

Abstract
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) inhibits not only hematopoietic cell proliferation but also fibroblast proliferation and collagen synthesis in vitro. Ac-SDKP also prevents collagen deposition and cell proliferation in the left ventricle (LV) in rats with renovascular hypertension (renin dependent). However, it is not clear whether Ac-SDKP has similar effects in a model of renin-independent hypertension (aldosterone-salt). Using a hypertensive rat model of cardiac and renal fibrosis created by chronic elevation of circulating aldosterone (ALDO) levels, we examined the effect of Ac-SDKP on blood pressure, cardiac and renal fibrosis and hypertrophy, and proliferating cell nuclear antigen (PCNA) expression in the LV and left kidney. Uninephrectomized rats were divided into 4 groups: (1) controls that received tap water, (2) rats that received ALDO (0.75 μg/h SC) and 1% NaCl/0.2% KCl in drinking water (ALDO-salt), (3) rats that received ALDO-salt plus Ac-SDKP 400 μg · kg–1 · day–1 SC, and (4) rats that received ALDO-salt plus Ac-SDKP 800 μg · kg–1 · d–1 SC. After 6 weeks of treatment, the ALDO-salt group was found to have significantly increased blood pressure with decreased body weight and plasma renin concentration (P<0.05), LV and renal hypertrophy as well as renal injury, significantly increased collagen content in both ventricles and kidney as well as increased collagen volume fraction in the LV (P<0.0001), and significantly increased interstitial and perivascular PCNA-positive cells in the LV and kidney (P<0.0001). Ac-SDKP at 800 μg · kg–1 · d–1 markedly prevented cardiac and renal fibrosis (P<0.005) without affecting blood pressure or organ hypertrophy. It also suppressed PCNA expression in the LV and kidney in a dose-dependent manner. We concluded that Ac-SDKP prevents increased collagen deposition and cell proliferation in the heart and kidney in ALDO-salt hypertensive rats. Because ACE inhibitors increase plasma and tissue Ac-SDKP and decrease cardiac and renal fibrosis, we speculate that Ac-SDKP may participate in the antifibrotic effect of ACE inhibitors.
Keywords: aldosterone, hypertension, mineralocorticoid, collagen, heart, kidney
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a natural inhibitor of pluripotent hematopoietic stem cell proliferation1,2 that is normally present in human plasma and circulating mononuclear cells3 and is cleaved to an inactive form by the NH2-terminal catalytic domain of ACE.4 There have been reports that administration of captopril, an ACE inhibitor (ACEi), prevented degradation of endogenous Ac-SDKP and raised its circulating and urine concentrations.4–6 However, the antiproliferative effects of Ac-SDKP are not limited to hematopoietic cells. We found that Ac-SDKP not only inhibited adult rat cardiac fibroblast proliferation and collagen synthesis in vitro but also reduced the number of proliferating cell nuclear antigen (PCNA)-positive cells and prevented enhanced collagen deposition in the left ventricle (LV) in 2-kidney, 1-clip hypertensive rats.7 In addition, Ac-SDKP at 10–7 to 10–5 mol/L significantly attenuated serum-induced renal fibroblast proliferation. The rate of proliferation was not altered by 100 nmol/L captopril (an ACEi) or 10–8 mol/L Ac-SDKP but was reduced significantly by combined treatment.8 In other studies, ACEi significantly attenuated cardiac fibrosis in rats with heart failure induced by myocardial infarction,9 in spontaneously hypertensive rats,10 and in aldosterone (ALDO)-salt–treated rats11 and prevented renal fibrosis in rats with deoxycorticosterone acetate-salt treatment12 and unilateral ureteral obstruction.13 One might therefore speculate that the antifibrotic effects of ACEi in the heart and kidney are partially mediated by Ac-SDKP; however, direct evidence of this is lacking.
ALDO-salt hypertension in rats is characterized by marked deposition of interstitial collagen in both the heart and kidney,14,15 associated with interstitial cell proliferation.16 To test our hypothesis that Ac-SDKP prevents collagen deposition and cell proliferation in the heart and kidney, we investigated its effect on blood pressure and renal injury as well as hypertrophy, collagen deposition, and cell proliferation (PCNA expression) in the heart and kidney in rats with ALDO-salt hypertension (renin independent).
Methods
Animals and Experimental Design
Twelve-week-old male Sprague-Dawley rats (SD; 350 to 375 g; Charles River) were anesthetized with methohexital sodium (50 mg/kg IP; Brevital, Lilly), and the left kidney was removed. d-Aldosterone (ALDO, 0.75 μg/h; Sigma Chemical Co) alone or combined with Ac-SDKP was infused subcutaneously for 6 weeks via osmotic minipump (Alzet 2 ML4). ALDO was dissolved in 0.9% NaCl plus 5 mL/L ethanol, and rats receiving ALDO were given 1% NaCl/0.2% KCl to drink. Control rats were also uninephrectomized. Animals were divided into 4 groups: (1) controls receiving tap water (n=12), (2) ALDO-salt plus vehicle (n=13), (3) ALDO-salt plus Ac-SDKP (400 μg · kg–1 · d–1) (n=13), and (4) ALDO-salt plus Ac-SDKP (800 μg · kg–1 · d–1) (n=12). In our previous study, Ac-SDKP at 400 μg · kg–1 · d–1 significantly prevented cardiac fibrosis in 2-kidney, 1-clip rats; moreover, plasma Ac-SDKP was nearly doubled in Ac-SDKP (400 and 800 μg· kg–1 · d–1)-treated rats compared with vehicle, reaching significance only at 800 μg · kg–1. d–1. Therefore, in the present study we chose these 2 dosages. This study was approved by the Henry Ford Hospital Care of Experimental Animals Committee.
Measurement of Systolic Blood Pressure and Sample Collection
Systolic blood pressure (SBP) was measured by tail cuff twice a week for 6 weeks. After 6 weeks of treatment, animals were anesthetized with 50 mg/kg pentobarbital sodium, and blood from the aorta was collected in a heparinized tube; it was centrifuged at 2000g for 15 minutes at 4°C, and plasma was stored at –70°C until plasma renin concentration (PRC) was measured. The heart was stopped at diastole with an intraventricular injection of 15% KCl and then rapidly excised along with the right kidney. The left ventricle plus septum (LV), right ventricle (RV), and left and right atria and kidney were weighed, and the LV was sectioned transversely into 3 slices from apex to base. One slice was rapidly frozen in isopentane precooled in liquid nitrogen and stored at –70°C; another was fixed in 10% formalin solution for morphometric studies; and the third was stored at –20°C until the hydroxyproline assay. A fourth of the kidney was fixed in 4% paraformaldehyde solution for histological studies, and ≈50 mg wet wt from the remaining three fourths was frozen and used to measure hydroxyproline.
PRC
Plasma (60 μL) was incubated with sheep angiotensinogen in buffer (0.1 mol/L sodium phosphate, 0.02 mol/L Na2EDTA, and 0.05% PMSF pH 6.5) at 37°C for 2 hours. The incubation was stopped by immediately boiling in water for 10 minutes, and the mixture was centrifuged at 1680g for 15 minutes, with the supernatants stored at –20°C until the assay. Generated angiotensin (Ang) I was measured with a clinical RIA kit (DiaSorin), and the results were expressed as ng Ang I · mL plasma–1 ·h–1.
Hydroxyproline Assay
The collagen content of myocardial and renal tissue was determined by hydroxyproline assay.17 For this, tissue was freeze-dried and weighed and then homogenized in 0.1 mol/L NaCl and 5 mmol/L NaHCO3, washed 5 times with the same solution, and hydrolyzed with 6N HCl for 16 hours at 110°C. Samples were filtered and vacuum-dried and then dissolved in distilled water. Hydroxyproline content was determined with a colorimetric assay and a standard curve of 0 to 5 μg hydroxyproline. Data were expressed as μg collagen/mg dry wt, with the assumption that collagen contains an average of 13.5% hydroxyproline.18
Histochemical Analysis of Interstitial Collagen Volume Fraction in the LV
Sections that were 10 μm thick were cut from each frozen slice and stained separately with (1) fluorescein-labeled peanut agglutinin (Vector Laboratories) after pretreatment with 3.3 U/mL neuroaminidase type V (Sigma Chemical Co) to delineate myocyte cross-sectional area (an indicator of myocyte volume) and the interstitial space (consisting of collagen and capillaries) and (2) rhodamine-labeled Griffonia simplicifolia lectin I (GSL I) to show only the capillaries, because GSL I selectively binds to capillaries.19,20 Two sections were cut from each slice, and 4 radially oriented microscopic fields were selected at random from each section and photographed with 35-mm film at a magnification of ×100. Total surface area (microscopic field), interstitial space (collagen plus capillaries), and area occupied by capillaries alone were measured with computer-assisted videodensitometry (JAVA; Jandel). Interstitial collagen fraction was calculated as percent total surface area occupied by the interstitial space minus percent total surface area occupied by the capillaries. The average interstitial collagen fraction was calculated with the use of data obtained from all 8 fields.21
Immunohistochemical Staining for PCNA in the LV and Renal Cortex
Sections that were 4 μm thick were deparaffinized, rehydrated, and boiled in 0.2% citric acid (pH 6.0) for 10 minutes for antigen retrieval. They were washed 3 times in PBS for 5 minutes each time, preincubated with blocking serum (1% normal serum) for 30 minutes, and then incubated with a mouse monoclonal antibody against PCNA (1:250 dilution; Chemicon) at room temperature for 30 minutes. Each section was washed 3 times in PBS, and PCNA was assayed with a Vectastain ABC kit (Vector Laboratories). Sections were treated with diaminobenzidine substrate (Vector) and counterstained with hematoxylin. For each sample, 12 randomly selected fields in the LV and cortical fields in the kidney were examined under high magnification (×400). Proliferating cells with dark-brown nuclei were counted and expressed as the number of PCNA-positive cells per field.
Renal Histology
The right kidney was fixed in buffered paraformaldehyde (4%) and stained with Masson trichrome and periodic acid–Schiff. Histological evaluation of coronal sections was performed by one of the authors (L.R.) in a blind fashion. Glomerular injury was evaluated with a semiquantitative scoring method as described previously, with severity graded from 0 to 4+ according to the percentage of glomerular involvement.22 The glomerular injury score (GIS) was calculated by adding together the products of the severity score and the percentage of glomeruli displaying the same degree of severity.22,23 Tubulointerstitial injury (TII) was also determined. Tubular dilatation, interstitial fibrosis, and mononuclear cell infiltration were graded semiquantitatively from 0 to 5+ in an average of 3 to 5 fields per section.22,23
Statistical Analysis
The groups given ALDO-salt plus vehicle, Ac-SDKP at 400 μg · kg–1 · d–1, and Ac-SDKP at 800 μg · kg–1 · d–1 were compared with the controls using Student’s t test for the variables systolic blood pressure, heart rate, PRC, body weight (BW), LV weight, atrial weight, RV weight, LV and RV collagen, collagen volume fraction, LV PCNA, and kidney PCNA. Because multiple comparisons were conducted, Hochberg’s method was used to adjust the α level of significance. Abnormally distributed variables were analyzed using a nonparametric method: Wilcoxon’s 2-sample test. These variables included kidney weight, kidney collagen, and the kidney injury scores. Again, the α level of significance was adjusted for multiple comparisons with Hochberg’s method. Values are expressed as mean±SEM.
Results
SBP, Heart Rate, PRC, BW, and Organ Weight
Two weeks after ALDO-salt treatment, SBP in the ALDO-salt/vehicle group (n=13) was significantly increased compared with the controls (n=12), reaching the highest level by the third week and remaining at this plateau for 3 weeks. Ac-SDKP at 400 (n=13) or 800 μg · kg–1 · d–1 (n=12) for 6 weeks had no effect on the development of hypertension in rats treated with ALDO-salt (Figure 1). Heart rate was unchanged in all 4 groups. After 6 weeks of treatment, PRC was dramatically decreased in the ALDO-salt–treated groups compared with the controls and was not affected by Ac-SDKP (Table). BW was significantly higher in the controls than in the other 3 groups (P<0.001) (Table). LV weight was significantly increased in the ALDO-salt/vehicle group, and Ac-SDKP did not suppress this increase. Kidney weight was also significantly increased in ALDO-salt hypertensive rats but was not affected by Ac-SDKP. There were no significant differences in atrial weight or RV weight among the 4 groups (Table).
Figure 1.

SBP in ALDO-salt hypertensive SD rats treated with Ac-SDKP. Ac-SDKP had no effect on increased SBP from 1 to 6 weeks after treatment. *P<0.001, †P<0.05 for all groups compared with ALDO-salt + vehicle.
Table.
SBP, LV Weight, RV Weight, Atrial Weight, Kidney Weight, PRC, and Body Weight Measured After 6 Weeks
| ALDO-Salt Group | ||||
|---|---|---|---|---|
| Parameter | Control Group | Vehicle | Ac-SDKP (400 μg · kg–1 · d–1) |
Ac-SDKP (800 μg · kg–1 · d–1) |
| SBP, mm Hg | 122.9±1.3 | 186.0±4.9* | 188.5±8.7* | 176.8±7.2* |
| LV weight, mg | 949.8±85.0 | 1139.2±133.3* | 1154.4±144.2* | 1134.8±90.9* |
| RV weight, mg | 245.4±44.0 | 211.0±31.3 | 215.7±31.4 | 212.6±38.4 |
| Atrial weight, mg | 63.7±10.1 | 63.9±9.6 | 64.3±16.2 | 64.9±11.3 |
| Kidney weight, mg | 2192.5±53.2 | 3618.0±192.9* | 3388.2±116.6* | 3383.0±123.3* |
| PRC, ng · mL−1 · h−1 | 12.8±2.2 | 3.5±0.8* | 2.1±0.6* | 4.0±0.4* |
| Body weight, g | 504.9±26.1 | 374.9±72.9* | 372.9±44.8* | 398.4±32.9* |
P<0.005 vs control.
Collagen Content of the LV, RV, and Kidney
Ventricular collagen was significantly increased in the ALDO-salt/vehicle group (LV 12.0±1.5 μg/mg dry LV, RV 11.0±1.1 μg/mg dry RV) compared with the controls (LV 7.1±1.1, RV 7.8±0.7) (P<0.0001), and this increase was significantly prevented by Ac-SDKP at 800 μg · kg–1 · d–1 (LV 9.4±2.1, RV 8.8±0.9; P<0.005) (Figure 2). We also observed a significant increase in renal collagen in the vehicle group (27.6±0.8 μg/mg dry kidney) compared with the controls (21.2±0.7) (P<0.005), which was completely prevented by Ac-SDKP at 800 μg· kg–1· d–1 (20.1±1.1) (P<0.005) (Figure 2).
Figure 2.

Collagen deposition in the (A) LV, (B) RV, and (C) kidney, as well as (D) collagen volume fraction of the LV, in controls and ALDO-salt hypertensive rats after 6 weeks of Ac-SDKP treatment. Collagen content in the LV, RV, and kidney was significantly increased in the vehicle group. Ac-SDKP at 800 μg · kg–1 · d–1 completely blocked increased collagen deposition in the kidney but only partially in the LV and RV. The effect of Ac-SDKP on LV collagen volume fraction showed the same pattern as LV collagen content. *P<0.005 vs controls. †P<0.005 vs ALDO-salt + vehicle. ‡P<0.05 vs ALDO-salt + vehicle.
Interstitial Collagen Volume Fraction in the LV
Histochemical analysis showed that the interstitial collagen volume fraction in the LV was significantly increased in untreated ALDO-salt rats (P<0.0001). It was significantly reduced in ALDO-salt rats treated with Ac-SDKP (800 μg · kg–1 · d–1) (P<0.0001) (Figure 2), confirming the collagen content data.
PCNA Expression in the LV and Renal Cortex
Few PCNA-positive cells were found in the LV interstitial space in the controls; however, in the ALDO-salt–treated rats they were distributed diffusely throughout both interstitial and perivascular spaces. Likewise in the renal cortex, few PCNA-positive cells could be detected in the renal tubular epithelium in the control group, whereas in the ALDO-salt–treated rats, they were easily found in both renal tubular epithelium and interstitium (Figure 3). We could not find PCNA-positive cells in the glomeruli. PCNA-positive cells were significantly increased in both LV and kidney in the ALDO-salt/vehicle group (P<0.0001). There were fewer PCNA-positive cells in the LV interstitial and perivascular spaces and renal cortex in the Ac-SDKP–treated groups (P<0.01), although they still occurred more frequently than in the controls (Figure 4).
Figure 3.
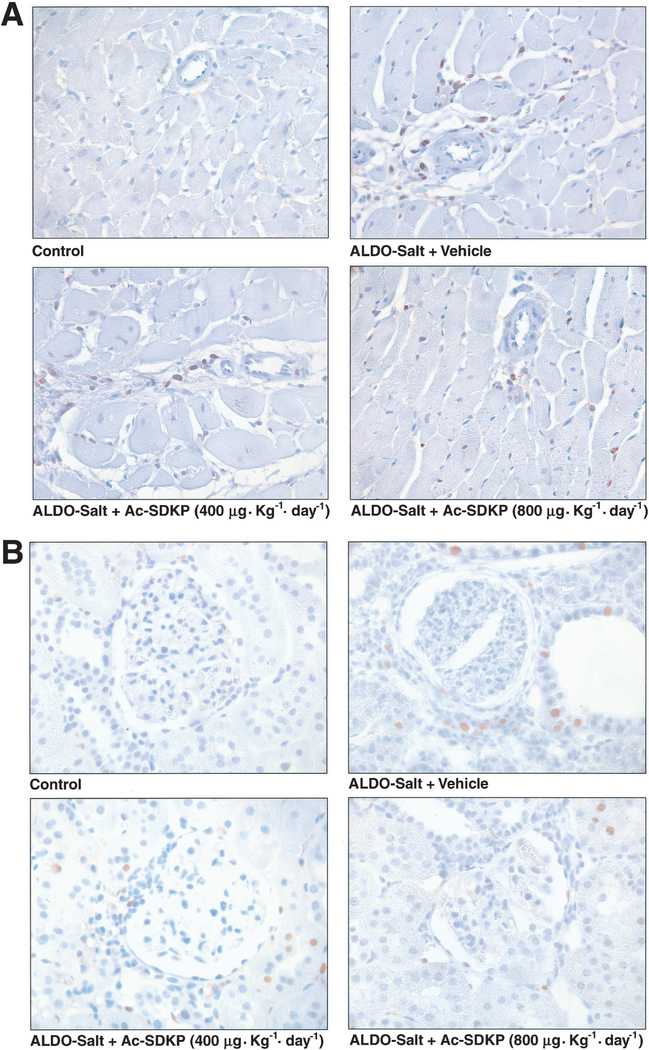
Immunohistochemical staining for PCNA-positive cells (brown) in the LV (A) and renal cortex (B) from controls, rats given ALDO-salt + vehicle, and rats given ALDO-salt + Ac-SDKP (400 or 800 μg · kg–1 · d–1) 6 weeks after treatment. Hematoxylin counterstaining, magnification ×400.
Figure 4.

Effect of Ac-SDKP on the number of PCNA-positive cells in the (A) LV interstitial space and (B) renal cortex in rats treated with ALDO-salt for 6 weeks. There were significantly more PCNA-positive cells in the ALDO-salt–treated LV and kidney than in the controls. Ac-SDKP lowered the number of PCNA-positive cells in a dose-dependent manner compared with ALDO-salt + vehicle. *P<0.0001 vs controls. †P<0.001 vs ALDO-salt + vehicle. ‡P<0.01 vs ALDO-salt + vehicle.
Glomerular and Tubulointerstitial Injury
Glomerular and tubulointerstitial injury were, respectively, 62- and 1.6-fold more severe in ALDO-salt/vehicle rats than in the controls (P<0.005). Ac-SDKP tended to prevent these injuries in a dose-dependent manner, although the difference was not significant (Figure 5).
Figure 5.

Effect of Ac-SDKP on (A) glomerular and (B) tubulointerstitial injury induced by ALDO-salt hypertension in rats. ALDO-salt induced glomerular and tubulointerstitial injury (*P<0.001 vs controls). Ac-SDKP tended to prevent ALDO-salt-induced injury, but the difference was not significant.
Discussion
Chronic excess ALDO in the circulation combined with a high-salt diet in uninephrectomized rats has been reported to cause hypertension, LV and renal hypertrophy, and marked collagen deposition in both ventricles and kidneys.15,16 In this model, which is characterized by extensive cardiac and renal fibrosis with a depressed circulating renin-angiotensin system, ACEi had an antifibrotic effect on the heart and kidney without an effect on blood pressure or cardiac and renal hypertrophy in rats,11,24 a finding attributed to blockade of the tissue renin-angiotensin system. However, other factors such as Ac-SDKP should not be excluded from this process, because Ac-SDKP inhibits fibroblast proliferation7,8; more-over, it is found in tissues in which ACE is present and is hydrolyzed almost exclusively by ACE.4,5,25 Nevertheless, direct evidence for involvement by Ac-SDKP is lacking.
In the present study, Ac-SDKP at 800 μg · kg–1 · d–1 significantly prevented both RV and LV fibrosis without affecting blood pressure or LV hypertrophy. The interstitial collagen volume fraction in the LV also confirmed our findings. Attenuated LV collagen deposition without changes in LV weight is not new; others have reported that spironolactone (an ALDO receptor antagonist),26 captopril (an ACEi), and candesartan (an AT1 receptor antagonist)24 were able to prevent cardiac fibrosis in mineralocorticoid-salt hypertension but could not prevent LV hypertrophy. Our results show that collagen represents only a minute fraction of dry LV weight, suggesting that changes in collagen content should not significantly affect LV weight. Brilla et al27 reported that after pressure overload, fibrosis was found not only in the hypertrophic LV but also in the normotensive, nonhypertrophic RV, thereby discounting blood pressure as contributing to increased collagen content in this model. In the present study, only high-dose Ac-SDKP (800 μg · kg–1 · d–1) prevented enhanced collagen deposition in the LV; this is twice the dose used to blunt increased LV collagen in 2-kidney, 1-clip hypertensive rats.7 The reason for this difference is not clear. Sun et al11 showed that after 4 weeks of ALDO-salt treatment, ACE binding was increased in the endothelium of the aorta, aortic valve, and intramyocardial coronary arteries of both ventricles. ACE activity was also markedly increased in discrete areas of the myocardium of both ventricles at 6 weeks; these sites corresponded to microscopic scars. These data suggest that Ac-SDKP degra-dation occurred not only in the vasculature but also in the interstitium. It is possible that the increased effective dosage could be due to heightened ACE activity in ALDO-salt hypertensive rats.11,28
As expected, renal hypertrophy and fibrosis were observed. Glomerular and tubulointerstitial injury in ALDO-salt/vehicle rats were, respectively, 62- and 1.6-fold more severe than in the controls. Ac-SDKP markedly inhibited collagen deposition in the kidney but had no effect on renal hypertrophy. Yoshioka et al8 reported that in vitro proliferation of renal fibroblasts was significantly attenuated by Ac-SDKP (10–7 to 10–5 mol/L). Thus, Ac-SDKP may be an endogenous modulator of renal fibroblast proliferation. The renal protective effect of Ac-SDKP could be attributed in part to inhibition of cell proliferation and collagen deposition in the kidney.
During the formation of fibrous tissue, fibroblasts and infiltrating mononuclear cells (macrophages) are abundant in the interstitium.16,29 In ALDO-salt hypertensive rats, interstitial cells proliferated in both ventricles at 3 to 6 weeks of ALDO infusion and were associated with subsequent fibrosis as early as 4 weeks after ALDO-salt treatment.16 Moreover, increased proliferation of renal interstitial cells has been reported after injury induced by ureteral obstruction, accompanied by increased collagen deposition.30 PCNA is a 36-kDa auxiliary protein to DNA polymerase-δ which is found in various concentrations within cells throughout the cell cycle, reaching a maximum during the S phase.31 Therefore, it is an indicator of cells in the S phase of cell growth, making it a marker of cell proliferation.32 In the present study, we observed diffuse interstitial and perivascular PCNA-positive cells in the LV in ALDO-salt–treated rats, whereas few positive cells were found in the controls. Similarly, few PCNA-positive cells were detected in the renal tubular epithelium in the control group, whereas in ALDO-salt–treated rats, PCNA-positive cells were easily found in both renal tubular epithelium and interstitium. Ac-SDKP decreased the number of nuclei that stained positive for PCNA in the LV and renal cortex of rats treated with ALDO-salt, perhaps an indication that it attenuates enhanced cell proliferation in the heart and kidney in this model. These results indicate that Ac-SDKP prevents the proliferation of not only pluripotent hematopoietic stem cells and hepatocytes33 but also cardiac fibroblasts and renal cells. Increased fibroblast proliferation was observed in the heart and kidney in ALDO-salt hypertensive rats and was associated with cardiac and renal fibrosis.16,32 The antifibrotic effect of Ac-SDKP could result from inhibition of both fibroblast proliferation and collagen synthesis, because in previous in vitro studies we found that Ac-SDKP not only prevented fibroblast proliferation (as assessed by 3H-thymidine incorporation assay) but also attenuated collagen synthesis as assessed by hydroxyproline assay (expressed as μg collagen/mg protein) and 3H-proline incorporation (expressed as percent collagen synthesis).7
In summary, cardiac and renal fibrosis resulting from chronic ALDO-salt treatment in rats was ameliorated by treatment with Ac-SDKP. In this ALDO-salt hypertensive rat model, increased interstitial and perivascular PCNA-positive cells were evident in the interstitium of the heart and kidney, whereas cardiac and renal fibrosis was prominently displayed. Ac-SDKP significantly decreased the number of PCNA-positive cells and prevented collagen deposition, similar to the beneficial effects found in 2-kidney, 1-clip hypertensive rats (renin-dependent hypertension). Our study strongly suggests that Ac-SDKP prevents cardiac and renal fibrosis by inhibiting collagen production and cell proliferation in ALDO-salt hypertension. Because ACEi increase plasma5 and tissue Ac-SDKP34 and decrease cardiac and renal fibrosis,9 –13 we speculate that Ac-SDKP may participate in the antifibrotic effect of ACEi.
Acknowledgments
This study was supported by AHA Scientist Development Grant 0130128N and NIH grant HL-2898219.
References
- 1.Lenfant M, Wdzieczak-Bakala J, Guittet E, Prome JC, Sotty D, Frindel E. Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci U S A. 1989;86: 779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonnet D, Lemoine FM, Pontvert-Delucq S, Baillou C, Najman A, Guigon M. Direct and reversible inhibitory effect of the tetrapeptide acetyl-N-Ser-Asp-Lys-Pro (Seraspenide) on the growth of human CD34+ subpopulations in response to growth factors. Blood. 1993;82:3307–3314. [PubMed] [Google Scholar]
- 3.Pradelles P, Frobert Y, Créminon C, Liozon E, Massé A, Frindel E. Negative regulator of pluripotent hematopoietic stem cell proliferation in human white blood cells and plasma as analysed by enzyme immunoassay. Biochem Biophys Res Commun. 1990;170:986–993. [DOI] [PubMed] [Google Scholar]
- 4.Azizi M, Rousseau A, Ezan E, Guyene T-T, Michelet S, Grognet J-M, Lenfant M, Corvol P, Ménard J. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J Clin Invest. 1996;97:839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azizi M, Ezan E, Nicolet L, Grognet JM, Menard J. High plasma level of N-acetyl-seryl-aspartyl-lysyl-proline: a new marker of chronic angiotensin-converting enzyme inhibition. Hypertension. 1997;30:1015–1019. [DOI] [PubMed] [Google Scholar]
- 6.Junot C, Menard J, Gonzales M-F, Michaud A, Corvol P, Ezan E. In vivo assessment of captopril selectivity of angiotensin I-converting enzyme inhibition: differential inhibition of acetyl-Ser-Asp-Lys-Pro and angiotensin I hydrolysis. J Pharmacol Exp Ther. 1999;289:1257–1261. [PubMed] [Google Scholar]
- 7.Rhaleb N-E, Peng H, Harding P, Yang X-P, Carretero OA. Ac-SDKP, a new inhibitor of cardiac fibroblast proliferation. Hypertension. 1998; 32:609 Abstract. [Google Scholar]
- 8.Yoshioka T, Iwamoto N, Nitta K, Irie K, Xiao H-J, Ito K, Muraki T. AcSDKP is a novel anti-proliferative peptide for renal fibroblasts. Naunyn-Schmiedebergs Arch Pathol. 1998;358:R742 Abstract. [Google Scholar]
- 9.Liu Y-H, Yang X-P, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure: role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99: 1926–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooks WW, Bing OH, Robinson KG, Slawsky MT, Chaletsky DM, Conrad CH. Effect of angiotensin-converting enzyme inhibition on myocardial fibrosis and function in hypertrophied and failing myocardium from the spontaneously hypertensive rat. Circulation. 1997;96: 4002–4010. [DOI] [PubMed] [Google Scholar]
- 11.Sun Y, Ratajska A, Zhou G, Weber KT. Angiotensin-converting enzyme and myocardial fibrosis in the rat receiving angiotensin II or aldosterone. J Lab Clin Med. 1993;122:395–403. [PubMed] [Google Scholar]
- 12.Kim S, Ohta K, Hamaguchi A, Omura T, Yukimura T, Miura K, Inada Y, Wada T, Ishimura Y, Chatani F, Iwao H. Role of angiotensin II in renal injury of deoxycorticosterone acetate-salt hypertensive rats. Hyper-tension. 1994;24:195–204. [DOI] [PubMed] [Google Scholar]
- 13.Kaneto H, Morrissey J, McCracken R, Reyes A, Klahr S. Enalapril reduces collagen type IV synthesis and expansion of the interstitium in the obstructed kidney of the rat. Kidney Int. 1994;45:1637–1647. [DOI] [PubMed] [Google Scholar]
- 14.Young M, Fullerton M, Dilley R, Funder J, Mineralocorticoids, hyper-tension, and cardiac fibrosis. J Clin Invest. 1994;93:2578–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Y, Zhang J, Zhang JQ, Ramires FJA. Local angiotensin II and transforming growth factor-β1 in renal fibrosis of rats. Hypertension. 2000;35:1078–1084. [DOI] [PubMed] [Google Scholar]
- 16.Campbell SE, Janicki JS, Weber KT. Temporal differences in fibroblast proliferation and phenotype expression in response to chronic administration of angiotensin II or aldosterone. J Mol Cell Cardiol. 1995;27: 1545–1560. [DOI] [PubMed] [Google Scholar]
- 17.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 18.Chiariello M, Ambrosio G, Cappelli-Bigazzi M, Perrone-Filardi P, Brigante F, Sifola C. A biochemical method for the quantitation of myocardial scarring after experimental coronary artery occlusion. J Mol Cell Cardiol. 1986;18:283–290. [DOI] [PubMed] [Google Scholar]
- 19.Hansen-Smith FM, Watson L, Lu DY, Goldstein I. Griffonia simplicifolia I: fluorescent tracer for microcirculatory vessels in nonperfused thin muscles and sectioned muscle. Microvasc Res. 1988;36:199–215. [DOI] [PubMed] [Google Scholar]
- 20.Laitinen L. Griffonia simplicifolia lectins bind specifically to endothelial cells and some epithelial cells in mouse tissues. Histochem J. 1987;19: 225–234. [DOI] [PubMed] [Google Scholar]
- 21.Sabbah HN, Sharov VG, Lesch M, Goldstein S. Progression of heart failure: a role for interstitial fibrosis. Mol Cell Biochem. 1995;147:29–34. [DOI] [PubMed] [Google Scholar]
- 22.Raij L, Azar S, Keane W. Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int. 1984;26: 137–143. [DOI] [PubMed] [Google Scholar]
- 23.Hayakawa H, Raij L. Nitric oxide synthase activity and renal injury in genetic hypertension. Hypertension. 1998;31:266–270. [DOI] [PubMed] [Google Scholar]
- 24.Brown L, Duce B, Miric G, Sernia C. Reversal of cardiac fibrosis in deoxycorticosterone acetate-salt hypertensive rats by inhibition of the renin-angiotensin system. J Am Soc Nephrol. 1999;10:S143–S148. [PubMed] [Google Scholar]
- 25.Rousseau A, Michaud A, Chauvet M-T, Lenfant M, Corvol P. The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J Biol Chem. 1995;270:3656–3661. [DOI] [PubMed] [Google Scholar]
- 26.Brilla CG, Matsubara LS, Weber KT. Antifibrotic effects of spironolactone in preventing myocardial fibrosis in systemic arterial hypertension. Am J Cardiol. 1993;71:12A–16A. [DOI] [PubMed] [Google Scholar]
- 27.Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res. 1990;67: 1355–1364. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa M, Nasjletti A. Plasma kinin concentration in deoxycorticosterone-salt hypertension. Hypertension. 1988;11:411–415. [DOI] [PubMed] [Google Scholar]
- 29.Ishidoya S, Morrissey J, McCracken R, Reyes A, Klahr S. Angiotensin II receptor antagonist ameliorates renal tubulointerstitial fibrosis caused by unilateral ureteral obstruction. Kidney Int. 1995;47:1285–1294. [DOI] [PubMed] [Google Scholar]
- 30.Klahr S, Pukerson ML. The pathophysiology of obstructive nephropathy: the role of vasoactive compounds in the hemodynamic and structural abnormalities of the obstructed kidney. Am J Kidney Dis. 1994;23: 219–223. [DOI] [PubMed] [Google Scholar]
- 31.Bravo R, Frank R, Blundell PA, Macdonald-Bravo H. Cyclin/PCNA is the auxiliary protein of DNA polymerase-delta. Nature. 1987;326: 515–517. [DOI] [PubMed] [Google Scholar]
- 32.Coltrera MD, Gown AM. PCNA/cyclin expression and BrdU uptake define different subpopulations in different cell lines. J Histochem Cytochem. 1991;39:23–30. [DOI] [PubMed] [Google Scholar]
- 33.Lombard M-N, Sotty D, Wdzieczak-Bakala J, Lenfant M. In vivo effect of the tetrapeptide, N-acetyl-Ser-Asp-Lys-Pro, on the G1-S transition of rat hepatocytes. Cell Tissue Kinet. 1990;23:99–103. [DOI] [PubMed] [Google Scholar]
- 34.Junot C, Nicolet L, Ezan E, Gonzales M-F, Menard J, Azizi M. Effects of angiotensin-converting enzyme inhibition on plasma, urine, and tissue concentrations of hemoregulatory peptide acetyl-Ser-Asp-Lys-Pro in rats. J Cardiovasc Pharmacol. 1999;291:982–987. [PubMed] [Google Scholar]