

ABSTRACT
Nutrient status affects brain development; however, the effects of nutrient availability on neural progenitor cell proliferation in vivo are poorly understood. Without food, Xenopus laevis tadpoles enter a period of stasis during which neural progenitor proliferation is drastically reduced, but resumes when food becomes available. Here, we investigate how neural progenitors halt cell division in response to nutrient restriction and subsequently re-enter the cell cycle upon feeding. We demonstrate that nutrient restriction causes neural progenitors to arrest in G2 of the cell cycle with increased DNA content, and that nutrient availability triggers progenitors to re-enter the cell cycle at M phase. Initiation of the nutrient restriction-induced G2 arrest is rapamycin insensitive, but cell cycle re-entry requires mTOR. Finally, we show that activation of insulin receptor signaling is sufficient to increase neural progenitor cell proliferation in the absence of food. A G2 arrest mechanism provides an adaptive strategy to control brain development in response to nutrient availability by triggering a synchronous burst of cell proliferation when nutrients become available. This may be a general cellular mechanism that allows developmental flexibility during times of limited resources.
KEY WORDS: Neurogenesis, Cell cycle, Nutrition, Stasis, G2 arrest, Optic tectum
Highlighted Article: The Xenopus tadpole brain adapts to limited nutrition by reversibly arresting neural progenitor cell division at G2, recovery from which requires insulin receptor and mTOR signaling downstream of nutrients.
INTRODUCTION
Access to nutrients is essential for normal development. Animals must be able to cope with variations in nutrient availability in order to survive and produce viable offspring. Nutrient status regulates cell proliferation throughout the body, especially in the brain where high levels of proliferation have large metabolic requirements (Agathocleous et al., 2012; Warburg, 1956). Across species, nutrient restriction (NR) affects brain development by decreasing cell proliferation, neuron numbers and neuronal connectivity (Benítez-Santana et al., 2017; Bolduc et al., 2016; Brown and Susser, 2008; Georgieff, 2007; Georgieff et al., 2015; Igarashi et al., 2015; Lanet et al., 2013; Metcalfe and Monaghan, 2001), and these effects are reversible in only limited cases. During Drosophila pupation, when animals are naturally deprived of external nutrients, cells in the imaginal discs undergo a form of temporary stasis where they pause in the cell cycle in order to synchronize proliferation (Milan et al., 1996). Studies in zebrafish indicate that neural cell proliferation is inhibited under NR conditions, and neural progenitor cells (NPCs) can resume proliferation when food is available (Benítez-Santana et al., 2017). We have shown that Xenopus laevis tadpoles that have been deprived of external nutrients cease proliferation in the developing brain, and that cell division resumes upon re-introduction of food (McKeown et al., 2017). Completely depriving younger tadpoles of food by surgically removing the yolk stores halts NPC proliferation in the developing retina (Love et al., 2014), indicating that proliferative stasis can occur across different neuronal tissues in the tadpole. Yet the cellular and molecular mechanisms by which nutrient status affects NPC proliferation in vivo remain unclear.
Control of cell cycle dynamics is a likely mechanism by which nutrient status affects proliferation. Cells exit the cell cycle for a variety of reasons, including G0 cell cycle exit for differentiation, or irreversible G2 arrest in response to DNA damage (Barnum and O'Connell, 2014; DiPaola, 2002; Duursma and Cimprich, 2010) or viral infection (Bressy et al., 2019; Davy and Doorbar, 2007). Dividing cells typically pause for some period in G0, pending signals that regulate progression into G1 and subsequent cell division or terminal differentiation. In general, healthy cells that enter the cell cycle complete mitosis; however, somatic cells temporarily arrest in G1/S in larval Caenorhabditis elegans in response to NR until food becomes available (Baugh et al., 2009). G1/S pausing has been described in somatic cells across species and is thought to be important for sensing environmental, metabolic and stress cues (Bouldin and Kimelman, 2014). In the germline, cells can temporarily arrest in G2 in order to synchronize cell division through meiosis (Seidel and Kimble, 2015). Moreover, cell division synchrony via G2 pausing has been described during embryogenesis in many species, including Drosophila, chordates and Xenopus (Bouldin and Kimelman, 2014; Kimura et al., 1997; Meserve and Duronio, 2017; Milan et al., 1996; Ogura et al., 2011; Ogura and Sasakura, 2016; Thuret et al., 2015). Although NR-induced reversible G2 arrest has been described in adult Hydra and developing Drosophila (Buzgariu et al., 2014; Otsuki and Brand, 2018), whether neural progenitors in the vertebrate brain enter a reversible G2 arrest in response to NR and whether mechanisms regulating G2 arrest are conserved in vertebrates have not yet been determined.
Several nutrient-sensing pathways may underlie cellular responses to nutrient status, including signaling via the insulin receptor, amino acid-sensing via G protein-coupled receptors, and glucose transport signaling, all of which converge on the mTOR signaling pathway, making mTOR a prime candidate for regulation of nutrient-dependent changes in cell proliferation (Agathocleous and Harris, 2013; Garelick and Kennedy, 2011; Hall, 2016; Jacinto and Hall, 2003; Laplante and Sabatini, 2009; Lee, 2015; Loewith and Hall, 2011). Indeed, in both nutrient-restricted adult zebrafish brains and nutrient-deprived Xenopus tadpole retinas, mTOR is required for resumption of cell proliferation following nutrient restriction (Benítez-Santana et al., 2017; Love et al., 2014). Interestingly, mTOR has been shown to both drive progression through G1 phase of the cell cycle by controlling cell size (Fingar et al., 2003), and to control G2 progression into mitosis, although whether mTOR promotes or blocks G2/M-phase entry may vary in different experimental systems (Proud, 2010; Ramirez-Valle et al., 2010). During G2 arrest in Drosophila embryos, cell cycle re-entry depends on mTOR signaling downstream of the insulin ligand and requires amino acids (Britton and Edgar, 1998; Britton et al., 2002; Chell and Brand, 2010; Lee et al., 2014; Nässel et al., 2015; Sousa-Nunes et al., 2011; Spéder and Brand, 2014). However, the specific role of nutrient sensors and mTOR signaling in regulating neural progenitor cell division in the vertebrate brain in vivo remains unclear.
We address these open questions in Xenopus; we have previously demonstrated that Xenopus laevis tadpoles enter a period of developmental stasis in response to nutrient restriction (McKeown et al., 2017). During stasis, Xenopus tadpoles continue to swim and exhibit visual avoidance behaviors, but rates of overall body growth and brain development decrease, and NPC proliferation halts. This stasis period can last for up to 9 days and is completely reversible if nutrients become available within the 9-day window. Here, we investigate the cellular mechanisms underlying nutrient restriction-induced stasis in the developing Xenopus brain. The developing Xenopus tectum provides an ideal system for these studies based on extensive prior characterization of cell proliferation and neuronal differentiation (Bestman and Cline, 2008; Bestman et al., 2015, 2012; Chiu et al., 2008; Cline, 2001; Ewald et al., 2008; Haas et al., 2006; Herrgen and Akerman, 2016; Lau et al., 2017; Peunova et al., 2001; Ruthazer et al., 2013; Sharma and Cline, 2010; Straznicky and Gaze, 1972; Tremblay et al., 2009; Van Keuren-Jensen and Cline, 2008; Wu and Cline, 2003). NPCs in the optic tectum are radial glial cells lining the ventricle (Bestman et al., 2012; Herrgen and Akerman, 2016; Peunova et al., 2001; Sharma and Cline, 2010). In vivo time-lapse imaging and single cell lineage analysis indicate that these NPCs are Sox2-expressing progenitors that undergo symmetrical regenerative proliferation, followed by asymmetric neurogenic divisions (Bestman et al., 2012). Here, we show that nutrient restriction causes neural progenitors to arrest in G2 and re-feeding triggers NPCs to re-enter the cell cycle at M phase. We demonstrate that, although initiation of G2 arrest is independent of mTOR, cell cycle re-entry after G2 arrest requires mTOR signaling. Furthermore, G2 arrest can be overridden by activating insulin receptor signaling even in the absence of food. This is the first demonstration of reversible G2 arrest in a vertebrate system in vivo induced by NR, suggesting that this mechanism may be a more widely used strategy of cellular adaptation to NR, or more generally, environmental stress, than previously recognized. The described mechanism of reversible stasis by G2 arrest provides cellular level flexibility when animals are confronted with adverse environmental conditions.
RESULTS
Nutrient restriction affects proliferative capacity of neural progenitor cells
During normal development in the Xenopus laevis midbrain, Sox2-expressing NPCs located along the optic tectal midline divide and differentiate to produce neuronal daughter cells that are displaced laterally to populate the optic tectum (Fig. 1A), while also retaining a self-renewing population of progenitors that continue to divide along the midline (Bestman et al., 2012; Peunova et al., 2001; Sharma and Cline, 2010; Straznicky and Gaze, 1972). Tectal NPCs are Sox2-expressing radial glial cells lining the ventricle that incorporate the thymidine analog 5-chloro-2′-deoxyuridine (CldU) and lack neuronal markers such as NeuroD, N-β-Tubulin and HuC/D (Fig. 1, Fig. S1) (Bestman et al., 2012; Sharma and Cline, 2010). In the tectum, these radial glia NPCs are the only dividing neural progenitors. Vascular endothelial cells also proliferate but can be distinguished from NPCs based on their distinct morphology and location (Lau et al., 2017; Rovainen and Kakarala, 1989; Sharma and Cline, 2010). We have previously shown that tectal NPCs stop dividing when the animal enters a period of developmental stasis in response to nutrient restriction (McKeown et al., 2017). To understand the cellular responses to nutrient availability in the brain, we first investigated NPC proliferation during nutrient-restricted and fed conditions at early stages of brain development. Stage 41 animals, approximately 3 days old, were given a 24-h pulse of CldU to label all actively dividing progenitors. Because CldU incorporates during the DNA replication cycle of S phase, and the tectal cell cycle is less than 24 h at this stage, this protocol is sufficient to label the entire dividing population (Herrgen and Akerman, 2016; Sharma and Cline, 2010; Thuret et al., 2015). As cells divide, the CldU-labeled DNA will also divide and becomes more diffuse over several rounds of cell division, whereas cells that retain bright CldU label have either ceased division along the ventricular midline or differentiated into neurons and migrated laterally from the midline (Bestman et al., 2012; Grandel et al., 2006; Sharma and Cline, 2010). By the end of the 24 h labeling period, animals have developed to stage 45 and will soon begin to forage for food. They were then either nutrient restricted (NR) or allowed to feed ad libitum (fed) for 6 days and then sacrificed and processed for CldU immunofluorescence (Fig. 1A,B). It is important to note that images of brains are shown at the same scale (Fig. 1Ci,Cii). After 6 days, the brains of fed animals were larger than the brains of NR animals (Fig. 1Ci,Cii), consistent with increased brain growth in fed animals. The optic tecta of fed animals displayed extensive CldU labeling throughout the tectum (Fig. 1Ci), and decreased fluorescence intensity of CldU labeling along the midline (Fig. 1Ciii), suggesting that the progenitors labeled 6 days prior continued to divide, differentiate into neurons, and densely populate the tectum lateral from the midline. In contrast, the smaller brains of nutrient-restricted animals had bright, densely packed CldU-labeled cells that remained at the midline along the ventricle (Fig. 1Cii,Civ), suggesting that the NPC division and neuronal differentiation were limited in NR conditions. To address this further, we labeled NR and fed animals with a shorter (2 h) pulse of CldU and co-labeled for progenitor and neuronal markers (Fig. 1D,E, Fig. S1). Sox2-expressing progenitors lining the ventricular midline incorporated CldU and immunolabeled for the cell proliferation marker phospho-histone H3 (pH3) (Fig. S1). Images of the tectal midline in both fed and NR animals show that Sox2+ NPCs are labeled with CldU (Fig. 1Ei,Eii). Although there were fewer CldU-incorporating cells under these conditions, Sox2+ NPCs were still present along the midline in the NR brains (Fig. 1Eii). Furthermore, labeling for HuC/D demonstrated that progenitor cells lining the ventricular midline were HuC/D negative (Fig. 1Eiii-Evi). The HuC/D-negative progenitor region was expanded in the fed animals (Fig. 1Eiii,Ev) compared with NR animals (Fig. 1Eiv,Evi), indicating an increase in progenitors in fed animals. Moreover, this labeling revealed CldU-labeled cells that included both HuC/D− progenitors and HuC/D+ neurons displaced laterally from the ventricle in both fed and NR groups, albeit increased in fed groups (Fig. 1Eiii-Eiv). pH3 labeling revealed high levels of HuC/D− mitotic cells lining the ventricle in fed animals (Fig. 1Ev) and at lower levels in NR animals (Fig. 1Evi). These data suggest that nutrients drive extensive proliferation of NPCs, resulting in neurogenic growth of the CldU-labeled NPC expanding progenitor pool. In contrast, nutrient restriction limits proliferation of NPCs, suggesting that the majority of progenitors are quiescent in the NR condition (Fig. 1C,E).
Fig. 1.

Nutrient restriction limits brain growth and NPC proliferation. (A) Diagram of the Xenopus optic tectum. NPCs reside along the ventricle at the midline (green), and their progeny are pushed laterally as NPCs divide and differentiate into neurons (red). The midline of the tectal ventricle is indicated with a dashed yellow line. (B) Experimental timeline for the data shown in C. Animals were treated for 24 h with CldU and then allowed to develop for 6 days in either fed or nutrient-restricted conditions. (C) Whole-mount confocal images of CldU immunofluorescence in the optic tectum in fed (i, iii) and nutrient-restricted (ii, iv) animals. 20× magnification 6.2 μm z-projections (i-ii) and 60× magnification 1.66 μm z-projections (iii-iv) are shown. Confocal images of brains were collected under identical settings and are shown at the same scale (i-ii and iii-iv). (D) Experimental timeline for the data shown in E. Animals were treated for 2 h with CldU and then allowed to develop for 2-3 days in either fed or nutrient-restricted conditions. (E) Whole-mount confocal images of Sox2 and CldU (i-ii), HuC/D and CldU (iii-iv), and HuC/D and pH3 (v-vi) immunofluorescence along the tectal midline in fed (i, iii, v) and nutrient-restricted (ii, iv, vi) animals. Single optical sections are shown. Scale bars: 100 μm (C); 20 μm (E).
Nutrients induce NPCs to divide 16 h after feeding
At stage 46/47, tadpoles exhaust their yolk stores and begin to forage for food (Nieuwkoop and Faber, 1956). Our previous work showed that during nutrient restriction-induced developmental stasis, animals reduce CNS NPC proliferation. Upon re-introduction of external nutrients, nutrient-restricted animals exit developmental stasis and resume development, and importantly, food re-initiates tectal NPC proliferation (McKeown et al., 2017). To understand the mechanisms by which nutrient-restricted progenitors re-initiate proliferation, we performed a series of experiments to test the proliferative response to re-introduction of food in a controlled manner. Animals were nutrient restricted at stage 47 for 2 days, and then food was re-introduced ad libitum in their rearing media. At different time points after re-introduction of food, animals were sacrificed, fixed, and brains were processed for pH3 immunofluorescence to label mitotic cells (Fig. 2A). Control animals were clutch mates, left nutrient-restricted for the duration of the experiment. Whole-mount brains were imaged and pH3+ cells were counted throughout the entire z-series of the tectum (Fig. 2B). The number of pH3+ cells did not change at 1, 2, 4, 8 and 12 h after feeding, but pH3+ cell numbers increased significantly 16 h after feeding, and continued to increase at 24 and 48 h (Fig. 2B,C). It is important to note that in this 16 h experimental window, the animals must find, consume, digest and metabolize the food before the nutrients are delivered to the brain. Nonetheless, these data demonstrate that nutrient-restricted progenitors are poised to divide and enter M phase 16 h after the re-introduction of food to the animal.
Fig. 2.

Nutrient-induced increase in cell proliferation occurs 16 h after feeding. (A) Experimental timeline. Animals were nutrient restricted for 2 days at Stage 47 and then food was re-introduced. Animals were sacrificed and processed for pH3 immunolabeling at the indicated time points after feeding. (B) Whole-mount confocal images of pH3 immunofluorescence in the optic tectum at the time points indicated. Fd, food. (C) Quantification of pH3 labeling showing total cell counts per tectum at each time point (mean±s.e.m.). Green data points indicate fed and gray are NR animals. At 16 h after feeding, pH3+ cells are significantly increased compared with NR controls, ****P<0.0001. n.s., not significant. n=6-47 animals per group per time point, from multiple independent clutches, for a total of 304 animals, across 16 groups from multiple independent clutches; see Table S1. Scale bars: 100 μm.
Nutrient-induced proliferation requires mTOR signaling
One of the molecular triggers that could be responsible for the nutrient-induced proliferation is the mTOR signaling pathway (Chantranupong et al., 2015; Cloetta et al., 2013; Garelick and Kennedy, 2011; Laplante and Sabatini, 2009; Lee, 2015; Loewith and Hall, 2011; Proud, 2010; Reiling and Sabatini, 2006). To test the role of mTOR signaling in nutrient-induced proliferation, we investigated mTOR activation and the activity of mTOR-activated proteins in brains of nutrient-restricted animals that were provided food in the presence or absence of rapamycin (Fig. 3A), which binds directly to mTOR and inhibits downstream mTOR signaling (Arriola Apelo and Lamming, 2016; Sabers et al., 1995). Western blots of brain tissue showed that food increases the fraction of phosphorylated mTOR (p-mTOR) compared with the nutrient-restricted condition (Fig. 3B, gray bar), indicating that feeding activates mTOR signaling in the brain (Acosta-Jaquez et al., 2009). Moreover, we found that this nutrient-induced increase in p-mTOR is blocked by rapamycin (Fig. 3B, blue bar). These data show that p-mTOR is increased in response to feeding, providing a link between nutrients and mTOR activation in the tadpole brain.
Fig. 3.

Nutrient-induced proliferation requires mTOR signaling. (A) Experimental timeline. Animals were nutrient restricted for 2 days after Stage 47 and then food was re-introduced concomitant with the mTOR-blocking drug rapamycin or DMSO vehicle for up to an additional 2 days. Brains were processed for pH3 immunolabeling 16 h, 24 h and 48 h after feeding, or for western blots 48 h after feeding as indicated. (B) Western blot analysis shows that feeding increased phospho-mTOR (p-mTOR) and that increase in p-mTOR was blocked by rapamycin. (C) Western blot analysis shows that feeding increased phospho-ribosomal protein S6 (p-rS6) and that increase was blocked by rapamycin. (D) Whole-mount confocal images of pH3 immunolabeling in the optic tectum 16 h, 24 h and 48 h after feeding, with or without rapamycin treatment. Fd, food. (E) Quantification of total pH3+ cells per tectum 16 h, 24 h and 48 h after feeding, with or without rapamycin showing that rapamycin blocks the feeding-induced increase in proliferation at all time points tests. Data for each condition are shown as individual data points and mean±s.e.m. are shown as black bars. Blue symbols signify rapamycin treatment and gray symbols are controls. For western blots, n=5 brains per treatment per time point, a minimum of 3 biological replicates was used for each quantification shown. For cell counts, n=8-18 animals per group per time point for a total of 150 animals across 12 groups from multiple independent clutches; see Table S1. **P<0.01, ***P<0.001, ****P<0.0001, n.s., not significant. Scale bars: 100 μm.
mTOR signaling activates ribosomal proteins and subsequent protein translation (Fingar et al., 2003; Hara et al., 1998). To probe further the mechanistic link between nutrients and mTOR signaling in Xenopus stasis, we tested whether phosphorylation of ribosomal protein S6 (p-rS6), a downstream target of mTOR activation, is affected by re-feeding after nutrient restriction in the tadpole brain. We found that p-rS6 levels were reduced in NR brains and significantly increased in the brain in response to feeding (Fig. 3C, gray bar). Furthermore, this nutrient-induced increase in p-rS6 in tadpole brain was blocked by rapamycin (Fig. 3C, blue bar). Moreover, p-rS6 immunofluorescence was decreased in NR tecta and increased in fed animals, particularly along the ventricle where progenitors reside (Fig. S2). We also tested whether signaling components upstream of mTOR were affected by nutrients. Both the upstream inhibitor phospho-PTEN (S380) and the upstream activator phospho-AKT (S473) were increased in NR animals compared with fed animals (Fig. S3), reflecting the complex feedback mechanisms known to be involved in the mTOR signaling pathway (Laplante and Sabatini, 2009; Lee, 2015; Loewith and Hall, 2011; Manning and Toker, 2017; Martin and Hall, 2005; Memmott and Dennis, 2009; O'Reilly et al., 2006; Palu and Thummel, 2015; Proud, 2004; Vadlakonda et al., 2013). These data indicate that nutrients affect components of the signaling pathway upstream of mTOR, and activate mTOR and its downstream targets in the tadpole brain. Together, the data suggest that mTOR signaling regulates feeding-induced NPC proliferation.
To test whether the NPC proliferation we see upon introduction of nutrients results from the increased mTOR signaling described above, we assayed proliferation in animals that were nutrient restricted for 2 days and then provided with food, with or without rapamycin in their rearing media for an additional 1-2 days. Control animals were treated with 1% DMSO vehicle. Brains were processed for pH3 immunolabeling 16, 24 and 48 h later (Fig. 3A). Rapamycin treatment did not appear to affect feeding behavior, based on the presence of food in their gut. Rapamycin had no effect on proliferation in the nutrient-restricted animals (Fig. 3Di,Diii,Dv,Dvii,E). Feeding significantly increased proliferation, and rapamycin blocked the robust feeding-induced proliferation in the tectum as early as 16 h after feeding (Fig. 3Dii,Div,Dvi,Dviii,E). The nutrient-regulated increase in neural cell proliferation and the inhibitory effects of rapamycin persisted up to 48 h (Fig. 3Dix-Dxii,E). These data indicate that nutrient-induced NPC proliferation is controlled by mTOR signaling in vivo. Taken together, these data show that re-feeding after nutrient restriction activates mTOR signaling pathway components that are required for feeding-induced proliferation of NPCs in the tadpole brain.
mTOR signaling is not required for the initiation of nutrient restriction-induced proliferative arrest
The experiments described above indicate that animals in stasis cease NPC proliferation, that cell division resumes 16 h after re-introduction of food and that this resumption of proliferation requires mTOR signaling. To investigate further the role of mTOR signaling in this nutrient-regulated neural cell proliferation, we tested whether mTOR acts as a molecular switch to control the cellular response to nutrients. To test whether mTOR is required for entry into proliferative arrest induced by nutrient restriction, animals were treated with rapamycin at stage 46, when they still have maternal yolk stores in their gut, and they remained in rapamycin for 2 days while they were nutrient restricted (Fig. 4A). Brains were processed for pH3 immunolabeling and the number of dividing cells was counted throughout the entire z-series of the tectum. Cell proliferation decreased over the 2-day course of the experiment, as expected for nutrient restriction; however, rapamycin did not affect this nutrient restriction-induced decline in proliferation (Fig. 4B). These data indicate that, although mTOR is required for NPCs to exit stasis, mTOR signaling is not required for NPCs to halt cell division as the animal enters stasis, and that mTOR is not acting as a nutrient-sensing bi-directional switch in the developing tadpole brain.
Fig. 4.
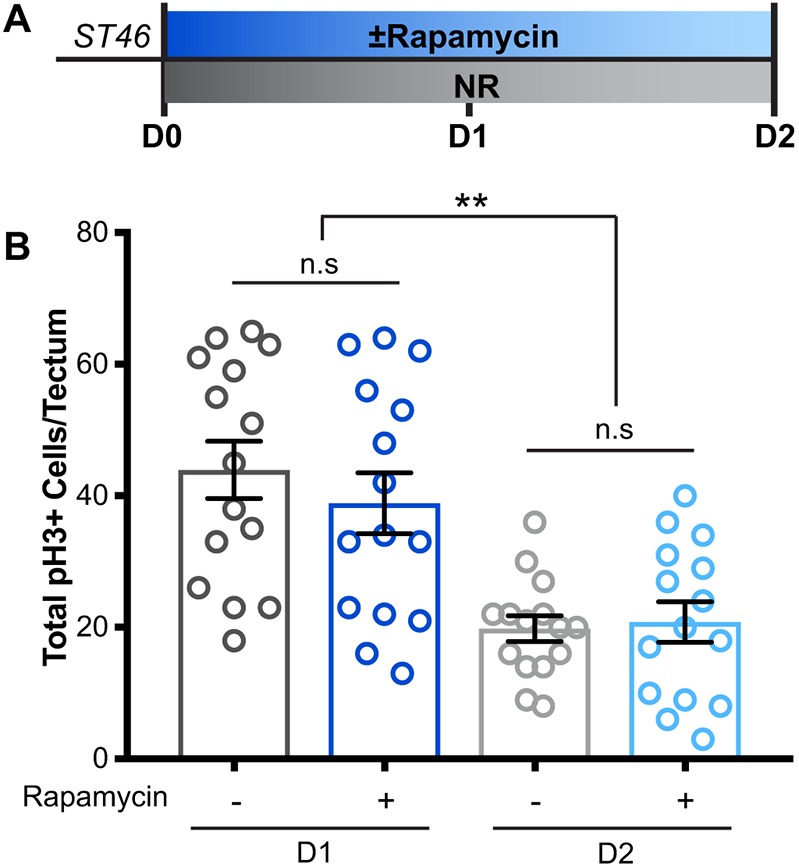
NR-induced stasis entry does not require mTOR. (A) Experimental timeline. Stage 46 animals were treated with rapamycin prior to the need for external food, during the nutrient-restriction period at the onset of stasis. Controls were treated with DMSO vehicle. Brains were processed for pH3 immunolabeling 1 and 2 days after rapamycin treatment. (B) Quantification of pH3+ cells showing that cell division decreases, and animals enter stasis even in the presence of the mTOR-blocking drug rapamycin. n=15 animals per group for a total of 60 animals across 4 groups from multiple independent clutches; see Table S1. ***P<0.01. n.s., not significant.
Nutrients trigger direct M-phase entry
In response to NR, the animal enters developmental stasis during which NPC proliferation is drastically decreased. Cells in the tectum resume proliferation 16 h after the re-introduction of food (Fig. 2). The cell cycle at this stage in Xenopus tectal NPCs is approximately 24 h (Fig. 5A), with G2 lasting 8 h in the normally fed condition (Fig. S4) (Herrgen and Akerman, 2016; Sharma and Cline, 2010). To investigate further how nutrient availability affects NPC progression through the cell cycle, we performed a series of CldU-pH3 pulse-chase experiments to label cells in S phase and M phase in the same animal. Animals were nutrient restricted for 2 days and then provided food or not. During the first hour of the ±food period, all animals were given a 1-h pulse of CldU to sparsely label cells in S phase. Animals from this treatment regime were sacrificed 1, 2, 4, 8, 16, 24 and 48 h later and processed for CldU and pH3 immunolabeling, as shown in the protocol in Fig. 5B. CldU+ and pH3+ cells were counted throughout the entire z-series of the tectum. Consistent with our earlier findings, nutrients induce a burst of pH3+ cells at 16 h after feeding (compare Fig. 2C and Fig. 5C, green). Surprisingly, nutrients only induce a significant increase in CldU+ cells at 24 h after feeding (Fig. 5C, red), consistent with a 24 h cell cycle. These data indicate that nutrients trigger NPCs to enter M phase prior to S phase, suggesting that NR-induced stasis causes NPCs to enter G2 arrest.
Fig. 5.

Nutrients trigger direct M-phase entry. (A) Cell cycle schematic with labeling methods indicated. The cell cycle in Xenopus neural progenitors at these stages is ∼24 h. CldU is incorporated into DNA during S phase (red) and PH3 is a terminal label of cells in M phase (green). (B) Experimental timeline. Stage 47 animals were subjected to 2 days of nutrient restriction and then treated with a 1 h pulse of CldU with or without food. Animals were sacrificed at 1, 2, 4, 8, 12, 16 and 24 h after feeding and brains were fixed and processed for CldU and pH3 immunolabeling. (C) Quantification of CldU+ (red) or pH3+ (green) cells from immunofluorescent z-stacks of the optic tectum at designated time points represented as a time course. Fd, food. n=7-16 animals per group for a total of 150 total animals across 15 groups from multiple independent clutches; see Table S1. ****P<0.0001.
To test further whether nutrients cause cells to re-enter the cell cycle directly in M phase, animals were nutrient restricted for 2 days and then treated with CldU for 24 h, the entire duration of the cell cycle (Herrgen and Akerman, 2016; Sharma and Cline, 2010; Fig. 5C), with or without food. At the end of the 24 h, brains were processed for CldU and pH3 immunolabeling (Fig. 6A). If nutrient-restricted NPCs arrest in G2 and nutrient availability triggers re-entry into the cell cycle directly into M phase, we predict we would detect pH3+ cells that are CldU−, even after 24 h of labeling. We found that in the absence of food, few cells were pH3+ and almost all pH3+ cells were also CldU+ (Fig. 6B-E,J,K). The number of pH3+CldU+ NPCs that remained in the cell cycle showed a small but significant decrease in the NR animals (Fig. 6J, yellow). These data indicate that although cell division is low in NR, a small pool of progenitors progresses through the cell cycle even in the absence of food, possibly representing a pool of self-renewing progenitors (Fig. 1D). In contrast, in the NR+food animals the majority of pH3+ cells were not labeled with CldU (Fig. 6F-I,J,K), indicating that nutrient-restricted cells resume proliferation by entering M phase directly without having gone through S phase during the prior 24 h. Moreover, even after 48 h, the CldU-labeled population failed to double in the NR brains, indicating that cells are accumulating somewhere after S phase in the absence of food (Fig. S5). Taken together, these data demonstrate that nutrient availability causes cells to exit arrest and resume proliferation, and that they do so by re-entering the cell cycle at M phase. Moreover, these data suggest that animals in stasis have NPCs that are paused during the G2 phase of the cell cycle.
Fig. 6.
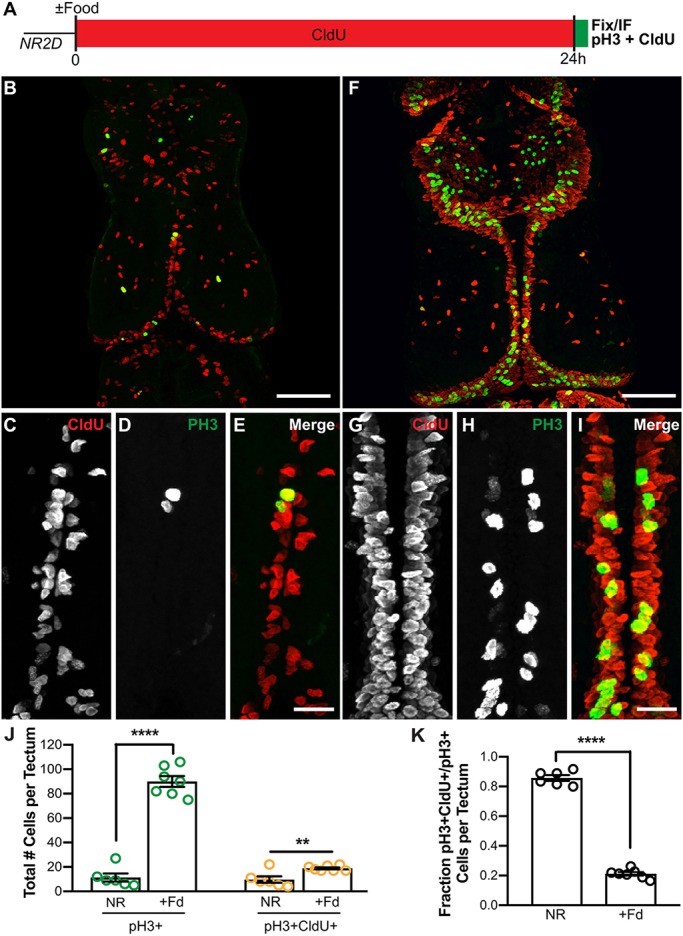
Feeding triggers NPCs to exit G2 arrest and enter the cell cycle at M phase. (A) Experimental timeline. Stage 47 animals were nutrient restricted for 2 days and then treated with CldU for 24 h with or without food. After 24 h, brains were processed for both pH3 and CldU immunolabeling. (B,F) Whole-mount confocal images of CldU (red) and pH3 (green) immunolabeling in the optic tectum in nutrient-restricted (B) and fed (F) animals. Images are 60 μm z-projections. (C-E,G-I) Digitally magnified images from the midline of the animals shown in B and F. All samples were collected under identical imaging parameters and post-processed identically. (J) Quantification of pH3+ cells (green) and pH3+CldU+ (yellow) cells in the 60 μm Z-series showing that food increases the number of pH3+ cells but not the pH3+CldU+ population. (K) Fraction of pH3+ cells that had also been previously labeled with CldU presented as average of total cells per tectum ±s.e.m. n=7 animals per group; see Table S1. **P<0.01, ****P<0.0001. Scale bars: 100 μm (B,F); 20 μm (C-E,G-I).
NR-NPCs pause in G2 with increased DNA content
Thus far, the data indicate that nutrient-restricted NPCs exit the cell cycle after S phase and upon re-introduction of food they re-enter the cell cycle just prior to M phase, suggesting that NR causes NPCs to pause in G2. If so, then NR-NPCs would have 4N DNA content during developmental stasis. We therefore tested whether the DNA content in tectal NPCs is greater in nutrient-restricted animals compared with fed animals. Because fewer than 100 NR-NPCs were induced to divide with feeding in a single tectum (Figs 2, 3, 5 and 6), we assessed DNA content by fluorescence, both on a population level with line scans and on an individual cell level. Animals were either continuously fed or nutrient restricted for 2 days and then fixed and stained for TOPRO-3, which uniformly labels DNA in a linear fashion (Martin et al., 2005; Milanovich et al., 1996). For the population analysis, confocal images through brains were collected and fluorescence intensity was measured across the midline by line scan analyses (Fig. 7A,B). Single line scan averages per animal were generated from line scans across 75 μm in the rostral-caudal axis from three different optical sections per brain, taken at identical z-depths across animals. Fig. 7C shows four examples of averaged line scans from tecta of nutrient-restricted animals and three examples of controls. TOPRO-3 fluorescence intensity shows a peak on each side of the midline, and the peaks from NR animals (blue) are about twice the intensity as those of fed animals (orange), suggesting higher DNA content in progenitors along the ventricle in NR animals (Fig. 7D). In addition, the area of fluorescent label is wider in fed animals demonstrating an increase in the number of nuclei in fed versus NR animals, consistent with an increase in proliferation in fed animals (Fig. 7E). We calculated a fluorescent index for each animal as the ratio of the peak TOPRO-3 fluorescent intensity normalized to the labeled area, to control for the increased number of cells in fed animals. The fluorescent index in tecta from nutrient-restricted animals was twice that of fed animals, indicating that the nutrient-restricted animals have roughly twice the DNA fluorescence as the fed animals, despite decreases in cell proliferation and total cell number (Fig. 7F). These data indicate that nutrient restriction increases cellular DNA content at a population level along the ventricle where NPCs reside.
Fig. 7.

Nutrient restriction increases DNA content in NPCs. (A-F) Whole tectum fluorescence intensity analysis. Brains from continuously fed (orange, A) and nutrient-restricted (blue, B) animals were stained with TOPRO-3 to label DNA. Boxes in A and B indicate the area analyzed by line scan. (C) Example of averaged line scans from four individual nutrient-restricted (blue) and three individual fed (orange) animals showing a higher peak of DNA labeling along the midline in the NR brains. (D) Average peak intensity measurements demonstrate increased fluorescence in the NR brains, consistent with increased DNA content. (E) Average area of TOPRO-3 labeling, indicating that there are more cells in the fed animals. (F) Fluorescent Index (ratio of peak intensity/area) indicates that DNA content is significantly greater in the tectal proliferative layer of nutrient-restricted animals than in fed animals. (G-I) Single cell fluorescent intensity analysis. Cell tracing of progenitor cells along the midline in fed (G) and nutrient-restricted (H) animals. Individually traced cells are outlined in yellow. Asterisk indicates an example neuronal cell body adjacent to the neuropil that was traced for baseline 2N DNA content. (I) Fluorescence/cell area ratio indicates that DNA content is higher in the progenitor population than in neurons (gray), and DNA content is further increased in NR-NPCs (blue) compared with fed NPCs (orange). For A-F, n=16-17 animals per group for a total of 33 animals from 2 independent clutches. For G-I, n=10 animals per group (50 cells per animal) for a total of 500 cells measured per condition. See Table S1 and Materials and Methods. ****P<0.0001. Scale bars: 100 μm (A,B); 20 μm (G,H).
To test further the DNA content of NR-NPCs, fluorescence intensity was measured from individual cell nuclei. NPCs were traced along the ventricle in single optical sections from continuously fed and NR animals (Fig. 7G,H), and compared with neurons as a control for 2N DNA-containing cells (Fig. 7G, asterisk). Although both groups of NPCs had increased fluorescence intensity over the mature neuron control cells, the NR-NPCs had an even higher fluorescence intensity at a cellular level than the fed NPCs (Fig. S6A). On average, Xenopus tectal NPCs in the NR animals were smaller than both fed NPCs and neurons (Fig. S6C), consistent with published reports in other species (Lloyd, 2013; Martin and Hall, 2005; Pérez-Hidalgo and Moreno, 2016), To correct for cell size, we calculated a ratio of fluorescence intensity over area, which demonstrates an increase in DNA content in NR-NPCs compared with both the fed NPCs and mature 2N neurons (Fig. 7I). Using the neuronal measurements to define a range for 2N-containing cells, we found an increase in the number of NR-NPCs that contained a higher than 2N fluorescent DNA content compared with the fed NPCs, indicating that there are more 4N-containing cells in the NR animals than in the fed animals, despite the fed animals having more cells actively dividing (Fig. 7I, Fig. S6A,B). These data demonstrate that tectal progenitors lining the ventricle in NR animals have increased DNA content compared with both mature neurons and NPCs in the fed animals. The increased DNA content in non-dividing NR progenitors further indicates that tectal progenitor cells pause at G2 in nutrient-restricted animals, leaving NPCs poised to re-enter the cell cycle when nutrients become available again.
Activation of insulin receptor signaling is sufficient to drive cell division of G2-arrested cells in the absence of nutrients
Data presented thus far indicate that nutrient-restricted NPCs are paused in G2, by an mTOR-dependent mechanism. In many systems, mTOR signaling is activated downstream of the insulin receptor (Laplante and Sabatini, 2009; Lee, 2015; Loewith and Hall, 2011; Rafalski and Brunet, 2011). Insulin has been identified in the tadpole pancreas at this stage (Shuldiner et al., 1991), and our previous studies have shown that the insulin receptor (InsR) is present and active in the tectum (Chiu et al., 2008). To investigate further the mechanisms underlying G2 arrest in NPCs in the absence of nutrients, we tested whether activation of the insulin signaling pathway could drive cell division of nutrient-restricted cells using the insulin-mimetic drug bPV(phen) (Bevan et al., 1995; Love et al., 2014; Posner et al., 1994). Animals were subjected to nutrient restriction for several days and then bPV(phen) or vehicle was injected directly into the brain ventricle (Fig. 8A). Animals were sacrificed and processed for pH3 immunofluorescence at time points from 90 min to 16 h after injection and cells in M phase were counted. Control animals were nutrient restricted without subsequent treatment or were fed ad libitum (Fig. 8B-D). Activation of insulin signaling by direct brain injection of the insulin mimetic bPV(phen) increased cell division by 4 h after injection in the absence of any external food (Fig. 8D,E). This level of proliferation was indistinguishable from that of clutch mates that were also nutrient restricted and then fed for 16 h (Fig. 8C,E), indicating that activation of insulin signaling is sufficient to drive cells out of G2 arrest and into mitosis. This 4 h time window to cell division is rapid compared with the 16 h required by feeding, indicating that the majority of the 16 h delay observed earlier is indeed due to the requirement that the animal find, consume, digest and metabolize the food. Importantly, the appearance of the M-phase marker pH3 at just 4 h after injection further supports the hypothesis that NR-NPCs had previously been paused in the G2 phase of the cell cycle. Indeed, 4 h prior to M phase would still find the cell in G2. A shorter 90 min period of bPV(phen) treatment did not increase cell proliferation in NR animals above baseline levels of NR and vehicle injections, though this does not rule out a peak time point between 1.5 and 4 h (Fig. 8E). Moreover, these data show that InsR activation is sufficient to reverse G2 arrest in nutrient-restricted NPCs; however, whether it is necessary remains unknown. Nevertheless, nutrient-restricted NPCs arrest in G2 and can be driven back into M phase by activation of InsR signaling in the absence of food. Taken together, these data demonstrate that Xenopus NPCs enter an mTOR-dependent reversible state of G2 arrest in response to nutrient restriction that is likely to be downstream of the insulin receptor.
Fig. 8.

Activation of insulin signaling is sufficient to push G2-arrested cells back into the cell cycle in the absence of nutrients. (A) Experimental timeline. Animals were nutrient restricted for 2 days and then 300 nM bPV(phen), or H2O control, was injected directly into the tectal ventricle. Brains were fixed and processed for pH3 immunofluorescence at 90 min and 4 h after injection. Control animals were nutrient restricted for 2 days and then fed for 16 h prior to fixation, as shown in the lower timeline. (B-D) Confocal images of pH3-labeled tecta from injected animals at 4 h and a control group that was given food to induce proliferation. (E) Quantification of pH3 labeling showing total cell counts per tectum in each condition (error bars represent s.e.m.). Gray data points are from NR and H2O-injected animals. Purple data points indicate bPV(phen) animals, and green data points represent fed controls. n=6-39 animals per group per time point for a total of 160 animals across 16 groups from multiple independent clutches, see Table S1. Fd, food. ****P<0.0001. n.s., not significant. Scale bar: 100 μm.
DISCUSSION
Cell proliferation in the brain demands significant energy consumption and is sensitive to nutrient restriction. Developing organisms are particularly sensitive to alterations in nutrient availability (Georgieff, 2007; Georgieff et al., 2015; Metcalfe and Monaghan, 2001), yet how NPCs in the vertebrate brain sense and respond to nutrient availability is not clear. We sought to uncover the cellular mechanisms underlying nutrient-responsive whole-animal stasis in the developing Xenopus laevis brain. We demonstrated that during stasis NPCs in tadpole brains arrest in the G2 phase of the cell cycle with increased DNA content upon nutrient restriction, and that re-introduction of food triggers cell cycle re-entry at M phase. Exit from G2 arrest requires mTOR signaling and NPCs can be forced out of G2 arrest by activating the insulin receptor signaling pathway. Our study is the first to describe a reversible G2 arrest in the vertebrate brain in vivo.
Nutrient restriction blocks neural progenitor cell proliferation
Nutrient restriction severely reduces NPC proliferation in the developing Xenopus brain, resulting in a significant decrease in the size of the brain and reduction in tadpole development as a whole (McKeown et al., 2017). Re-introduction of food causes a rapid and robust recovery of proliferation, suggesting that developmental stasis results from a reversible cell cycle arrest. Medaka fishes also exhibit reversible cell cycle arrest in response to environmental cues when exposed to cold temperatures; however, in this case each cell arrests in whatever stage of the cell cycle it happens to be in at the time of exposure to cold (Sampetrean et al., 2009). We demonstrated that nutrient-restricted Xenopus tectal progenitors synchronously enter M phase 16 h after the animal resumes eating. The synchronous progression to M phase after feeding produced an almost tenfold increase in proliferation, shown with the M-phase marker pH3, in vast excess of the expected doubling seen at 24 h with the S-phase marker CldU (Fig. 5). Interestingly, even in the absence of food, a small population of cells still progressed through the cell cycle. We propose that NR causes NPCs to become quiescent and that upon re-introduction of food these quiescent progenitor cells enter M phase en masse. We further propose that these nutrient-responsive cells are expanding progenitor cells, responsible for populating the brain with neurons (Fig. 1C), and that the basally dividing cells are self-renewing progenitors, which are relatively insensitive to nutrient status. These data support a model for cell cycle arrest during developmental stasis, in which a small number of self-renewing progenitors are protected from environmental stress, whereas the vast majority of nutrient-responsive NPCs are poised to resume proliferation and populate the brain with neurons upon the re-introduction of food.
Nutrient restriction arrests Xenopus neural progenitors in G2 phase of the cell cycle
We demonstrated that during stasis, Xenopus NPCs halt cell division but quickly resume proliferation upon re-introduction of food. This finding suggests that during stasis, NPCs are arrested in the cell cycle. Canonical cell cycle checkpoints are obvious candidates for the temporary block in cell division seen in these cells. We reasoned that cells were not arresting at the G1 ‘restriction’ checkpoint because cells stopped at this checkpoint are thought to either exit the cell cycle completely or enter G0 and terminally differentiate (Barnum and O'Connell, 2014), neither of which is consistent with our findings that arrested NPCs divide after feeding. Although, a recent study shows that Drosophila embryonic neuroblasts arrested in G0 can resume proliferation in response to feeding (Otsuki and Brand, 2018), data presented here argue against a model for G0 arrest during Xenopus stasis. The metaphase or ‘spindle checkpoint’ was dismissed because of the 24 h doubling of CldU+ cells after feeding, which ruled out problems with daughter cell separation at the spindle checkpoint. We considered the recently described S/G2 checkpoint (Saldivar et al., 2018); however, problems at this checkpoint result in premature mitosis, which is opposite to our observations. Our findings that arrested Xenopus NPCs have 4N DNA content and enter M phase directly upon re-introduction of food without having gone through S phase in the previous 24 h (Figs 6 and 7) indicate that the cells are likely blocked during G2. Although it is possible that NR-NPCs are stuck at the G2/M checkpoint, traditionally the DNA-damage checkpoint, this checkpoint has not been reported to be reversible and results in cell death (Barnum and O'Connell, 2014; Shaltiel et al., 2015; Stark and Taylor, 2004), and we do not observe any increase in cell death during Xenopus stasis (McKeown et al., 2017).
Why might NPCs arrest at G2 in response to NR when other cell cycle checkpoints more typically halt division? One possibility is that cells in G2 may be relatively protected from environmental insult and resulting cell death. This has been demonstrated in Hydra and in mammalian cells in culture, where G2-arrested cells are resistant to pro-apoptotic agents (Buzgariu et al., 2014; Harper et al., 2010; Reiter et al., 2012). On the other hand, the majority of programmed cell death occurs as a result of the DNA-damage checkpoint at the G2/M transition, so lengthening G2 may allow for DNA repair in cells that were nutritionally stressed during S-phase replication (Shaltiel et al., 2015). Nonetheless, by arresting at G2, NPCs are uniquely poised to initiate mitosis rapidly once nutrients are available. This mechanism of cell cycle modulation during G2 has been employed in several developmental systems, allowing for bursts and waves of synchronous division (Ogura et al., 2011; Ogura and Sasakura, 2016; Ueno et al., 2011). Indeed, G2 quiescence has been described in the developing Drosophila nervous system, when external nutrients are naturally unavailable (Chell and Brand, 2010; Egger et al., 2008; Otsuki and Brand, 2018; Spéder and Brand, 2014). In our model of nutrient restriction, accumulation of G2-arrested NPCs and the burst of cell proliferation once food is available is expected to increase newly generated neurons that incorporate into brain circuits. This may be a significant advantage, allowing the animal to ‘catch up’ in brain development. Additionally, newborn neurons have high energy requirements (Birket et al., 2011; Ochocki and Simon, 2013; Rafalski and Brunet, 2011; Rafalski et al., 2012; Vander Heiden et al., 2009), so arresting in G2 until nutrient conditions are more favorable could ensure successful division and circuit assembly. Our finding that NR-NPCs have decreased cell size points to a change in energy consumption consistent with decreased mTOR signaling, although, in Drosophila, G2-arrested neuroblasts have an increased cell size (Otsuki and Brand, 2018), indicating that that further investigation is necessary. Interestingly, in the chick retina, progenitors committed to generate horizontal cells arrest in G2 prior to their terminal mitosis (Boije et al., 2009); however, the continued doubling in CldU+ cells at 24 and 48 h seen in our system (Figs 2 and 5) distinguished this type of G2 arrest from our observations in the tadpole brain. Nonetheless, reversible G2 arrest in Xenopus neural progenitors allows for a mechanism by which a developing animal can adapt during times of environmental uncertainty, quickly recovering proliferative capacity to continue neuronal development.
mTOR and insulin receptor operate in the nutrient-sensing pathway
This study begins to dissect the underlying molecular mechanisms by which neuronal progenitor cells become G2 arrested in response to a lack of external nutrients. The process by which nutrient-restricted tectal progenitors enter arrest does not depend on mTOR signaling, but mTOR is required for cells to exit cell cycle arrest and proliferate. Although mTOR signaling has been well-studied in promoting G1 phase of the cell cycle across species (Laplante and Sabatini, 2009; Proud, 2010), the role of mTOR during G2/M progression is less understood and the data appear contradictory. In developing Drosophila imaginal discs, mTOR activation inhibits the G2/M transition and blocking mTOR with rapamycin accelerates M-phase entry and proliferation (Wu et al., 2007). However, in yeast and mammals, mTOR activity promotes cell cycle progression through G2/M phases and rapamycin blocks cell division at the G2/M transition (Garelick and Kennedy, 2011; Ramirez-Valle et al., 2010). Our study shows that Xenopus neural progenitors exiting G2 arrest are similar to yeast and mammals in that mTOR is required for cells to progress through G2/M.
mTOR activity is downstream of several nutrient-sensing signaling pathways, including amino acid-sensing G protein-coupled receptors, stress receptors, glucose transporters and insulin receptor signaling (Ochocki and Simon, 2013; Proud, 2004; Rafalski and Brunet, 2011). In both C. elegans and Drosophila, insulin signaling plays a role in developmental arrest (Baugh, 2013; Chell and Brand, 2010; Colombani et al., 2012; Munoz and Riddle, 2003; Spéder and Brand, 2014), suggesting that insulin signaling may play a conserved role in species that undergo stasis. In contrast, in an adult zebrafish model of hyperglycemia, stem cells in the midbrain stop proliferating (Dorsemans et al., 2017), suggesting that the capacity for modulating nutrient adaptability decreases with development. We tested whether insulin receptor signaling could act as a nutrient-sensor upstream of mTOR-dependent stasis. We found that activating the insulin receptor signaling pathway with the drug bPV(phen) is sufficient to drive cells out of G2 arrest and into M phase even in the absence of food. Although the insulin mimetic bPV(phen) has also been reported to act as a PTEN inhibitor (Bevan et al., 1995; Posner et al., 1994), the increase in p-PTEN in our NR animals (Fig. S3) suggests that PTEN is already inhibited in NR-NPCs and bPV(phen) is not acting in this capacity in our system. Activation of InsR signaling by bPV(phen) is sufficient to pull NR-NPCs out of G2 arrest; however, whether InsR is necessary for this process remains unknown. InsR signaling is just one of multiple pathways that converge onto mTOR, including glucose, stress and amino acid-sensing receptors (Jacinto and Hall, 2003; Manning and Toker, 2017; Martin and Hall, 2005; Ochocki and Simon, 2013; Proud, 2004; Rafalski and Brunet, 2011; Rafalski et al., 2012; Reiling and Sabatini, 2006). In Drosophila, amino acids have been shown to be required for neuroblasts to exit cell cycle arrest (Britton and Edgar, 1998; Britton et al., 2002; Chell and Brand, 2010; Lee et al., 2014; Nässel et al., 2015; Sousa-Nunes et al., 2011; Spéder and Brand, 2014). Future studies are required to determine what specific component of the food is responsible for NR-NPCs to exit stasis in the Xenopus tadpole. Our data suggest that insulin receptor signaling and mTOR cooperate in a signaling pathway to sense and respond to nutrient availability and initiate cell cycle progression. In combination with previous studies from our group showing that insulin receptor signaling is involved in tectal circuit function and neuronal plasticity (Chiu et al., 2008), these studies indicate a key role for insulin receptor signaling during tadpole brain development.
In summary, we have discovered a mechanism by which neural progenitor cells can adapt in response to nutrient availability in an awake, behaving vertebrate. Reversible G2 arrest in Xenopus neural progenitors allows for a mechanism by which a developing animal can adapt during times of environmental uncertainty, quickly recovering proliferative capacity to continue neuronal development.
MATERIALS AND METHODS
Animals and feeding protocols
All animal protocols were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute. Albino Xenopus laevis tadpoles of either sex (bred in-house or purchased from Xenopus Express, Brooksville, FL, USA, RRID:XEP_Xla200) were reared in 0.1× Steinberg's Solution or vivarium water (pH 7.0) in a 22°C incubator with a 12 h:12 h light:dark cycle. Each clutch of tadpoles is the product of a single independent breeding pair. Tadpoles were staged according to Nieuwkoop and Faber (1956). For each experiment, stage 41 tadpoles were removed from the breeding tank and placed as a group into a large bowl of 0.1× Steinberg's solution or vivarium tank water without supplemental food. Once animals reached stage 47, they were randomly divided into equal groups treated as either continuously fed animals, which were fed once a day and allowed to feed ad libitum throughout the experiment, nutrient restricted (NR) animals reared without supplemental food, or delayed feeding animals that were NR for 2 days and then fed ad libitum starting at the time points indicated in each experiment. All fed animals were given 500 μl of a 30% slurry of Xenopus Express Tadpole Food (Xenopus Express) added daily to the rearing medium as described (McKeown et al., 2017). Xenopus Express Tadpole Food is composed of a mixture of Brewer's yeast, spirulina and vitamins. All animals were anesthetized in 0.02% MS222 (3-aminobenzoic acid ethyl ester, Sigma-Aldrich) before surgical procedures, and were terminally euthanized in 0.1% MS222 at the end of the experiment. Upon dissection, animals from the fed groups that did not have visible food in their guts were not included in further analysis.
Drug treatments
mTOR was blocked with a bath solution containing 10 mM rapamycin (Sigma-Aldrich) diluted in rearing media for up to 48 h. Control animals were treated similarly with drug vehicle, 0.1% dimethyl sulfoxide (DMSO). Insulin signaling was activated with the insulin mimetic, potassium bisperoxo(1,10-phenanthroline) oxovanadate (V) hydrate referred to hereafter as bPV(phen) (Sigma-Aldrich), injected at 300 nM in H2O directly into the brain ventricle.
CldU labeling
Proliferating cells were labeled in S phase with the thymidine analog 5-chloro-2′-deoxyuridine (CldU) (MP Biomedicals). Cells were labeled by transferring animals to rearing solution containing 10 mM CldU for 1-24 h as indicated (McKeown et al., 2013; Sharma and Cline, 2010). For immunolabeling, tadpoles were terminally anesthetized in 0.1% MS222 solution and fixed with 4% paraformaldehyde in PBS (pH 7.4) overnight at 4°C. Brains were processed for immunofluorescence as described below.
Immunofluorescence labeling
Fixed tadpoles were washed with PBS and brains were dissected into PBS with 0.01% Triton X-100 (PTx). Brains were permeabilized with 0.1% Triton X-100 in PBS, and then placed in blocking buffer (4% bovine serum albumin and 1% normal goat serum in PTx) for 24 h at 4°C. Primary antibodies were incubated for 1-2 days at 4°C diluted in blocking buffer, followed by additional washes and detection with Alexa Fluor dye-conjugated secondary antibodies (1:500, Life Technologies, A21202, A21121, A11008, A21208, A11004, A11010, A21434, A21094, A21071 and A21052). For CldU labeling, samples were treated with 2 N HCl at 37°C for 10 min after permeabilization, followed by three additional PTx washes prior to blocking. For HuC/D labeling, an epitope retrieval step of 10 mM citrate buffer pH 6.0 for 10 min in boiling water was added after permeabilization, followed by three additional PTx washes prior to blocking. For nuclear labeling, TOPRO-3 (1:1000, Life Technologies, T3605) was added in lieu of primary antibodies for 15 min at room temperature and then brains were washed three times in PTx. All samples were mounted with Fluoroshield Gel Mount (Accurate Chemical). Primary antibodies used for immunolabeling were rat anti-CldU [BU1/75(ICR1) monoclonal, OBT0030G, Accurate Chemical, RRID:AB_609567, 1:500], rabbit anti-Sox2 (D6D9 monoclonal, 3579, Cell Signaling Technology, RRID:AB_2195767, 1:400), mouse anti-HuC/D (16A11 monoclonal, ThermoFisher, A21271, RRID:AB_221448, 1:500), rabbit anti-phospho-histone H3 (ser10) (D2C8 monoclonal, 3475, Cell Signaling Technology, RRID:AB_10694639, 1:1000) and rabbit anti-phospho-ribosomal S6 (polyclonal 07-433, EMD, RRID:AB_310612, 1:500).
Microscopy and presentation
All samples within an experiment were prepared, imaged and analyzed in parallel using identical acquisition and analysis settings. For whole-mount immunofluorescence, confocal stacks were collected on an Olympus Fluoview FV500 laser-scanning confocal microscope equipped with 20×/0.8NA and 60×/1.42NA oil-immersion objectives. Brains were imaged to 60 μm depth starting just below the dorsal surface of the skin. Images were acquired under identical settings within an experiment and pixel saturation was minimal during image acquisition. No post-acquisition alterations were made to the intensity or levels of any images. Analysis was performed in the z-dimension but for presentation purposes, maximum intensity z-projections were made using ImageJ, where indicated. Figures were compiled using Adobe Illustrator.
Cell counting
For cell counting, the anterior border of the tectum was defined as the anterior commissure, and the posterior border was defined as the caudal extent of the third ventricle. We counted the number of labeled cells from optical stacks of whole imaged tecta in ImageJ using the same borders defined for the largest cross-sectional area from 51 optical sections (60 μm in depth), starting from the most dorsal portion of the brain. pH3 only labels nuclei in M phase, so any background is non-specific or due to minor secondary antibody binding. All pH3+ cells in the tectum were included. CldU is incorporated into the DNA during S phase, which results in more variability in levels of labeling. Thus, for CldU+ cell counts, cells that met a threshold fluorescence intensity of at least three times the intensity of the background tissue were included.
DNA content analysis
All animals were imaged under identical conditions on a Fluoview500 laser scanning microscope. Imaging parameters were established to avoid saturation. Animals were labeled with TOPRO-3 DNA dye, which has been shown to stain DNA linearly and accurately report DNA content in the far-red channel (Martin et al., 2005; Milanovich et al., 1996). For line scan analysis, a region of interest (ROI) box of 500 μm wide and 75 μm deep was centered on the midline of the tectum just caudal to the anterior commissure at a depth of approximately 18 μm beneath the surface of the tectum. ImageJ was used to determine pixel intensities across the ROI in the y-dimension and report average fluorescence intensity for the optical section: at 1.24 μm/pixel resolution, a 75 μm ROI box will contain 60 individual line scans, the average of which is reported in the shown traces. To control for variations in penetration of the dye and photobleaching, three different optical sections were measured for each animal within a 10 μm z-depth ventral from the first. Both average peak intensity and area under the curve were determined from each series of line scans. The Fluorescent Index was defined as the ratio of the average peak intensity to the total fluorescent area within the ROI per animal. Two different experiments from two different clutches were pooled (normalized and determined to be not significantly different; area under curve measurements determined not to be significantly different as raw numbers and pooled). n=16 NR animals, n=17 fed animals, (average of three optical sections per animal with 60 line scans per optical section averaged). For individual cell analysis, cell outlines from a single optical section at identical z-depths were highlighted with a watershed mask and then individually traced in ImageJ. Fluorescence intensity for each individual cell trace was calculated as a Raw Internal Density in ImageJ. Because the tectum is a complex three-dimensional tissue, nuclear outlines in a single optical section could represent any z-depth of a given cell, so to control for cell size we measured the area of each cross-sectional tracing and calculated a fluorescence/area ratio. Fifty cells were traced per animal, 45 cells lining the ventricle where the progenitors reside and five cells at the periphery of the cell body layer adjacent to the neuropil where mature neurons reside. Ten brains from both NR and fed groups were analyzed for a total of 450 progenitors from each group. Fluorescence measurements from post-mitotic neurons were considered to be the baseline range for 2N DNA.
Western blots
Whole brains were dissected and immediately frozen on dry ice and stored at −80°C (n=5 brains per treatment per time point). Samples were thawed on ice in RIPA buffer plus protease inhibitor cocktail (1× Roche Complete), supplemented with phosphatase inhibitors NaFl (10 mM) and NaVO3 (1 mM), and homogenized. Protein concentrations were determined by BCA reaction read in triplicate on a Nanodrop spectrophotometer. Proteins were denatured by boiling in Laemmli sample buffer and 2 μg were loaded per well. Proteins were separated on 4-20% gradient SDS-PAGE and transferred to nitrocellulose. Primary antibodies used were rabbit anti-mTOR (7C10 monoclonal, 2983, Cell Signaling Technology, RRID:AB_2105622, 1:1000), rabbit anti-phospho-mTOR (ser2448) (polyclonal, 2971, Cell Signaling Technology, RRID:AB_330970, 1:1000), rabbit anti-phospho-ribosomal S6 (polyclonal 07-433, EMD, RRID:AB_310612, 1:1000), rabbit anti-phospho-PTEN (S380) (polyclonal, 9551, Cell Signaling Technology, RRID:AB_331407, 1:1000), rabbit anti-phospho-AKT (S473) (D9E monoclonal, 4060, Cell Signaling Technology, RRID:AB_2315049, 1:1000), rabbit anti-panAKT (C67E7 monoclonal, 4691, RRID:AB_915783, 1:1000), and mouse anti-actin (C4 monoclonal MAB1501, Millipore, RRID:AB_2223041, 1:5000). HRP-conjugated secondary antibodies (BioRad, 172-1011 and 172-1019; 1:5000) were used for detection, and bands were visualized by enhanced chemiluminescence. Densitometry measurements were performed in ImageJ. In addition to loading equal amounts of protein, bands were normalized to total protein levels determined by Ponceau S staining and further normalized to a second antibody loading control, where indicated. A minimum of three biological replicates was used for each quantification shown.
Statistical analyses
To analyze data for statistical differences between multiple groups in Figs 2, 3, 4, 5, Figs S4 and S5, we used analysis of variance (ANOVA), and post-hoc comparisons across treatment groups were made with Tukey's multiple comparisons test. For pairwise comparisons between two groups in Figs 6, 7 and Fig. S6, two-tailed non-parametric t-tests were used. For pairwise comparisons of normalized western blot data in Fig. S3, the Wilcoxon signed-rank test was used. Data from experimental replicates of independent clutches were pooled when shown to be normally distributed and not significantly different by a two-tailed non-parametric t-test. Data are represented as mean±s.e.m. Statistical analyses and graph presentations were performed with Prism 8.1 (GraphPad Software). Details of statistical analyses are summarized in Table S1.
Supplementary Material
Acknowledgements
We thank Natalie McLain for animal maintenance and Evan Fitchett for assistance with western blots. We are grateful to members of the Cline lab, both past and present, for helpful discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: C.R.M., H.T.C.; Methodology: C.R.M., H.T.C.; Validation: C.R.M.; Formal analysis: C.R.M.; Investigation: C.R.M.; Writing - original draft: C.R.M.; Writing - review & editing: C.R.M., H.T.C.; Visualization: C.R.M.; Supervision: H.T.C.; Funding acquisition: H.T.C.
Funding
This research is supported by National Institutes of Health grants (EY011261 and EY027437), Dart NeuroScience LLC, and an endowment from the Hahn Family Foundation to H.T.C. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.178871.supplemental
References
- Acosta-Jaquez H. A., Keller J. A., Foster K. G., Ekim B., Soliman G. A., Feener E. P., Ballif B. A. and Fingar D. C. (2009). Site-specific mTOR phosphorylation promotes mTORC1-mediated signaling and cell growth. Mol. Cell. Biol. 29, 4308-4324. 10.1128/MCB.01665-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agathocleous M. and Harris W. A. (2013). Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol. 23, 484-492. 10.1016/j.tcb.2013.05.004 [DOI] [PubMed] [Google Scholar]
- Agathocleous M., Love N. K., Randlett O., Harris J. J., Liu J., Murray A. J. and Harris W. A. (2012). Metabolic differentiation in the embryonic retina. Nat. Cell Biol. 14, 859-864. 10.1038/ncb2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriola Apelo S. I. and Lamming D. W. (2016). Rapamycin: An InhibiTOR of Aging Emerges From the Soil of Easter Island. J. Gerontl. A Biol. Sci. Med. Sci. 71, 841-849. 10.1093/gerona/glw090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnum K. J. and O'Connell M. J. (2014). Cell cycle regulation by checkpoints. Methods Mol. Biol. 1170, 29-40. 10.1007/978-1-4939-0888-2_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh L. R. (2013). To grow or not to grow: nutritional control of development during Caenorhabditis elegans L1 arrest. Genetics 194, 539-555. 10.1534/genetics.113.150847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh L. R., DeModena J. and Sternberg P. W. (2009). RNA pol II accumulates at promoters of growth genes during developmental arrest. Science 324, 92-94. 10.1126/science.1169628 [DOI] [PubMed] [Google Scholar]
- Benítez-Santana T., Simion M., Corraze G., Médale F. and Joly J.-S. (2017). Effect of nutrient availability on progenitor cells in zebrafish (Danio Rerio). Dev. Neurobiol. 77, 26-38. 10.1002/dneu.22406 [DOI] [PubMed] [Google Scholar]
- Bestman J. E. and Cline H. T. (2008). The RNA binding protein CPEB regulates dendrite morphogenesis and neuronal circuit assembly in vivo. Proc. Natl. Acad. Sci. USA 105, 20494-20499. 10.1073/pnas.0806296105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestman J. E., Lee-Osbourne J. and Cline H. T. (2012). In vivo time-lapse imaging of cell proliferation and differentiation in the optic tectum of Xenopus laevis tadpoles. J. Comp. Neurol. 520, 401-433. 10.1002/cne.22795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestman J. E., Huang L.-C., Lee-Osbourne J., Cheung P. and Cline H. T. (2015). An in vivo screen to identify candidate neurogenic genes in the developing Xenopus visual system. Dev. Biol. 408, 269-291. 10.1016/j.ydbio.2015.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan A. P., Drake P. G., Yale J.-F., Shaver A. and Posner B. I. (1995). Peroxovanadium compounds: biological actions and mechanism of insulin-mimesis. Mol. Cell. Biochem. 153, 49-58. 10.1007/BF01075918 [DOI] [PubMed] [Google Scholar]
- Birket M. J., Orr A. L., Gerencser A. A., Madden D. T., Vitelli C., Swistowski A., Brand M. D. and Zeng X. (2011). A reduction in ATP demand and mitochondrial activity with neural differentiation of human embryonic stem cells. J. Cell Sci. 124, 348-358. 10.1242/jcs.072272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boije H., Edqvist P.-H. D. and Hallböök F. (2009). Horizontal cell progenitors arrest in G2-phase and undergo terminal mitosis on the vitreal side of the chick retina. Dev. Biol. 330, 105-113. 10.1016/j.ydbio.2009.03.013 [DOI] [PubMed] [Google Scholar]
- Bolduc F. V., Lau A., Rosenfelt C. S., Langer S., Wang N., Smithson L., Lefebvre D., Alexander R. T., Dickson C. T., Li L. et al. (2016). Cognitive enhancement in infants associated with increased maternal fruit intake during pregnancy: results from a birth cohort study with validation in an animal model. EBioMedicine 8, 331-340. 10.1016/j.ebiom.2016.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouldin C. M. and Kimelman D. (2014). Cdc25 and the importance of G2 control: insights from developmental biology. Cell Cycle 13, 2165-2171. 10.4161/cc.29537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressy C., Droby G. N., Maldonado B. D., Steuerwald N. and Grdzelishvili V. Z. (2019). Cell cycle arrest in G2/M phase enhances replication of interferon-sensitive cytoplasmic RNA viruses via inhibition of antiviral gene expression. J. Virol. 93, e01885-18 10.1128/JVI.01885-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton J. S. and Edgar B. A. (1998). Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development 125, 2149-2158. [DOI] [PubMed] [Google Scholar]
- Britton J. S., Lockwood W. K., Li L., Cohen S. M. and Edgar B. A. (2002). Drosophila's insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev. Cell 2, 239-249. 10.1016/S1534-5807(02)00117-X [DOI] [PubMed] [Google Scholar]
- Brown A. S. and Susser E. S. (2008). Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr. Bull. 34, 1054-1063. 10.1093/schbul/sbn096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzgariu W., Crescenzi M. and Galliot B. (2014). Robust G2 pausing of adult stem cells in Hydra. Differentiation 87, 83-99. 10.1016/j.diff.2014.03.001 [DOI] [PubMed] [Google Scholar]
- Chantranupong L., Wolfson R. L. and Sabatini D. M. (2015). Nutrient-sensing mechanisms across evolution. Cell 161, 67-83. 10.1016/j.cell.2015.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chell J. M. and Brand A. H. (2010). Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell 143, 1161-1173. 10.1016/j.cell.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S.-L., Chen C.-M. and Cline H. T. (2008). Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 58, 708-719. 10.1016/j.neuron.2008.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline H. T. (2001). Dendritic arbor development and synaptogenesis. Curr. Opin. Neurobiol. 11, 118-126. 10.1016/S0959-4388(00)00182-3 [DOI] [PubMed] [Google Scholar]
- Cloetta D., Thomanetz V., Baranek C., Lustenberger R. M., Lin S., Oliveri F., Atanasoski S. and Ruegg M. A. (2013). Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J. Neurosci. 33, 7799-7810. 10.1523/JNEUROSCI.3294-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani J., Andersen D. S. and Leopold P. (2012). Secreted peptide Dilp8 coordinates Drosophila tissue growth with developmental timing. Science 336, 582-585. 10.1126/science.1216689 [DOI] [PubMed] [Google Scholar]
- Davy C. and Doorbar J. (2007). G2/M cell cycle arrest in the life cycle of viruses. Virology 368, 219-226. 10.1016/j.virol.2007.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPaola R. S. (2002). To arrest or not to G2-M cell-cycle arrest. Clin. Cancer Res. 8, 3311-3314. [PubMed] [Google Scholar]
- Dorsemans A. C., Soulé S., Weger M., Bourdon E., Lefebvre d'Hellencourt C., Meilhac O. and Diotel N. (2017). Impaired constitutive and regenerative neurogenesis in adult hyperglycemic zebrafish. J. Comp. Neurol. 525, 442-458. 10.1002/cne.24065 [DOI] [PubMed] [Google Scholar]
- Duursma A. M. and Cimprich K. A. (2010). Checkpoint recovery after DNA damage: a rolling stop for CDKs. EMBO Rep. 11, 411-412. 10.1038/embor.2010.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger B., Chell J. M. and Brand A. H. (2008). Insights into neural stem cell biology from flies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 39-56. 10.1098/rstb.2006.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald R. C., Van Keuren-Jensen K. R., Aizenman C. D. and Cline H. T. (2008). Roles of NR2A and NR2B in the development of dendritic arbor morphology in vivo. J. Neurosci. 28, 850-861. 10.1523/JNEUROSCI.5078-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingar D. C., Richardson C. J., Tee A. R., Cheatham L., Tsou C. and Blenis J. (2003). mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 24, 200-216. 10.1128/MCB.24.1.200-216.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garelick M. G. and Kennedy B. K. (2011). TOR on the brain. Exp. Gerontol. 46, 155-163. 10.1016/j.exger.2010.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieff M. K. (2007). Nutrition and the developing brain: nutrient priorities and measurement. Am. J. Clin. Nutr. 85, 614S-620S. 10.1093/ajcn/85.2.614S [DOI] [PubMed] [Google Scholar]
- Georgieff M. K., Brunette K. E. and Tran P. V. (2015). Early life nutrition and neural plasticity. Dev. Psychopathol 27, 411-423. 10.1017/S0954579415000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandel H., Kaslin J., Ganz J., Wenzel I. and Brand M. (2006). Neural stem cells and neurogenesis in the adult zebrafish brain: origin, proliferation dynamics, migration and cell fate. Dev. Biol. 295, 263-277. 10.1016/j.ydbio.2006.03.040 [DOI] [PubMed] [Google Scholar]
- Haas K., Li J. and Cline H. T. (2006). AMPA receptors regulate experience-dependent dendritic arbor growth in vivo. Proc. Natl. Acad. Sci. USA 103, 12127-12131. 10.1073/pnas.0602670103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. N. (2016). TOR and paradigm change: cell growth is controlled. Mol. Biol. Cell 27, 2804-2806. 10.1091/mbc.e15-05-0311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K., Yonezawa K., Weng Q.-P., Kozlowski M. T., Belham C. and Avruch J. (1998). Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 273, 14484-14494. 10.1074/jbc.273.23.14484 [DOI] [PubMed] [Google Scholar]
- Harper L. J., Costea D. E., Gammon L., Fazil B., Biddle A. and Mackenzie I. C. (2010). Normal and malignant epithelial cells with stem-like properties have an extended G2 cell cycle phase that is associated with apoptotic resistance. BMC Cancer 10, 166 10.1186/1471-2407-10-166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrgen L. and Akerman C. J. (2016). Mapping neurogenesis onset in the optic tectum of Xenopus laevis. Dev. Neurobiol. 76, 1328-1341. 10.1002/dneu.22393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi M., Santos R. A. and Cohen-Cory S. (2015). Impact of maternal n-3 polyunsaturated fatty acid deficiency on dendritic arbor morphology and connectivity of developing Xenopus laevis central neurons in vivo. J. Neurosci. 35, 6079-6092. 10.1523/JNEUROSCI.4102-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E. and Hall M. N. (2003). Tor signalling in bugs, brain and brawn. Nat. Rev. Mol. Cell Biol. 4, 117-126. 10.1038/nrm1018 [DOI] [PubMed] [Google Scholar]
- Kimura K., Usui-Ishihara A. and Usui K. (1997). G2 arrest of cell cycle ensures a determination process of sensory mother cell formation in Drosophila. Dev. Genes Evol. 207, 199-202. 10.1007/s004270050108 [DOI] [PubMed] [Google Scholar]
- Lanet E., Gould A. P. and Maurange C. (2013). Protection of neuronal diversity at the expense of neuronal numbers during nutrient restriction in the Drosophila visual system. Cell Rep 3, 587-594. 10.1016/j.celrep.2013.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M. and Sabatini D. M. (2009). mTOR signaling at a glance. J. Cell Sci. 122, 3589-3594. 10.1242/jcs.051011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau M., Li J. and Cline H. T. (2017). In vivo analysis of the neurovascular niche in the developing Xenopus brain. eNeuro 4, ENEURO.0030-17.2017 10.1523/ENEURO.0030-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. Y. (2015). Roles of mTOR signaling in brain development. Exp. Neurobiol. 24, 177-185. 10.5607/en.2015.24.3.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. P., Jang T. and Davidowitz G. (2014). Exploring the nutritional basis of starvation resistance in Drosophila melanogaster. Funct. Ecol. 28, 1144-1155. 10.1111/1365-2435.12247 [DOI] [Google Scholar]
- Lloyd A. C. (2013). The regulation of cell size. Cell 154, 1194-1205. 10.1016/j.cell.2013.08.053 [DOI] [PubMed] [Google Scholar]
- Loewith R. and Hall M. N. (2011). Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189, 1177-1201. 10.1534/genetics.111.133363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love N. K., Keshavan N., Lewis R., Harris W. A. and Agathocleous M. (2014). A nutrient-sensitive restriction point is active during retinal progenitor cell differentiation. Development 141, 697-706. 10.1242/dev.103978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning B. D. and Toker A. (2017). AKT/PKB signaling: navigating the network. Cell 169, 381-405. 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. E. and Hall M. N. (2005). The expanding TOR signaling network. Curr. Opin. Cell Biol. 17, 158-166. 10.1016/j.ceb.2005.02.008 [DOI] [PubMed] [Google Scholar]
- Martin R. M., Leonhardt H. and Cardoso M. C. (2005). DNA labeling in living cells. Cytometry A 67A, 45-52. 10.1002/cyto.a.20172 [DOI] [PubMed] [Google Scholar]
- McKeown C. R., Sharma P., Sharipov H. E., Shen W. and Cline H. T. (2013). Neurogenesis is required for behavioral recovery after injury in the visual system of Xenopus laevis. J. Comp. Neurol. 521, 2262-2278. 10.1002/cne.23283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown C. R., Thompson C. K. and Cline H. T. (2017). Reversible developmental stasis in response to nutrient availability in the Xenopus laevis central nervous system. J. Exp. Biol. 220, 358-368. 10.1242/jeb.151043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memmott R. M. and Dennis P. A. (2009). Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell. Signal. 21, 656-664. 10.1016/j.cellsig.2009.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meserve J. H. and Duronio R. J. (2017). A population of G2-arrested cells are selected as sensory organ precursors for the interommatidial bristles of the Drosophila eye. Dev. Biol. 430, 374-384. 10.1016/j.ydbio.2017.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe N. B. and Monaghan P. (2001). Compensation for a bad start: grow now, pay later? Trends Ecol. Evol. 16, 254-260. 10.1016/S0169-5347(01)02124-3 [DOI] [PubMed] [Google Scholar]
- Milan M., Campuzano S. and Garcia-Bellido A. (1996). Cell cycling and patterned cell proliferation in the wing primordium of Drosophila. Proc. Natl. Acad. Sci. USA 93, 640-645. 10.1073/pnas.93.2.640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanovich N., Suh M., Jankowiak R., Small G. J. and Hayes J. M. (1996). Binding of TO-PRO-3 and TOTO-3 to DNA: Fluorescence and hole-burning studies. J. Phys. Chem. 100, 9181-9186. 10.1021/jp9600625 [DOI] [Google Scholar]
- Munoz M. J. and Riddle D. L. (2003). Positive selection of Caenorhabditis elegans mutants with increased stress resistance and longevity. Genetics 163, 171-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nässel D. R., Liu Y. and Luo J. (2015). Insulin/IGF signaling and its regulation in Drosophila. Gen. Comp. Endocrinol. 221, 255-266. 10.1016/j.ygcen.2014.11.021 [DOI] [PubMed] [Google Scholar]
- Nieuwkoop P. D. and Faber J. (1956). Normal table of Xenopus laevis (Daudin); a systemical and chronological survey of the development from fertilized egg till the end of metamorphosis, Amsterdam: North-Holland Publishing Co. [Google Scholar]
- Ochocki J. D. and Simon M. C. (2013). Nutrient-sensing pathways and metabolic regulation in stem cells. J. Cell Biol. 203, 23-33. 10.1083/jcb.201303110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y. and Sasakura Y. (2016). Developmental control of cell-cycle compensation provides a switch for patterned mitosis at the onset of chordate neurulation. Dev. Cell 37, 148-161. 10.1016/j.devcel.2016.03.013 [DOI] [PubMed] [Google Scholar]
- Ogura Y., Sakaue-Sawano A., Nakagawa M., Satoh N., Miyawaki A. and Sasakura Y. (2011). Coordination of mitosis and morphogenesis: role of a prolonged G2 phase during chordate neurulation. Development 138, 577-587. 10.1242/dev.053132 [DOI] [PubMed] [Google Scholar]
- O'Reilly K. E., Rojo F., She Q.-B., Solit D., Mills G. B., Smith D., Lane H., Hofmann F., Hicklin D. J., Ludwig D. L. et al. (2006). mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500-1508. 10.1158/0008-5472.CAN-05-2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuki L. and Brand A. H. (2018). Cell cycle heterogeneity directs the timing of neural stem cell activation from quiescence. Science 360, 99-102. 10.1126/science.aan8795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palu R. A. S. and Thummel C. S. (2015). Linking nutrients to growth through a positive feedback loop. Dev. Cell 35, 265-266. 10.1016/j.devcel.2015.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Hidalgo L. and Moreno S. (2016). Nutrients control cell size. Cell Cycle 15, 1655-1656. 10.1080/15384101.2016.1172471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peunova N., Scheinker V., Cline H. and Enikolopov G. (2001). Nitric oxide is an essential negative regulator of cell proliferation in Xenopus brain. J. Neurosci. 21, 8809-8818. 10.1523/JNEUROSCI.21-22-08809.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner B. I., Faure R., Burgess J. W., Bevan A. P., Lachance D., Zhang-Sun G., Fantus I. G., Ng J. B., Hall D. A., Lum B. S. et al. (1994). Peroxovanadium compounds. A new class of potent phosphotyrosine phosphatase inhibitors which are insulin mimetics. J. Biol. Chem. 269, 4596-4604. [PubMed] [Google Scholar]
- Proud C. G. (2004). The multifaceted role of mTOR in cellular stress responses. DNA Repair (Amst) 3, 927-934. 10.1016/j.dnarep.2004.03.012 [DOI] [PubMed] [Google Scholar]
- Proud C. G. (2010). mTORC1 and cell cycle control. In The Enzymes Vol. XXVII, pp. 129-146. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- Rafalski V. A. and Brunet A. (2011). Energy metabolism in adult neural stem cell fate. Prog. Neurobiol. 93, 182-203. 10.1016/j.pneurobio.2010.10.007 [DOI] [PubMed] [Google Scholar]
- Rafalski V. A., Mancini E. and Brunet A. (2012). Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J. Cell Sci. 125, 5597-5608. 10.1242/jcs.114827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Valle F., Badura M. L., Braunstein S., Narasimhan M. and Schneider R. J. (2010). Mitotic raptor promotes mTORC1 activity, G(2)/M cell cycle progression, and internal ribosome entry site-mediated mRNA translation. Mol. Cell. Biol. 30, 3151-3164. 10.1128/MCB.00322-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiling J. H. and Sabatini D. M. (2006). Stress and mTORture signaling. Oncogene 25, 6373-6383. 10.1038/sj.onc.1209889 [DOI] [PubMed] [Google Scholar]
- Reiter S., Crescenzi M., Galliot B. and Buzgariu W. (2012). Hydra, a versatile model to study the homeostatic and developmental functions of cell death. Int. J. Dev. Biol. 56, 593-604. 10.1387/ijdb.123499sr [DOI] [PubMed] [Google Scholar]
- Rovainen C. M. and Kakarala M. H. (1989). Angiogenesis on the optic tectum of albino Xenopus laevis tadpoles. Brain Res. Dev. Brain Res. 48, 197-213. 10.1016/0165-3806(89)90076-X [DOI] [PubMed] [Google Scholar]
- Ruthazer E. S., Schohl A., Schwartz N., Tavakoli A., Tremblay M. and Cline H. T. (2013). In vivo time-lapse imaging of neuronal development in Xenopus. Cold Spring Harb. Protoc. 2013, 804-809. 10.1101/pdb.top077156 [DOI] [PubMed] [Google Scholar]
- Sabers C. J., Martin M. M., Brunn G. J., Williams J. M., Dumont F. J., Wiederrecht G. and Abraham R. T. (1995). Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 270, 815-822. 10.1074/jbc.270.2.815 [DOI] [PubMed] [Google Scholar]
- Saldivar J. C., Hamperl S., Bocek M. J., Chung M., Bass T. E., Cisneros-Soberanis F., Samejima K., Xie L., Paulson J. R., Earnshaw W. C. et al. (2018). An intrinsic S/G2 checkpoint enforced by ATR. Science 361, 806-810. 10.1126/science.aap9346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampetrean O., Iida S.-I., Makino S., Matsuzaki Y., Ohno K. and Saya H. (2009). Reversible whole-organism cell cycle arrest in a living vertebrate. Cell Cycle 8, 620-627. 10.4161/cc.8.4.7785 [DOI] [PubMed] [Google Scholar]
- Seidel H. S. and Kimble J. (2015). Cell-cycle quiescence maintains Caenorhabditis elegans germline stem cells independent of GLP-1/Notch. eLife 4, e10832 10.7554/eLife.10832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltiel I. A., Krenning L., Bruinsma W. and Medema R. H. (2015). The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 128, 607-620. 10.1242/jcs.163766 [DOI] [PubMed] [Google Scholar]
- Sharma P. and Cline H. T. (2010). Visual activity regulates neural progenitor cells in developing xenopus CNS through musashi1. Neuron 68, 442-455. 10.1016/j.neuron.2010.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuldiner A. R., de Pablo F., Moore C. A. and Roth J. (1991). Two nonallelic insulin genes in Xenopus laevis are expressed differentially during neurulation in prepancreatic embryos. Proc. Natl. Acad. Sci. USA 88, 7679-7683. 10.1073/pnas.88.17.7679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa-Nunes R., Yee L. L. and Gould A. P. (2011). Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature 471, 508-512. 10.1038/nature09867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spéder P. and Brand A. H. (2014). Gap junction proteins in the blood-brain barrier control nutrient-dependent reactivation of Drosophila neural stem cells. Dev. Cell 30, 309-321. 10.1016/j.devcel.2014.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark G. R. and Taylor W. R. (2004). Analyzing the G2/M checkpoint. In Methods in Molecular Biology (ed. Schönthal A. H.), pp. 51-82. Totowa, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- Straznicky K. and Gaze R. M. (1972). The development of the tectum in Xenopus laevis: an autoradiographic study. J. Embryol. Exp. Morphol. 28, 87-115. [PubMed] [Google Scholar]
- Thuret R., Auger H. and Papalopulu N. (2015). Analysis of neural progenitors from embryogenesis to juvenile adult in Xenopus laevis reveals biphasic neurogenesis and continuous lengthening of the cell cycle. Biol. Open 4, 1772-1781. 10.1242/bio.013391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay M., Fugere V., Tsui J., Schohl A., Tavakoli A., Travencolo B. A. N., Costa L. F. and Ruthazer E. S. (2009). Regulation of radial glial motility by visual experience. J. Neurosci. 29, 14066-14076. 10.1523/JNEUROSCI.3542-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S., Ueno T. and Iwao Y. (2011). Role of the PI3K-TOR-S6K pathway in the onset of cell cycle elongation during Xenopus early embryogenesis. Dev. Growth Differ. 53, 924-933. 10.1111/j.1440-169X.2011.01297.x [DOI] [PubMed] [Google Scholar]
- Vadlakonda L., Dash A., Pasupuleti M., Anil Kumar K. and Reddanna P. (2013). The Paradox of Akt-mTOR interactions. Front. Oncol. 3, 165 10.3389/fonc.2013.00165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Keuren-Jensen K. R. and Cline H. T. (2008). Homer proteins shape Xenopus optic tectal cell dendritic arbor development in vivo. Dev. Neurobiol. 68, 1315-1324. 10.1002/dneu.20659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M. G., Cantley L. C. and Thompson C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029-1033. 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. (1956). On the origin of cancer cells. Science 123, 309-314. 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- Wu G. Y. and Cline H. T. (2003). Time-lapse in vivo imaging of the morphological development of Xenopus optic tectal interneurons. J. Comp. Neurol. 459, 392-406. 10.1002/cne.10618 [DOI] [PubMed] [Google Scholar]
- Wu M. Y. W., Cully M., Andersen D. and Leevers S. J. (2007). Insulin delays the progression of Drosophila cells through G2/M by activating the dTOR/dRaptor complex. EMBO J. 26, 371-379. 10.1038/sj.emboj.7601487 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.