined with a 20× objective. The total number of bundles was counted and divided by the total area of each tissue sample (bundle density). Ann image of each bundle encountered was captured at appropriate magnification. Major radius (rM) and minor radius (rm) were measured. The bundles were modeled as ellipses; the area was calculated as the area of an ellipse (A = rM × rm × π). The total number of parasympathetic and sympathetic nerves in each bundle was counted. Lastly, the tissue was divided into 4 quadrants. From each quadrant, a random sample of five 40× views of myocardium were taken, for a total of 20 images per section. Myocardial parasympathetic and sympathetic nerve fiber density was determined in each image. Parasympathetic fibers stained dark brown ,while sympathetic fibers stained navy blue. These 20 samples were used to estimate the mean myocardial fiber density and coefficient of variation for each section.
Quantification of neuronal cell content in GP and LSG.
Atrial sections were examined for presence of GPs and a micrograph of each GP was obtained at 10× or 20×. Quantification was performed with ImageJ; the GP area and the area of each postganglionic neuron cell body within the GP was delineated. LSG sections were imaged with a TissueGnostics microscope to create tiled micrographs. Six ganglionic areas were delineated in each LSG, and the area of each postganglionic neuron cell body within those ganglions was delineated as above. Results are expressed as total cellular area divided by ganglion area.
In vitro tachypacing
Atrial cardiomyocyte isolation.
While the dog was still deeply anesthetized, the hearts was quickly removed and processed as detailed above. The ventricles were cut away, the left circumflex coronary artery were cannulated, and the left atrium and right atrium were dissected free. The atria were slowly perfused with cold cardioplegia, while leaks from arterial branches were ligated with suture to assure adequate perfusion. The atria were then perfused with Tyrode’s at 37°C for 5 minutes to remove cardioplegia solution and assess for viability (i.e., the reestablishment of beating). If viable, the atria were then perfused at ~12 ml/min with Ca2+-free Tyrode’s solution for about 20 minutes, followed by about 40 minutes of perfusion with the same solution containing Liberase (Liberase TH Research Grade, Roche Diagnostics, 05401151001) and 1% BSA, all at 37°C. Thereafter, the atrial tissue was transferred to dish and cut into small pieces (~0.5 cm2). These pieces were then transferred to conical plastic tubes, and fresh enzyme solution (37 °C) was added, followed by trituration for 5–15 minutes. The triturated tissue suspension was then filtered through nylon mesh (800 μm). The filtered cell tissue suspension was centrifuged at 500 g for less than 1 minute, enzyme solution was poured off, and cell pellet was resuspended in Tyrode’s solution containing 250 μM Ca2+ and 0.1 % BSA. This resuspension was then filtered through a nylon mesh (210 μm), centrifuged at 500 g for less than 1 minute, and again resuspended in Tyrode’s solution containing 250 μM Ca2+ and 0.1% BSA to isolate dispersed cells. After cells settled for about 30 minutes, the solution was suctioned off and gradually replaced with a HEPES-buffered solution containing (mM) NaCl 137, KCl 5.4, MgC12 1.0, CaCl2 1.8, HEPES 10, glucose 11, and 0.1% BSA; pH 7.4. After isolating the cardiomyocytes, Ca2+ concentration was raised to 1.8 mM in 1× Tyrode’s Solution.
HL-1 cell and primary canine atrial cardiomyocyte culture and in vitro tachypacing.
HL-1 is a cell line derived from the AT-1 mouse atrial cardiomyocyte tumor lineage (obtained from MilliporeSigma). Cells were cultured on gelatin-fibronectin–coated 6-well plates with Complete Claycomb Medium (MilliporeSigma) supplemented with 10% FBS, 100 μM norepinephrine, 4 mM L-glutamine, 50 IU/ml penicillin, and 50 μg/ml streptomycin, as previously described (49). Freshly isolated atrial cardiomyocytes from control dogs were plated onto laminin-coated multiwell plates with M199 plating media (Invitrogen) supplemented with 10% FBS, 50 IU/ml penicillin, 50 μg/ml streptomycin, and ITS (Insulin-transferring-Sodium Selenite, MilliporeSigma) in mammalian tissue culture incubator at 37°C, 95% O2/5% CO2. Cells were either unpaced (control) or paced at 1, 2, 3, or 6 Hz for 8 hours using square wave 5 ms, 18V pulses with a C-dish (IonOptix). Alternatively, cells were paced at 6 Hz with 0, 25%, or 50% variability. After in vitro tachypacing, RNA was extracted for subsequent qPCR analysis, as detailed above.
Mathematical modeling of parasympathetic and sympathetic stimulation on atrial tissue
A spatially distributed atrial cell model.
To model the spatiotemporal distribution of Ca2+ in atrial myocytes, we implemented an established mathematical model by Restrepo et al. (50, 51), which was recently adapted by us to describe atrial myocytes (21). In this model, the cell interior is divided into a 3-D array of compartments that represent distinct intracellular spaces. The Ca2+ concentration within these compartments is treated as spatially uniform, and neighboring compartments are diffusively coupled. We refer to the space in the vicinity of a RyR cluster as a Ca2+ release unit (CRU). Following our previous study (21), we distinguish between compartments that are close to or far from the cell membrane. CRUs in the cell can therefore be divided into 2 groups: (a) junctional CRUs where the RyR cluster is close to the cell membrane and can interact with membrane-bound LCCs and NCX, and (b) nonjunctional CRUs with RyR clusters far from the cell membrane. For nonjunctional CRUs, we simply remove LCC and NCX transporters from the model. We recently described that atrial myocytes have a poorly developed t-tubule system with the bulk of Ca2+ signaling occurring at the cell boundary (52). To account for this architecture, we let all boundary CRUs be junctional CRUs, while all nonjunctional CRUs reside in the cell interior. In this study, our 3-D cell model is composed of an array of 40 × 16 × 16 CRUs. To model voltage dynamics, our spatially distributed model is coupled with an established atrial AP model by Grandi et al. (53).
Ionic model of parasympathetic and sympathetic activation.
To model cardiac tissue, we applied a recently developed phenomenological model of Ca2+ cycling in atrial myocytes (54). In this model, we keep track only on the population of Ca2+ sparks at junctional and nonjunctional CRUs in the cell. This is achieved by developing phenomenological rate equations governing the recruitment and extinguishing of Ca2+ sparks. Model parameters are determined by fitting rate constants to the population number dynamics observed in the detailed spatially distributed model described above. To model sympathetic stimulation, we apply our Ca2+cycling model, which accounts for stochastic Ca2+ wave propagation in the atrial cell interior (54). To control the onset of Ca2+ wave propagation, we vary a model parameter pb, which represents the threshold for Ca2+ wave propagation from the atrial cell boundary to the interior. In our computational model, the frequency of Ca2+ waves increases with decreasing threshold pb. Thus, we can model sympathetic stimulation, which is associated with an increased frequency of Ca2+ waves, by reducing pb. To model parasympathetic stimulation, we include the contribution of IKACh based on experimental data from Koumi et al. (55). This current is modeled as the following:
(Equation 2),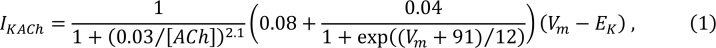
where [ACh] is the concentration of acetycholine, Vm is the membrane potential, and EK is the reversal potential for K+. Following Grandi et al. (56), we vary [ACh] in the range of 1 × 10–4 to 1 × 10–2 μmol/l so that IKACh conductance increases as [ACh] increases within this range. We can vary [ACh] and pb independently to explore the electrophysiological effect of parasympathetic and sympathetic stimulation, respectively.
Modeling wave propagation in tissue.
We apply our computational model to explore the effect of parasympathetic and sympathetic stimulation on AP wave propagation in 2-D cardiac tissue. To model electrical wave propagation, we apply the cable equation:
(Equation 3),
where Cm = 1 μF/cm2 is the membrane capacitance, DV = 5 × 10–4 cm2/ms is the effective voltage diffusion coefficient, and Iion is the total transmembrane current. The cable equation is integrated using an operator splitting approach (57), with a space step Δx = 0.015 cm and with a variable time step in the range dt = 0.01–0.1 ms. To explore the dynamics of a paced 2-D tissue, we simulate a 2-D sheet of L × L cells, where L = 160.
Statistics
Unless specified below, all data were analyzed as follows: All values are expressed as mean ± SEM or median ± interquartile range with interquartile range for nonparametric data. When comparing 1 factor between 2 conditions in parametric data, a 2-tailed Student’s t test was performed. When comparing 1 factor between 3 regions (PLA vs. LAFW vs. LAA), a 1-way ANOVA or Kruskal-Wallis 1-way ANOVA on ranks was performed, with Holm-Sidak method for pairwise comparison. When results were paired, a 1-way repeated measures ANOVA with Bonferroni method for pairwise comparison was used. When comparing 2 factors of 3 regions, a 2-way ANOVA with Holm-Sidak method for pairwise comparison was performed. If required, statistical analysis was performed after log transformation for nonparametric values. All statistics considered α ≤ 0.05.
Assuming all bundle sizes are independent, a 2-way ANOVA was performed on individual bundle sizes. Mean AF characteristic changes with autonomic blockade was performed with a mixed linear model using a fixed effect for time and random intercept effects for dogs. This model accounts for the repeated data over time within animal and also accounts for the fact that some animals had only time 1 and time 2 data, while others had all 3 time points.
Study approval
Dogs used in this study were maintained in accordance to Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011), and the study was approved by the IACUC of Northwestern University.
Author contributions
GG, AP, WZ, and RA conceived and designed experiments. GG, AP, LW, MG, WZ, SY, AB, and BB conducted the experiments and acquired data. GG, AP, LW, MG, DAJ, and GK analyzed the data. YS conducted mathematical simulations. GG, AP, MG, and RA wrote the manuscript. GG, AP, and MG prepared the figures. SY, DAJ, BPK, RP, JJG, GA, JAW, and YS contributed to the Discussion section.
Supplementary Material
Acknowledgments
AP is a recipient of the Kenneth M. Rosen Fellowship in Cardiac Pacing and Electrophysiology from the Heart Rhythm Society, funded via an unrestricted research grant by Medtronic. RA has received grants R01 HL093490 and R01 HL140061, the AHA Strategically Focused Research Networks AF Center grant, and a grant from the NIH Center for Accelerated Innovations at Cleveland Clinic (NCAI-CC). YS has received grant R01-119095.
Version 1. 09/10/2019
In-Press Preview
Version 2. 10/17/2019
Electronic publication
Version 3. 11/14/2019
Updated issue information.
Footnotes
Conflict of interest: RA has ownership interest in Rhythm Therapeutics Inc.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(20):e130532.https://doi.org/10.1172/jci.insight.130532.
Contributor Information
Georg Gussak, Email: g-gussak@northwestern.edu.
Anna Pfenniger, Email: anna.pfenniger@northwestern.edu.
Mehul Gilani, Email: mehul.gilani@my.rfums.org.
Wenwei Zhang, Email: wenwei.zhang@northwestern.edu.
Shin Yoo, Email: shinyoo@northwestern.edu.
David A. Johnson, Email: daj@northwestern.edu.
Amy Burrell, Email: amy.burrell@northwestern.edu.
Brandon Benefield, Email: b-benefield@northwestern.edu.
Bradley P. Knight, Email: bradley.knight@nm.org.
Rod Passman, Email: r-passman@northwestern.edu.
Jeffrey J. Goldberger, Email: j-goldberger@miami.edu.
Gary Aistrup, Email: glais@mmrl.edu.
J. Andrew Wasserstrom, Email: ja-wasserstrom@northwestern.edu.
Yohannes Shiferaw, Email: yohannes.shiferaw@csun.edu.
Rishi Arora, Email: r-arora@northwestern.edu.
References
- 1.Benjamin EJ, et al. Prevention of atrial fibrillation: report from a national heart, lung, and blood institute workshop. Circulation. 2009;119(4):606–618. doi: 10.1161/CIRCULATIONAHA.108.825380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piccini JP, Fauchier L. Rhythm control in atrial fibrillation. Lancet. 2016;388(10046):829–840. doi: 10.1016/S0140-6736(16)31277-6. [DOI] [PubMed] [Google Scholar]
- 3.Waks JW, Zimetbaum P. Antiarrhythmic Drug Therapy for Rhythm Control in Atrial Fibrillation. J Cardiovasc Pharmacol Ther. 2017;22(1):3–19. doi: 10.1177/1074248416651722. [DOI] [PubMed] [Google Scholar]
- 4.Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res. 2014;114(9):1500–1515. doi: 10.1161/CIRCRESAHA.114.303772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jayachandran JV, Sih HJ, Winkle W, Zipes DP, Hutchins GD, Olgin JE. Atrial fibrillation produced by prolonged rapid atrial pacing is associated with heterogeneous changes in atrial sympathetic innervation. Circulation. 2000;101(10):1185–1191. doi: 10.1161/01.CIR.101.10.1185. [DOI] [PubMed] [Google Scholar]
- 6.Chang CM, et al. Nerve sprouting and sympathetic hyperinnervation in a canine model of atrial fibrillation produced by prolonged right atrial pacing. Circulation. 2001;103(1):22–25. doi: 10.1161/01.CIR.103.1.22. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen BL, Fishbein MC, Chen LS, Chen PS, Masroor S. Histopathological substrate for chronic atrial fibrillation in humans. Heart Rhythm. 2009;6(4):454–460. doi: 10.1016/j.hrthm.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fioranelli M, et al. Analysis of heart rate variability five minutes before the onset of paroxysmal atrial fibrillation. Pacing Clin Electrophysiol. 1999;22(5):743–749. doi: 10.1111/j.1540-8159.1999.tb00538.x. [DOI] [PubMed] [Google Scholar]
- 9.Bettoni M, Zimmermann M. Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation. 2002;105(23):2753–2759. doi: 10.1161/01.CIR.0000018443.44005.D8. [DOI] [PubMed] [Google Scholar]
- 10.Choi EK, et al. Intrinsic cardiac nerve activity and paroxysmal atrial tachyarrhythmia in ambulatory dogs. Circulation. 2010;121(24):2615–2623. doi: 10.1161/CIRCULATIONAHA.109.919829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arora R. Recent insights into the role of the autonomic nervous system in the creation of substrate for atrial fibrillation: implications for therapies targeting the atrial autonomic nervous system. Circ Arrhythm Electrophysiol. 2012;5(4):850–859. doi: 10.1161/CIRCEP.112.972273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arora R, et al. Neural substrate for atrial fibrillation: implications for targeted parasympathetic blockade in the posterior left atrium. Am J Physiol Heart Circ Physiol. 2008;294(1):H134–H144. doi: 10.1152/ajpheart.00732.2007. [DOI] [PubMed] [Google Scholar]
- 13.Hou Y, et al. Ganglionated plexi modulate extrinsic cardiac autonomic nerve input: effects on sinus rate, atrioventricular conduction, refractoriness, and inducibility of atrial fibrillation. J Am Coll Cardiol. 2007;50(1):61–68. doi: 10.1016/j.jacc.2007.02.066. [DOI] [PubMed] [Google Scholar]
- 14.Patterson E, Po SS, Scherlag BJ, Lazzara R. Triggered firing in pulmonary veins initiated by in vitro autonomic nerve stimulation. Heart Rhythm. 2005;2(6):624–631. doi: 10.1016/j.hrthm.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 15.Schauerte P, et al. Focal atrial fibrillation: experimental evidence for a pathophysiologic role of the autonomic nervous system. J Cardiovasc Electrophysiol. 2001;12(5):592–599. doi: 10.1046/j.1540-8167.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- 16.Di Biase L, et al. Left atrial appendage: an underrecognized trigger site of atrial fibrillation. Circulation. 2010;122(2):109–118. doi: 10.1161/CIRCULATIONAHA.109.928903. [DOI] [PubMed] [Google Scholar]
- 17.Romero J, et al. Benefit of left atrial appendage electrical isolation for persistent and long-standing persistent atrial fibrillation: a systematic review and meta-analysis. Europace. 2018;20(8):1268–1278. doi: 10.1093/europace/eux372. [DOI] [PubMed] [Google Scholar]
- 18.Lee RJ, et al. Percutaneous alternative to the Maze procedure for the treatment of persistent or long-standing persistent atrial fibrillation (aMAZE trial): Rationale and design. Am Heart J. 2015;170(6):1184–1194. doi: 10.1016/j.ahj.2015.09.019. [DOI] [PubMed] [Google Scholar]
- 19.Zhou S, et al. Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res. 2004;95(1):76–83. doi: 10.1161/01.RES.0000133678.22968.e3. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen BL, et al. Acute myocardial infarction induces bilateral stellate ganglia neural remodeling in rabbits. Cardiovasc Pathol. 2012;21(3):143–148. doi: 10.1016/j.carpath.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shiferaw Y, Aistrup GL, Wasserstrom JA. Mechanism for Triggered Waves in Atrial Myocytes. Biophys J. 2017;113(3):656–670. doi: 10.1016/j.bpj.2017.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aistrup GL, et al. Triggered intracellular calcium waves in dog and human left atrial myocytes from normal and failing hearts. Cardiovasc Res. 2017;113(13):1688–1699. doi: 10.1093/cvr/cvx167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh S, et al. Hypertrophy of neurons within cardiac ganglia in human, canine, and rat heart failure: the potential role of nerve growth factor. J Am Heart Assoc. 2013;2(4):e000210. doi: 10.1161/JAHA.113.000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volders PG. Novel insights into the role of the sympathetic nervous system in cardiac arrhythmogenesis. Heart Rhythm. 2010;7(12):1900–1906. doi: 10.1016/j.hrthm.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Patterson E, et al. Spontaneous pulmonary vein firing in man: relationship to tachycardia-pause early afterdepolarizations and triggered arrhythmia in canine pulmonary veins in vitro. J Cardiovasc Electrophysiol. 2007;18(10):1067–1075. doi: 10.1111/j.1540-8167.2007.00909.x. [DOI] [PubMed] [Google Scholar]
- 26.Scherlag BJ, Patterson E, Po SS. The neural basis of atrial fibrillation. J Electrocardiol. 2006;39(4 Suppl):S180–S183. doi: 10.1016/j.jelectrocard.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 27.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003;107(18):2355–2360. doi: 10.1161/01.CIR.0000065578.00869.7C. [DOI] [PubMed] [Google Scholar]
- 28.Choi EK, et al. Triggered firing and atrial fibrillation in transgenic mice with selective atrial fibrosis induced by overexpression of TGF-β1. Circ J. 2012;76(6):1354–1362. doi: 10.1253/circj.CJ-11-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan AY, Li H, Wachsmann-Hogiu S, Chen LS, Chen PS, Fishbein MC. Autonomic innervation and segmental muscular disconnections at the human pulmonary vein-atrial junction: implications for catheter ablation of atrial-pulmonary vein junction. J Am Coll Cardiol. 2006;48(1):132–143. doi: 10.1016/j.jacc.2006.02.054. [DOI] [PubMed] [Google Scholar]
- 30.Ng GA. Neuro-cardiac interaction in malignant ventricular arrhythmia and sudden cardiac death. Auton Neurosci. 2016;199:66–79. doi: 10.1016/j.autneu.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Kolman JM, Minár J, Horák null Serologic examination of birds from the area of Southern Moravia for the presence of antibodies against arboviruses of the groups alfa, flavo, Bunyamwera supergroup, and the virus Yaba 1-Lednice 110. I. Domestic fowls. Zentralbl Bakteriol Orig A. 1975;233(3):279–287. [PubMed] [Google Scholar]
- 32.Ogawa M, et al. Left stellate ganglion and vagal nerve activity and cardiac arrhythmias in ambulatory dogs with pacing-induced congestive heart failure. J Am Coll Cardiol. 2007;50(4):335–343. doi: 10.1016/j.jacc.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 33.Raman D, et al. Polysomnographic Heart Rate Variability Indices and Atrial Ectopy Associated with Incident Atrial Fibrillation Risk in Older Community-dwelling Men. JACC Clin Electrophysiol. 2017;3(5):451–460. doi: 10.1016/j.jacep.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyauchi Y, et al. Altered atrial electrical restitution and heterogeneous sympathetic hyperinnervation in hearts with chronic left ventricular myocardial infarction: implications for atrial fibrillation. Circulation. 2003;108(3):360–366. doi: 10.1161/01.CIR.0000080327.32573.7C. [DOI] [PubMed] [Google Scholar]
- 35.Li Z, Wang M, Zhang Y, Zheng S, Wang X, Hou Y. The effect of the left stellate ganglion on sympathetic neural remodeling of the left atrium in rats following myocardial infarction. Pacing Clin Electrophysiol. 2015;38(1):107–114. doi: 10.1111/pace.12513. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, et al. Increase of Autonomic Nerve Factors in Epicardial Ganglionated Plexi During Rapid Atrial Pacing Induced Acute Atrial Fibrillation. Med Sci Monit. 2017;23:3657–3665. doi: 10.12659/MSM.902621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JH, et al. Catheter Ablation of Atrial Fibrillation Raises the Plasma Level of NGF-β Which Is Associated with Sympathetic Nerve Activity. Yonsei Med J. 2015;56(6):1530–1537. doi: 10.3349/ymj.2015.56.6.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Afzal MR, et al. Impact of left atrial appendage exclusion using an epicardial ligation system (LARIAT) on atrial fibrillation burden in patients with cardiac implantable electronic devices. Heart Rhythm. 2015;12(1):52–59. doi: 10.1016/j.hrthm.2014.09.053. [DOI] [PubMed] [Google Scholar]
- 39.Lakkireddy D, et al. Left Atrial Appendage Ligation and Ablation for Persistent Atrial Fibrillation: The LAALA-AF Registry. JACC Clin Electrophysiol. 2015;1(3):153–160. doi: 10.1016/j.jacep.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 40.Fink T, et al. Combination of Left Atrial Appendage Isolation and Ligation to Treat Nonresponders of Pulmonary Vein Isolation. JACC Clin Electrophysiol. 2018;4(12):1569–1579. doi: 10.1016/j.jacep.2018.09.022. [DOI] [PubMed] [Google Scholar]
- 41.Di Biase L, et al. Left Atrial Appendage Isolation in Patients With Longstanding Persistent AF Undergoing Catheter Ablation: BELIEF Trial. J Am Coll Cardiol. 2016;68(18):1929–1940. doi: 10.1016/j.jacc.2016.07.770. [DOI] [PubMed] [Google Scholar]
- 42.Benussi S, et al. Thoracoscopic appendage exclusion with an atriclip device as a solo treatment for focal atrial tachycardia. Circulation. 2011;123(14):1575–1578. doi: 10.1161/CIRCULATIONAHA.110.005652. [DOI] [PubMed] [Google Scholar]
- 43.Starck CT, et al. Epicardial left atrial appendage clip occlusion also provides the electrical isolation of the left atrial appendage. Interact Cardiovasc Thorac Surg. 2012;15(3):416–418. doi: 10.1093/icvts/ivs136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Everett TH, Kok LC, Vaughn RH, Moorman JR, Haines DE. Frequency domain algorithm for quantifying atrial fibrillation organization to increase defibrillation efficacy. IEEE Trans Biomed Eng. 2001;48(9):969–978. doi: 10.1109/10.942586. [DOI] [PubMed] [Google Scholar]
- 45.Everett TH, Moorman JR, Kok LC, Akar JG, Haines DE. Assessment of global atrial fibrillation organization to optimize timing of atrial defibrillation. Circulation. 2001;103(23):2857–2861. doi: 10.1161/01.CIR.103.23.2857. [DOI] [PubMed] [Google Scholar]
- 46.Ng J, et al. Measuring the complexity of atrial fibrillation electrograms. J Cardiovasc Electrophysiol. 2010;21(6):649–655. doi: 10.1111/j.1540-8167.2009.01695.x. [DOI] [PubMed] [Google Scholar]
- 47.Ng J, Gordon D, Passman RS, Knight BP, Arora R, Goldberger JJ. Electrogram morphology recurrence patterns during atrial fibrillation. Heart Rhythm. 2014;11(11):2027–2034. doi: 10.1016/j.hrthm.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ng J, et al. Autonomic remodeling in the left atrium and pulmonary veins in heart failure: creation of a dynamic substrate for atrial fibrillation. Circ Arrhythm Electrophysiol. 2011;4(3):388–396. doi: 10.1161/CIRCEP.110.959650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Claycomb WC, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95(6):2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Restrepo JG, Weiss JN, Karma A. Calsequestrin-mediated mechanism for cellular calcium transient alternans. Biophys J. 2008;95(8):3767–3789. doi: 10.1529/biophysj.108.130419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Restrepo JG, Karma A. Spatiotemporal intracellular calcium dynamics during cardiac alternans. Chaos. 2009;19(3):037115. doi: 10.1063/1.3207835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arora R, et al. Regional distribution of T-tubule density in left and right atria in dogs. Heart Rhythm. 2017;14(2):273–281. doi: 10.1016/j.hrthm.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 2010;48(1):112–121. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shiferaw Y, Aistrup GL, Wasserstrom JA. Synchronization of Triggered Waves in Atrial Tissue. Biophys J. 2018;115(6):1130–1141. doi: 10.1016/j.bpj.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koumi S, Backer CL, Arentzen CE. Characterization of inwardly rectifying K+ channel in human cardiac myocytes. Alterations in channel behavior in myocytes isolated from patients with idiopathic dilated cardiomyopathy. Circulation. 1995;92(2):164–174. doi: 10.1161/01.CIR.92.2.164. [DOI] [PubMed] [Google Scholar]
- 56.Grandi E, et al. Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circ Res. 2011;109(9):1055–1066. doi: 10.1161/CIRCRESAHA.111.253955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qu Z, Garfinkel A. An advanced algorithm for solving partial differential equation in cardiac conduction. IEEE Trans Biomed Eng. 1999;46(9):1166–1168. doi: 10.1109/10.784149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.