

Abstract
In traditional oligodeoxynucleotide (ODN) synthesis, phosphate groups are protected with 2-cyanoethyl group, and amino groups are protected with acyl groups. At the end of ODN synthesis, deprotection is achieved with strong bases and nucleophiles. Therefore, traditional technologies are not suitable for the synthesis of ODNs containing sensitive functionalities. To address the problem, we report the use of Dim and Dmoc groups, which are based on the 1,3‐dithian‐2‐yl-methyl function, for phosphate and amine protection for solid phase ODN synthesis. Using the new Dim-Dmoc protection, deprotection was achieved under mild oxidative conditions without using any strong bases and nucleophiles. As a result, the new technology is suitable for the synthesis of ODNs containing sensitive functions. To demonstrate feasibility, seven 20-mer ODNs including four that contain the sensitive ester and alkyl chloride groups were synthesized, purified with RP HPLC and characterized with MALDI-TOF MS and enzyme digestion essays. High purity ODNs were obtained.
Keywords: Dim, Dmoc, oligonucleotides, protecting group, solid phase synthesis
Graphical Abstract
Introduction
Synthetic oligodeoxynucleotides (ODNs) and their analogs have found wide applications in many areas. Examples include antisense drug development,1 DNA-protein interactions,2 nanotechnology,3–4 bioconjugation,5 CRISPR genome editing,6 DNA damage and repair,7 DNA methylation and demethylation,8 DNA data storage,9 and synthetic biology.10 It is projected that ODN analogs that contain sensitive functional groups have the potential to greatly expand the scope of the applications, and bring about new research directions. Examples of sensitive ODN analogs include those containing functional groups such as alkyl halides, benzyl halide, allyl halides, α-halo amides, esters, activated esters, carbonates, thioesters, tosylates, sulfonic esters, sultones, phosphates, α,β-unsaturated carbonyl compounds, epoxides, aziridines, maleimides, vinyl arenes, methides, vinyl ethers, acetals, and hemiacetals. These groups are generally stable under typical chemical and biological conditions, and can co-exist with functional groups of natural ODNs. However, they cannot survive the harsh acidic and basic conditions used in traditional ODN synthesis and deprotection. Therefore, traditional ODN synthesis technologies cannot be used to synthesize such sensitive ODNs. Some efforts have been made to address the problem, but limited success has been achieved.11–20 These have been discussed in detail in our previous publications,21–22 Owing to the high potential of modified ODNs to bring transformative impact to many research areas, it is therefore significant to develop synthetic technologies that can be used to install any sensitive functional groups that are compatible with natural ODNs into any positions of ODNs.
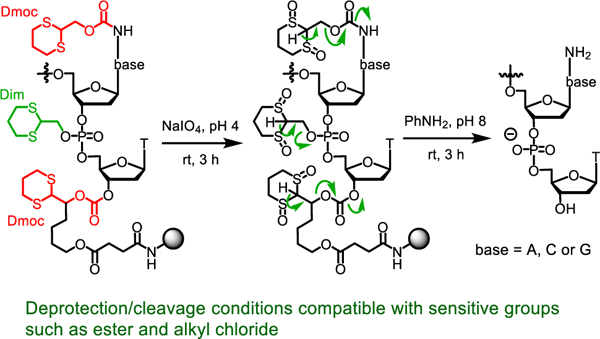
To achieve this goal, we recently introduced the 1,3‐dithian‐2‐yl-methoxycarbonyl function (Dmoc) as amino protection groups and cleavable linker for solid phase ODN synthesis.21–23 Mainly due to the concern of low efficiency of inorganic oxidizing agents to penetrate into the relatively hydrophobic fully protected ODNs to oxidize the dithioketals for deprotection and cleavage, we used the 2-cyanoethyl group for phosphate protection. At the end of synthesis, deprotection and cleavage were achieved in three steps. First, the 2-cyanoethyl groups were removed with the non-nucleophilic organic base DBU, which is compatible with some electrophilic groups, in acetonitrile (Scheme 1). This converted the hydrophobic fully protected ODN 1 into the hydrophilic 2. The hydrophilic anionic phosphate groups were believed to be beneficial for the inorganic oxidizing agent in water to penetrate into ODN in the next step. Second, the dithioketals in 2 were oxidized with sodium periodate to give 3. This drastically increased the acidity of H-2 in the 1,3-dithane function. Third, after washing away the inorganic materials, β-eliminations were induced with the weak base aniline, and the ODNs were cleaved from the solid support and fully deprotected to give 4. In this paper, we report our results on the study of the feasibility of using the Dim instead of 2-cyanoethyl function to protect the phosphate group. ODNs synthesized with this protection strategy should appear as 5 (Scheme 1). Deprotection and cleavage can then be achieved in two steps by oxidation of the dithioketals to give 6 followed by β-elimination. Besides reducing the number of steps for deprotection from three to two, another advantage is that the use of the strong base DBU is avoided, which is expected to expand the scope of sensitive groups that can be incorporated into ODNs. Indeed, our results showed that the new protecting strategy was feasible, and the concern of inefficient oxidation of dithioketals in the relatively hydrophobic 5 was unnecessary. Using the new Dim-Dmoc technology, ODNs including those that contain sensitive functions can be synthesized in good yields and high purity under finely tuned but reliable conditions.
Scheme 1.

Deprotection of ODNs assembled with 2-cyanoethyl-Dmoc and Dim-Dmoc phosphoramidite monomers.
Results and Discussion
To use the Dim-Dmoc technology to synthesize ODNs, the phosphoramidite monomers 7a-d and the solid support with Dmoc linker 8 were needed (Figure 1). Preparation of 8 was reported previously.21 The synthesis of 7a-d is shown in Scheme 2. Compound 9 in toluene was reacted with commercially available bis(diisopropylamino)chlorophosphine (10) in the presence of the amine base diisopropylamine at room temperature overnight under an inert atmosphere.24–25 This gave the intermediate 11 along with insoluble diisopropylamine hydrochloride. The intermediate was not isolated. A nucleoside with 5’-OH protected with a 4,4’-dimethoxytrityl (DMTr) and amino group protected with the Dmoc group (12a-d or e), which was prepared following procedures reported earlier,21–22 and the activator diisopropylammonium tetrazolide (13) were dissolved in DCM. The intermediate 11 in the supernatant was transferred via a cannula with its inflow end wrapped with a copper wire-secured filter paper into the solution of 12 and activator. The insoluble diisopropylamine hydrochloride was not transferred due to the filter paper. The reaction was allowed to proceed at room temperature overnight. The crude product was purified with flash column chromatography without aqueous workup. Good to excellent yields of the Dim-Dmoc phosphoramidites 7a-d were obtained (Scheme 2).
Figure 1.

Dim-Dmoc phosphoramidite monomers and CPG with Dmoc linker.
Scheme 2.

Synthesis of Dim-Dmoc phosphoramidite monomers.
With the Dim-Dmoc phosphoramidites in hand, we tested the feasibility of using them as building blocks for ODN synthesis under weakly nucleophilic and weakly basic deprotection and cleavage conditions by the synthesis of the unmodified ODNs 14a-c and modified sensitive ODNs 14d-g (Figure 2). These ODNs were not designed for any specific applications. Instead, they were used as model ODNs for the identification of specific conditions for the new ODN synthesis technology and for demonstrating its feasibility for the synthesis of sensitive ODNs. During our studies, 14a was first synthesized. CPG with a Dmoc linker (8) was used as the solid support.21 The phosphoramidites 7a-d were used as nucleoside monomers. The syntheses were conducted on a MerMade 6 DNA synthesizer using typical scripts with some modifications. Briefly, detritylation was achieved with 3% DCA in DCM. For coupling, 0.1 M solutions of 7a-d in acetonitrile were used with 5-(ethylthio)-1H-tetrazole as activator. Capping was accomplished using 2-cyanoethyl N,N,N’,N’-tetraisopropylphosphorodiamidite with the same activator for coupling instead of the commonly used acetic anhydride. Typical conditions involving iodine was used for oxidation. In the last coupling step, the 5’-trityl protected instead of 5’-DMTr protected phosphoramidite 7e was used to incorporate the nucleotide at the 5’-end of the ODN. The reasons for using the unusual capping and 5’-tagging methods are explained later in the paper. The synthesis of 7e is shown in Scheme 2, and was similar as the synthesis of 7a-d. Deprotection and cleavage of ODN was achieved in two steps (Scheme 3). First, the dithioketal bonds in the Dim and Dmoc functions in the fully protected ODN 15 were oxidized with a solution of sodium periodate in water at room temperature to give 16. Excess oxidizing agents and other materials were simply removed by washing the CPG with water. Second, the CPG was suspended in a solution of aniline in water. This induced β-eliminations of the oxidized Dim and Dmoc functions in 16, and cleaved the ODN from CPG to give ODN 17. We also tested to use the completely non-nucleophilic base potassium carbonate for this step, but complex mixture was obtained as revealed by HPLC analysis indicating random additions of the 2-methylene-1,3-dithiane 1,3-dioxide deprotection side product to different locations of the ODN. Therefore in this step, aniline not only served as a base but also served as a scavenger for the Michael acceptor side product to prevent it from reacting with the ODN. After treating with aniline, 17 was fully deprotected except for a trityl tag at its 5’-end, which was desirable for the purpose of assisting RP HPLC purification of the ODN. To remove small organic molecules, the ODN was precipitated from water with butanol. The residue was injected into RP HPLC to generate the profile of crude ODN, in which the tagged full-length ODN was well separated from other materials (Figure 3). The trityl-tagged ODN was collected, and analyzed with HPLC giving a single peak (profile in supporting information). Removing the tag was achieved with 80% acetic acid, which is the typical condition for detritylation of DMTr-tagged ODNs. The ODN with trityl tag removed was purified with RP HPLC, and the purified fully deprotected ODN was analyzed again with RP HPLC. As shown in Figure 3, a single sharp peak was observed. The pure ODN 14a was analyzed with MALDI-TOF MS. Correct molecular peak was found (Figure 4). No adducts of ODNs and Michael acceptor side product from ODN deprotection were observed. In addition, the ODN was digested to nucleosides using the Nucleoside Digestion Mix from New England BioLabs. The resulting mixture was analyzed with RP HPLC. Four peaks were observed, which matched perfectly with the peaks from control experiments using ODNs synthesized with typical commercial phosphoramidites (dABz, dCAc, dGiBu and dT), and deprotected and cleaved using concentrated ammonium hydroxide. The amount of pure ODN obtained was estimated by UV, and a OD260 of 2.15 was given for a 0.52 µmol synthesis (Supporting Information). Based on the value of OD260, the percentage yield was calculated to be 2%, which was close to the yield of a control 20-mer ODN synthesized using standard commercial phosphoramidites and CPG 8, and cleaved and deprotected under standard conditions using concentrated ammonium hydroxide (Table 1).23 Besides 14a, two additional unmodified ODNs 14b-c were synthesized and analyzed under similar conditions to further confirm the viability of the new technology. ODN analysis data and yields are summarized in Table 1. Detailed information are given in the Supporting Information.
Figure 2.
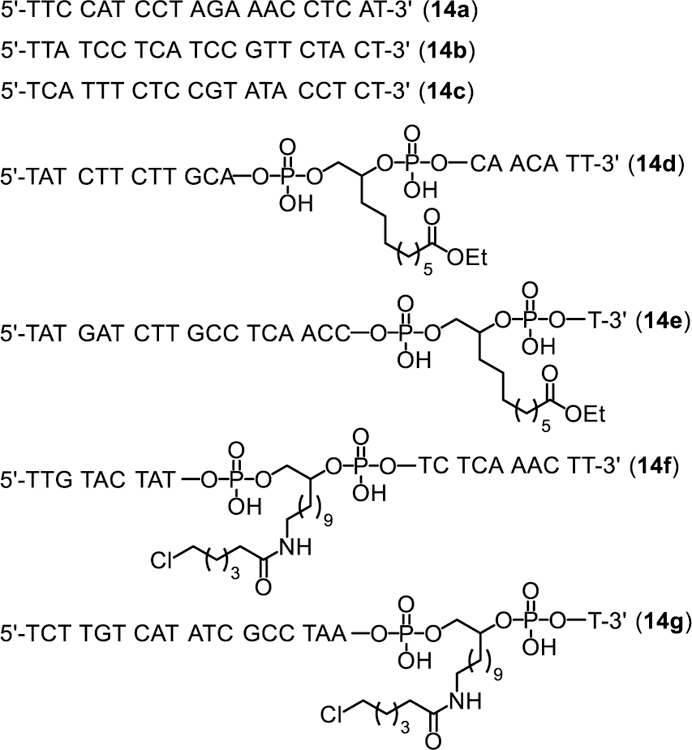
ODN sequences.
Scheme 3.

ODN deprotection and cleavage.
Figure 3.

HPLC profiles. In the profiles of crude ODNs, the major peak at around 40 minutes is the fully deprotected ODN with a 5’-trityl tag. In the profiles of pure ODNs, the single sharp peak is the fully deprotected ODN without a 5’-trityl tag.
Figure 4.

MALDI-TOF MS of ODNs 14a, 14d and 14f.
Table 1.
Characterization data and yields of ODNs
| entry | ODN | MALDI-TOF MS |
RP HPLC retention timea | ODN purityb | ODN OD260c | ODN yield | |
|---|---|---|---|---|---|---|---|
| calculated | found | ||||||
| 1 | 14a | 5992.0 | 5992.0 | 19.2 min | 99% | 2.15 | 2.0% |
| 2 | 14b | 5965.0 | 5965.1 | 19.5 min | 98% | 2.34 | 2.5% |
| 3 | 14c | 5965.0 | 5965.0 | 19.5 min | 98% | 2.34 | 2.5% |
| 4 | 14d | 6017.1 | 6017.1 | 22.3 min | 100% | 4.79 | 4.8% |
| 5 | 14e | 6018.1 | 6018.2 | 27.8 min | 94% | 1.14 | 1.2% |
| 6 | 14f | 6135.2 | 6135.6 | 26.1 min | 93% | 1.84 | 1.9% |
| 7 | 14g | 6136.2 | 6136.1 | 25.0 min | 100% | 1.35 | 1.4% |
| 8 | control | 5965.0 | - | 19.5 min | 100% | 8.30 | 8.8%d |
HPLC conditions are given in the Experimental Section.
Purity is estimated from HPLC profile.
The values are for 0.52 µmol synthesis.
The sequence is 5’-TTA TCC ACT TCC GTT CTA CT-3’.
The control ODN was synthesized, deprotected, cleaved, and purified under standard conditions using commercial phosphoramidite monomers and CPG 8. More details are given in our previous report.23
Next, we tested the feasibility of the Dim-Dmoc technology for the synthesis of sensitive ODNs by incorporating the ester and alkyl chloride functions into ODNs. The synthesis of the required Dim phosphoramidite monomers (18a-b) is shown in Scheme 4. The known compound 19 was converted to 24 in five simple steps. Compounds 19 and 24 were then converted to their corresponding Dim phosphoramidites 18a-b, respectively using the similar conditions for the synthesis of 7a-e. The phosphoramidites 18a-b contain the sensitive ethyl ester and alkyl chloride groups, which are sensitive to traditional ODN cleavage and deprotection conditions involving heating in a concentrated ammonium hydroxide solution. The alkyl chloride is also sensitive to bases via β-elimination. The ODN sequences 14d-g (Figure 2) were selected for the studies, of which the long chain chloroalkane-containing 14f-g could provide a means to prepare protein-DNA conjugates via the bioorthogonal reaction between haloalkane dehalogenase and chloroalkanes.26 The ODNs were synthesized under the same conditions described for the synthesis of unmodified ODNs. Deprotection and cleavage conditions were also the same. RP HPLC profiles of crude and pure ODNs 14d and 14f are in Figure 3. Their MALDI-TOF MS spectra are in Figure 4. All other analytical data are in the Supporting Information. ODN analysis data and yields are summarized in Table 1. The data proved that the Dim-Dmoc technology can be used to synthesize the sensitive ODNs that contain the ester and alkyl chloride functionalities in high yields and purity.
Scheme 4.

Synthesis of Dim phosphoramidites 18a-b that contain sensitive groups.
The use of the Dim group to protect the phosphate groups in ODN synthesis has two advantages over the method in our previous reports,21–23 in which the 2-cyanoethyl group was used. First, the number of steps in deprotection and cleavage is reduced from three to two, which significantly simplifies the procedure. Second, the use of the strong organic base DBU to remove the 2-cyanoethyl groups is avoided, which can expand the scope of sensitive groups to be incorporated into ODNs. Earlier, our decision to use the 2-cyanoethyl group instead of Dim for phosphate protection was based on several considerations including the complex nature of chemical ODN synthesis, difficulty to make highly pure Dim phosphoramidites (7a-d) required for repeated use in a multistep linear synthesis with satisfactory overall yield, and as mentioned earlier the concern of inefficient oxidation of dithioketals during ODN deprotection and cleavage. ODN synthesis is a highly complex process. After careful engineering by many chemists in several decades, the standard procedure is robust. However, slight modification of the procedure can cause significant problems, and those problems are usually very difficult to diagnose and address. For the synthesis of Dim phosphoramidites 7a-d, it was a concern too. Unlike their 2-cyanoethyl counterparts, which can be synthesized using the commercially available 2-cyanoethyl-N,N,N’,N’-tetraisopropylphosphordiamidite as the phosphitylation agent, these compounds have to be synthesized using a new phosphitylation agent such as 11, which is sensitive to moisture and oxygen, and therefore was predicted to be difficult to prepare and purify. For concerns on the inefficiency of oxidation of the dithioketals during ODN deprotection and cleavage, the fully protected ODNs on the CPG are relatively hydrophobic; which excludes the use of oxidizing agents that can only function in water. However, to oxidize multiple dithioketals with complete conversion, the reaction must be highly efficient, and thus the broadness of the scope of oxidizing agents that can be tested is important for the technology to be successful. Therefore, during our initial studies, we chose to use the much simpler and well established 2-cyanoethyl protection chemistry.
Indeed, we met many problems during the studies. For example, at the beginning of the project, our RP HPLC profiles were messy. After testing many hypotheses, we finally found that one of the problems was cap exchange, in which the Dmoc groups used for amino protection were replaced by acyl groups during capping involving using reagents such as acetic anhydride under traditional capping conditions. This was counter intuitive because the donation of the lone pair of electrons from the oxygen atom to the carbonyl carbon in the Dmoc function would make the Dmoc protection more stable than acyl protections. Once the problem was diagnosed, it was solved elegantly by using the phosphorylation chemistry instead of the acylation chemistry for capping. The synthesis of 7a-d and making them highly pure for ODN synthesis were indeed difficult too. We screened many conditions, and were finally able to identify a procedure involving using toluene as the solvent and diisopropylamine as base to prepare the phosphitylation agent 11. With toluene and diisopropylamine, 11 was soluble while the side product diisopropylammonium salt was not. This allowed us to obtain 11 with sufficient purity for the phosphitylation reaction without aqueous workup and chromatography purification.24–25 Of surprise was that the oxidation of dithioketals in the fully protected relatively hydrophobic 5 during ODN deprotection and cleavage was achieved with ease using the aqueous solution of sodium periodate. The efficiency was similar to the oxidation of 2, which was highly hydrophilic due to the anionic phosphate groups. Probably, the fully protected ODNs had limited but sufficient solubility in water for the oxidation reaction to occur at the outermost sphere of the ODN coated CPG. Once the dithioketals in the outermost layer were oxidized, the solubility increased, and the reaction gradually penetrated into the inner layer and all dithioketals were oxidized efficiently. However, one observation still puzzles us. We synthesized a simple model oligosulfoxide compound, which contained six sulfoxide groups. We thought that this compound would be highly soluble in water. To our surprise, it had very limited solubility in any solvents including water.27
During the development of the technology, we found that using the trityl group instead of the DMTr group as the 5’-tag to assist RP HPLC purification was needed. When DMTr group was used, the tag was unstable in the sodium periodate oxidation step. The trityl group was found stable under the oxidation conditions. Importantly, we found that the trityl group could be removed efficiently with 80% acetic acid under similar conditions used for removing the DMTr group after HPLC purification, which was inconsistent to the report that deprotection of trityl group required two days at room temperature.23,28 One concern on developing the Dim-Dmoc technology was the difficulty to identify selective oxidative conditions for the oxidation of phosphite triesters to phosphate triesters during ODN synthesis and for oxidation of dithioketals during deprotection and cleavage. For the former, we were gratified to find that the standard iodine oxidation conditions were highly selective, and premature oxidation of the dithioketals had never been observed. For the latter, the sodium periodate solution elegantly accomplished an otherwise highly challenging task, which required highly efficient and selective oxidation of the multiple dithioketal groups while not damaging the ODNs via oxidation of the nucleobases and other portions of the molecules.23
Conclusions
In conclusion, a new method for solid phase ODN synthesis has been developed. The method uses Dim for phosphate protection, Dmoc for amino protection, and a Dmoc linker for anchoring the ODN to solid support. With the new protection and linking strategy, ODN deprotection and cleavage can be achieved under oxidative conditions without using any strong bases and nucleophiles. Therefore the new method is suitable for the synthesis of ODN analogs containing base labile and electrophilic groups, a task that cannot be accomplished or is highly challenging to accomplish using traditional technologies. We expect that the new method will be able to provide a wide range of sensitive ODN analogs to researchers in research areas such as antisense drug development, DNA-protein interaction studies, nanotechnology and bioconjugation.
Experimental Section
All reactions were performed in oven-dried glassware under argon using standard Schlenk techniques. Reagents and solvents available from commercial sources were used as received unless otherwise noted. THF, toluene, and CH2Cl2 were dried using an Innovative Technology Pure-Solv™ system. Pyridine and diisopropylamine were distilled over CaH2 under nitrogen. Lcaa-CPG (pore size 497 Å) was purchased from Prime Synthesis. Compounds 12a-e were prepared according to reported procedure.21–22,29 Thin layer chromatography (TLC) was performed using Sigma-Aldrich TLC plates, silica gel 60F-254 over glass support, 250 μm thickness. Flash column chromatography was performed using SiliCycle silica gel, particle size 40–63 μm. 1H, 13C and 31P NMR spectra were measured on a Varian UNITY INOVA spectrometer at 400, 100 and 162 MHz, respectively; chemical shifts (δ) were reported in reference to solvent peaks (residue CHCl3 at δ 7.24 ppm for 1H and CDCl3 at δ 77.00 ppm for 13C) and to H3PO4 (δ 0.00 ppm for 31P). HRMS was obtained on a Thermo HR-Orbitrap Elite Mass Spectrometer. LRMS was obtained on a Thermo Finnigan LCQ Advantage Ion Trap Mass Spectrometer. MALDI-TOF MS were obtained on a Bruker’s microflex™ LRF MALDI-TOF System. ODNs were synthesized on a MerMade 6 solid phase synthesizer. RP HPLC was performed on a JASCO LC-2000Plus System: pump, PU-2089Plus Quaternary Gradient; detector UV-2075Plus. A C-18 reversed phase analytical column (5 μm diameter, 100 Å, 250 × 3.20 mm) was used. Solvent A: 0.1 M triethylammonium acetate, 5% acetonitrile. Solvent B: 90% acetonitrile. All profiles were generated by detecting absorbance at 260 nm using the linear gradient solvent system: solvent B (0%–45%) in solvent A over 60 min followed by solvent B (45%–100%) in solvent A over 20 min at a flow rate of 1.0 mL/min.
5’-O-(4,4’-Dimethoxytriyl)thymidine-3’-O-[O-[(1,3-dithian-2-yl)methyl]-N,N-diisopropylphosphoramidite (7a).
To a solution of 9 (1.57 g, 10.48 mmol, 1.5 eq.) and freshly distilled diisopropyl amine (9.85 mL, 69.9 mmol, 10 eq.) in dry toluene (25 mL) was added bis(diisopropylamino)chlorophosphine (10, 2.80 g, 10.48 mmol, 1.5 eq.) at rt under argon. After stirring overnight, a cloudy solution containing the soluble intermediate 11 and insoluble diisopropylamine hydrochloride side product was formed. The intermediate in the supernatant was transferred into a solution of 12a (3.80 g, 6.99 mmol, 1 eq.) and diisopropylammonium tetrazolide (13, 1.80 g, 10.48 mmol, 1.5 eq.) in dry DCM (50 mL) via a cannula with its inflow end wrapped with a copper wire-secured filter paper. The insoluble diisopropylamine hydrochloride was not transferred due to the filter paper. The reaction mixture was stirred overnight, and then concentrated to dryness. The residue was dissolved in a mixture of solvents (1:1 hexanes/EtOAc with 5% Et3N) and loaded directly on a column for flash column chromatography (SiO2, 1:1 hexanes/EtOAc with 5% Et3N). Compound 7a was obtained as a white foam (5.04 g, 88%): Mixture of two diastereoisomers; Rf = 0.2 and 0.3 (SiO2, 1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.04–1.16 (m, 12H), 1.38 (s, 3H), 1.77–1.87 (m, 1H), 1.96–2.07 (m, 1H), 2.28–2.42 (m, 1H), 2.45–2.58 (m, 1H), 2.60–2.69 (m, 2H), 2.65–2.84 (m, 4H), 3.29–3.46 (m, 2H), 3.47–3.69 (m, 2H), 3.76 (s, 6H), 3.80–3.89 (m, 1H), 4.04–4.23 (m, 1H), 4.74–4.77 (m, 1H), 6.38 (t, J = 5.8 Hz, 1H), 6.81 (dd, J = 8.8, 3.2 Hz, 4H), 7.20–7.29 (m, 7H), 7.40 (d, J = 7.6 Hz, 2H), 7.60 (s, 0.5H), 7.63 (s, 0.5 H), 8.84 (brs, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 12.1, 24.81, 24.88, 24.95, 25.0, 26.1, 26.2, 28.8 (d, Jcp = 9.2 Hz), 29.0 (d, Jcp = 17.2 Hz), 40.5 (d, Jcp = 5.4 Hz), 40.6 (d, Jcp = 1.8 Hz), 43.4 (d, Jcp = 3.4 Hz), 43.5 (d, Jcp = 3.4 Hz), 47.1 (d, Jcp = 7.0 Hz), 47.8 (d, Jcp = 6.8 Hz), 55.5, 63.3, 63.7, 64.8 (d, Jcp = 18.2 Hz), 65.0 (d, Jcp = 18.9 Hz), 73.6 (d, Jcp = 15.6 Hz), 74.1 (d, Jcp = 15.2 Hz), 84.8, 85.0, 85.4 (d, Jcp = 6.7 Hz), 86.0 (d, Jcp = 2.8 Hz), 87.0, 87.1, 111.2, 113.4, 127.2, 128.1, 128.4, 130.4, 135.5, 135.6, 135.7, 136.0, 136.1, 144.5, 144.6, 150.4, 158.8, 164.0; 31P{1H} NMR (162 MHz, CDCl3) δ 149.4, 149.6 ppm; HRMS (ESI) m/z calcd for C42H55N3O8PS2 [M+H]+ 824.3168, found 824.3170.
N-[(1,3-Dithian-2-yl)methoxy)carbonyl]-5’-O-(4,4’-dimethoxytrityl)-2’-deoxycytidine-3’-O-[O-[(1,3-dithian-2-yl)methyl]-N,N-diisopropylphosphoramidite (7b).
The same procedure for 7a was used. Flash column chromatography (SiO2, 1:1 hexanes/EtOAc with 5% Et3N) gave 7b as a white foam (1.25 g, 52%): Mixture of two diastereoisomers; Rf = 0.2 and 0.3 (SiO2, 1:2 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.04–1.26 (m, 12H), 1.72–1.84 (m, 2H), 1.90–2.09 (m, 4H), 2.30–2.47 (m, 2H), 2.59–2.74 (m, 6H), 2.85–2.93 (m, 2H), 3.39–3.60 (m, 4H), 3.61–3.89 (m, 1H), 3.77 (s, 6H), 3.91–4.16 (m, 2H), 4.17–4.22 (m, 1H), 4.40–4.49 (m, 1H), 6.18–6.22 (m, 1H), 6.81 (d, J = 7.4 Hz, 4H), 7.18–7.29 (m, 7H), 7.7.39 (d, J = 7.6 Hz, 2H), 8.27–8.29 (m, 0.5H), 8.34–8.35 (m, 0.5H); 13C{1H} NMR (100 MHz, CDCl3) δ 23.3 (d, Jcp = 2.2 Hz), 23.4 (d, Jcp = 1.6 Hz), 24.85, 24.89, 24.92, 24.98, 25.1, 25.7, 25.9, 26.1, 26.2, 27.5, 27.6, 28.7 (d, Jcp = 13.0 Hz), 29.1 (d, Jcp = 21.5 Hz), 41.2 (d, Jcp = 5.7 Hz), 41.5, 43.1, 43.4, 43.5, 45.4, 45.5, 47.1 (d, Jcp = 6.9 Hz), 47.7 (d, Jcp = 8.3 Hz), 55.5, 61.9, 62.4, 64.7 (d, Jcp = 19.9 Hz), 64.8 (d, Jcp = 18.5 Hz), 65.8, 65.9, 71.4 (d, Jcp = 9.3 Hz), 71.9 (d, Jcp = 10.1 Hz), 85.2 (d, Jcp = 7.3 Hz), 86.1, 87.0, 94.5, 113.4, 127.2, 128.1, 128.4, 130.2, 130.3, 135.5, 135.6, 135.7, 135.8, 144.3, 144.4, 144.9, 145.0, 151.9, 155.0, 158.7, 161.9, 162.0; 31P{1H} NMR (162 MHz, CDCl3) δ 149.2, 149.5; HRMS (ESI) m/z calcd for C47H62N4O9PS4 [M+H]+ 985.3137, found 985.3130.
N-[(1,3-Dithian-2-yl)methoxy)carbonyl]-5’-O-(4,4’-dimethoxytrityl)-2’-deoxyadenosine-3’-O-[O-[(1,3-dithian-2-yl)methyl]-N,N-diisopropylphosphoramidite (7c).
The same procedure for 7a was used. Flash column chromatography (SiO2, 1:1 hexanes/EtOAc with 5% Et3N) gave 7c as a white foam (1.30 g, 68%): Mixture of two diastereoisomers; Rf = 0.3 and 0.4 (SiO2, 1:2 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.09–1.25 (m, 12H), 1.75–1.84 (m, 2H), 1.95–2.04 (m, 4H), 2.59–2.75 (m, 6H), 2.87–2.98 (m, 4H), 3.31–4.00 (m, 4H), 3.75 (s, 6H), 4.00 (t, J = 6.5 Hz, 0.5H), 4.05–4.18 (m, 1.5H), 4.21–4.27 (m, 0.5H), 4.30–4.39 (m, 0.5H), 4.55 (d, J = 7.1 Hz, 2H), 4.80–4.88 (m, 1H), 6.46 (t, J = 6.5 Hz, 1H), 6.74–6.77 (m, 4H), 7.14–7.30 (m, 7H), 7.36 (d, J = 11.9 Hz, 2H), 8.16 (s, 0.5H), 8.19 (s, 0.5H), 8.68 (s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 23.27 (d, Jcp = 2.6 Hz), 23.33 (d, Jcp = 2.0 Hz), 24.86, 24.92, 24.99, 25.8, 26.1, 27.6, 28.8 (d, Jcp = 11.5 Hz), 29.1 (d, Jcp = 14.5 Hz), 40.1 (d, Jcp = 14.8 Hz), 43.3, 43.4, 43.5, 45.45, 45.51, 47.2 (d, Jcp = 7.5 Hz), 47.7 (d, Jcp = 7.6 Hz), 55.5, 63.4, 63.7, 64.7 (d, Jcp = 13.8 Hz), 65.4 (d, Jcp = 18.5 Hz), 65.6, 73.9 (d, Jcp = 13.7 Hz), 74.0 (d, Jcp = 15.3 Hz), 84.8, 85.1, 85.9, 86.4, 86.6, 86.7, 113.3, 122.6, 127.0, 128.0, 128.3, 130.2, 135.78, 135.85, 141.6, 141.7, 144.67, 144.72, 149.2, 150.5, 151.06, 151.12, 152.8, 158.6; 31P{1H} NMR (162 MHz, CDCl3) δ 149.4, 149.6; HRMS (ESI) m/z calcd for C48H62N6O8PS4 [M+H]+ 1009.3249, found 1009.3255.
N-[(1,3-Dithian-2-yl)methoxy)carbonyl]-5’-O-(4,4’-dimethoxytrityl)-2’-deoxyguanosine-3’-O-[O-[(1,3-dithian-2-yl)methyl]-N,N-diisopropylphosphoramidite (7d).
The same procedure for 7a was used. Flash column chromatography (SiO2, 8:1:1 EtOAc/ACN/Et3N) gave 7d as a white foam (1.30 g, 68%): Mixture of two diastereoisomers; Rf = 0.2 and 0.3 (SiO2, 8:1:1 EtOAc/ACN/Et3N). 1H NMR (400 MHz, CDCl3) δ 1.07–1.16 (m, 12H), 1.77–1.86 (m, 2H), 1.97–2.08 (m, 4H), 2.59–2.94 (m, 10H), 3.25–3.31 (m, 2H), 3.52–3.58 (m, 2H), 3.75 (s, 6H), 3.58–4.21 (m, 2.5H), 4.29–4.32 (m, 0.5H), 4.50 (d, J = 3.5 Hz, 1H), 4.52 (d, J = 3.4 Hz, 1H), 4.72–4.81 (m, 1H), 6.18–6.23 (m, 1H), 6.72–6.78 (m, 4H), 7.16–7.30 (m, 7H), 7.37 (d, J = 7.0 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.8 (s, 0.5H), 7.82 (s, 0.5H); 13C{1H} NMR (100 MHz, CDCl3) δ 24.86, 24.91, 24.94, 24.98, 25.6, 26.1, 27.1, 28.88 (d, Jcp = 11.1 Hz), 29.16 (d, Jcp = 8.5 Hz), 39.9, 42.5, 43.4, 43.5, 47.2 (d, Jcp = 6.9 Hz), 47.6 (d, Jcp =7.4 Hz), 55.5, 63.6, 63.9, 64.8 (d, Jcp = 6.6 Hz), 65.0 (d, Jcp = 6.5 Hz), 66.0, 73.9 (d, Jcp = 11.1 Hz), 74.1 (d, Jcp = 16.5 Hz), 84.3, 84.4, 85.7 (d, Jcp = 6.6 Hz), 86.2 (d, Jcp = 2.9 Hz), 86.6, 113.3, 121.6, 127.0, 128.0, 128.3, 128.4, 130.18, 130.24, 135.8, 135.9, 137.4, 137.5, 144.6, 144.7, 146.3, 148.30, 148.32, 153.11, 153.13, 155.7, 158.6; 31P{1H} NMR (162 MHz, CDCl3) δ 148.9, 149.6; HRMS (ESI) m/z calcd for C48H62N6O9PS4 [M+H]+ 1025.3198, found 1025.3205.
5’-O-Triyl-thymidine-3’-O-[O-[(1,3-dithian-2-yl)methyl]-N,N-diisopropylphosphoramidite (7e).
The same procedure for 7a was used. Flash column chromatography (SiO2, 1:1 hexanes/EtOAc with 5% Et3N) gave 7e as a white foam (233 mg, 87%): Mixture of two diastereoisomers; Rf = 0.2 and 0.3 (SiO2, 1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.04–1.27 (m, 12H), 1.40 (s, 3H), 1.78–1.86 (m, 1H), 1.96–2.05 (m, 1H), 2.29–2.98 (m, 8H), 3.30–3.99 (m, 5H), 4.05–4.25 (m, 1H), 4.74–4.81 (m, 1H), 6.38 (t, J = 7.1 Hz, 1H), 7.18–7.35 (m, 9H), 7.36–7.45 (m, 6H), 7.56 (s, 0.5H), 7.60 (s, 0.5H), 9.11 (brs, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 12.1, 24.84, 24.88, 24.91, 24.95, 25.01, 26.1, 26.2, 28.8 (d, Jcp = 8.4 Hz), 29.0 (d, Jcp = 17.3 Hz), 40.4 (d, Jcp = 5.1 Hz), 40.6, 43.4, 43.5, 47.0 (d, Jcp = 7.2 Hz), 47.5 (d, Jcp = 7.4 Hz), 63.5, 63.9, 64.8 (d, Jcp = 17.9 Hz), 65.0 (d, Jcp = 18.4 Hz), 73.6 (d, Jcp = 15.3 Hz), 73.9 (d, Jcp = 14.4 Hz), 84.8, 85.0, 85.3 (d, Jcp = 6.7 Hz), 85.9, 87.55, 87.61, 111.1, 111.2, 127.5, 128.1, 128.9, 135.9, 136.0, 143.5, 143.6, 150.5, 164.1; 31P{1H} NMR (162 MHz, CDCl3) δ 149.4, 149.7; HRMS (ESI) m/z calcd C40H51N3O6PS2 [M+H]+ 764.2956, found 764.2960.
11-[Bis(4-methoxyphenyl)(phenyl)methoxy]undecane-1,10-diol (20).
To a suspension of lithium aluminum hydride (1.15 g, 30.29 mmol, 5 eq.) in dry THF (25 mL) was added a solution of 19 (3.15 g, 6.06 mmol, 1 eq.) in dry THF (50 mL) dropwise via cannula at 0 °C under nitrogen. The reaction mixture was stirred for 3 h, and then quenched by dropwise addition of H2O (1.15 mL), 15% NaOH (1.15 mL), and H2O (3.45 mL), sequentially. The white precipitate was removed by filtration over Celite. The filtrate was concentrated to dryness. Flash column chromatography (SiO2, 1:1 hexanes/EtOAc with 5% Et3N) gave 20 as a colorless oil (2.45 g, 80%): Rf = 0.2 (SiO2, 1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.20–1.42 (m, 14H), 1.53 (p, J = 5.8 Hz, 2H), 1.65 (brs, 1H), 2.45 (brs, 1H), 3.02 (dd, J = 9.3, 7.6 Hz, 1H), 3.16 (dd, J = 9.6, 3.6 Hz, 1H), 3.59 (t, J = 6.6 Hz, 2H), 3.73–3.75 (m, 1H), 3.76 (s, 6H), 6.82 (d, J = 8.9 Hz, 4H), 7.20 (tt, J = 7.4, 1.2 Hz, 1H), 7.28 (t, J = 7.2 Hz, 2H), 7.32 (d, J = 9.9 Hz, 2H), 7.43 (d, J = 9.6 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 25.8, 26.1, 29.72, 29.78, 29.8, 29.9, 33.0, 33.7, 55.5, 63.2, 67.9, 71.2, 86.2, 113.3, 126.9, 127.9, 128.3, 130.2, 136.2, 145.0, 158.5; HRMS (ESI) m/z calcd for C32H43O5 [M+H]+ 507.3110, found 507.3122.
11-(Bis(4-methoxyphenyl)(phenyl)methoxy)-10-hydroxyundecyl 4-methylbenzenesulfonate (21).
To a solution of 20 (2.06 g, 4.07 mmol, 1 eq.) in freshly distilled pyridine (50 mL) was added TsCl (0.814 g, 1.05 eq.) at 0 °C under nitrogen. The mixture was stirred at the same temperature for 8 h. The majority of pyridine was evaporated on a rotary evaporator under vacuum generated by an oil pump. The remaining content was poured into a separatory funnel containing 5% NaHCO3 (100 mL) and extracted with EtOAc (50 mL × 3). The extracts were dried over anhydrous Na2SO4, filtered, and concentrated. Flash column chromatography (SiO2, 2:1 hexanes/EtOAc with 5% Et3N) gave 21 as a pale-yellow oil (1.37 g, 51%): Rf = 0.4 (SiO2, 1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.12–1.41 (m, 14H), 1.61 (p, J = 6.7 Hz, 2H), 2.42 (s, 3H), 3.01 (t, J = 9.2 Hz, 1H), 3.16 (dd, J = 9.4, 3.3 Hz, 1H), 3.70–3.74 (m, 1H), 3.76 (s, 6H), 4.00 (t, J = 6.5 Hz, 2H), 6.81 (d, J = 8.8 Hz, 4H), 7.20 (t, J = 7.1 Hz, 1H), 7.26 (t, J = 4.8 Hz, 2H), 7.31 (d, J = 8.8 Hz, 6H), 7.43 (d, J = 7.3 Hz, 2H), 7.77 (d, J = 8.3 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 21.9, 25.6, 25.7, 29.1, 29.2, 29.60, 29.67, 29.8, 33.6, 55.5, 67.8, 70.9, 71.2, 86.2, 113.3, 126.9, 127.9, 128.3, 129.9, 130.2, 133.4, 136.2, 144.7, 145.0, 158.5; HRMS (ESI) m/z calcd for C39H49O7S [M+H]+ 661.3199, found 661.3204.
12-(Bis(4-methoxyphenyl)(phenyl)methoxy)-11-hydroxydodecanenitrile (22).
To a solution of 21 (6.78 g, 10.28 mmol, 1 eq.) in dry DMSO (25 mL) was added KCN (0.802 g, 12.34 mmol, 1.2 eq.) at rt under nitrogen. The reaction mixture was stirred at 60 °C overnight. After cooling to rt, EtOAc (100 mL) was added, and the organic phase was washed with brine (100 ml), dried over anhydrous Na2SO4, filtered, and concentrated. Flash column chromatography (SiO2, 4:1 hexanes/EtOAc with 5% Et3N) gave 22 as a colorless oil (4.20 g, 79%): Rf = 0.2 (SiO2, 4:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.22–1.26 (m, 10H), 1.37–1.42 (m, 4H), 1.61 (p, d = 7.1 Hz, 2H), 2.27 (t, J = 7.1 Hz, 2H), 2.41 (brs, 1H), 3.02 (dd, J = 9.2, 7.5 Hz, 1H), 3.16 (dd, J = 9.3, 3.3 Hz, 1H), 3.76 (s, 6H), 6.81 (d, J = 8.9 Hz, 4H), 7.20 (t, J = 7.4 Hz, 1H), 7.28 (t, J = 7.8 Hz, 2H), 7.32 (d, J = 8.5 Hz), 7.43 (d, J = 8.2 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ 17.4, 25.7, 25.8, 28.9, 29.0, 29.5, 29.7, 29.9, 33.7, 55.5, 67.9, 71.2, 86.3, 113.3, 120.0, 126.9, 127.95, 127.98, 128.3, 130.2, 136.3, 145.0, 158.6; HRMS (ESI) m/z calcd for C33H42NO4 [M+H]+ 516.3113, found 516.3120.
12-Amino-1-(bis(4-methoxyphenyl)(phenyl)methoxy)dodecan-2-ol (23).
To a suspension of lithium aluminum hydride (1.55 g, 40.8 mmol, 5 eq.) in dry THF (50 mL) was added a solution of 22 (4.20 g, 8.16 mmol, 1 eq.) in dry THF (50 mL) dropwise via cannula at 0 °C under nitrogen. The mixture was stirred overnight while warming to rt gradually. The reaction was then quenched by dropwise addition of H2O (1.55 mL), 15% aq. NaOH (1.55 mL), and H2O (4.65 mL), sequentially. The white precipitate was removed by filtration over Celite and the filtrate was concentrated to dryness. Flash column chromatography (SiO2, 8:1:1 EtOAc/MeOH/Et3N) gave 23 as a pale-yellow oil (2.50 g, 60%): Rf = 0.2 (SiO2, 8:1:1 EtOAc/MeOH/Et3N); 1H NMR (400 MHz, CDCl3) δ 1.15–1.49 (m, 18H), 2.11 (brs, 2H), 2.67 (t, J = 7.1 Hz, 2H), 2.99 (dd, J = 9.1, 7.7 Hz, 1H), 3.14 (dd, J = 9.3, 3.1 Hz, 1H), 3.70–3.73 (m, 1H), 3.77 (s, 6H), 6.81 (d, J = 8.8 Hz, 4H), 7.19 (t, J = 6.6 Hz, 1H), 7.27 (t, J = 7.2 Hz, 2H), 7.30 (d, J = 8.6 Hz, 4H), 7.41 (d, J = 7.4 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 25.8, 27.2, 29.7, 29.82 (2C), 29.88, 29.9, 33.7 (2C), 42.4, 55.5, 67.9, 71.2, 86.3, 113.3, 126.9, 128.0, 128.3, 130.2, 136.2, 145.0, 158.6; HRMS (ESI) m/z calcd for C33H46NO4 [M+H]+ 520.3426, found 520.3429.
N-[12-[bis(4-methoxyphenyl)(phenyl)methoxy]-11-hydroxydodecyl]-6-chlorohexanamide (24).
To a solution of 23 (220 mg, 0.423 mmol, 1 eq.) and triethylamine (88 µL, 0.635 mmol, 1.5 eq.) in dry DCM (15 mL) was added 6-chlorohexanoyl chloride (0.051 mL, 0.423 mmol, 1 eq.) at −78 °C under nitrogen. The mixture was stirred for 1 h while warming to rt slowly. Water (15 mL) was added and the organic contents were extracted with DCM (15 mL × 3). The extracts were combined and dried over anhydrous Na2SO4, filtered, and concentrated. Flash column chromatography (SiO2, 2:1 hexanes/EtOAc with 5% Et3N) gave 24 as a pale-yellow oil (0.134 g, 49%): Rf = 0.5 (SiO2, 1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.19–1.51 (m, 20H), 1.64 (p, J = 8.8 Hz, 2H), 1.75 (p, J = 6.7 Hz, 2H), 2.14 (t, J = 7.4 Hz, 2H), 2.35 (brs, 1H), 3.00 (dd, J = 9.3, 7.6 Hz, 1H), 3.14 (dd, J = 9.3, 3.3 Hz, 1H), 3.20 (q, J = 7.1 Hz, 2H), 3.50 (t, J = 6.6 Hz, 2H), 3.70–3.74 (m, 1H), 3.76 (s, 6H), 5.49 (brs, 1H), 6.80 (d, J = 8.9 Hz, 4H), 7.19 (tt, J = 7.2, 2.1 Hz, 1H), 7.26 (t, J = 7.8 Hz, 2H), 7.30 (d, J = 8.8 Hz, 4H), 7.41 (d, J = 8.7 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 25.3, 25.8, 26.8, 27.2, 29.6, 29.77, 29.79 (2C), 29.89, 29.97, 32.6, 33.7, 36.9, 39.8, 45.1, 55.5, 67.9, 71.2, 86.3, 113.3, 126.9, 127.9, 128.3, 130.2, 136.3, 145.0, 158.6, 172.6; HRMS (ESI) m/z calcd for C39H55ClNO5 [M+H]+ 652.3768, found 652.3770.
1-(Bis(4-methoxyphenyl)(phenyl)methoxy)-11-ethoxy-11-oxoundecan-2-yl-(1,3-dithian-2-yl)methyl-N,N-diisopropylphosphoramidite (18a).
The same procedure for 7a was used. Flash column chromatography (SiO2, 9:1 hexanes/EtOAc with 5% Et3N) gave 18a as a colorless oil (412 mg, 79%): Mixture of two diastereoisomers; Rf = 0.6 and 0.7 (SiO2, 3:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.05 (d, J = 6.8 Hz, 3H), 1.11–1.35 (m, 23H), 1.45–1.79 (m, 3H), 1.79–1.95 (m, 1H), 1.95–2.12 (m, 1H), 2.259 (t, J = 7.7 Hz, 1H), 2.263 (t, J = 7.5 Hz, 1H), 2.57–2.68 (m, 1H), 2.69–2.89 (m, 3H), 2.96 (q, J = 2.9 Hz, 1H), 3.06 (q, J = 5.8 Hz, 1H), 3.22 (q, J = 5.2 Hz, 1H), 3.22 (q, J = 5.0 Hz), 3.47–3.65 (m, 2H), 3.65–3.80 (m, 1H), 3.766 (s, 3H), 3.773 (s, 3H), 3.84–3.92 (m, 1H), 3.92–4.05 (m, 1H), 4.11 (q, J = 7.1 Hz, 2H), 4.10–4.21 (m, 1H), 6.78 (d, J = 11.7 Hz, 2H), 6.81 (d, J = 7.5 Hz, 2H), 7.13–7.21 (m, 1H), 7.24 (t, J = 7.9 Hz, 1H), 7.26 (t, J = 7.2 Hz, 1H), 7.33 (d, J = 8.6 Hz, 2H), 7.35 (dd, J = 8.0, 1.6 Hz, 2H), 7.45 (d, J = 5.1 Hz, 1H), 7.46 (d, J = 5.2 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 14.6, 24.84, 24.86, 24.91, 24.94, 25.00, 25.04, 25.07, 25.11, 25.17, 25.31, 25.34, 25.39, 26.30, 26.34, 28.6 (d, Jcp = 7.0 Hz), 28.9 (d, Jcp = 9.0 Hz), 29.47, 29.51, 29.59, 29.72, 29.76, 29.91, 30.02, 33.76, 33.9 (d, Jcp = 6.3 Hz), 34.7, 43.2 (d, Jcp = 4.2 Hz), 43.4 (d, Jcp = 4.0 Hz), 46.9 (d, Jcp = 5.5 Hz), 47.3 (d, Jcp = 7.0 Hz), 55.5, 60.4, 64.9 (d, Jcp = 7.4 Hz), 65.1 (d, Jcp = 18.5 Hz), 66.3 (d, Jcp = 1.8 Hz), 66.4 (d, Jcp = 3.3 Hz), 73.7 (d, Jcp = 15.0 Hz), 74.3 (d, Jcp = 18.7 Hz), 85.9, 113.1, 126.7, 127.8, 128.45, 128.53, 130.30, 130.37, 136.6, 136.7, 145.3, 145.4, 158.4, 174.0; 31P{1H} NMR (162 MHz, CDCl3) δ 149.0, 149.2; HRMS (ESI) m/z calcd for C45H67NO7PS2 [M+H]+ 828.4096, found 828.4099.
(1,3-Dithian-2-yl)methyl-(1-(bis(4-methoxyphenyl)(phenyl)methoxy)-12-(6-chlorohexanamido)dodecan-2-yl)-N,N-diisopropylphosphoramidite (18b).
The same procedure for 7a was used. Flash column chromatography (SiO2, 1:1 hexanes/EtOAc with 5% Et3N) gave 18b as a pale-yellow oil (294 mg, 86%): Mixture of two diastereoisomers; Rf = 0.2 and 0.3 (SiO2, 1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.03 (d, J = 6.8 Hz, 2H), 1.10–1.35 (m, 22H), 1.40–1.51 (m, 4H), 1.56–1.69 (m, 4H), 1.77 (p, J = 7.1 Hz), 1.81–1.94 (m, 1H), 1.95–2.10 (m, 3H), 2.15 (t, J = 7.4 Hz, 2H), 2.56–3.15 (m, 6H), 3.21 (t, J = 6.8 Hz, 1H), 3.22 (t, J = 6.5 Hz, 1H), 3.52 (t, J = 6.6 Hz, 2H), 3.55–4.18 (m, 4H), 3.76 (s, 1H), 3.77 (s, 3H), 4.35–4.57 (m, 2H), 5.47 (brs, 1H), 6.78 (d, J = 8.8 Hz, 2H), 6.80 (d, J = 7.3 Hz, 2H), 7.13–7.21 (m, 1H), 7.21–7.28 (m, 2H), 7.32 (dd, J = 6.5, 2.6 Hz, 2H), 7.34 (dd, J = 8.4, 1.7 Hz, 2H), 7.44 (dd, J = 5.4, 1.6 Hz, 1H), 7.46 (dd, J = 7.2, 1.6 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ 24.84, 24.86, 24.90, 24.94, 25.00, 25.07, 25.11, 25.17, 25.28, 25.38, 25.6, 25.9, 26.1, 26.30, 26.34, 26.7, 26.8, 27.0, 27.2, 27.3, 28.6, 28.7, 28.85, 28.88, 29.6, 29.81, 29.85, 29.88, 29.93, 29.99, 30.03, 32.6, 33.79 (d, Jcp = 3.0 Hz), 33.86 (d, Jcp = 5.2 Hz), 36.9, 39.8, 43.2, 43.4, 45.1, 46.93 (d, Jcp = 7.1 Hz), 47.29 (d, Jcp = 7.3 Hz), 55.5, 64.9 (d, Jcp = 17.4 Hz), 66.3 (d, Jcp = 6.8 Hz), 74.4, 85.9, 113.2, 126.7, 127.8, 128.45, 128.53, 130.3, 136.6, 136.7, 145.3, 145.4, 158.4, 172.6; 31P{1H} NMR (162 MHz, CDCl3) δ 149.0, 149.2; HRMS (ESI) m/z calcd for C50H77ClN2O6PS2 [M+H]+ 931.4649, found 931.4650.
ODN synthesis, cleavage and deprotection, and analysis.
All ODNs were synthesized on dT-Dmoc-CPG (26 µmol/g loading, 20 mg, 0.52 µmol) using a MerMade 6 Synthesizer. Dim-Dmoc phosphoramidites were used as monomers. The conditions suggested by synthesizer manufacturer for 1 μmol synthesis were used except that coupling was optionally increased from 2 to 3 times and capping was achieved using 2-cyanoethyl-N,N,N’,N’-tetraisopropylphosphordiamidite instead of acetic anhydride. Briefly, detritylation, DCA (3%, DCM), 90 sec × 2; coupling, phosphoramidite (7a-e, 18a or 18e, 0.1 M, MeCN), 5-(ethylthio)-1H-tetrazole (0.25 M, MeCN), 60 sec × 2 (or 3); capping, 2-cyanoethyl-N,N,N’,N’-tetraisopropylphosphordiamidite (0.1 M, MeCN) and 5-(ethylthio)-1H-tetrazole (0.25 M, MeCN), 60 sec × 3; oxidation, I2 (0.02 M, THF/pyridine/H2O, 70/20/10, v/v/v), 40 sec. For incorporating the last nucleoside, 7e instead of 7a was used. At the end of synthesis, the 5’-trityl group was kept on. The CPG was divided into 5 equal portions. One portion was gently shaken in a solution of aqueous NaIO4 (0.4 M, 1 mL, pH 4) at rt for 3 h. The supernatant was removed with a pipette, and the CPG was rinsed briefly with water (1 mL × 4). To the CPG was added aqueous aniline solution (3%, 1 mL, pH 8) and the mixture was shaken at rt for 3 h. The supernatant was transferred into a centrifugal tube, which was concentrated to ~100 μL. To the tube was added 1-butanol (900 μL). The tube was vortexed briefly and centrifuged (14.5K rpm, 5 min). The supernatant was removed with a pipette carefully without sucking the ODN precipitate. The ODN was dissolved in H2O (100 μL) and ~35 μL was injected into RP HPLC to generate the crude ODN. Fractions of the major ODN peak at ~39 min were collected, concentrated to ~100 μL, and injected into HPLC to give the profile of purified trityl-tagged ODN. To the dried trityl-tagged ODN was added 1 mL of 80% AcOH, and the mixture was shaken gently at rt for 3 h. Volatiles were evaporated. The residue was dissolved in ~100 μL water and injected into RP HPLC. The major peak of de-tritylated ODN at ~21 min was collected and concentrated to dryness. The residue was the pure de-tritylated ODN, which was dissolved in 100 μL water and injected into HPLC to generate the profile of pure de-tritylated ODN. The pure ODN was analyzed MALDI-TOF MS. Digestion of the ODN was performed using the Nucleoside Digestion Mix from New England BioLabs, which is a mixture of enzymes that provides a convenient one-step method to generate single nucleosides from DNA or RNA. Following the protocol provided by the manufacturer, suitable amount of an ODN (for 14a, 14b and 14c, 3 µg was used; for 14d, 14e, 14f and 14g, 2 µg was used), Nucleoside Digestion Mix Reaction Buffer (10X; 6 µL for 3 µg ODN, and 4 µL for 2 µg ODN), Nucleoside Digestion Mix (3 µL for 3 µg ODN, and 2 µL for 2 µg ODN), and water (60 µL for 3 µg ODN, and 40 µL for 2 µg ODN) were combined and incubated at 37 °C for 3–4 h. After cooling to rt, volatiles were evaporated until dryness using a centrifugal vacuum evaporator. The residue was re-dissolved in 50 µL water. The solution (for 14b, 25 µL; for others, 50 µL) was analyzed with RP HPLC. The conditions were the same as for ODN analysis except that a different gradient system was used: solvent A (100%) for 5 min, followed by solvent B (0–10%) in solvent A over 30 min. No purification of the nucleosides was necessary before the analysis. The HPLC profiles of the nucleosides from the ODNs 14a-g and two control ODNs synthesized using commercial dABz, dCAc, dGiBu, and dT 5’-DMTr-2-cyanoethyl-diisopropylphosphoramidites and deprotected under standard conditions using concentrated NH4OH are in the Supporting Information. Information about OD260 of the ODNs are provided in the UV spectra section of the Supporting Information.
Supplementary Material
Acknowledgements
Financial support from NIH (GM109288), Robert and Kathleen Lane Endowed Fellowship (S.S. and B.H.), David and Valeria Pruett Fellowship (D.E. and B.H.), and PHF Graduate Assistantship (S.S. and B.H.); the assistance from D. W. Seppala (electronics), J. L. Lutz (NMR), L. R. Mazzoleni (MS), M. Khaksari (MS), and A. Galerneau (MS); and NSF equipment grants (CHE1048655, CHE9512455, AGS1531454); are gratefully acknowledged.
Footnotes
Associated Contents
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
HPLC profiles, MALDI-TOF MS, UV and OD260 of ODNs; HPLC profiles of nucleosides from enzymatic digestion of ODNs, and images of 1H, 13C and 31P NMR spectra of new compounds.
A patent on the oligonucleotide synthesis technology is pending at USPTO.
References
- 1.Benizri S; Gissot A; Martin A; Vialet B; Grinstaff MW; Barthelemy P. Bioconjugated oligonucleotides: Recent developments and therapeutic applications. Bioconjugate Chem 2019, 30 (2), 366–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel AB; Louder RK; Greber BJ; Gruberg S; Luo J; Fang J; Liu YT; Banish J; Hahn S; Nogales E. Structure of human TFIID and mechanism of TBP loading onto promoter DNA. Science 2018, 362 (6421), doi: 10.1126/science.aau8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nedorezova DD; Fakhardo AF; Nemirich DV; Bryushkova EA; Kolpashchikov DM. Towards DNA nanomachines for cancer treatment: Achieving selective and efficient cleavage of folded RNA. Angew. Chem. Int. Edit 2019, 58 (14), 4654–4658. [DOI] [PubMed] [Google Scholar]
- 4.Woods D; Doty D; Myhrvold C; Hui J; Zhou F; Yin P; Winfree E. Diverse and robust molecular algorithms using reprogrammable DNA self-assembly. Nature 2019, 567 (7748), 366–372. [DOI] [PubMed] [Google Scholar]
- 5.Wang GL; Li Z; Ma N. Next-generation DNA-functionalized quantum dots as biological sensors. ACS Chem. Biol 2018, 13 (7), 1705–1713. [DOI] [PubMed] [Google Scholar]
- 6.Jiang DJ; Xu CL; Tsang SH. Revolution in gene medicine therapy and genome surgery. Genes 2018, 9 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bednarski JJ; Sleckman BP. At the intersection of DNA damage and immune responses. Nat. Rev. Immunol 2019, 19 (4), 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez-Fierro A; Duenas-Gonzalez A. Emerging DNA methylation inhibitors for cancer therapy: Challenges and prospects. Expert Rev. Precis. Med. Drug Dev 2019, 4 (1), 27–35. [Google Scholar]
- 9.Chen KK; Kong JL; Zhu JB; Ermann N; Predki P; Keyser UF. Digital data storage using DNA nanostructures and solid-state nanopores. Nano Lett 2019, 19 (2), 1210–1215. [DOI] [PubMed] [Google Scholar]
- 10.Erb TJ Back to the future: Why we need enzymology to build a synthetic metabolism of the future? Beilstein J. Org. Chem 2019, 15 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meher G; Meher NK; Iyer RP. Nucleobase protection of deoxyribo- and ribonucleosides. Current Protocols in Nucleic Acid Chemistry 2017, 2.1.1–2.1.40. doi: 10.1002/cpnc.1032. [DOI] [PubMed]
- 12.Virta P Solid-phase synthesis of base-sensitive oligonucleotides. ARKIVOC 2009. 54–83.
- 13.Garcia RG; Brank AS; Christman JK; Marquez VE; Eritja R. Synthesis of oligonucleotide inhibitors of DNA (cytosine-C5) methyltransferase containing 5-azacytosine residues at specific sites. Antisense Nucleic Acid Drug Dev 2001, 11 (6), 369–378. [DOI] [PubMed] [Google Scholar]
- 14.Hayakawa Y; Wakabayashi S; Kato H; Noyori R. The allylic protection method in solid-phase oligonucleotide synthesis - an efficient preparation of solid-anchored DNA oligomers. J. Am. Chem. Soc 1990, 112 (5), 1691–1696. [Google Scholar]
- 15.Avino AM; Eritja R. Use of NPE-protecting groups for the preparation of oligonucleotides without using nucleophiles during the final deprotection. Nucleos. Nucleot 1994, 13 (10), 2059–2069. [Google Scholar]
- 16.Johnsson RA; Bogojeski JJ; Damha MJ. An evaluation of selective deprotection conditions for the synthesis of RNA on a light labile solid support. Bioorg. Med. Chem. Lett 2014, 24 (9), 2146–2149. [DOI] [PubMed] [Google Scholar]
- 17.Avino A; Garcia RG; Marquez VE; Eritja R. Preparation and properties of oligodeoxynucleotides containing 4-O-butylthymine, 2-fluorohypoxanthine and 5-azacytosine. Bioorg. Med. Chem. Lett 1995, 5 (20), 2331–2336. [Google Scholar]
- 18.Eritja R; Robles J; Avino A; Albericio F; Pedroso E. A synthetic procedure for the preparation of oligonucleotides without using ammonia and its application for the synthesis of oligonucleotides containing O-4-alkyl thymidines. Tetrahedron 1992, 48 (20), 4171–4182. [Google Scholar]
- 19.Matray TJ; Greenberg MM. Site-specific incorporation of the alkaline labile, oxidative stress product (5R)-5,6-dihydro-5-hydroxythymidine in an oligonucleotide. J. Am. Chem. Soc 1994, 116 (15), 6931–6932. [Google Scholar]
- 20.Ohkubo A; Ezawa Y; Seio K; Sekine M. O-Selectivity and utility of phosphorylation mediated by phosphite triester intermediates in the N-unprotected phosphoramidite method. J. Am. Chem. Soc 2004, 126 (35), 10884–10896. [DOI] [PubMed] [Google Scholar]
- 21.Lin X; Chen JS; Shahsavari S; Green N; Goyal D; Fang SY. Synthesis of oligodeoxynucleotides containing electrophilic groups. Org. Lett 2016, 18 (15), 3870–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halami B; Shahsavari S; Nelson Z; Prehoda L; Eriyagama D. N. a. M.; Fang SY. Incorporation of sensitive ester and chloropurine groups into oligodeoxynucleotides through solid-phase synthesis. ChemistrySelect 2018, 3 (31), 8857–8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shahsavari S; Eriyagama D. N. a. M.; Halami B; Begoyan V; Tanasova M; Chen J; Fang S. Electrophilic oligodeoxynucleotide synthesis using dM-Dmoc for amino protection. Beilstein J. Org. Chem 2019, 15, 1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cieslak J; Grajkowski A; Livengood V; Beaucage SL. The 4-methylthio-1-butyl group for phosphate/thiophosphate protection in oligodeoxyribonucleotide synthesis. Current Protocols in Nucleic Acid Chemistry 2004. 3.11.11–13.11.14. doi: 10.1002/0471142700.nc0311s19 [DOI] [PubMed]
- 25.Cieslak J; Grajkowski A; Livengood V; Beaucage SL. Thermolytic 4-methylthio-1-butyl group for phosphate/thiophosphate protection in solid-phase synthesis of DNA oligonucleotides. J. Org. Chem 2004, 69 (7), 2509–2515. [DOI] [PubMed] [Google Scholar]
- 26.Zeng KZ; Li Q; Wang J; Yin GW; Zhang YJ; Xiao CN; Fan TP; Zhao XF; Zheng XH. One-step methodology for the direct covalent capture of GPCRs from complex matrices onto solid surfaces based on the bioorthogonal reaction between haloalkane dehalogenase and chloroalkanes. Chem. Sci 2018, 9 (2), 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halami B; Eriyagama D. N. a. M.; Chillar K; Nelson Z; Prehoda L. A linear oligosulfoxide, synthesis and solubility studies. 2019. manuscript to be submitted (uploaded with this manuscript submission). [DOI] [PMC free article] [PubMed]
- 28.Gilham PT; Khorana HG. Studies on polynucleotides. I. A new and general method for the chemical synthesis of the C5’-C3’ internucleotidic linkage. Syntheses of deoxyribo-dinucleotides. J. Am. Chem. Soc 1958, 80 (23), 6212–6222. [Google Scholar]
- 29.Shahsavari S; Chen JS; Wigstrom T; Gooding J; Gauronskas A; Fang SY. Tritylation of alcohols under mild conditions without using silver salts. Tetrahedron Lett 2016, 57 (34), 3877–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.