

Supplemental Digital Content is available in the text.
Keywords: blood coagulation factor inhibitors, enoxaparin, mendelian randomization analysis, odds ratio, risk, thrombosis
Background and Purpose—
Coagulation factor XI (FXI) is a novel target for antithrombotic therapy addressed by various therapeutic modalities currently in clinical development. The expected magnitude of thrombotic event reduction mediated by targeting FXI is unclear.
Methods—
We analyzed the association of 2 common genetic variants, which alter levels of FXI, with a range of human phenotypes. We combined variants into a genetic score standardized to a 30% increase in relative activated partial thromboplastin time, equivalent to what can be achieved with pharmacological FXI reduction. Using data from 371 695 participants in the United Kingdom Biobank and 2 large-scale genome-wide association studies, we examined the effect of this FXI score on thrombotic and bleeding end points.
Results—
Genetic disposition to lower FXI levels was associated with reduced risks of venous thrombosis (odds ratio, 95% CI; P value; odds ratio=0.1, 0.07–0.14; P=3×10−43) and ischemic stroke (odds ratio=0.47, 0.36–0.61; P=2×10−8) but not with major bleeding (odds ratio=0.7, 0.45–1.04; P=0.0739). The observed relative risk reductions were consistent within a range of subgroups that were at high risk for thrombosis. Consistently, we observed higher absolute risk reductions conferred by genetically lower FXI levels in high-risk subgroups, such as patients with atrial fibrillation.
Conclusions—
Human genetic data suggest that pharmacological inhibition of FXI may achieve considerable reductions in ischemic stroke risk without clear evidence for an associated risk of major bleeding. The quantitative framework developed can be used to support the estimation of achievable risk reductions with pharmacological modulation of FXI.
Current therapeutic strategies for preventing thrombosis focus on inhibition of components of the central part of the coagulation cascade (common pathway), including targets such as coagulation factor 10 (eg, rivaroxaban or apixaban), coagulation factor 2 (eg, dabigatran), or both (heparins, including low molecular weight heparins such as enoxaparin). Although these inhibitors are effective at preventing thrombosis, they also compound specific risks for bleeding complications.1,2 There is, however, emerging evidence that selectively targeting factor XI (FXI), another serine protease in the coagulation cascade, might have antithrombotic effects without an increase in bleeding risk.3
Data from a recent clinical trial demonstrated that pharmacological lowering of FXI by a second-generation antisense oligonucleotide leads to an increase in activated partial thromboplastin time (aPTT; ≈1.4-fold on the day of surgery, 36 days after application) and a reduction in venous thromboembolic events in patients undergoing total knee arthroplasty. However, this proof-of-concept trial was too small to assess the effect on other thrombotic end points.4
DNA sequence variants in therapeutic target genes represent naturally occurring, lifelong variation of gene activity (ie, experiments of nature).5,6 Consequently, if a genetic predisposition to reduced FXI levels associates with a reduced risk of thrombotic events, these results would support the therapeutic hypothesis that pharmacological inhibition of FXI will also reduce the occurrence of thrombotic end points.
Individuals harboring a naturally occurring deficiency in Factor XI have been studied in detail and show a reduced occurrence of cardiovascular and venous thromboembolism events,7 with only a relatively mild increase in bleeding compared with other coagulation factor deficiencies such as Hemophilia A and B.8 However, because full FXI deficiency due to loss-of-function mutations is a relatively rare condition, all of the studies to date have been conducted in founder populations, including the Ashkenazi Jewish community.7 These individuals may differ in a range of factors when compared with individuals without FXI deficiency, and it is unclear to what extent such other factors (eg, additional coagulation factor deficiencies) might impact the phenotype of these populations. Recently, an association of a genetic score of FXI levels and ischemic stroke was reported in a Mendelian randomization study.9
In this study, we expand on this by using both common and rare variants with known impact on FXI10,11 to (1) determine the effects of a genetic disposition to lower FXI levels on a range of thrombotic and bleeding outcomes, across a range of relevant patient groups (eg, patients with atrial fibrillation) and (2) to combine clinical and genetic data to anticipate and quantify risk reductions achievable with pharmacological inhibition of FXI.
Methods
Access to individual level data used in this study can be requested from the United Kingdom Biobank (UKB). Summary level information on analysis performed is available from the corresponding authors on reasonable request. For detailed descriptions of all the methodologies applied, please refer to the online-only Data Supplement.
Samples and Data
The UKB is a population cohort of ≈500 000 subjects from the United Kingdom.1 It combines deep phenotyping in the form of electronic health records with genome-wide genotyping and imputation. After quality control of genotype data (online-only Data Supplement), we retained 371 695 samples for further analysis in the UKB Imputation V3 dataset. Whole-exome sequencing data of FXI variants was available for 50k UKB subjects (called using the UKB FE variant calling pipeline). To enhance power for the analysis of ischemic stroke and MI, we supplemented our results with summary level data from 2 large genomics consortia, MEGASTROKE12 (www.megastroke.org) and CARDIoGRAMplusC4D13 (http://www.cardiogramplusc4d.org). All analyses of individual level data conducted in this study were covered by the informed consent of the UKB, and no additional ethics committee approval was required.
Outcome Definitions in UKB
When available predefined outcome definitions provided as part of the UKB data were used (ischemic stroke and myocardial infarction). Additional clinical end points were defined based on UKB electronic health records, which include ICD10- and ICD9-based hospital admission records, OPCS-based (Office of Population Censuses and Surveys Classification of Interventions and Procedures, version 4) operating procedure codes, and information derived from questionnaires at study entry. Prospective survival analyses considered only clinical end points occurring during follow up. For a full list of phenotypic definitions, refer to Table I in the online-only Data Supplement.
Definition of a Genetic Score Using Common Genetic Variants in FXI
We leveraged common variants in the FXI gene to characterize the effects of a genetic disposition to reduced FXI levels. We selected 2 common variants as instruments for a genetic predisposition to reduced FXI levels: rs4253417 (minor allele frequency: 40.4%) and rs1593 (minor allele frequency: 12.6%), which are both intronic variants in FXI, located in or near regions annotated by ENCODE data14,15 to be of regulatory relevance in liver tissue, the primary site of synthesis of FXI (see Figure 1). These variants were selected because they (1) have previously been shown to have robust and independent effects on circulating FXI levels,10 (2) have been robustly associated with activated partial thromboplastin time (aPTT in seconds, a measure of coagulation speed).11 Generally, as FXI levels decrease, the aPTT will increase.
Figure 1.
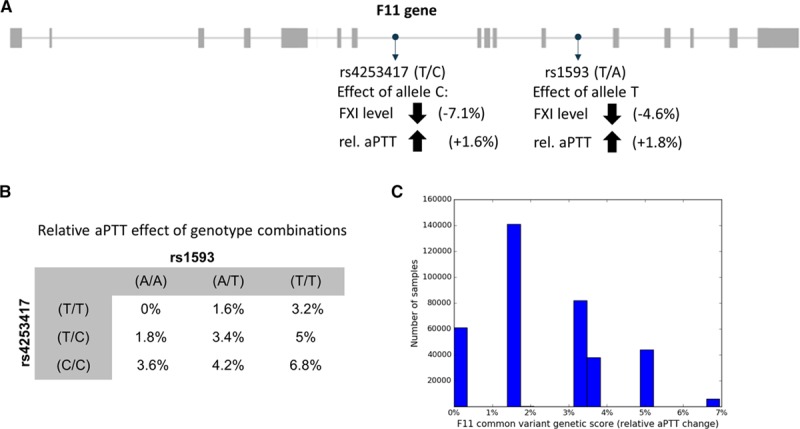
Derivation of a common variant genetic factor XI (FXI) score. A, The genetic FXI score was derived based on 2 common, intronic variants (rs4253417 and rs1593) with previously described effects on FXI levels10 and aPTT.11 B, Possible combinations of genotypes for the 2 variants showed a range from 0% to 6.8% genetically determined relative activated partial thromboplastin time (aPTT) increase. C, Distribution of score values in the UK Biobank.
To examine the effects of reduced FXI levels on cardiovascular risk factors and thrombotic and bleeding end points, we counted the number of FXI-lowering alleles of rs4253417 and rs1593 carried by each individual (ie, individuals carried 0–4 effect alleles). The weight of the effect alleles in the score was given by the change in aPTT relative to the global mean of 29s in the ARIC aPTT study.11 The score was defined with respect to the aPTT increasing (ie, FXI lowering) alleles: C (aPTT effect of +1.6% and FXI level effect of −7.1% per C allele) for rs4253417 and T (aPTT effect +1.8% and FXI level effect of −4.6% per T allele) for rs1593. The score value for each UKB individual was then the sum of the effects of each effect allele carried. For example, an individual with a (C/C) genotype for rs4253417 and a (A/T) genotype for rs1593 would carry 3 effect alleles with a combined score of 1.6%+1.6%+1.8%=5% aPTT change. In total, there were 9 possible genotype combinations for the 2 SNPs, each with a distinct associated score value (Figure 1B). To enable the comparison of genetic and clinical risk estimates, we standardized the genetic FXI score to reflect a 30% relative increase in aPTT, which is within the range of the presurgery (day 36 after application) effects of a 200 mg (+20% relative aPTT) and 300 mg (+40% relative aPTT) dose of a FXI-anti sense oligonucleotide measured in a Phase 2a proof-of-concept study.4 This level of FXI-mediated aPTT prolongation was shown to be efficacious for reduction of venous thromboembolism after total knee arthroplasty in that study.4 To achieve this standardization to a 30% relative aPTT increase in the UKB FXI score, the score was rescaled such that a 30% increase in relative aPTT corresponded to a score value of 1.0; for studies where only summary level results were available, we first divided the reported effect (eg, the log odds ratio for ischemic stroke in MEGASTROKE) with the reported absolute aPTT change per allele for each SNP and then multiplied with the absolute aPTT change corresponding to a 30% relative increase. To assess the fit of the assumption of an additive score, we also defined a categorical version of the FXI score where each level was defined by a genotype combination (Figure 1B). Only the 6 genotype combinations with >500 observed subjects were included (Figure 1C).
Analysis of Rare FXI Loss-of-Function Variants
Putative FXI loss-of-function variants, in the genotype and whole exome sequencing data were predicted using the Loftee tool (https://github.com/konradjk/loftee; see the online-only Data Supplement for more details). In total, we identified 477 putative FXI loss-of-function allele carriers (120 and 357 in the whole exome sequencing and genotype datasets, respectively). Putative F11 loss-of-function variants were analyzed separately in the whole exome sequencing and genotype datasets and subsequently integrated using a fixed-effect meta-analysis.
Statistical Methods
The cross-sectional analysis of clinical end points was performed using logistic regression, for the survival analyses, a Cox proportional hazards model was applied. Regression analyses were stratified by sex, with age at baseline, genotyping platform, and the first 10 principal components of the genotypes (provided as part of the UKB dataset) as covariates. To combine standardized risk estimates (UKB with CARDIoGRAMplusC4D or MEGASTROKE), inverse variance weighted fixed effect meta-analysis was used.
Association analyses were performed using the R statistical programming language v3.4.3. (https://www.r-project.org/).16 UKB genotypes were parsed using PLINK v2.0.0 (https://www.cog-genomics.org/plink2) and bgenix v1.1.3 (https://bitbucket.org/gavinband/bgen/wiki/bgenix). Multiple testing was accounted for by (1) control of the familywise error rate with the stringent Bonferroni correction for tests of the primary end points and (2) by control of the false discovery rate with the Benjamini-Hochberg procedure17 for the exploratory interaction and PheWAS analyses.17,18 Estimates from trials of enoxaparin versus placebo and FXI inhibition versus enoxaparin were combined using an indirect comparison approach as previously described.19 To investigate potential differences in the effect of genetically lower FXI levels in certain subgroups, we performed an interaction analysis for ischemic stroke and venous thromboembolism (VT) and the FXI genetic score. For this purpose, we first identified significant risk factors for VT or ischemic stroke by conducting Cox proportional hazard regressions of ischemic stroke/VT and separately each phenotype with at least 500 cases on our panel (Table I in the online-only Data Supplement). For significant risk factors (FDR <5%), we constructed separate Cox proportional hazard regression models for the genetic score and VT or ischemic stroke within each subgroup (defined by presence or absence of the risk factor) to quantify the effect of genetically lower FXI within each subgroup. To formally test for a difference in effects of genetically lower FXI levels, we evaluated a model that included the risk factor as a covariate and an interaction term between the FXI genetic score and the presence or absence of the risk factor.
Results
Effect of Genetic Predisposition to Lower FXI on Cardiovascular Risk Factors
The starting point for our analyses was the development of a genetic score for FXI activity. The 5928 individuals carrying the full 4 effect alleles of the genetic score are estimated to have a 21.9% reduction in FXI levels, and a 6.8% increase in relative aPTT compared with the 60 881 individuals carrying zero effect alleles (Figure 1C). This means that subjects with zero effect alleles have on average a 21.9% increased FXI level compared with subjects with 4 effect alleles. Akin to a comparison of baseline characteristics between intervention groups in randomized clinical trials, we assessed the effect of the FXI genetic score on a range of cardiovascular risk factors, including diabetes mellitus, blood pressure, and body mass index (Table). We noted no significant associations, suggesting there were no systematic imbalances in the prevalence of cardiovascular risk factors in individuals with a genetic predisposition for lower FXI levels.
Table.
Cross-Sectional Correlates of the FXI Genetic Score

Effect of Genetic Predisposition to Lower FXI on Thrombotic and Bleeding Events
To investigate the effect of genetic predisposition to lower FXI activity with thrombotic and bleeding end points, we performed association analyses of the FXI genetic score with myocardial infarction, ischemic stroke, venous thromboembolism, and major bleeding end points in the UKB (see Table I in the online-only Data Supplement for definitions). The genetic FXI score was significantly associated with 2 out of the 4 primary thrombotic and bleeding end points. Genetically lower FXI levels were associated with lower risk of ischemic stroke in the UKB+MEGASTROKE data (odds ratio [OR]=0.47, 95% CI, 0.36–0.61; P=1.5×10−8), yielding a combined 53% reduction in ischemic stroke risk (Figure 2A). Consistent with clinical data on FXI inhibition,4 genetically lower FXI levels were associated with lower risk of venous thromboembolism (OR, 0.1; 95% CI, 0.07–0.14; P=3.03×10−43) in the UKB. We did not observe a significant association of reduced FXI levels with risk of major bleeding (OR, 0.69; 0.45–1.04; P=0.0739) in UKB participants. There was also no association of genetically lower FXI levels with risk of myocardial infarction in the CARDIoGRAMplusC4D Consortium data nor in UKB participants, yielding a combined estimate of (OR=1.0; 95% CI, 0.79–1.25; P=0.969; Figure 2A). Information on subtypes of ischemic stroke was only available in the MEGASTROKE consortium data,12 where we found a significant risk reduction only for the cardioembolic stroke subtype (OR=0.16; 95% CI, 0.09–0.28; P=2.08×10−10; Figure 2B). To check the validity of the log-linear extrapolation to a 30% relative aPTT increase, we (1) examined the effects of groups defined by individual genotype combinations of rs4253417 and rs1593 and how well they adhered to the linear model and (2) extended the genetic score with estimates based on 477 predicted FXI loss-of-function variants identified in whole-exome sequencing of 50k UKB participants (see Methods, Table II, and Figure I in the online-only Data Supplement). We observed for all 4 outcomes a good adherence of the individual genotype group estimates to the log-linear model and estimates that are broadly consistent with the linear model from the loss-of-function analysis (Figure 3; Figure II in the online-only Data Supplement).
Figure 2.
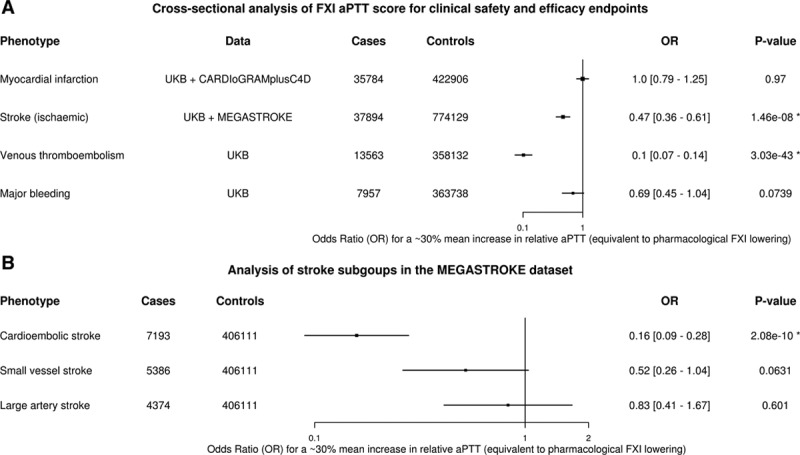
Cross-sectional analyses of the genetic factor XI (FXI) score for clinical end points and ischemic stroke subtypes. A, Association of the FXI genetic score with 4 primary safety and efficacy outcomes (United Kingdom Biobank [UKB], external genetics consortia). After correcting for testing 4 primary outcomes (Bonferroni threshold=0.05/4=0.01), we observe significant associations of the FXI genetic score, expressed as a 30% relative increase in activated partial thromboplastin time (aPTT), with venous thromboembolism (odds ratio [OR]=0.1 [0.07–0.14], P=3.03×10−43) and ischemic stroke (OR=0.47 [0.36–0.61]; P=1.5×10−8) and (B) cross-sectional analysis of CCS ischemic stroke subgroups from the MEGASTROKE dataset. Integration of MEGASTROKE12 effect sizes for 2 common FXI SNPs (rs4253417, rs1593) by fixed-effect meta-analysis showed significant risk reduction for the cardioembolic stroke subtype (OR=0.16 [0.09–0.28]; P=2.08×10−10).
Figure 3.
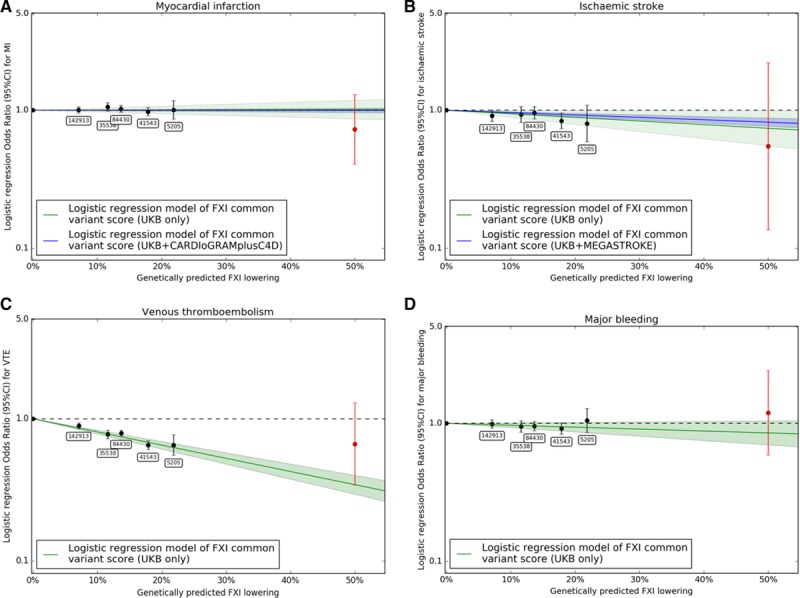
Analysis of fit of effect estimates of individual genotype groups to the log-linear model. Effects for (A) myocardial infarction, (B) ischemic stroke, (C) venous thromboembolism, and (D) major bleeding. Effect estimates for the categorical version of the factor XI (FXI) score where each level corresponds to a specific combination of genotypes of rs4253417 and rs1593 (black) show good fit to the log-linear model (green). Effects of FXI loss-of-function variants in 50k whole-exome sequences (red) were broadly consistent with the linear model, although CIs were wide because of the limited sample size of 477 FXI LOF variant carriers.
To limit the potential impact of misclassification because of self-reported outcomes, we conducted sensitivity analyses in UKB, in which we only considered thrombotic or bleeding events occurring during follow-up (and hence captured through electronic health records). Time-to-event analyses conducted on those events occurring during follow-up yielded very comparable results (Figure III in the online-only Data Supplement).
In an exploratory analysis of 46 additional end points available within UKB, we did not observe any additional associations after correcting for multiple testing (Figure IV in the online-only Data Supplement). Similarly, we did not observe a significant effect of the FXI score on all-cause mortality (Figure V in the online-only Data Supplement).
Consistency of Effects of Genetically Lower FXI-Levels in Subpopulations With High Thrombosis Risk
Akin to subgroup analyses within a randomized clinical trial, we sought to evaluate whether there were particular subgroups within UKB with relatively greater or reduced benefits resulting from genetic predisposition to low FXI levels. We therefore systematically identified high-risk subgroups within UKB that had a significantly increased risk of venous thrombosis or ischemic stroke. Through this approach, we were able to identify known high-risk groups such as patients with cancer for venous thrombosis, and patients with atrial fibrillation for ischemic stroke (Figure VI in the online-only Data Supplement). We then calculated the relative risk reductions for venous thrombosis or ischemic stroke conferred by genetically lower FXI levels within each of the high risk groups and compared it to all other participants using a Wald test for interaction (see Figure 4A and 4B for 2 example risk factors, Tables III and IV in the online-only Data Supplement). After accounting for multiple testing by controlling the false discovery rate, there were no high-risk subgroups with a significantly different relative risk reduction conferred by genetically lower FXI levels (see Tables III and IV in the online-only Data Supplement). For example, predisposition to genetically lower FXI was associated with similar relative risk reductions for ischemic stroke in those individuals with a diagnosis of atrial fibrillation (hazard ratio [HR], 0.18; 95% CI, 0.04–0.76; P=0.02, N=14 276), as in those without atrial fibrillation (HR, 0.26; 95% CI, 0.1–0.65; P=0.00378, N=357 419; Pinteraction=0.657). Conversely, this suggests that the absolute risk reductions conferred by genetically lower FXI levels are higher in high-risk patient groups, such as patients with atrial fibrillation, atherosclerotic vascular diseases, or cancer. For example, comparing the extremes of our genetic score in prospective analyses in UKB, the difference in absolute ischemic stroke rates was considerably higher in individuals with atrial fibrillation compared with those without (Figure 4C): comparing, for example, ischemic stroke event rates 6 years after baseline, we note that the absolute difference in event rates between the extreme groups of our genetic score was 0.0149 events/patient for patients with atrial fibrillation and 0.0016 events/patient for patients without (derived based on unadjusted Kaplan-Meier curves, P = 2 × 10−6, see the online-only Data Supplement for details regarding the methodology). This finding was also confirmed qualitatively in a covariate-adjusted Cox regression model.
Figure 4.

Effects of genetically lower factor XI (FXI) levels in high-risk subgroups in the UK Biobank. Hazard ratios (HR) of subgroup analyses of relevant risk factors for (A) ischemic stroke and (B) venous thromboembolism and the FXI genetic score. We observed no significant interactions between any risk factors and the FXI genetic score. C, Survival analysis of differences in ischemic stroke rate for participants with the strongest observed genetic effect on FXI (red) and participants with no genetic effect (black) stratified by atrial fibrillation (AF) presence. While we observed similar relative risk reduction conferred by genetically lower FXI in the presence (dashed) and absence (solid) of AF, it can be seen that the absolute risk reduction in the AF subgroup is markedly higher.
Benchmarking Effects on Clinical End Points From Genetically Lowered Against Pharmacologically Lowered FXI Levels
Our analyses compared individuals with a genetic predisposition to lower FXI levels with those individuals without such a predisposition, and hence can be considered a natural, placebo-controlled clinical experiment. As the clinical trial of pharmacological FXI inhibition for prevention of venous thrombosis4 compared against enoxaparin (rather than placebo), we estimated a placebo comparison, to be able to directly compare genetic and clinical trial results. We did this by correcting the calculated effect of the FXI antisense compared with enoxaparin (OR, 0.84; 95% CI, 0.68–1.03 and OR, 0.1; 95% CI, 0.05–0.23), for the 200 and 300 mg dosage, respectively, see the online-only Data Supplement) for the effect attributable to enoxaparin (OR, 0.27; 95% CI, 0.19–0.38), Figure VII in the online-only Data Supplement), yielding an overall combined relative risk reduction for venous thrombosis of OR=0.086 (CI, 0.06–0.13, see the online-only Data Supplement) at an relative aPTT FXI increase of 30%. This estimate is compatible with, but slightly smaller than, the estimate observed for genetically lower FXI and venous thrombosis risk (OR, 0.1; 95% CI, 0.07–0.14), by a log-scale factor of 0.93 (SE, 0.11; calibration factor). Hence, to provide a more reliable estimate of the potential clinically achievable effects, we applied this calibration factor to the relative risk reduction in ischemic stroke observed with genetically lower FXI levels (Figure VIII in the online-only Data Supplement). From this, we obtain an estimated risk reduction of ischemic stroke of OR=0.44 (CI 0.31–0.62), which we would expect to observe in a placebo-compared clinical trial of a FXI-lowering therapy, which increases aPTT by 30% over baseline, pretreatment values.
Discussion
A genetic predisposition to lower FXI levels was associated with increased aPTT and lower risk for efficacy end points venous thrombosis and ischemic stroke, while there was no evidence for an association with major bleeding events. The observed relative reductions in efficacy end points were consistent across a range of relevant subgroups that are at high risk of thrombotic events (eg, participants with atrial fibrillation).
Our study is consistent with, but also provides additional insights compared with, previous work which reported reduced occurrence of venous thrombosis, ischemic stroke, and overall cardiovascular outcomes (including myocardial infarction events) in FXI-deficient population isolates.7,20,21 As FXI-deficient population isolates also differ with regard to important confounding factors (eg, diabetes mellitus and hypertension),7 our study, where we did not observe any difference in the distribution of these potential confounders, provides more robust evidence that the observed effects on efficacy and safety end points can be attributed to FXI. As the CIs for the association with myocardial infarction would still be compatible with clinically relevant risk reductions, more evidence is needed to accurately estimate any potential impact of FXI on myocardial infarction. This could be achieved through study of loss-of-function variants (reflecting higher grades of FXI lowering), by combining evidence from large sequencing consortia of myocardial infarction with sequencing data currently emerging in UKB.
The robust relative risk reductions for venous thrombosis and ischemic stroke that we observed for genetically lower FXI levels were consistent across a number of relevant high-risk subpopulations. While in several cases our analyses were limited in statistical power (eg, because of small size of the high-risk populations), our results did not highlight any particular subgroup where the relative risk reduction resulting from genetically lower FXI activity is considerably different. This supports the notion that (1) observed relative risk reductions are transferable across different patient populations and (2) that by targeting FXI to high-risk populations, where the expected absolute risk is increased, absolute risk reductions with FXI modulation should be higher, as observed for patients with atrial fibrillation.
Finally, we benchmarked the effect of genetically lower FXI level on venous thrombosis against the effects of pharmacological FXI lowering on the same end point.4 We used this comparison to calculate a more reliable estimate for clinically achievable risk reductions for ischemic stroke, an end point not yet studied in clinical trials involving reduction of FXI. Our approach suggests a relative risk reduction versus placebo of ≈56% (OR, 0.44; 95% CI, 0.31–0.62) for a FXI-mediated aPTT increase of 30% could be achievable, which is broadly in the same magnitude of effect as the 60% to 70% relative risk reductions observed for warfarin compared with placebo.22 As data for additional FXI modulating agents at different doses will become available, our model can become helpful in complementing traditional approaches for estimating the expected clinical effect size and consequently inform dosing and sample size considerations of large and costly Phase III studies of ischemic stroke end points.
Our study has several limitations relating to the following key assumptions. First, that measurements of aPTT, the outcome used to standardize our genetic score, are comparable between the genetic study11 and the FXI clinical trial.4 Although the overall distributions of the measurements seem comparable, we cannot rule out subtle differences between these measurements. This limitation should be addressed in the future by measuring effects of genetic variants and pharmacological agents in the same study and participants. Second, that risk estimates obtained from the clinical trial population (total knee arthroplasty patients) and UKB (general population) are comparable. Although the low number of participants undergoing total knee arthroplasty in UKB prevented us from conducting meaningful subgroup analyses, our observation that relative risk reductions for genetically lower FXI were similar in other relevant subgroups supports the validity of this assumption. Third, that end point definitions are comparable between UKB electronic health records and clinical trials. While the thrombotic outcomes (Stroke, MI) have shown reasonable concordance with clinically validated outcomes,23,24 clinical validation for the VT and major bleeding end points is lacking at this stage. The forthcoming release of GP records in UKB could enable future validation efforts. Fourth, that the relationship between genetic and trial estimates, reflected by the calibration factor, is transferable across different end points. Our current understanding of the differences between genetic and pharmacological drug target modulation highlights differences in the start and duration of exposure (from birth versus later in life and lifelong versus short term) as a plausible reason. We would not expect these factors to impact differently on venous thrombosis or ischemic stroke; however, this assumption can ultimately only be verified once data from a clinical trial of FXI modulation for stroke prevention become available. Fifth, that results are not biased by horizontal pleiotropy. As MR-Egger25 requires >2 variants, we could not test for this directly, but the lack of reported association with other unrelated cardiovascular risk factors for the included SNPs makes this assumption plausible. Finally, the extrapolation of pharmacologically achievable effect sizes based on the observed relationship between genetically lower FXI levels and clinical end points assumes a log-linear relationship. While this assumption is compatible with the observed data of modest FXI lowering through common genetic variants, it may not hold at greater levels of FXI lowering. Our analysis of putative FXI loss-of-function variants in 50k whole-exome sequences did yield estimates at ≈50% FXI activity that are consistent with the log-linear model, but the analysis was underpowered. The expected emergence of sequencing data for the remaining ≈450k UKB participants should provide further clarity with regard to possible nonlinear effects. The emergence of additional clinical trials in the future should also enable the study of higher degrees of FXI lowering through pharmacological modulation. Overall, these assumptions and resulting limitations emphasize the need for a more integrative approach in clinical development, such that learnings from human genetic studies can be leveraged in a more systematic manner.
In summary, genetic predisposition to lower FXI levels was associated with reduced risks of VTE and ischemic stroke, providing further evidence that pharmacological lowering of FXI may become a relevant clinical approach for prevention of these conditions. The quantitative relationships and statistical models derived can be applied to guide the clinical development programs of FXI-lowering therapies.
Acknowledgments
This research has been conducted using the UK Biobank Resource under application 28807. The MEGASTROKE project received funding from sources specified at http://www.megastroke.org/acknowledgements.html. Additional data on coronary artery disease/myocardial infarction have been contributed by CARDIoGRAMplusC4D investigators and have been downloaded from www.CARDIOGRAMPLUSC4D.ORG.
Sources of Funding
The study was funded by Bayer AG. Dr Ellinor is supported by the National Institutes of Health (1RO1HL092577, R01HL128914, K24HL105780), by the American Heart Association (18SFRN34110082) and by the Fondation Leducq (14CVD01).
Disclosures
Drs. Khera, Ellinor, Kathiresan, and Mr Chaffin receive grant support from Bayer AG to the Broad Institute. Dr Ellinor has consulted for Bayer AG, Novartis, and Quest Diagnostics. Drs. Georgi, Mielke, Gelis, Mundl, van Giesen, Ziegelbauer, and Freitag are full-time employees of Bayer AG. Dr Kathiresan is a full-time employee of Verve Therapeutics.
Supplementary Material
Footnotes
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/STROKEAHA.119.026545.
References
- 1.LaPointe NM, Chen AY, Alexander KP, Roe MT, Pollack CV, Jr, Lytle BL, et al. Enoxaparin dosing and associated risk of in-hospital bleeding and death in patients with non ST-segment elevation acute coronary syndromes. Arch Intern Med. 2007;167:1539–1544. doi: 10.1001/archinte.167.14.1539. doi: 10.1001/archinte.167.14.1539. [DOI] [PubMed] [Google Scholar]
- 2.McDonald SB, Renna M, Spitznagel EL, Avidan M, Hogue CW, Jr, Moon MR, et al. Preoperative use of enoxaparin increases the risk of postoperative bleeding and re-exploration in cardiac surgery patients. J Cardiothorac Vasc Anesth. 2005;19:4–10. doi: 10.1053/j.jvca.2004.11.002. doi: 10.1053/j.jvca.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Wheeler AP, Gailani D. The intrinsic pathway of coagulation as a target for antithrombotic therapy. Hematol Oncol Clin North Am. 2016;30:1099–1114. doi: 10.1016/j.hoc.2016.05.007. doi: 10.1016/j.hoc.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Büller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, et al. FXI-ASO TKA Investigators. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372:232–240. doi: 10.1056/NEJMoa1405760. doi: 10.1056/NEJMoa1405760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12:581–594. doi: 10.1038/nrd4051. doi: 10.1038/nrd4051. [DOI] [PubMed] [Google Scholar]
- 6.Cardon LR, Harris T. Precision medicine, genomics and drug discovery. Hum Mol Genet. 2016;25(R2):R166–R172. doi: 10.1093/hmg/ddw246. doi: 10.1093/hmg/ddw246. [DOI] [PubMed] [Google Scholar]
- 7.Preis M, Hirsch J, Kotler A, Zoabi A, Stein N, Rennert G, et al. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017;129:1210–1215. doi: 10.1182/blood-2016-09-742262. doi: 10.1182/blood-2016-09-742262. [DOI] [PubMed] [Google Scholar]
- 8.Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol. 2016;9:629–637. doi: 10.1080/17474086.2016.1191944. doi: 10.1080/17474086.2016.1191944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gill D, Georgakis MK, Laffan M, Sabater-Lleal M, Malik R, Tzoulaki I, et al. Genetically determined FXI (factor XI) levels and risk of stroke. Stroke. 2018;49:2761–2763. doi: 10.1161/STROKEAHA.118.022792. doi: 10.1161/STROKEAHA.118.022792. [DOI] [PubMed] [Google Scholar]
- 10.Sennblad B, Basu S, Mazur J, Suchon P, Martinez-Perez A, van Hylckama Vlieg A, et al. Genome-wide association study with additional genetic and post-transcriptional analyses reveals novel regulators of plasma factor XI levels. Hum Mol Genet. 2017;26:637–649. doi: 10.1093/hmg/ddw401. doi: 10.1093/hmg/ddw401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang W, Schwienbacher C, Lopez LM, Ben-Shlomo Y, Oudot-Mellakh T, Johnson AD, et al. Genetic associations for activated partial thromboplastin time and prothrombin time, their gene expression profiles, and risk of coronary artery disease. Am J Hum Genet. 2012;91:152–162. doi: 10.1016/j.ajhg.2012.05.009. doi: 10.1016/j.ajhg.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. AFGen Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium; International Genomics of Blood Pressure (iGEN-BP) Consortium; INVENT Consortium; STARNET; BioBank Japan Cooperative Hospital Group; COMPASS Consortium; EPIC-CVD Consortium; EPIC-InterAct Consortium; International Stroke Genetics Consortium (ISGC); METASTROKE Consortium; Neurology Working Group of the CHARGE Consortium; NINDS Stroke Genetics Network (SiGN); UK Young Lacunar DNA Study; MEGASTROKE Consortium. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–537. doi: 10.1038/s41588-018-0058-3. doi: 10.1038/s41588-018-0058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–D934. doi: 10.1093/nar/gkr917. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.R Core Team. R: A language and environment for statistical computing. Available at: https://www.r-project.org/. Accessed March, 2019.
- 17.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann. Statist. 2001;29:1165–1188. [Google Scholar]
- 18.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 19.Hasselblad V, Kong DF. Statistical methods for comparison to placebo in active-control trials. Drug Inf J. 2001;35:435–449. [Google Scholar]
- 20.Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105:269–273. doi: 10.1160/TH10-05-0307. doi: 10.1160/TH10-05-0307. [DOI] [PubMed] [Google Scholar]
- 21.Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111:4113–4117. doi: 10.1182/blood-2007-10-120139. doi: 10.1182/blood-2007-10-120139. [DOI] [PubMed] [Google Scholar]
- 22.Diener HC, Easton JD, Hankey GJ, Hart RG. Novel oral anticoagulants in secondary prevention of stroke. Best Pract Res Clin Haematol. 2013;26:131–139. doi: 10.1016/j.beha.2013.07.007. doi: 10.1016/j.beha.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Schnier C, Bush K, Nolan J, Sudlow C. Definitions of stroke for uk biobank phase 1 outcomes adjudication. 2017. Available at: http://biobank.ctsu.ox.ac.uk/crystal/crystal/docs/alg_outcome_stroke.pdf. Accessed March, 2019.
- 24.Schnier C, Bush K, Nolan J, Sudlow C. Definitions of acute myocardial infarction and main myocardial infarction pathological types uk biobank phase 1 outcomes adjudication. 2017. Available at: http://biobank.ctsu.ox.ac.uk/crystal/crystal/docs/alg_outcome_mi.pdf. Accessed March, 2019.
- 25.Burgess S, Thompson SG. Interpreting findings from mendelian randomization using the MR-egger method. Eur J Epidemiol. 2017;32:377–389. doi: 10.1007/s10654-017-0255-x. doi: 10.1007/s10654-017-0255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]