

Abstract
Key points
Muscle mitochondrial networks changed from a longitudinal, fibre parallel orientation to a perpendicular configuration during postnatal development.
Mitochondrial dynamics, mitophagy and calcium uptake proteins were abundant during early postnatal development.
Mitochondrial biogenesis and oxidative phosphorylation proteins were upregulated throughout muscle development.
Postnatal muscle mitochondrial network formation is accompanied by a change in protein expression profile from mitochondria designed for co‐ordinated cellular assembly to mitochondria highly specialized for cellular energy metabolism.
Abstract
Striated muscle mitochondria form connected networks capable of rapid cellular energy distribution. However, the mitochondrial reticulum is not formed at birth and the mechanisms driving network development remain unclear. In the present study, we aimed to establish the network formation timecourse and protein expression profile during postnatal development of the murine muscle mitochondrial reticulum. Two‐photon microscopy was used to observe mitochondrial network orientation in tibialis anterior (TA) muscles of live mice at postnatal days (P) 1, 7, 14, 21 and 42, respectively. All muscle fibres maintained a longitudinal, fibre parallel mitochondrial network orientation early in development (P1–7). Mixed networks were most common at P14 but, by P21, almost all fibres had developed the perpendicular mitochondrial orientation observed in mature, glycolytic fibres. Tandem mass tag proteomics were then applied to examine changes in 6869 protein abundances in developing TA muscles. Mitochondrial proteins increased by 32% from P1 to P42. In addition, both nuclear‐ and mitochondrial‐DNA encoded oxidative phosphorylation (OxPhos) components were increased during development, whereas OxPhos assembly factors decreased. Although mitochondrial dynamics and mitophagy were induced at P1–7, mitochondrial biogenesis was enhanced after P14. Moreover, calcium signalling proteins and the mitochondrial calcium uniporter had the highest expression early in postnatal development. In conclusion, mitochondrial networks transform from a fibre parallel to perpendicular orientation during the second and third weeks after birth in murine glycolytic skeletal muscle. This structural transition is accompanied by a change in protein expression profile from mitochondria designed for co‐ordinated cellular assembly to mitochondria highly specialized for cellular energy metabolism.
Keywords: postnatal muscle development, mitochondrial development, proteomics
Key points
Muscle mitochondrial networks changed from a longitudinal, fibre parallel orientation to a perpendicular configuration during postnatal development.
Mitochondrial dynamics, mitophagy and calcium uptake proteins were abundant during early postnatal development.
Mitochondrial biogenesis and oxidative phosphorylation proteins were upregulated throughout muscle development.
Postnatal muscle mitochondrial network formation is accompanied by a change in protein expression profile from mitochondria designed for co‐ordinated cellular assembly to mitochondria highly specialized for cellular energy metabolism.
Introduction
Skeletal muscle development is characterized by a robust increase in the size and contractile performance of muscle fibres and occurs mostly within the first month after birth in mice (Gokhin et al. 2008; White et al. 2010). During this postnatal period, there is also a major reorganization of the contractile elements, as well as myocellular organelles, including the t‐tubules and sarcoplasmic reticulum (SR) (Luff & Atwood, 1971). Similarly, mitochondrial structure undergoes a large transformation in developing muscle (Mishra et al. 2015). Previously, it was shown that mitochondria in the rat diaphragm are arranged individually and parallel to the muscle fibre axis at birth but, by 2 months of age, a highly connected, grid‐like network is formed (Bakeeva et al. 1981). The mature muscle mitochondrial reticulum was demonstrated to provide a pathway for rapid cellular energy distribution in the form of the mitochondrial membrane potential (Glancy et al. 2015), as hypothesized several decades earlier (Skulachev, 1969). However, the mechanisms driving the postnatal formation of the muscle mitochondrial reticulum remain unclear.
Mitochondria are dynamic organelles that change their morphological and biochemical properties in response to physiological stimuli (Kim et al. 2017; Pickles et al. 2018). Mitochondrial fusion occurs via co‐operative regulation of the mitofusins (i.e. MFN1 and MFN2) and OPA1 that exert GTPase activity to fuse the outer and inner mitochondrial membranes, respectively. Mitochondrial fission is probably also regulated by another GTPase family member, Drp1, which is recruited by adaptor proteins such as Fis1, Mff and Mid49/51 (Mishra & Chan, 2016). Despite the importance of mitochondrial dynamics for mitochondrial quality control (Twig et al. 2008), only a few studies have examined how those fusion and fission proteins work in muscle development. For example, it has been documented that both MFN 1 and MFN2 are required for healthy mitochondrial development in tibialis anterior (TA) muscle through maintaining mtDNA stability (Chen et al. 2010). Additionally, in a study using mice with muscle‐specific overexpression of Drp1, Touvier et al. (2015) also demonstrated the importance of a fine‐tuned fission process during postnatal muscle growth because overexpressed Drp1 not only retards muscle growth, but also significantly impairs mitochondrial network formation in skeletal muscle. Conversely, the role of the mitochondrial fission adaptor proteins in muscle development is not well described.
Mitophagy also plays an important role for maintaining mitochondrial quality control via the degradation of malfunctional or damaged mitochondria by lysosomes after being engulfed by autophagosomes (Sin et al. 2016; Pickles et al. 2018). This mitochondria‐specific autophagy is governed by the PINK1/Parkin pathway, resulting in mitochondrial remodelling and turnover (Yang et al. 2008; Lazarou et al. 2015). In a recent study, impaired Parkin led to dysregulated mitochondrial function, as well as smaller fibre size (Peker et al. 2018), suggesting that it has an essential role in muscle development. A loss of PINK1 and Parkin has also been recently associated with a strong inflammatory phenotype after exhaustive exercise in mice (Sliter et al. 2018). However, more studies are warranted to better understand mitophagic regulation in postnatally developing skeletal muscle.
In addition to controlling mitochondrial degradation, the maintenance of optimal cellular function also requires the capacity to increase mitochondrial quantity. Mitochondrial biogenesis is regulated via the action of peroxisome proliferator‐activated receptor‐γ coactivator (PGC)‐1α, which interacts with various transcription factors and enhances mitochondrial oxidative capacity by stimulating nuclear DNA‐ and mitochondrial DNA‐encoded mitochondrial protein components. Although the contribution of PGC‐1α to mitochondrial structural and functional development has been shown in cardiac and skeletal muscle (Lehman et al. 2000; Czubryt et al. 2003; Zechner et al. 2010; Martin et al. 2014), the specific roles of the various transcription factors downstream of PGC‐1 in muscle development are less clear.
It has been suggested that mitochondrial network development is directly related to the cellular configuration of the SR and t‐tubules because those subcellular components are tightly aligned with mitochondria in mature skeletal muscle after undergoing dynamic changes during early postnatal development (Schiaffino & Margreth, 1969; Walker & Edge, 1971; Takekura et al. 2001). Indeed, the mitochondria, SR and t‐tubules all play integral roles in calcium signalling in the muscle, and calcium signalling proteins such as calcineurin and calmodulin kinase (CaMK) have been demonstrated as key factors for mitochondrial biogenesis in skeletal muscle (Wu et al. 2002). Moreover, mitochondrial calcium uniporter (MCU) has been identified as an important regulator of muscle function in mice, especially during strenuous exercise (Pan et al. 2013; Gherardi et al. 2018). However, the regulation of mitochondrial calcium uptake during muscle development remains unknown.
To better understand how the muscle mitochondrial reticulum is formed, we first aimed to establish the timecourse of mitochondrial network development in mice. Second, we explored the global changes in protein expression in skeletal muscle of mice during postnatal development. In particular, we focused on the protein composition of the mitochondria with specific attention on (i) mitochondrial dynamics and turnover; (ii) mitochondrial energetic pathways; and (iii) calcium handling and signalling in TA muscles of mice at postnatal days (P) 1, 7, 14, 21 and 42. We hypothesized that mitochondrial dynamics and calcium signalling proteins would be most abundant early in development, whereas mitochondrial energetics and calcium handling protein expression would increase throughout postnatal muscle development.
Methods
Ethical approval
All procedures were approved by the National Heart, Lung, and Blood Institute Animal Care and Use Committee and performed in accordance with the guidelines described in the Animal Care and Welfare Act (7 USC 2142 § 13).
Animals
C57BL6/N mice, aged 6–8 weeks old, were purchased from Taconic Biosciences (Rensselaer, NY, USA) and set up as breeding pairs. MitoDendra mice (Pham et al. 2012), aged 2–4 months old, were purchased from The Jackson Laboratory (strain #018397) and also set up as breeding pairs (The Jackson Laboratory, Bar Harbor, ME, USA). Their progeny were randomly selected for each experimental group: postnatal (P) 1, 7, 14, 21 and 42. Animals were given free access to food and water and pups were weaned at P21–28. As a result of the difficulty in using anogenital distance to reliably determine sex in newborn pups, we did not group mice depending on sex but, instead, randomly used both male and female mice. For proteomic analyses, TA muscles (n = 4 per group) were excised when animals were anaesthetized under isoflurane inhalation (1–2% with oxygen) and, for P1 and P7, pooled samples (i.e. one sample = 3–5 animal tissues) were used because of their small tissue size, respectively. Mice were then killed by exsanguination. Excised tissues were immediately frozen on dry ice and saved at −80°C for further processing.
Two‐photon microscopy
Two‐photon excitation microscopy (TCS SP8; Leica Microsystems, Wetzlar, Germany) was used to detect mitochondrial networks in the TA of live mice, as described previously (Glancy et al. 2014). In brief, mice (n = 3 per group) were placed under 1–2% isoflurane anaesthesia through a nosecone, skin and fascia of the TA muscles were gently removed using fine forceps, and the leg was coupled to the upright 25×, 1.1 NA objective (Nikon, Tokyo, Japan) with an optical coupling gel (Rothstein et al. 2006). Endogenous NAD(P)H fluorescence was excited at 750 nm and emission collected in the range 414–538 nm. The pixel sizes in the NAD(P)H images were in the range 150–300 nm, providing a resolution of ∼300–600 nm according to the Nyquist limit. With MitoDendra mice, we took images with pixels sizes in the range 40–100 nm aiming to maximize the resolution obtained by our system and to obtain greater clarity of the mitochondrial network structures (the diffraction limit of resolution is ∼200–250 nm). Mitochondrial network orientation was determined by a blind tester who scored either 0 or 1 for each category for a given fibre. Fibres were scored as fibre parallel (mitochondria running parallel to the contractile axis of the muscle), perpendicular (mitochondria oriented 90° relative to the contractile axis) or mixed (mitochondria oriented both parallel and perpendicular to the contractile axis). For example, if perpendicular mitochondria were observed, the tester scored the perpendicular section with ‘1’ and marked the others with ‘0’. At least 30 fibres per group were tested and data are shown as percentages. To further confirm mitochondrial network orientation changes during muscle development, we also performed in situ MitoTracker Red (Thermo Fisher Scientific, Waltham, MA, USA) staining in TA muscles of C57BL/6 mice.
Proteomics analysis
Preparation of protein extracts
Frozen TA muscles (n = 4 per group; 3–5 pooled tissues for P1 and P7) were transferred to a urea‐based lysis buffer (6 m urea, 2 m thiourea, 50 mm triethylammonium bicarbonate) at a ratio of 1:5 and homogenized by ceramic beads using two steps of 20 s at 5590 g (5500 rpm) and 4°C (Precellys® Cryolys Evolution; Bertin Technologies, Montigny‐le‐Bretonneux, France). Tissue lysates were further homogenized by microcentrifuge spin columns (QIAshredder; Qiagen, Valencia, CA, USA) to reduce viscosity and clarified by centrifugation (16 000 g for 20 min at 4°C). The supernatants were transferred into new tubes for further processing. Protein concentration was estimated by the Bradford assay (Thermo Fisher Scientific) and 100 μg of each lysate was digested with trypsin and labelled with Tandem Mass Tag (TMT) 10‐plex labelling reagent kit in accordance with the manufacturer's instructions (Thermo Fisher Scientific).
Offline HPLC peptide fractionation
High pH reversed‐phase liquid chromatography was performed on an offline Model 1200 series HPLC system (Agilent Technologies Inc., Santa Clara, CA, USA). The desalted peptides were resuspended in 0.1 mL of 10 mm triethyl ammonium bicarbonate with 2% (v/v) acetonitrile. Peptides were loaded onto an Xbridge C18 HPLC column (Waters, Milford, MA, USA; 2.1 mm inner diameter × 100 mm, 5 μm particle size) and profiled with a linear gradient of 5–35% buffer B (90% acetonitrile, 10 mm triethyl ammonium bicarbonate) over 60 min at a flow rate of 0.25 mL min−1. The chromatographic performance was monitored by sampling the eluate with a diode array detector (Model 1200 series HPLC system; Agilent Technologies Inc.) scanning between wavelengths of 200 and 400 nm. Fractions were collected at intervals of 1 min followed by fraction concatenation. Fifteen concatenated fractions were dried and resuspended in 0.01% formic acid, 2% acetonitrile. Approximately 500 ng of peptide mixture was loaded per liquid chromatography‐mass spectrometry run.
Mass spectrometry
All fractions were analysed on an Ultimate 3000‐nLC coupled to an Orbitrap Fusion Lumos Tribrid instrument (Thermo Fisher Scientific) equipped with a nanoelectrospray source. Peptides were separated on an EASY‐Spray C18 column (Thermo Fisher Scientific; 75 μm × 25 cm inner diameter, 2 μm particle size and 100 Å pore size). Peptide fractions were placed in an autosampler and separation was achieved by a 120 min gradient from 5–25% buffer B (100% acetonitrile and 0.1% formic acid) at a flow rate of 300 nL min−1. An electrospray voltage of 1.9 kV was applied to the eluent via the EASY‐Spray column electrode. The Lumos was operated in positive ion data‐dependent mode, using synchronous precursor selection (SPS) (MS3 method) (Ting et al. 2011). Full scan MS1 was performed in the Orbitrap with a precursor selection range of m/z 375–1275 at nominal resolution of 1.2 × 105. The automatic gain control (AGC) target and maximum accumulation time settings were set to 2 × 105 and 50 ms, respectively. MS2 was triggered by selecting the most intense precursor ions above an intensity threshold of 5 × 103 for collision induced dissociation‐MS2 fragmentation with an AGC target and maximum accumulation time settings of 1 × 104 and 60 ms, respectively. Mass filtering was performed by the quadrupole with a transmission window of m/z 0.7, followed by collision induced dissociation fragmentation in the linear ion trap with 35% normalized collision energy in rapid scan mode and the parallelizable time option was selected. SPS was applied to co‐select 10 fragment ions for high energy collision induced dissociation (HCD)‐MS3 analysis. SPS ions were all selected within the range m/z 400–1200 and were set to preclude selection of the precursor ion and complement TMT ion series (Wuhr et al. 2012). The AGC target and maximum accumulation time were set to 1 × 105 and 125 ms, respectively, and parallelizable time option was selected. Co‐selected precursors for SPS‐MS3 underwent HCD fragmentation with 65% normalized collision energy and were analysed in the Orbitrap with nominal resolution of 5 × 104. The number of SPS‐MS3 spectra acquired between full scans was restricted to a duty cycle of 3 s.
Data processing
Raw data files were processed using Proteome Discoverer, version 2.2 (Thermo Fisher Scientific), using both Mascot, version 2.5.1 (Matrix Science, London, UK) and Sequest HT (Thermo Fisher Scientific) search algorithms. Mascot and Sequest HT searches were performed against SwissProt human database (July 2017; 20,202 sequences), with carbamidomethylation of cysteine, and TMT 10‐plex modification of lysine and peptide N‐terminus set as static modifications and oxidation of methionine as the dynamic modification. For SPS‐MS3, the precursor and fragment ion tolerances of 10 ppm and 0.6 Da were applied, respectively. Up to two‐missed tryptic cleavages were permitted. Percolator algorithm was used to calculate the false discovery rate of peptide spectrum matches, set to a q value of 0.05 (Kall et al. 2007; Kall et al. 2008; Brosch et al. 2009; Spivak et al. 2009).
TMT 10‐plex quantification was also performed by Proteome Discoverer by calculating the sum of centroided ions within 20 ppm window around the expected m/z for each of the 10 TMT reporter ions. Quantification was performed at the MS3 level where the median of all quantifiable peptide spectrum matches for each protein group was used for protein ratios.
General pathway changes during postnatal development were assessed using the Reactome Pathway Browser (reactome.org). First, identified proteins were sorted into five groups in accordance with their expression profile across time: proteins whose abundance increased at every time point (up); proteins whose abundance decreased at every time point (down); proteins whose abundance increased from P1 to P7 and P14 then decreased at P21 and P42 (up then down); proteins whose abundance decreased from P1 to P7 and P14 then increased at P21 and P42 (down then up); and the remaining proteins (other). The Uniprot Accession IDs for each group were then loaded into Reactome Analysis interface and analysed using the Project‐to‐Human and Include Interactors options. Pathways with an entities false discovery rate < 0.05 were considered to be significantly enhanced.
Western blotting
Using a glass tissue grinder, TA muscles (n = 3 per group; 3–5 pooled tissues for P1 and P7) were homogenized in RIPA buffer at a ratio (catalogue no. 20‐188; Millipore, Billerica, MA, USA) of 100 mg per 0.1 ml including protease inhibitor cocktail (P8340; Sigma, St Louis, MO, USA). After leaving on ice for 15 min, samples were centrifuged at 16,000 g for 10 min at 4°C and then the supernatant was transferred into a clean 1.7 mL tube. Protein concentration was determined by the Bradford assay. Loading samples were then prepared by mixing with dithiothreitol‐included SDS protein loading solution (351‐082‐661; Quality Biological, Gaithersburg, MD, USA) and boiling at 95°C for 5 min. Using a XCell SureLock Mini‐Cell Electrophoresis system (Invitrogen, Carlsbad, CA, USA), 25–30 μg of proteins were separated in NuPAGE Bis‐Tris Gels (10% or 12%, 1.0 mm; Invitrogen) at 150 V. Subsequently, proteins in the gels were transferred onto polyvinylidene fluoride membranes via a XCell Blot Module system (Invitrogen) at 25 V for 2 h at 4°C. After blocking with Odyssey Blocking Buffer (927‐40000; Li‐Cor, Lincoln, NE, USA) for 1 h at room temperature, membranes were incubated with primary antibodies (OPA1, BD Biosciences, San Jose, CA, 612606, dilution 1:1000; PINK1, Abcam, Cambridge, MA, USA, ab23707, dilution 1:1000; Parkin, Cell Signaling Technology, Beverly, MA, USA, 4211, dilution 1:1000; PGC‐1α, Millipore, AB3242, dilution 1:1000; MCU, Abcam, ab121499, dilution 1:1000) overnight at 4°C, with agitation on a rotator. The next day, membranes were washed with PBS buffer (dilution 1:1000, Tween 20) for 3 × 10 min, and then incubated with corresponding secondary antibodies for 1 h at room temperature. After washing again with PBS buffer for 3 × 10 min, blots were detected by an Azure imaging system (c600; Azure Biosystems, Dublin, CA, USA) following a brief incubation with Pierce ECL Western Blotting substrate (32106; Thermo Fisher Scientific). ImageJ (NIH, Bethesda, MD, USA) was used to quantify band densities of each protein.
Blue native (BN) page
TA muscles (50–150 mg) (n = 3 per group; 3–5 pooled tissues for P1 and P7) were homogenized in 0.5‐1.5 mL of 1 × NativePAGE sample buffer (BN2003; Invitrogen) that included 1 × protease inhibitor cocktail and 1% n‐dodecyl‐β‐d‐maltoside. The homogenates were incubated on ice for 30 min and then centrifuged at 16,000 g for 30 min at 4°C. After transferring into clean 1.7 mL tubes, supernatants were sonicated for 3 × 15 s and protein concentrations were determined by the Bradford assay. Next, 15 μg of proteins including 5% G‐250 Sample Additive (BN2004; Invitrogen) were separated by a 4–16% NativePAGE Gel System (BN1002; Invitrogen) at 150 V at 4°C. Then, gels were incubated in fixing solution (50% methanol; 10% acetic acid; ddH2O) for 10 min at room temperature, with gentle shaking on a rotator. Using a Colloidal Blue Staining Kit (LC6025; Invitrogen), the gels were stained overnight at temperature, with gentle shaking on a rotator. The next day, gels were washed with ddH2O until clear bands could be detected with minimum background. Using an Azure imaging system (c600; Azure Biosystems), protein bands on gels were imaged and captured in black and white.
Statistical analysis
All data are shown as the mean ± SEM. Significant differences between groups were determined by a one‐way ANOVA with Dunnett's post hoc test. P < 0.05 was considered statistically significant.
Results
Mitochondrial network configuration in skeletal muscle dramatically changes during postnatal development
To understand the timecourse of mitochondrial network formation during postnatal skeletal muscle development, we used multiphoton microscopy to image endogenous mitochondrial NAD(P)H fluorescence in the TA muscles of live mice. As shown in Fig. 1 A–E, fibre parallel mitochondrial networks were noted during the initial phase of postnatal muscle development (P1–7). At P14, we observed mitochondrial networks that were composed of parallel, perpendicular and mixed mitochondrial structures in 22.15 ± 3.49%, 32.74 ± 4.62% and 43.92 ± 4.69% of muscles, respectively. Here, the term ‘mixed’ refers to the shape of a network, where a similar magnitude of both parallel and perpendicular mitochondrial orientations was observed. After P21, fibre perpendicular mitochondrial networks were the predominant configuration in the TA muscle.
Figure 1. Change in mitochondrial network configuration in skeletal muscle during postnatal development.
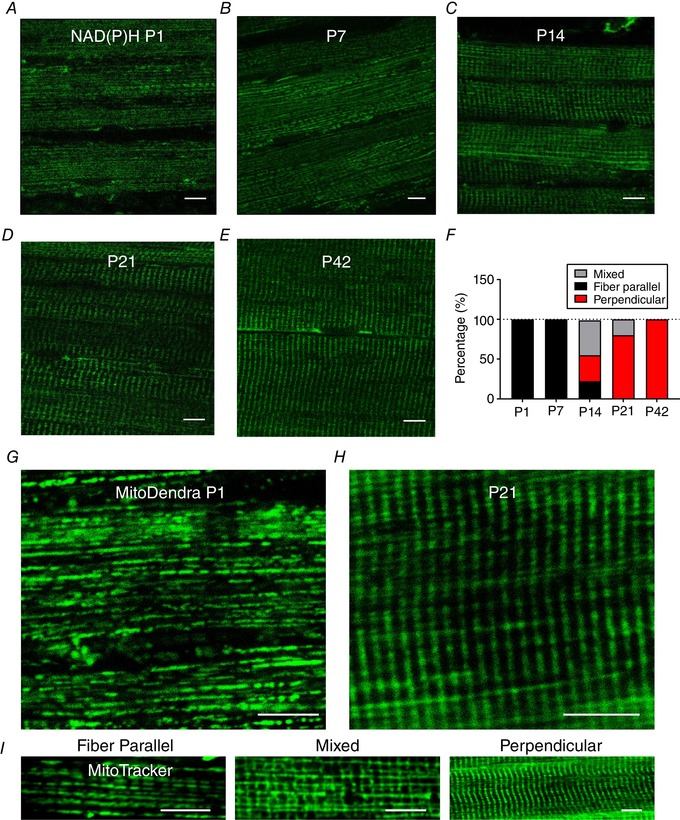
A–E, using two‐photon microscopy of endogenous NAD(P)H fluorescence, mitochondrial configuration was observed in TA muscles of live C57BL/6 mice at P1–42. Pixel sizes of the NAD(P)H images were in the range 150–300 nm. Captured images of each group are displayed in individual panels (n = 3 per group). F, mitochondrial directionality was determined by at least 30 fibres per group and data are shown as a percentage (%). G–H, representative images (40–100 nm pixel sizes) of two‐photon microscopy of live MitoDendra mouse TA muscles at P1 and P21. I, representative two‐photon microscopy images of in situ MitoTracker Red fluorescence in C57BL/6 mouse TA muscles showing fibre parallel, mixed and fibre perpendicular mitochondrial network orientations. Scale bars = 10 μm.
To confirm our results, we further examined mitochondrial orientations in TA muscles of live MitoDendra mice (P1 and P21) (Fig. 1 G and H) and also observed in situ MitoTracker Red fluorescence in C57BL/6 mouse TA muscles (Fig. 1 I). Taken together, a dramatic shift in mitochondrial network orientation from a fibre parallel to a fibre perpendicular configuration occurred over the course of postnatal muscle development.
Global skeletal muscle and mitochondrial proteome changes during postnatal muscle development
Next, we were interested in which proteins may be involved in the observed changes in mitochondrial orientation in the growing muscle. Using a TMT‐based proteomics approach (Chaves et al. 2013), we examined changes in the abundances of 6869 proteins in the TA muscle across P1–42 (see Supporting information, Data S1). Analysis of the protein expression profiles across time revealed that 3340 (48.6%) of the identified muscle proteins continually decreased in abundance across postnatal muscle development, whereas only 247 (3.6%) proteins continually increased across time. Reactome pathway analysis found that 18 of the 26 pathways identified from the developmentally decreasing proteins were related to transcription or translation (see Supporting information, Data S1). Consistent with this pathway analysis, nuclear and ribosomal proteins saw a more than three‐fold decrease in abundance from P1 to P42 (Table 1). Conversely, 16 of 18 Reactome pathways identified from the developmentally increasing proteins were related to metabolism (see Supporting information, Data S1), which was in accordance with the 31.7% and 125.8% increases in mitochondrial and glycolytic protein abundances, respectively (Table 1). Contractile proteins were the most abundant muscle constituent throughout postnatal development, making up 57.1% of all proteins at P1 and increasing by an additional 31.3% by P42. There were also large increases in the abundance of SR proteins (53.5%), as well as in creatine kinase (212.8%) and, particularly, myoglobin (3747.6%) across muscle development.
Table 1.
Major change of the muscle proteome during postnatal development
| Percentage change relative to P1 | ||||||
|---|---|---|---|---|---|---|
| Number of proteins | Percentage abundance (P1) of total muscle proteins | P7 | P14 | P21 | P42 | |
| Contractile | 85 | 57.0 | 10.0 | 24.4 | 26.2 | 31.3 |
| Nucleus | 973 | 7.8 | −35.1 | −86.6 | −148.5 | −202.2 |
| Mitochondria | 529 | 5.1 | 10.1 | 12.2 | 20.0 | 31.7 |
| Glycolysis | 30 | 2.3 | 34.3 | 62.5 | 101.9 | 125.8 |
| Cytoskeletal | 152 | 6.7 | −9.2 | −21.3 | −30.1 | −28.1 |
| Sarcoplasmic reticulum | 82 | 2.7 | 17.3 | 35.4 | 63.3 | 53.5 |
| Ribosome | 80 | 1.9 | −21.0 | −60.7 | −137.5 | −264.6 |
| Creatine kinase | 3 | 0.9 | 49.1 | 124.2 | 190.2 | 212.8 |
| T‐tubules | 13 | 0.2 | 14.8 | 35.8 | 41.2 | 26.0 |
| Lysosome | 62 | 0.1 | −36.5 | −108.8 | −180.6 | −197.9 |
| Carbonic anhydrase | 8 | 0.1 | −2.7 | −45.2 | −26.4 | 28.1 |
| Pentose phosphatase | 7 | 0.1 | −8.9 | −43.0 | −100.9 | −130.1 |
| Myoglobin | 1 | 0.0 | 136.9 | 299.0 | 639.3 | 3747.6 |
| Other | 4655 | 14.9 | −12.9 | −45.0 | −64.3 | −89.8 |
Similar to the whole muscle proteome, there was a large increase in metabolism‐related protein abundances within the mitochondria during muscle development (Table 2). OxPhos, which was the most abundant group of mitochondrial proteins even at P1, increased by 43.7% from P1 to P42, whereas lipid metabolism, tricarboxylic acid cycle and fuel transport proteins increased by 16.4%, 59.6% and 79.6%, respectively. Despite the increase in overall mitochondrial protein abundance across development, mitochondrial transport proteins (e.g. TIM and TOM) and transcription/translation protein expression was downregulated by 17.8% and 26.0% from P1 to P42, respectively.
Table 2.
Change of the mitochondrial composition during postnatal muscle development
| Percentage change relative P1 | ||||||
|---|---|---|---|---|---|---|
| Number of proteins | Percentage abundance (P1) of total mitochondrial proteins | P7 | P14 | P21 | P42 | |
| Oxidative phosphorylation | 139 | 41.37 | 14.5 | 20.6 | 27.3 | 43.7 |
| Tricarboxylic acid cycle | 24 | 17.42 | 19.8 | 27.2 | 41.5 | 59.6 |
| Lipid metabolism | 35 | 10.46 | 16.4 | 8.3 | 21.2 | 16.4 |
| Transcription/translation | 126 | 5.48 | −8.7 | −16.3 | −23.0 | −26.0 |
| Protein import | 29 | 2.40 | −4.2 | −11.1 | −17.4 | −17.8 |
| Fuel transporters | 10 | 1.74 | 27.2 | 40.2 | 64.9 | 79.6 |
| Protease | 16 | 0.99 | −14.8 | −21.9 | −26.6 | −28.1 |
| Fusion | 3 | 0.40 | −5.1 | −12.5 | −11.3 | −8.5 |
| Fission | 9 | 0.84 | 6.5 | −2.6 | −15.2 | −28.2 |
| Calcium transport | 7 | 0.48 | −23.4 | −44.0 | −53.9 | −57.5 |
| MICOS | 7 | 1.46 | 6.8 | 10.1 | 8.0 | 12.2 |
| Motility | 2 | 0.05 | −15.1 | −27.3 | −39.7 | −49.8 |
| Total (mitochondria) | 529 | 10.1 | 12.2 | 20.0 | 31.7 | |
MICOS, mitochondrial contact site and cristae organizing system.
Postnatal development of mitochondrial OxPhos complexes
To further determine how mitochondrial oxidative capacity is regulated during postnatal muscle development, we examined chronological changes in OxPhos protein abundances in TA muscles from P1 to P42. For all five OxPhos complexes, we found increase in nuclear DNA (nDNA)‐encoded OxPhos protein levels during postnatal muscle growth (Fig. 2 A). Additionally, we were able to detect 12 of the 13 mitochondrial DNA (mtDNA)‐encoded proteins, only missing ND6 and, similar to the nDNA‐encoded subunits, expression levels increased across muscle development (Fig. 2 B). By contrast to those OxPhos components, OxPhos assembly and chaperone proteins were downregulated over the course of postnatal muscle development (Fig. 2 C) and ∼30% fewer OxPhos assembly factors were found at P42 compared to P1. To better evaluate the assembly of OxPhos complexes, we used BN‐PAGE, which enabled us to determine abundances of the OxPhos complexes as a whole rather than as individual subunits. As shown in Fig. 2 D, complex III (i.e. ubiquinone‐cytochrome c oxidoreductase)‐ and complex IV (cytochrome c oxidase)‐associated protein abundances were significantly upregulated in the growing muscles because the protein abundances in P42 were ∼2.9‐ and ∼1.9‐fold higher compared to P1 (P < 0.05) (Fig. 2 E), respectively. However, other complex proteins were shown to have no significant changes between groups, although there was a non‐significant trend toward an increase in the supercomplexes and complex V (ATP synthase). Also of note, a discrete band at ∼70 kDa in the BN‐PAGE gel was detected in the TA muscles of P1 and P7 mice. We conducted additional mass spectrometric analysis and found that this band probably corresponds to a cytoskeletal (tubulin) complex that decreases in abundance across muscle development.
Figure 2. Developmental change of OxPhos components in skeletal muscle.
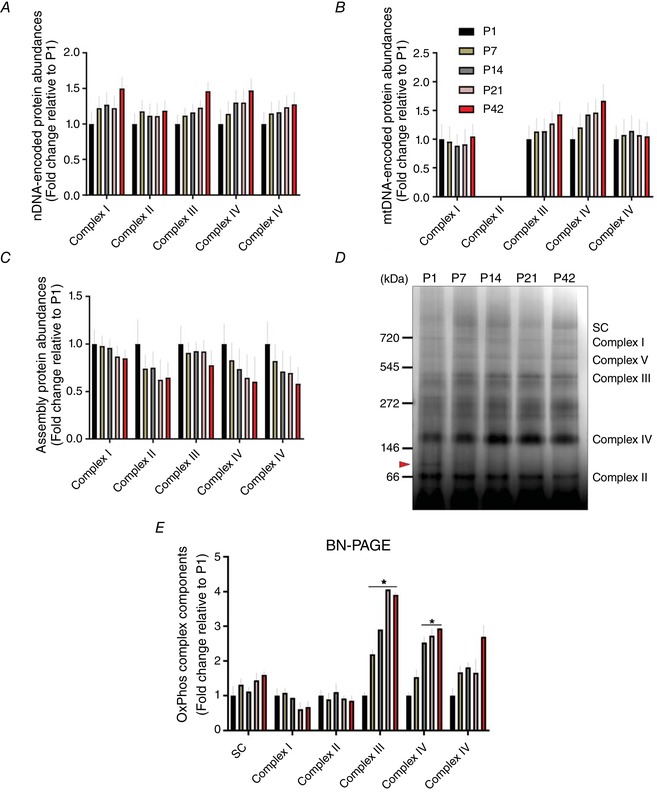
Fold changes of protein abundances for nDNA‐ (A) and mtDNA‐ (B) encoded OxPhos components across postnatal development, as well as (C) OxPhos assembly proteins compared to P1 (n = 4 per group; 3–5 pooled tissues for P1 and P7 groups, respectively). D, representative image of BN‐PAGE shows change in OxPhos protein subunits in TA muscles along with OxPhos supercomplexes (SC). An arrow between 66 and 146 kDa indicates a discrete band in P1. E, results of densitometry for BN‐PAGE. Data are shown as fold changes relative to P1 and are depicted as the mean ± SEM (* P < 0.05 vs. P1; n = 3 per group; 3–5 pooled tissues for P1 and P7 groups, respectively). [Color figure can be viewed at wileyonlinelibrary.com]
Given that cytochrome c oxidase plays a pivotal role for mitochondrial energetics (Viscomi et al. 2011; Larsen et al. 2012), we took a more detailed look at the developmental changes of individual cytochrome c oxidase subunits and assembly factors (Fig. 3 A and B). We detected 16 different cytochrome c oxidase subunits in our proteome, 13 of which increased from P1 to P42 including each of the mtDNA‐encoded subunits (Fig. 3 A). The three notable exceptions occurred as a result of subunit isoform switches during postnatal development. Cox4 has two isoforms, with isoform 1 (Cox4i1) comprising 99.5% of the total pool at p1 and 99.9% at P42. Conversely, Cox4i2 made up 0.5% at P1 and decreased to 0.1% at P42. At P1, isoform 1 of Cox6a, also known as the liver isoform, made up 76.8% of total Cox6a, whereas isoform 2 (heart) made up the remainder. However, by P42, the heart isoform increased in abundance to become the dominant isoform, making up 65.6% of the total Cox6a. Finally, Cox7a also has a heart (Cox7a1) and liver (Cox7a2) isoform. Although the heart isoform increases 2.7‐fold and the liver isoform decreases 1.4‐fold from P1 to P42, the liver isoform remains the dominant isoform at both time points, making up 88.3% and 66.7% of the total Cox7a at P1 and P42, respectively.
Figure 3. Individual profiling of cytochrome c oxidase proteins in developing muscle.
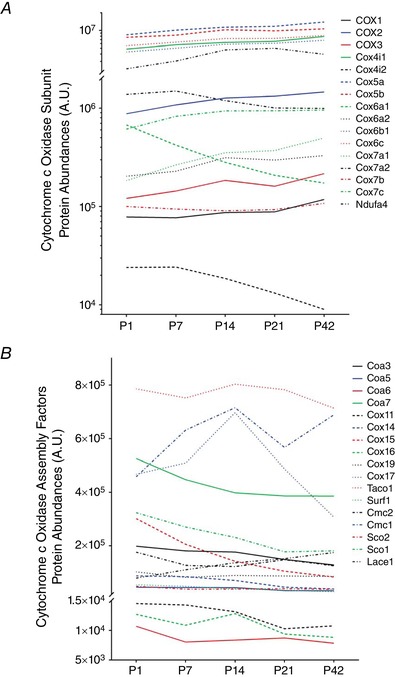
Changes of individual cytochrome c oxidase subunit proteins (A) and assembly factor proteins (B) during postnatal TA muscle development (n = 4 per group; 3–5 pooled tissues for P1 and P7 groups, respectively). [Color figure can be viewed at wileyonlinelibrary.com]
A closer look at the assembly factors for cytochrome c oxidase (Fig. 3 B) revealed that 14 of the 17 detected assembly and chaperone proteins generally decreased in abundance from P1 to P42. Of the three exceptions, COX assembly mitochondrial protein homologue (Cmc1) was the most abundant and increased its expression by over 50% by P14. The cytochrome c oxidase copper chaperone, Cox17, had a similar abundance profile to Cmc1 from P1 to P14 but decreased in expression by more than one‐half from P14 to P42. Also, Lace1, which has been shown to participate in the degradation of Cox4 and Cox6a (Cesnekova et al. 2016), increased its abundance by more than two‐fold from P1 to P42.
Mitochondrial anti‐oxidant‐related proteins are increased during postnatal muscle development
As reactive oxygen species are an important byproduct of OxPhos, we were also curious about how the expression of anti‐oxidant‐related proteins changed during postnatal musculature. As depicted in Fig. 4 A, we observed that, although cytoplasmic superoxide dismutase (SOD)1 and glutathione peroxidase (GPX) protein levels were decreased from P1 to P42, mitochondrial SOD2 and GPX protein abundances were largely upregulated, reaching ∼2.2‐ and ∼1.3‐fold increase at P42 relative to P1, respectively. Although glutathione S‐transferase (GST) protein levels were highly elevated during development, protein abundances for both glutathione reductase (GSR) and synthase (GSS), as well as catalase (CAT), were continuously decreased after birth. In addition, NADPH producing proteins such as isocitrate dehydrogenase (IDH2) and NAD(P) transhydrogenase (NNT) appeared to decrease across postnatal muscle growth.
Figure 4. Change of anti‐oxidant‐related protein abundances during postnatal muscle development.
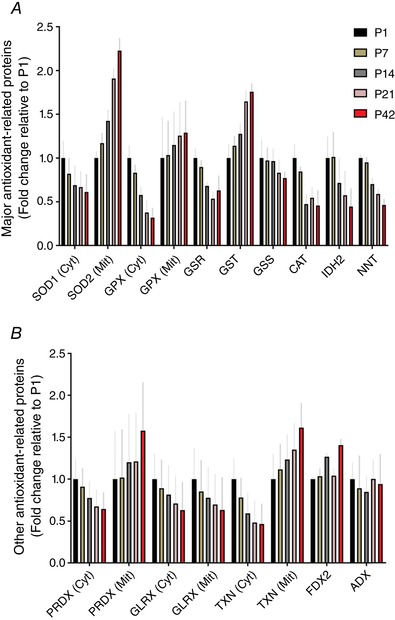
Fold changes of (A) major anti‐oxidant‐related and (B) other anti‐oxidant‐related cytoplasmic (Cyt) and mitochondrial (Mit) proteins in TA muscles compared to P1 (n = 4 per group; 3–5 pooled tissues for P1 and P7 groups, respectively). [Color figure can be viewed at wileyonlinelibrary.com]
Our proteome data also revealed that other anti‐oxidant‐related protein levels were differentially regulated by subcellular locations (i.e. cytoplasmic vs. mitochondria), except for glutaredoxin (GLRX). As shown in Fig. 4 B, mitochondrial peroxiredoxin (PRDX) and thioredoxin reductase (TXN) proteins were all ∼1.6‐fold upregulated by P42 compared to P1, whereas the corresponding cytoplasmic protein levels were decreased from P1 to P42.
Mitochondrial dynamics are upregulated during early postnatal muscle development
Next, we investigated how mitochondrial fusion and fission proteins are regulated in skeletal muscle during postnatal development as a result of the importance of mitochondrial dynamics in muscle mitochondrial network remodelling (Chan, 2006; Romanello & Sandri, 2015). Inner membrane fusion protein OPA1 increased at P42 compared to P1 (Fig. 5 A). However, the ratio of the short and long forms of OPA1 (Anand et al. 2014; Wai et al. 2015) increased almost three‐fold from P1 to P42 (Fig. 5 B), suggesting a lower inner membrane fusion potential in the relatively matured muscle. The combined protein abundances of mitochondrial outer membrane fusion proteins, the mitofusins, decreased across postnatal development (Fig. 5 A), although MFN2 expression increased at P7 and P14 before decreasing up to P42.
Figure 5. Change in mitochondrial dynamic proteins during postnatal muscle development.
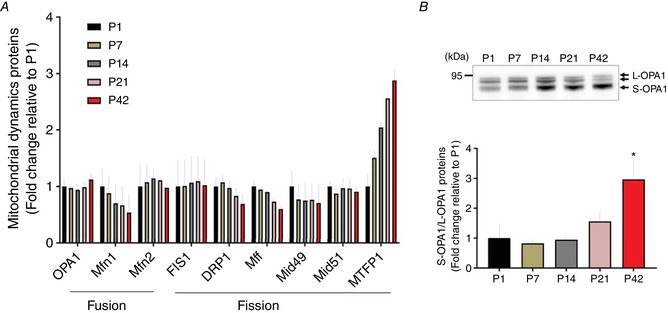
A, fold changes of protein abundance of fusion and fission proteins are depicted (n = 4 per group; 3–5 pooled tissues for P1 and P7 groups, respectively) compared to P1. B, representative image of western blotting for OPA1. Long (L)‐ and short (S)‐OPA1 were separated by molecular weights (top). The ratio of S‐OPA1 to L‐OPA1 was examined and data (fold change relative to P1) are shown as the mean ± SEM. A significant difference was found between groups (* P < 0.05 vs. P1, n = 3 per group; pooled tissues for P1 and P7 groups, respectively). [Color figure can be viewed at wileyonlinelibrary.com]
The most abundant mitochondrial fission protein, Drp1, decreased almost two‐fold from P1 to P42 (Fig. 5 A). Expression of fission adaptor proteins FIS1and Mid51 showed little change, if not a slight decrease, during muscle development. However, fission adaptors Mid49 and Mff decreased by 29.5% and 40.3% across development, respectively. Conversely, mitochondrial fission process1 (MTFP1) increased by 2.9‐fold from P1 to P42.
Mitophagy is induced during the initial muscle development
Given that autophagosome‐associated mitochondrial turnover (mitophagy) is crucial for healthy mitochondrial development (Hill et al. 2012; Hood et al. 2019), we aimed to identify how autophagy and mitophagy‐related proteins change across postnatal development. As shown in Fig. 6 A, autophagosomal degradation proteins were notably upregulated during the early muscle growth (P1–14) and the more mature muscles (P42) were shown to have around one‐half the LC3, p62 and ATG7 expression compared to P1.
Figure 6. Change in muscle autophagic and mitophagic proteins across postnatal period.
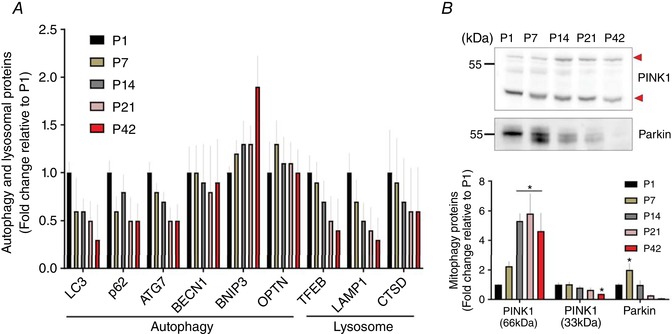
A, changes of protein abundances for autophagic and lysosomal protein markers (n = 4 per group; 3–5 pooled tissues for P1 and P7 groups, respectively). B, representative images of western blotting for PINK1 and Parkin (top). In the results of PINK1, short forms of PINK1 (33 kDa; lower arrow) were downregulated from P1 to P42, whereas full‐length forms of PINK1 (66 kDa; upper arrow) were increased. Parkin protein expressions in TA muscles appeared to peak at P7 (* P < 0.05 vs. P1, n = 3 per group; 3–5 pooled tissues for P1 and P7 groups, respectively). [Color figure can be viewed at wileyonlinelibrary.com]
In the current proteomics dataset, neither PINK1, nor Parkin, which are both key players for mitophagy (Matsuda et al. 2010; Youle & Narendra, 2011), were detected, probably as a result of a relatively low abundance. As in previous studies (Anichtchik et al. 2008; Gatliff et al. 2014), western blotting detected two prominent bands for PINK1 protein at ∼66 kDa (full length) and ∼33 kDa (truncated form) and showed that, although the full length of PINK1 increased from P1 to P42, the fragmented PINK1 protein levels were progressively downregulated (Fig. 6 B) (P < 0.05). In addition, Parkin was also significantly elevated during the first 2 weeks after birth (P1–14), peaking at P7 (Fig. 6 B) (P < 0.05) before decreasing up to P42.
In addition, protein abundances for the lysosomal system, the cellular site of autophagosomal degradation, showed expression patterns similar to those of the autophagic and mitophagic proteins. Lysosomal proteins including TFEB, LAMP1 and cathepsin D were downregulated by ∼64%, 66% and 38% at P42 compared to P1, respectively (Fig. 6 A).
Postnatal muscle development promotes mitochondrial biogenesis
We next investigated how mitochondrial biogenesis‐related proteins are regulated during postnatal muscle development. Although transcription coactivator PGC‐1α is a primary regulator for mitochondrial biogenesis, it was not detected in our muscle proteome, possibly because of its relatively low abundance. Instead, western blotting revealed that PGC‐1α protein levels were significantly increased during the muscle development, especially during P14–21, and the levels were ∼1.6‐fold higher compared to P1 (Fig. 7 A) (P < 0.05).
Figure 7. Change in mitochondrial biogenesis‐associated proteins in postnatal muscle growth.
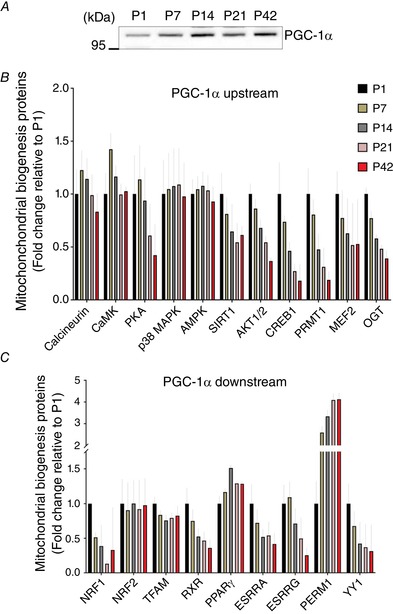
A, representative image of western blotting for PGC‐1α (n = 3 per group). Fold changes of muscle proteomic data are shown for PGC‐1α (B) upstream and (C) downstream proteins compared to P1 (n = 4 per group; 3–5 pooled tissues for P1 and P7 groups, respectively). [Color figure can be viewed at wileyonlinelibrary.com]
Because PGC‐1α is affected by various molecular signalling markers and regulates multiple transcription factors for mitochondrial biogenesis, our proteomics data were profiled depending on whether proteins are either upstream (Fig. 7 B) or downstream (Fig. 7 C) of PGC‐1α signalling pathways. Our data show that, overall, PGC‐1α upstream proteins appeared to be increased early in postnatal muscle development (P1–14): CaMK, protein kinase A (PKA), cAMP‐responsive element‐binding protein (CREB1), protein arginine methyltransferase 1 (PRMT1) and myocyte enhance factor 2 (MEF2) (Fig. 7 B). However, other signalling proteins, such as p38 mitogen‐activated protein kinase (p38MAPK), 5'‐AMP‐activated protein kinase (AMPK) and Sirtuin 1/2 (SIRT1/2), showed little change during muscle development.
As shown in Fig. 7 C, PGC‐1α downstream proteins appeared to have mixed expression patterns. Although nuclear respiratory factor 2 (NRF2) showed no change during muscle development, we observed significantly elevated NRF1 protein abundances during the early developmental phase (P1–7) before falling sharply. Mitochondrial transcription factor (TFAM) was probably largely upregulated at P1 and then maintained lesser abundances during the rest of the developmental periods. Although protein abundances for retinoid X receptor (RXR), oestrogen‐related receptor alpha (ESRRA), oestrogen‐related receptor gamma (ESRRG) and transcription factor YY1 were all elevated during early time points (P1–7), PGC‐1α and ERR‐induced regulator in muscle protein 1(PERM1) was ∼4.1‐fold increased by P42 compared to P1.
Calcium regulatory protein expression during postnatal mitochondrial development in skeletal muscle
Calcium handling is important for mitochondrial function and biogenesis in skeletal muscle (Wu et al. 2002) and its necessity during postnatal development was also reported in a recent study (Brinegar et al. 2017). Thus, we investigated overall changes of calcium handling proteins in our muscle proteome. It appeared that calcium signalling proteins, including calcineurin, CaMK and calpain, were upregulated during the first 2 weeks of postnatal development (P1–14) (Fig. 8 A). However, other calcium signalling proteins, such as calmodulin and phospholipase C, were shown to be more highly expressed during the later developmental phase (P14–42), and a noted increase was found in parvalbumin for which protein abundance was ∼82‐fold higher at P42 compared to P1 (Fig. 8 A).
Figure 8. Change of calcium handling protein abundances during postnatal muscle development.
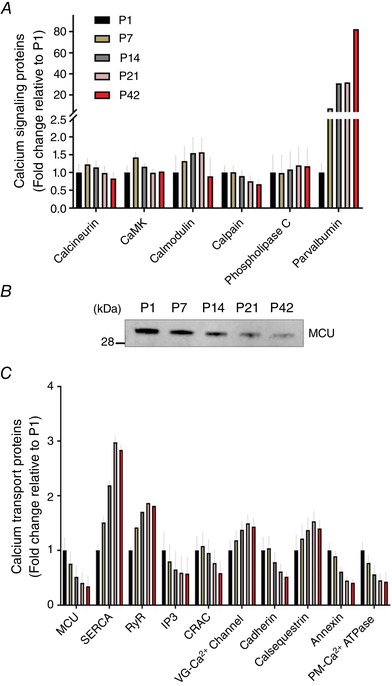
Fold changes of (A) calcium signalling‐ and (C) calcium transport‐associated protein abundances in TA muscles compared to P1 (n = 4 per group; 3–5 pooled tissues for P1 and P7 groups, respectively). B, representative image of western blotting for MCU (n = 3 per group; 3–5 pooled tissues for P1 and P7 groups, respectively). [Color figure can be viewed at wileyonlinelibrary.com]
Because the calcium concentration is tightly regulated by multiple calcium transport proteins, our proteomics data were examined further, aiming to determine how calcium transport proteins are regulated during postnatal muscle development. As shown in Fig. 8 B and C, many calcium transport proteins, including MCU, calcium release activated channel (CRAC), cadherin, and annexin, were highly upregulated during the initial muscle development (P1–7). In particular, because MCU is a mitochondria‐specific channel and has been identified to play an important role for muscle mitochondrial capacity (Pan et al. 2013; Mammucari et al. 2015), we further confirmed its protein expressions with western blotting and found that MCU protein levels at P1 were ∼3.9‐fold higher compared to P42 (P < 0.05) (Fig. 8 B). Micu1 and Micu2 protein expression followed a similar pattern to MCU (see Supporting information, Data S1). Nevertheless, we observed that muscle contraction‐associated calcium transporting proteins such as sarcoplasmic reticulum calcium transport ATPases (SERCA) and ryanodine receptors (RyR) were considerably upregulated along with the postnatal muscle growth. SERCA and RyR protein abundances were increased by ∼3.0‐ and ∼1.9‐fold by P21 respectively, and then plateaued by P42 (Fig. 8 B).
Discussion
The present study demonstrates a considerable change in mitochondrial network configuration in the skeletal muscle of mice during postnatal development (1, 7, 14, 21 and 42 days) and explores changes of total protein abundances in the growing muscle. Particularly, the present study characterizes proteomic changes for muscle mitochondrial development and also profiles protein markers for mitochondrial remodelling systems. Given that mitochondrial structures and functions are tightly regulated by calcium regulatory factors, we also attempted to understand changes of calcium handling proteins during the muscle growth.
Our in vivo microscopic observations suggest that postnatal mitochondrial development results in a dramatic structural transition in skeletal muscle. As reported previously (Boncompagni et al. 2009), we detected a fibre parallel mitochondrial orientation in the TA muscles of 1–7‐day‐old mice (Fig. 1 A–B and F). Conversely, a fibre perpendicular mitochondrial formation is dominant in mature skeletal muscle (Boncompagni et al. 2009; Mishra et al. 2015) and, in the present study, we show that the major structural transition occurs between P7 and P21 in mice (Fig. 1 C–F). Although these observations provide an overview of the change in mitochondrial network configuration across postnatal development, future studies are warranted to better understand the changes in 3D physical and functional connectivity among individual mitochondria and with other organelles (Bleck et al. 2018) during muscle development.
Postnatal muscle development is a dynamic process in which extensive changes in protein abundances occur to support muscle structural and functional development. As shown in the present study (Table 1), it was previously reported that contractile and cytoskeletal protein levels are significantly increased during postnatal TA muscle development in rats (Sun et al. 2009) and that glycolysis (Sperl et al. 1992) and creatine kinase (Watchko et al. 1996) are robustly upregulated, at least in part, to adapt to a growing intramuscular environment and increasing hindlimbs activity. Notably, Garry et al. (1996) reported exponentially increased myoglobin mRNA and protein levels during postnatal muscle development along with mitochondrial proteins. Nevertheless, transcriptional and translational protein groups were continuously downregulated during postnatal period, probably because of the notion that fetal myogenesis is accomplished during the prenatal phase (Buckingham et al. 2003).
To our knowledge, the present study is the first to describe the change of total OxPhos protein levels over the course of postnatal muscle development demonstrating a gradual increase in OxPhos elements encoded by both nuclear and mitochondrial genomes (Figs 2 and 3). Similarly, Kim et al. (1995) revealed that mRNA levels of complex IV subunits in skeletal muscles of mice were significantly elevated by 3 weeks after birth. It has been also shown that mtDNA contents were upregulated during postnatal muscle development (Minai et al. 2008). Although we did not directly measure mtDNA contents, robustly increased mtDNA‐encoded OxPhos components in our dataset may be indicative of elevated mtDNA content despite a decrease in transcription and translation protein levels. In addition, it can be suggested that upregulated OxPhos protein abundances may be indicative of increased mitochondrial respiratory capacity, although we did not measure mitochondrial respiration here. As shown with BN‐PAGE (Fig. 2 D), we detected considerably increased protein abundances for complex III and IV, which corroborates previous findings that, although complex I (i.e. NADH‐ubiquinone oxidoreductase) and complex II (succinate‐ubiquinone oxidoreductase) enzyme activity levels of postnatal skeletal muscles remained similar to prenatal muscles, complex III‐ and IV‐associated enzyme activities were more robustly upregulated during postnatal muscle development (Minai et al. 2008). The lack of increase in complex I detected by BN‐PAGE may be also the result of a large portion of the complex participating in a supercomplex structure as shown previously in mouse TA muscle (Garcia et al. 2017). Similar to the results reported in the present study, cardiac muscle oxidative phosphorylation capacity also appears to undergo a major developmental transition (Piquereau et al. 2010). However, direct comparisons of the timelines between cardiac and hindlimb skeletal muscles during development may be skewed by the requirement for a heart capable of pumping blood throughout the body at the time of birth, whereas the demand for hindlimb muscle function is relatively lower. Nevertheless, the general change in protein expression profile from mitochondria supporting cellular assembly to mitochondria specialized to support muscle contraction is probably similar between heart and skeletal muscle despite differences in the developmental timelines.
The present study reveals that nuclear‐encoded COX4 isoforms have different expression patterns across postnatal muscle development. As a convincing marker for mitochondrial aerobic capacity, muscle COX4i1 mRNA and protein abundances have been reported to increase in response to exercise training (Eivers et al. 2010) and chronic contractile activity (Parousis et al. 2018), and we also observed elevated protein levels during postnatal period (Fig. 3 A). Conversely, COX4i2 appears to move to the opposite direction compared to COX4i1 because its mRNA or protein abundance was downregulated in the trained skeletal muscle (Eivers et al. 2010), as also shown in the results of the present study. Meanwhile, in accordance with the present study, Boczonadi et al. (2015) depicted that isoforms of both COX6a and COX7a were changed during postnatal muscle development of mice. For example, isoforms 2 (heart) of COX6a and COX7a were exponentially upregulated within 4 weeks after birth, whereas liver isoforms were not significantly changed (Boczonadi et al. 2015). Despite this temporal change, the protein abundance of COX7a isoform 1 (Liver) appeared to be consistently dominant during the whole postnatal period, which may contribute to a tight linkage between complex III and IV (Cogliati et al. 2016).
In addition, our proteomics data show that mitochondrial OxPhos assembly and chaperone proteins in skeletal muscle were continuously downregulated after birth (Figs 2 C and 3 B). Given that mitochondrial OxPhos assembly is suggested to be fully accomplished during the fetal phase (Minai et al. 2008), those proteins may be important mostly during prenatal or early postnatal muscle development. However, in complex IV, two abundant assembly proteins, Cox17 and Cmc1, showed discrete expression patterns and peaked at P14. Because these proteins play a vital role for mitochondrial copper metabolism (Banci et al. 2008; Horn et al. 2008), it could be plausible that they may be upregulated to support copper handling and redox control in the developing muscle.
Along with the robust changes in OxPhos components, we also identified that mitochondrial anti‐oxidant‐related proteins, including SOD2, GPX, PRDX and TXN, are highly upregulated during the postnatal period (Fig. 4). Those increased anti‐oxidant proteins probably contribute toward maintaining redox homeostasis in the growing muscle, as shown in a previous study investigating the neonatal diaphragm muscles of lambs (Song & Pillow, 2013). Furthermore, it has also been reported that mitochondria‐specific anti‐oxidant enzymes are necessary for animal development because a loss of SOD2 led to early postnatal lethality in mice (Li et al. 1995). Moreover, muscle‐specific SOD2 knockout resulted in mice with oxidative and muscle damage, as well as exercise intolerance and lower activity of OxPhos components (Kuwahara et al. 2010). Thus, anti‐oxidant function probably plays a vital role for muscle mitochondrial development, although more studies will be needed to examine specific roles of individual proteins.
We next investigated how mitochondrial dynamics is regulated during postnatal skeletal muscle development. OPA1, the inner mitochondrial fusion protein, has been identified to be crucial for muscle development (Rai et al. 2014; Sin et al. 2016) and, in the present study, OPA1 was changed from fusion‐ to fission‐preferred format across postnatal muscle growth (Fig. 5 B). In addition, we observed an overall upregulation of outer mitochondrial fusion proteins (MFN1 and MFN2) during the early postnatal phase (Fig. 5 A), indicating that mitochondrial fusion may be promoted during initial muscle growth. Indeed, Papanicolaou et al. (2012) showed that the double deletion of MFN1 and MFN2 led to an abnormal cardiac muscle development in which irregular mitochondrial structures were also found along with decreased mtDNA content. As shown in the fusion proteins, we observed temporal changes of muscle fission proteins during postnatal development. In particular, Drp1 and Mff proteins were highly expressed during early developmental period and then downregulated in the relatively matured muscle (Fig. 5 A), suggesting an importance of timely regulated fission process. Touvier et al. (2015) revealed that the fine‐tuning fission is required for postnatal muscle growth because atypical skeletal muscle growth and dysregulated mitochondrial networks were reported in animals for which the Drp1 gene was designed to overexpress in skeletal muscle. Furthermore, it may be also critical to maintain a balance between fusion and fusion proteins because the muscle‐specific deletion of Drp1 resulted in not only a repression of mitochondrial fusion protein expressions, but also a dysregulated mitochondrial structural and functional development during neonatal muscle growth (Ishihara et al. 2015).
Along with mitochondrial fusion and fission proteins, autophagic proteins were regulated in the same direction, and they were mostly elevated in skeletal muscle during the early postnatal phase (Fig. 6). Recently, it was shown that muscle specific deletion of Atg7, a key upstream marker for the autophagic regulation, led to a severe retardation of postnatal skeletal muscle development (Zecchini et al. 2019), suggesting an important role of autophagic system for neonatal muscle growth. Furthermore, ablation of either fusion (Papanicolaou et al. 2012) or fission (Ishihara et al. 2015) proteins was shown to result in an abnormal regulation of LC3 and p62 proteins during cardiac muscle development, indicating a possible co‐operative action of those mitochondrial control systems across postnatal muscle growth. Mitochondria‐specific autophagy, mitophagy, has been also discussed as a central system for healthy muscle mitochondrial development (Dorn et al. 2015), which also appears to have a notable linkage between mitochondrial dynamics and turnover. In an in vitro study, siRNA‐mediated Parkin knockdown was shown to have considerably diminished protein levels of both MFN1/2 and Drp1 in C2C12 myoblasts (Peker et al. 2018) for which the initial differentiation process into myotubes requires the upregulation of autophagic proteins (Sin et al. 2016). Moreover, Gong et al. (2015) reported a significant interaction between MFN2 and PINK1/Parkin for cardiomyocyte development, where the elevated protein abundances for autophagy and mitophagy in the present study may exert an important role during the initial muscle growth. However, more studies are warranted to confirm the causal relationships in the postnatal skeletal muscle development.
Transcriptional coactivator PGC‐1α has been well accepted as a master regulator of mitochondrial biogenesis (Baar et al. 2002; Arany et al. 2005; Uguccioni & Hood, 2011). In addition, various transcription factors, as well as other cofactors, lead to mitochondrial biogenesis, metabolic regulation and other cellular signalling pathways in skeletal muscle by interacting with PGC‐1α (Knutti et al. 2001; Wu et al. 2002; Lira et al. 2010b). Meanwhile, PGC‐1α has been also demonstrated to have a vital role for muscle mitochondrial development because cardiac muscle‐specific PGC‐1α/β deletion led to a significant impairment of structural and functional mitochondrial development during the early postnatal phase (Martin et al. 2014). In the present study, various PGC‐1α upstream protein markers, including CnA, CaMK, PKA and PRMT1, were elevated during early postnatal muscle growth, and PGC‐1α protein levels were subsequently increased after P14 (Fig. 7). Moreover, CREB, a key downstream marker of both CaMK (Zuloaga et al. 2013) and PKA (Bruno et al. 2014), and MEF2, a CnA‐associated transcription factor (Dunn et al. 2000), were all upregulated during this early developmental phase (Fig. 7 B), which collectively implies that those PGC‐1α‐related cellular signalling may be important for the initial muscle development. Indeed, Franko et al. (2008) showed that skeletal muscle cell differentiation can be accomplished by a recruitment of CREB1 into the cytochrome c promoter, which is dependent on the presence of PGC‐1α (Puigserver et al. 1999). Nevertheless, although p38MAPK and AMPK‐associated post‐translational modification of PGC‐1α has been shown to lead to mitochondrial biogenesis following exercise or other physiological stress (Wright et al. 2007; Lira et al. 2010a), the present study revealed that it may be not the case in the postnatal muscle growth (Fig. 7 B).
PGC‐1α also exerts its tasks by interacting with multiple downstream transcription factors. In particular, PGC‐1α has been shown to stimulate a transcriptional regulation of NRF1, as well as TFAM, by which mitochondrial biogenesis and oxidative capacity are augmented in skeletal muscle (Wu et al. 1999; Gleyzer et al. 2005; Uguccioni & Hood, 2011). Paradoxically, we observed that both NRF1 and TFAM protein levels were greater right after birth and then were maintained at low levels during the subsequent muscle growth (Fig. 7 C) and it appears that, during the early postnatal development, their protein expression could be regulated without a significant effect of PGC‐1α. In a study of comparing fetal and adult heart muscle proteomes, Pohjoismäki et al. (2013) revealed significantly lower TFAM protein abundances in the adult heart muscle compared to the fetal samples. Hence, it may be plausible that these nuclear encoded transcription factors could be regulated by PGC‐1α independent mechanisms during fetal and early postnatal phases. On the other hand, oestrogen related receptors were found to be significantly upregulated during the transition of late fetal to early postnatal muscle development phase (Byrne et al. 2010), supporting the findings of the present study (Fig. 7 C). Also of note, PERM1 is necessary for PGC‐1α‐related mitochondrial biogenesis and respiratory capacity in skeletal muscle (Cho et al. 2013) and it showed the same expression pattern of PGC‐1α across postnatal muscle growth (Fig. 7 C).
Given the close structural relationship between mitochondria and SR (Boncompagni et al. 2009), as well as the significant effects of calcium signalling on mitochondrial biogenesis (Wu et al. 2002), we investigated changes in calcium‐related protein abundances in our dataset. Furthermore, a recent transcriptome study by Brinegar et al. (2017) suggested that alternative splicing is highly active during the early postnatal days (P1–14) and that it is important for calcium regulatory function in the growing skeletal muscle. In the present study, protein abundances of calcium signalling proteins including calcineurin, CaMK and calpain were higher in the skeletal muscles of 1–7‐day‐old mice (Fig. 8 A), which also suggests a possible important role of calcium regulator factors during postnatal muscle development. As reported in a previous study where both quantity and volume of SR were exponentially increased during postnatal muscle development (Luff & Atwood, 1971), our muscle proteome revealed consistently increasing protein levels of SR, as well as other muscle contraction‐associated calcium proteins (i.e. RyR), during postnatal growth (Fig. 8 C), although other calcium transport proteins that are more involved in muscle differentiation and development were highly upregulated during early postnatal days: CRAC (Stiber et al. 2008), cadherin (Kaufmann et al. 1999) and annexin (Rahman et al. 1997). Furthermore, we examined the change of mitochondria‐specific calcium channel proteins (i.e. MCU) and found that MCU protein abundances were sharply downregulated during initial muscle growth (Fig. 8 B and C). Accordingly, it was reported that mitochondrial calcium uptake rate peaked right after birth and then sharply declined in cardiac muscle (Bassani et al. 1998). Collectively, these data suggest that MCU may play an important function during the initial muscle development, although future studies are required to better confirm its role in muscle development.
In the present study, we conducted a large‐scale proteomic study on postnatally developing skeletal muscle. Because we aimed to understand the mitochondrial proteome in relation to the formation of the mitochondrial reticulum, we collected tissue samples up to P42 when mitochondrial network orientation was determined to be stable (Fig. 1). Because a 42‐day‐old mouse has not reached full maturity, some of the cellular and mitochondrial processes described in the present study have probably also not reached full maturation. However, the general trend for several mitochondrial processes of a mitochondrial protein expression profile moving from supporting co‐ordinated cellular assembly toward specializing for energy metabolism will probably not change by assessing later time points. Additionally, although we did not separate our groups by sex, there is probably an effect of sex on the development of the muscle mitochondrial proteome. Indeed, although the TA muscle in male and female mice is of similar size at birth, male muscles develop more quickly and grow bigger than female TA muscles (Rowe & Goldspink, 1969). However, although the magnitude and specific timing of the changes described in the present study may be altered by sex, the overall drive to shift the mitochondrial proteome from assisting assembly toward supporting muscle contraction during maturity is probably similar between male and females. Furthermore, although protein abundance does not provide a direct measure of protein function, protein expression levels probably provide an index of capacity for function. Indeed, mitochondrial composition can vary greatly across cell types and correlates well with various mitochondrial functions (Johnson et al. 2007; Glancy & Balaban, 2011; Phillips et al. 2012). Within this functional capacity, it is important to note that post‐translational modifications are often used as regulators of protein activity. As such, future studies into the role of post‐translational modifications with respect to regulating mitochondrial network development may offer critical insight. Our proteomic analyses in the present study focused primarily on the OxPhos, mitochondrial dynamics and biogenesis, mitophagy, anti‐oxidant, and calcium signalling proteins that may be involved in the development of the muscle mitochondrial reticulum. However, our dataset (see Supporting information, Data S1) contains over 6000 muscle proteins at five time points across postnatal development which includes detailed information on other cellular processes, such as transcription/translation, cytoskeletal or vascular development, that were beyond the scope of the present study. Thus, these data may serve as a significant resource with respect to our understanding of postnatal muscle development in general.
In conclusion, muscle mitochondrial network formation changes from a fibre parallel to a fibre perpendicular configuration during postnatal development in the largely glycolytic TA muscle and this is accompanied by extensive changes in the muscle proteome from P1 to P42. Mitochondrial OxPhos components encoded by both nuclear and mitochondrial DNA are continuously upregulated over the course of postnatal muscle growth. In particular, mitochondrial dynamics and mitophagic systems are more highly induced during the early developmental phase, whereas mitochondrial biogenesis is upregulated during the subsequent muscle maturation. Moreover, initial postnatal muscle development correlates well with the expression of calcium signalling factors, as well as MCU. Thus, near birth, muscle mitochondria are comprised of proteins supporting the co‐ordinated cellular assembly that takes place in early development, whereas mitochondria become largely specialized for energy metabolism as the muscle moves toward adulthood.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All experiments were performed at the National Institutes of Health, and the proteomics data collection was prepared at the NHLBI Proteomics Core. YK and BG designed and performed the in vivo imaging analyses. YK and PK designed and performed the blind test with the results of the imaging analyses. YK and BG designed and YK performed the proteomic analysis. YK and BG designed and YK performed the BN‐PAGE experiments. YK and DSY performed the western blotting experiments. YK and BG wrote the manuscript. YK, DSY, PK and BG edited the manuscript. All of the authors approved the final version of the manuscript submitted for publication. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Division of Intramural Research of the National Heart Lung, and Blood Institute (ZIA HL006221‐02) and the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (ZIA HL006221‐02). Daniel S. Yang is a recipient of Amgen Scholar Summer Internship via NIH Office of Intramural Training and Education Summer Program.
Supporting information
Data S1. Protein quantification for all 6869 detected proteins. Sheet1: All proteins. Sheet2: Up‐regulated reactome pathways. Sheet3: Proteins that increased continuously during muscle development. Sheet4: Proteins that decreased continuously. Sheet5: Proteins that increased then decreased. Sheet6: Proteins that decreased then increased. Sheet7: Remaining proteins.
Acknowledgements
We thank Angel Aponte of the NHLBI Proteomics Core for technical and theoretical support.
Biography
Yuho Kim is a Postdoctoral Fellow in the Muscle Energetics Laboratory at the National Heart, Lung, and Blood Institute. He obtained his doctoral degree in Exercise Science (Science Education) under the guidance of Dr Keith C. DeRuisseau at Syracuse University. After his postdoctoral training under Dr David A. Hood at York University, he is currently continuing his postdoctoral research under the guidance of Dr Brian Glancy. His current projects aim to understand mitochondrial reticulum development in skeletal muscle.

Edited by: Scott Powers & Russell Hepple
Linked articles: This article is highlighted in a Perspectives article by Ryan. To read this article, visit https://doi.org/10.1113/JP278057.
References
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E & Langer T (2014). The i‐AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204, 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anichtchik O, Diekmann H, Fleming A, Roach A, Goldsmith P & Rubinsztein DC (2008). Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J Neurosci 28, 8199–8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin, II , Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel‐Duby R, Rosenzweig A, Ingwall JS & Spiegelman BM (2005). Transcriptional coactivator PGC‐1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metabolism 1, 259–271. [DOI] [PubMed] [Google Scholar]
- Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP & Holloszy JO (2002). Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC‐1. FASEB J 16, 1879–1886. [DOI] [PubMed] [Google Scholar]
- Bakeeva LE, Chentsov YS & Skulachev VP (1981). Ontogenesis of mitochondrial reticulum in rat diaphragm muscle. Eur J Cell Biol 25, 175–181. [PubMed] [Google Scholar]
- Banci L, Bertini I, Ciofi‐Baffoni S, Hadjiloi T, Martinelli M & Palumaa P (2008). Mitochondrial copper(I) transfer from Cox17 to Sco1 is coupled to electron transfer. Proc Natl Acad Sci U S A 105, 6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani RA, Fagian MM, Bassani JW & Vercesi AE (1998). Changes in calcium uptake rate by rat cardiac mitochondria during postnatal development. J Mol Cell Cardiol 30, 2013–2023. [DOI] [PubMed] [Google Scholar]
- Bleck CKE, Kim Y, Willingham TB & Glancy B (2018). Subcellular connectomic analyses of energy networks in striated muscle. Nat Commun 9, 5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczonadi V, Giunta M, Lane M, Tulinius M, Schara U & Horvath R (2015). Investigating the role of the physiological isoform switch of cytochrome c oxidase subunits in reversible mitochondrial disease. Int J Biochem Cell Biol 63, 32–40. [DOI] [PubMed] [Google Scholar]
- Boncompagni S, Rossi AE, Micaroni M, Beznoussenko GV, Polishchuk RS, Dirksen RT & Protasi F (2009). Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Biol Cell 20, 1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinegar AE, Xia Z, Loehr JA, Li W, Rodney GG & Cooper TA (2017). Extensive alternative splicing transitions during postnatal skeletal muscle development are required for calcium handling functions. eLife 6, e27192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosch M, Yu L, Hubbard T & Choudhary J (2009). Accurate and Sensitive Peptide Identification with Mascot Percolator. J Proteome Res 8, 3176–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno NE, Kelly KA, Hawkins R, Bramah‐Lawani M, Amelio AL, Nwachukwu JC, Nettles KW & Conkright MD (2014). Creb coactivators direct anabolic responses and enhance performance of skeletal muscle. EMBO J 33, 1027–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D & Relaix F (2003). The formation of skeletal muscle: from somite to limb. J Anat 202, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne K, Vuocolo T, Gondro C, White JD, Cockett NE, Hadfield T, Bidwell CA, Waddell JN & Tellam RL (2010). A gene network switch enhances the oxidative capacity of ovine skeletal muscle during late fetal development. BMC Genomics 11, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesnekova J, Rodinova M, Hansikova H, Houstek J, Zeman J & Stiburek L (2016). The mammalian homologue of yeast Afg1 ATPase (lactation elevated 1) mediates degradation of nuclear‐encoded complex IV subunits. Biochem J 473, 797–804. [DOI] [PubMed] [Google Scholar]
- Chan DC (2006). Mitochondria: dynamic organelles in disease, aging, and development. Cell 125, 1241–1252. [DOI] [PubMed] [Google Scholar]
- Chaves DF, Carvalho PC, Lima DB, Nicastro H, Lorenzeti FM, Siqueira‐Filho M, Hirabara SM, Alves PH, Moresco JJ, Yates JR, 3rd & Lancha AH, Jr (2013). Comparative proteomic analysis of the aging soleus and extensor digitorum longus rat muscles using TMT labeling and mass spectrometry. J Proteome Res 12, 4532–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HC, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM & Chan DC (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Hazen BC, Russell AP & Kralli A (2013). Peroxisome proliferator‐activated receptor gamma coactivator 1 (PGC‐1)‐ and estrogen‐related receptor (ERR)‐induced regulator in muscle 1 (Perm1) is a tissue‐specific regulator of oxidative capacity in skeletal muscle cells. J Biol Chem 288, 25207–25218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Calvo E, Loureiro M, Guaras AM, Nieto‐Arellano R, Garcia‐Poyatos C, Ezkurdia I, Mercader N, Vazquez J & Enriquez JA (2016). Mechanism of super‐assembly of respiratory complexes III and IV. Nature 539, 579–582. [DOI] [PubMed] [Google Scholar]
- Czubryt MP, McAnally J, Fishman GI & Olson EN (2003). Regulation of peroxisome proliferator‐activated receptor gamma coactivator 1 alpha (PGC‐1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci U S A 100, 1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW, 2nd , Vega RB & Kelly DP (2015). Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 29, 1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn SE, Chin ER & Michel RN (2000). Matching of calcineurin activity to upstream effectors is critical for skeletal muscle fiber growth. J Cell Biol 151, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eivers SS, McGivney BA, Fonseca RG, MacHugh DE, Menson K, Park SD, Rivero JL, Taylor CT, Katz LM & Hill EW (2010). Alterations in oxidative gene expression in equine skeletal muscle following exercise and training. Physiol Genomics 40, 83–93. [DOI] [PubMed] [Google Scholar]
- Franko A, Mayer S, Thiel G, Mercy L, Arnould T, Hornig‐Do HT, Wiesner RJ & Goffart S (2008). CREB‐1alpha is recruited to and mediates upregulation of the cytochrome c promoter during enhanced mitochondrial biogenesis accompanying skeletal muscle differentiation. Mol Cell Biol 28, 2446–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CJ, Khajeh J, Coulanges E, Chen EI & Owusu‐Ansah E (2017). Regulation of mitochondrial complex I biogenesis in drosophila flight muscles. Cell Reports 20, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garry DJ, Bassel‐Duby RS, Richardson JA, Grayson J, Neufer PD & Williams RS (1996). Postnatal development and plasticity of specialized muscle fiber characteristics in the hindlimb. Dev Genet 19, 146–156. [DOI] [PubMed] [Google Scholar]
- Gatliff J, East D, Crosby J, Abeti R, Harvey R, Craigen W, Parker P & Campanella M (2014). TSPO interacts with VDAC1 and triggers a ROS‐mediated inhibition of mitochondrial quality control. Autophagy 10, 2279–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherardi G, Nogara L, Ciciliot S, Fadini GP, Blaauw B, Braghetta P, Bonaldo P, De Stefani D, Rizzuto R & Mammucari C (2018). Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ 26, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B & Balaban RS (2011). Protein composition and function of red and white skeletal muscle mitochondria. Am J Physiol Cell Physiol 300, C1280–C1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, Hartnell LM, Malide D, Yu ZX, Combs CA, Connelly PS, Subramaniam S & Balaban RS (2015). Mitochondrial reticulum for cellular energy distribution in muscle. Nature 523, 617–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, Hsu LY, Dao L, Bakalar M, French S, Chess DJ, Taylor JL, Picard M, Aponte A, Daniels MP, Esfahani S, Cushman S & Balaban RS (2014). In vivo microscopy reveals extensive embedding of capillaries within the sarcolemma of skeletal muscle fibers. Microcirculation 21, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleyzer N, Vercauteren K & Scarpulla RC (2005). Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF‐1 and NRF‐2) and PGC‐1 family coactivators. Mol Cell Biol 25, 1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin DS, Ward SR, Bremner SN & Lieber RL (2008). Quantitative analysis of neonatal skeletal muscle functional improvement in the mouse. J Exp Biol 211, 837–843. [DOI] [PubMed] [Google Scholar]
- Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ & Dorn GW (2015). Parkin‐mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350, aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill BG, Benavides GA, Lancaster JR, Jr. , Ballinger S, Dell'Italia L, Jianhua Z & Darley‐Usmar VM (2012). Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem 393, 1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood DA, Memme JM, Oliveira AN & Triolo M (2019). Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu Rev Physiol 81, 19–41. [DOI] [PubMed] [Google Scholar]
- Horn D, Al‐Ali H & Barrientos A (2008). Cmc1p is a conserved mitochondrial twin CX9C protein involved in cytochrome c oxidase biogenesis. Mol Cell Biol 28, 4354–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Ban‐Ishihara R, Maeda M, Matsunaga Y, Ichimura A, Kyogoku S, Aoki H, Katada S, Nakada K, Nomura M, Mizushima N, Mihara K & Ishihara N (2015). Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol Cell Biol 35, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DT, Harris RA, French S, Blair PV, You J, Bemis KG, Wang M & Balaban RS (2007). Tissue heterogeneity of the mammalian mitochondrial proteome. Am J Physiol Cell Physiol 292, C689–C697. [DOI] [PubMed] [Google Scholar]
- Kall L, Canterbury JD, Weston J, Noble WS & MacCoss MJ (2007). Semi‐supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4, 923–925. [DOI] [PubMed] [Google Scholar]
- Kall L, Storey JD, MacCoss MJ & Noble WS (2008). Posterior error probabilities and false discovery rates: Two sides of the same coin. J Proteome Res 7, 40–44. [DOI] [PubMed] [Google Scholar]
- Kaufmann U, Kirsch J, Irintchev A, Wernig A & Starzinski‐Powitz A (1999). The M‐cadherin catenin complex interacts with microtubules in skeletal muscle cells: implications for the fusion of myoblasts. J Cell Sci 112(Pt 1), 55–68. [DOI] [PubMed] [Google Scholar]
- Kim K, Lecordier A & Bowman LH (1995). Both nuclear and mitochondrial cytochrome c oxidase mRNA levels increase dramatically during mouse postnatal development. Biochem J 306, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Triolo M & Hood DA (2017). Impact of aging and exercise on mitochondrial quality control in skeletal muscle. Oxid Med Cell Longev 2017, 3165396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutti D, Kressler D & Kralli A (2001). Regulation of the transcriptional coactivator PGC‐1 via MAPK‐sensitive interaction with a repressor. Proc Natl Acad Sci U S A 98, 9713–9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara H, Horie T, Ishikawa S, Tsuda C, Kawakami S, Noda Y, Kaneko T, Tahara S, Tachibana T, Okabe M, Melki J, Takano R, Toda T, Morikawa D, Nojiri H, Kurosawa H, Shirasawa T & Shimizu T (2010). Oxidative stress in skeletal muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radic Biol Med 48, 1252–1262. [DOI] [PubMed] [Google Scholar]
- Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F & Hey‐Mogensen M (2012). Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 590, 3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI & Youle RJ (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM & Kelly DP (2000). Peroxisome proliferator‐activated receptor gamma coactivator‐1 promotes cardiac mitochondrial biogenesis. J Clin Invest 106, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC & Epstein CJ (1995). Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11, 376–381. [DOI] [PubMed] [Google Scholar]
- Lira VA, Benton CR, Yan Z & Bonen A (2010a). PGC‐1alpha regulation by exercise training and its influences on muscle function and insulin sensitivity. Am J Physiol Endocrinol Metab 299, E145–E161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira VA, Brown DL, Lira AK, Kavazis AN, Soltow QA, Zeanah EH & Criswell DS (2010b). Nitric oxide and AMPK cooperatively regulate PGC‐1 in skeletal muscle cells. J Physiol 588, 3551–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luff AR & Atwood HL (1971). Changes in the sarcoplasmic reticulum and transverse tubular system of fast and slow skeletal muscles of the mouse during postnatal development. J Cell Biol 51, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, Zentilin L, Sandri M, De Stefani D, Protasi F, Lanfranchi G & Rizzuto R (2015). The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep 10, 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, Attie AD, Muoio DM & Kelly DP (2014). Role for peroxisome proliferator‐activated receptor. Coactivator‐1 in the control of mitochondrial dynamics during postnatal cardiac growth. CircRes 114, 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N & Tanaka K (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minai L, Martinovic J, Chretien D, Dumez F, Razavi F, Munnich A & Rotig A (2008). Mitochondrial respiratory chain complex assembly and function during human fetal development. Mol Genet Metab 94, 120–126. [DOI] [PubMed] [Google Scholar]
- Mishra P & Chan DC (2016). Metabolic regulation of mitochondrial dynamics. J Cell Biol 212, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Varuzhanyan G, Pham AH & Chan DC (2015). Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab 22, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira, II , Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E & Finkel T (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC & Walsh K (2012). Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res 111, 1012–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parousis A, Carter HN, Tran C, Erlich AT, Mesbah Moosavi ZS, Pauly M & Hood DA (2018). Contractile activity attenuates autophagy suppression and reverses mitochondrial defects in skeletal muscle cells. Autophagy 14, 1886–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peker N, Donipadi V, Sharma M, McFarlane C & Kambadur R (2018). Loss of Parkin impairs mitochondrial function and leads to muscle atrophy. Am J Physiol Cell Physiol 315, C164–C185. [DOI] [PubMed] [Google Scholar]
- Pham AH, McCaffery JM & Chan DC (2012). Mouse lines with photo‐activatable mitochondria to study mitochondrial dynamics. Genesis 50, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D, Covian R, Aponte AM, Glancy B, Taylor JF, Chess D & Balaban RS (2012). Regulation of oxidative phosphorylation complex activity: effects of tissue‐specific metabolic stress within an allometric series and acute changes in workload. Am J Physiol Regul Integr Comp Physiol 302, R1034–R1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Vigie P & Youle RJ (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquereau J, Novotova M, Fortin D, Garnier A, Ventura‐Clapier R, Veksler V & Joubert F (2010). Postnatal development of mouse heart: formation of energetic microdomains. J Physiol 588, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohjoismäki JLO, Krüger M, Al‐Furoukh N, Lagerstedt A, Karhunen PJ & Braun T (2013). Postnatal cardiomyocyte growth and mitochondrial reorganization cause multiple changes in the proteome of human cardiomyocytes. Mol Biosyst 9, 1210–1219. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Adelmant G, Wu Z, Fan M, Xu J, O'Malley B & Spiegelman BM (1999). Activation of PPARgamma coactivator‐1 through transcription factor docking. Science 286, 1368–1371. [DOI] [PubMed] [Google Scholar]
- Rahman MM, Iida H & Shibata Y (1997). Expression and localization of annexin V and annexin VI during limb bud formation in the rat fetus. Anat Embryol (Berl) 195, 31–39. [DOI] [PubMed] [Google Scholar]
- Rai M, Katti P & Nongthomba U (2014). Drosophila Erect wing (Ewg) controls mitochondrial fusion during muscle growth and maintenance by regulation of the Opa1‐like gene. J Cell Sci 127, 191–203. [DOI] [PubMed] [Google Scholar]
- Romanello V & Sandri M (2015). Mitochondrial quality control and muscle mass maintenance. Front Physiol 6, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein EC, Nauman M, Chesnick S & Balaban RS (2006). Multi‐photon excitation microscopy in intact animals. J Microsc 222, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RW & Goldspink G (1969). Muscle fibre growth in five different muscles in both sexes of mice. J Anat 104, 519–530. [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S & Margreth A (1969). Coordinated development of the sarcoplasmic reticulum and T system during postnatal differentiation of rat skeletal muscle. J Cell Biol 41, 855–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin J, Andres AM, Taylor DJR, Weston T, Hiraumi Y, Stotland A, Kim BJ, Huang C, Doran KS & Gottlieb RA (2016). Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 12, 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP (1969). Energy accumulation in the cell. Nauka Press, Moscow (in Russian).
- Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C & Youle RJ (2018). Parkin and PINK1 mitigate STING‐induced inflammation. Nature 561, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Song Y & Pillow JJ (2013). Developmental regulation of molecular signalling in fetal and neonatal diaphragm protein metabolism. Exp Biol Med (Maywood) 238, 913–922. [DOI] [PubMed] [Google Scholar]
- Sperl W, Sengers RCA, Trijbels JMF, Ruitenbeek W, Degraaf R, Terlaak H, Vanlith T, Kerkhoff C & Janssen A (1992). Postnatal‐development of pyruvate oxidation in quadriceps muscle of the rat. Biol Neonate 61, 188–200. [DOI] [PubMed] [Google Scholar]
- Spivak M, Weston J, Bottou L, Kall L & Noble WS (2009). Improvements to the percolator algorithm for peptide identification from shotgun proteomics data sets. J Proteome Res 8, 3737–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiber J, Hawkins A, Zhang ZS, Wang S, Burch J, Graham V, Ward CC, Seth M, Finch E, Malouf N, Williams RS, Eu JP & Rosenberg P (2008). STIM1 signalling controls store‐operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol 10, 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Zhu T, Ding F, Hu N & Gu X (2009). Proteomic studies of rat tibialis anterior muscle during postnatal growth and development. Mol Cell Biochem 332, 161–171. [DOI] [PubMed] [Google Scholar]
- Takekura H, Flucher BE & Franzini‐Armstrong C (2001). Sequential docking, molecular differentiation, and positioning of T‐tubule/SR junctions in developing mouse skeletal muscle. Dev Biol 239, 204–214. [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP & Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touvier T, De Palma C, Rigamonti E, Scagliola A, Incerti E, Mazelin L, Thomas JL, D'Antonio M, Politi L, Schaeffer L, Clementi E & Brunelli S (2015). Muscle‐specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis 6, e1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Hyde B & Shirihai OS (2008). Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta 1777, 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uguccioni G & Hood DA (2011). The importance of PGC‐1alpha in contractile activity‐induced mitochondrial adaptations. Am J Physiol Endocrinol Metab 300, E361–E371. [DOI] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C & Zeviani M (2011). In vivo correction of COX deficiency by activation of the AMPK/PGC‐1 alpha axis. Cell Metabolism 14, 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, García‐Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Rupérez FJ, Barbas C, Ibañez B & Langer T (2015). Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350, aad0116. [DOI] [PubMed] [Google Scholar]
- Walker SM & Edge MB (1971). The sarcoplasmic reticulum and development of Z lines in skeletal muscle fibers of fetal and postnatal rats. Anat Rec 169, 661–678. [DOI] [PubMed] [Google Scholar]
- Watchko JF, Daood MJ & LaBella JJ (1996). Creatine kinase activity in rat skeletal muscle relates to myosin phenotype during development. Pediatr Res 40, 53–58. [DOI] [PubMed] [Google Scholar]
- White RB, Bierinx AS, Gnocchi VF & Zammit PS (2010). Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev Biol 10, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DC, Han DH, Garcia‐Roves PM, Geiger PC, Jones TE & Holloszy JO (2007). Exercise‐induced mitochondrial biogenesis begins before the increase in muscle PGC‐1alpha expression. J Biol Chem 282, 194–199. [DOI] [PubMed] [Google Scholar]
- Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel‐Duby R & Williams RS (2002). Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 296, 349–352. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC & Spiegelman BM (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC‐1. Cell 98, 115–124. [DOI] [PubMed] [Google Scholar]
- Wuhr M, Haas W, McAlister GC, Peshkin L, Rad R, Kirschner MW & Gygi SP (2012). Accurate multiplexed proteomics at the MS2 level using the complement reporter ion cluster. Anal Chem 84, 9214–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H & Lu B (2008). Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci USA 105, 7070–7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ & Narendra DP (2011). Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchini S, Giovarelli M, Perrotta C, Morisi F, Touvier T, Di Renzo I, Moscheni C, Bassi MT, Cervia D, Sandri M, Clementi E & De Palma C (2019). Autophagy controls neonatal myogenesis by regulating the GH‐IGF1 system through a NFE2L2‐ and DDIT3‐mediated mechanism. Autophagy 15, 58–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, Collia D, Chen Z, Wozniak DF, Leone TC & Kelly DP (2010). Total skeletal muscle PGC‐1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metabol 12, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuloaga R, Fuentes EN, Molina A & Valdes JA (2013). The cAMP response element binding protein (CREB) is activated by insulin‐like growth factor‐1 (IGF‐1) and regulates myostatin gene expression in skeletal myoblast. Biochem Biophys Res Commun 440, 258–264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Protein quantification for all 6869 detected proteins. Sheet1: All proteins. Sheet2: Up‐regulated reactome pathways. Sheet3: Proteins that increased continuously during muscle development. Sheet4: Proteins that decreased continuously. Sheet5: Proteins that increased then decreased. Sheet6: Proteins that decreased then increased. Sheet7: Remaining proteins.