

Abstract
Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) is implicated in cardiovascular disease by modulating apoptosis and oxidative stress. We hypothesized that LOX-1 may be involved in pathophysiology of stroke by mediating ischaemia/reperfusion (I/R)-dependent cell death. Transient middle cerebral artery occlusion (tMCAO) was performed in wild-type (WT) mice, endothelial-specific LOX-1 transgenic mice (eLOX-1TG) and WT animals treated with LOX-1 silencing RNA (siRNA). In WT mice exposed to tMCAO, LOX-1 expression and function were increased in the MCA. Compared to WT animals, eLOX-1TG mice displayed increased stroke volumes and worsened outcome after I/R. Conversely, LOX-1-silencing decreased both stroke volume and neurological impairment. Similarly, in HBMVECs, hypoxia/reoxygenation increased LOX-1 expression, while LOX-1 overexpressing cells showed increased death following hypoxia reoxygenation. Increased caspase-3 activation was observed following LOX-1 overexpression both in vivo and in vitro, thus representing a likely mediator. Finally, monocytes from ischaemic stroke patients exhibited increased LOX-1 expression which also correlated with disease severity. Our data unequivocally demonstrate a key role for LOX-1 in determining outcome following I/R brain damage. Our findings could be corroborated in human brain endothelial cells and monocytes from patients, underscoring their translational relevance and suggesting siRNA-mediated LOX-1 knockdown as a novel therapeutic strategy for stroke patients.
Keywords: Cell death, ischaemia/reperfusion, lectin-like oxidized low-density lipoprotein receptor-1, middle cerebral artery occlusion, stroke
Introduction
Annually, stroke accounts for about 1 million deaths in Europe. This makes it the second most common cause of mortality, surpassed only by ischaemic heart disease.1 Importantly, stroke is the leading cause of permanent disability in western countries, having caused the loss of more than 17 million disability-adjusted life years in 2015.1 Thus, apart from the human toll, stroke poses a major health-economic burden worldwide.2
Several pathophysiological components concur to determine the extent of brain damage after an ischaemic insult. While the ischaemic core is characterised by a complete lack of perfusion leading to an irreversible activation of neuronal necrosis signalling, the surrounding area (i.e. penumbra) undergoes a mitigated hypoperfusion which leaves room for potential therapeutic interventions.3 Indeed, this area is insufficiently perfused, yet still viable and thus therapeutically accessible by timely reperfusion interventions.3 In the penumbra, the surge of reactive oxygen species (ROS) alongside the establishment of a pro-inflammatory environment drives cells towards different death mechanisms including excitotoxicity and apoptosis.4 This is particularly evident when ischaemia is followed by an early restoration of blood flow, usually referred to as ischaemia/reperfusion (I/R) injury.5 Thus, the available treatments for stroke (thrombolysis and thrombectomy) aimed at early reperfusion do not simply limit lesion size and improve neurological outcome but also hold an intrinsic potential to damage cells which could be reduced by novel interventions aimed at targeting molecular pathways activated by I/R.
Originally described as the receptor for oxidized low-density lipoprotein (OxLDL) in endothelial cells,6 lectin-like oxLDL receptor-1 (LOX-1) is nowadays known to be sensitive to several mediators, such as ROS, angiotensin II and many other inflammatory signals.7 All of these agonists are deeply involved in the determination of cardiovascular risk8,9 and confer an important detrimental role to LOX-1 in the pathogenesis of different cardiovascular diseases such as myocardial infarction,10 atherosclerosis,11 and arterial thrombosis.12 In basal conditions, endothelial cells express low levels of LOX-1; however, following exposure to an inflammatory environment, its expression increases.13 Upon activation, LOX-1 induces endothelial apoptosis by stimulating free radicals production through nicotinamide adenine dinucleotide phosphate-oxidase and activation of the apoptotic B-cell lymphoma 2/cytochrome c/caspase-3 pathway.14
Despite considerable evidence linking inflammation and free radical production to a poor outcome after stroke, the specific role of endothelial LOX-1 in the setting of I/R-induced brain injury is still unknown. Thus, in this study, we investigated whether endothelial LOX-1 modulation affects cerebral damage in a mouse model of stroke. Furthermore, we tested the relevance of LOX-1 in primary human brain microvascular endothelial cells (HBMVECs) exposed to hypoxia/reoxygenation. Finally, we assessed LOX-1 expression in ischaemic stroke patients and correlated it to the National Institute of Health Stroke Scale (NIHSS).
Methods
Animals
Animal experiments were performed on 12-week-old male endothelial-specific LOX-1 transgenic (eLOX-1TG) mice and compared to age- and bodyweight-matched wild-type (WT) littermates or on 12-week-old male LOX-1-silenced (siLOX-1) C57BL/6 WT mice. eLOX-1TG mice were generated on a C57BL/6 background using a murine tyrosine kinase receptor Tie2 promoter as published previously.11 Endothelial specificity of LOX-1 overexpression with respect to other cell types in this strain was previously assessed.11 Targeted LOX-1 gene expression in endothelial cells was achieved using the coding sequence for the murine LOX-1 gene inserted into the expression vector pSP14/15, which contains the murine 2 kb Tie2 promoter together with 10 kb Tie2 enhancer originated from intron 1 of the endogenous murine Tie2 gene (the pSP14/15 vector was a kind gift of Thomas N. Sato, MD, PhD, University of Texas, TX, USA). In vivo LOX-1 silencing was performed as previously described.15 Briefly, pre-designed small interfering RNA (siRNA) targeting LOX-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were injected intravenously in a randomized way. Twenty-four hours before the surgical procedure, 1.6 nmol of siRNA was incubated with a mixture of 150 mM NaCl solution–jetPEI® (Polyplus TransfectionTM, New York, NY, USA) delivery reagent for 15 min at room temperature (RT) and injected into the tail vein. As a negative control, scrambled siRNA was used. All procedures were approved by the ‘Kanton Zurich Gesundheitsdirektion Veterinaeramt’. Animal experiments were performed conform to the Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes. Animal experiment were reported according to Animal Research: Reporting in Vivo Experiments guidelines.
Transient middle cerebral artery occlusion
To induce I/R brain injury, transient middle cerebral artery (MCA) occlusion (tMCAO) was performed as previously described.16 Briefly, mice were anaesthetized using isoflurane 3% and 1.5% for induction and maintenance, respectively, while body temperature was kept at 37℃. For analgesia, 0.5% bupivacaine was infiltrated at the incision side. Ischaemia was induced by inserting a 6-0 silicone-coated filament (Doccol Corporation, Sharon, MA, USA) into the common carotid artery until the origin of the left MCA after the dissection of common, internal and external carotid arteries. After 45 min, reperfusion was allowed for 24 h (eLOX-1TG mice) or 48 h (siLOX-1 mice), followed by euthanasia with carbon dioxide. Sham-operated mice underwent the same procedure without advancing the thread to the MCA origin. During anesthesia, regional cerebral blood flow in the area of the cortex supplied by the MCA was measured using laser Doppler flowmetry (PeriFluxSystem 5000 with probe model no. 418-1, Perimed AB, Jărfălla, Sweden). The microtip probe was positioned and glued ∼2 mm posterior and 6 mm lateral to the bregma. After euthanasia, peripheral blood was collected by intracardial puncture. Then, the animal was perfused with 10 mL cold PBS before harvesting brain and vessels. MCA origin was identified using a dissecting microscope and excised in total. Post-stroke mortality rate was below 10% in all animal groups.
Stroke volume
For determination of stroke volumes, murine brains were cut into five (1 mm thick) coronal sections and immersed in a 2% solution of 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich, Chemie GmbH, Buchs, Switzerland) at 37℃ for 20 min. Stroke areas were quantified using ImageJ. The following formula was applied in order to compensate for cerebral swelling (oedema) and subsequent overestimation of the infarct volume as previously described:17 corrected infarct volume =contralateral hemisphere volume−(ipsilateral hemisphere volume−infarct volume).
Neurological deficit assessment
Neurological status was assessed 2, 24 and 48 h after tMCAO by a four-point scale neurological score based on Bederson et al.18 as follows: grade 0, normal neurological function; grade 1, forelimb and torso flexion to the contralateral side upon lifting the animal by the tail; grade 2, circling to the contralateral side; grade 3, leaning to the contralateral side at rest; grade 4, no spontaneous motor activity, as previously described.15 Neurological performance was determined by the RotaRod test: animals were placed on a rotating rod at increasing speeds (4–44 revolutions/min) and latency to fall was recorded.15 An experimenter blinded to the group allocation evaluated the neurological deficit at 2 h, 24 h and 48 h after tMCAO.
Cell culture experiments
Primary HBMVECs (Cell Systems, Kirkland, WA, USA) between passages 6 and 8 were used for in vitro experiments. Endothelial cells were cultured in EBM-2 medium, supplemented with EGM-2 bullet kit (Lonza, Bettlach, Switzerland) and 10% fetal bovine serum. Cells were grown to 80% confluence before being transfected with LOX-1 expression construct (RC204704; Origene, Rockville, MD, USA) or empty vector (EV) (PS100001; Origene, Rockville, MD, USA) for 24 h using the Lipofectamine® 3000 transfection kit according to manufacturer’s recommendations (Invitrogen, Carlsbad, CA, USA). Next, cells were exposed to hypoxia (0.2% oxygen) for 4 h followed by 4 h of normoxia (21% oxygen) (reoxygenation) or kept at normoxic conditions (21% oxygen) for 8 h, as previously shown.15 Hypoxia was induced using a gas-controlled glove hypoxia workstation (Invivo2 400, Baker Ruskinn, Sanford, ME, USA).
Western blotting
Protein expression was determined by Western blot analysis. Brains and endothelial cells were lysed (Tris 50 mM, NaCl 150 mM, EDTA 1 mM, NaF 1 mM, DTT 1 mM, aprotinin 10 mg/mL, leupeptin 10 mg/mL, Na3VO4 0.1 mM, phenylmethylsulfonyl fluoride 1 mM and NP-40 0.5%) and total protein concentration was determined according to the manufacturer’s recommendations (Bio-Rad Laboratores AG, Fribourg, Switzerland); 20–30 µg of total protein lysates were separated on an 8 or 10% SDS-PAGE before being transferred to a polyvinylidene fluoride membrane by wet transfer (Bio-Rad Laboratores AG, Fribourg, Switzerland). Membranes were incubated with primary antibodies against LOX-1 (ab60178; 1:2000; Abcam, Cambridge, UK), anti-cleaved caspase-3 (#9661S; 1:1000; Cell Signalling, Beverly, MA, USA), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (MAB374; 1:40,000; Merck Millipore, Billerica, MA, USA) over night at 4℃ on a shaker. Secondary antibodies anti-mouse (1031-05) and anti-rabbit (4050-05) were obtained from Southern Biotechnology (Birmingham, AL, USA) and applied for 1 h at room temperature. Densitometric analyses were performed (Amersham Imager 600, General Electric; Healthcare Europe GmbH, Glattbrugg, Switzerland) and protein expression was normalized to GAPDH.
Cytotoxicity detection
Lactate dehydrogenase (LDH) release was determined in supernatant from cultured HBMVECs according to manufacturer’s recommendations (Cytotoxicity Detection Kit; Roche, Basel, Switzerland). Briefly, enzymatic activity of cytoplasmic LDH, released from dead cells, was detected by conversion of NAD+ to NADH/H+ and subsequent reduction of iodotetrazolium chloride to formazan. Absorbance was determined at 490 nm using an enzyme-linked immunosorbent assay (ELISA) plate reader (Infinite M200 pro; Tecan Group Ltd, Männedorf, Switzerland).
Oxidized LDL measurements
OxLDL concentrations in mouse serum and MCA homogenates isolated from 12 to 14-week-old WT males subjected to tMCAO was measured by a mouse oxLDL ELISA Kit (Cusabio Biotech, Wuhan, P. R. China) according to the manufacturer’s instructions.
Patients
A total of 22 ischaemic stroke patients admitted to the emergency room of San Raffaele Hospital (OSR, Milan, Italy) within 6 h after symptom onset were enrolled in the current study. Fourteen sex- and age-matched subjects (relatives or visitors of in-hospital patients), without history of cardio- or cerebrovascular diseases, were included as controls. Venous blood was collected 6 and 24 h after initial stroke symptoms. Of the 22 ischaemic stroke patients, 11 received thrombolytic treatment within 4.5 h from initial symptom onset. Ischaemic strokes were diagnosed based on clinical history, neurological examination and a brain computed tomography scan; classification was done according to the Oxford Community Stroke Project classification19 and stroke aetiology according to the Trial of ORG 10172 in Acute Stroke Treatment criteria.20 Moreover, stroke severity was assessed, using NIHSS on hospital admission. The study was approved by the local Ethics Committee at San Raffaele Scientific Institute, Milan, Italy and was performed conform to the declaration of Helsinki. All participants (or their representative relatives) provided signed informed consent.
Isolation of peripheral monocytes from study subjects
Monocytes from whole blood were isolated using anti-CD14-coated MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) on a magnetic separator (Miltenyi Biotec, Bergisch Gladbach, Germany), as previously described.21
Real-time PCR
Total RNA was extracted from murine MCAs or patients’ monocytes using TRI Reagent (Merck KGaA, Darmstadt, Germany) according to the manufacturer’s recommendations. Conversion of the total cellular RNA to cDNA was performed with Moloney murine leukaemia virus reverse transcriptase and random hexamers (GE Healthcare, Little Chalfont, UK) in a final volume of 35 µL, using 2 µg of total RNA according to manufacturer’s recommendations. RT-PCR was performed in a QuantStudio 7 Flex RT-PCR cycler (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. All RT-PCR experiments were performed using the SYBR Select Master Mix provided by Applied Biosystems (Life Technologies, Carlsbad, CA, USA). Each reaction (20 µL) contained 2 µL cDNA, 400 fmol of each primer and 10 µL of Master Mix. For murine tissue, the following primers were used: LOX-1: forward 5′-AAG TCA TGT GGC AAG AAG CC-3′; reverse 5′-TGG CGT AAT TGT GTC CAC TG-3′ and for GAPDH: forward 5′-TCG TCC CGT AGA CAA AAT GG-3′; reverse 5′-TTG AGG TCA ATG AAG GGG TC-3′. For human tissue, the following primers were used: LOX-1: forward 5′-GCA AAT GGA ACT TCA CCA CC-3′, reverse 5′-CAG TTA AAT GAG CCC GAG GA-3′; for TATA-binding protein: forward: 5′-ACA ACA GCC TGC CAC CTT AC-3′ and reverse: 5′-GTT CTG AAT AGG CTG TGC GG-3′. The amplification program consisted of 1 cycle at 95˚C for 10 min, followed by 40 cycles with a denaturing phase at 95˚C for 15 s, an annealing/elongation phase at 60˚C for 1 min. A melting curve analysis was performed after amplification to verify the accuracy of the amplicon. Cycle threshold (Ct) values for each gene were obtained for each sample and analysed with Graph Pad Prism 6 software (GraphPad Software, Inc, La Jolla, CA, USA). Differences in CT values between a test gene and endogenous controls (ΔCT) were calculated and used for statistical analyses.
Statistical analysis
Data are expressed as mean ± SEM. All statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software, Inc, La Jolla, CA, USA). Data were analysed by using one-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons or unpaired two-tailed Student’s t-test as appropriate. For repeated measurements, two-way ANOVA with Sidak post hoc test was used. Fisher’s exact test was used for comparison of categorical data between study subjects, and Pearson’s correlation analysis was used to test the correlation between two quantitative variables. A probability value (P) below or equal 0.05 was considered as statistically significant. Experimenters were blinded to group allocation during all experimental steps (from tMCAO to data analysis).
Results
Brain ischaemia/reperfusion induces LOX-1 expression and increases oxLDL uptake in cerebral vessels
To test the responsiveness of vascular LOX-1 to stroke in vivo, tMCAO was performed for 45 min, allowing 24 h of reperfusion in WT mice and sham-operated animals were used as negative controls. During tMCAO, the reduction in cerebral blood flow was assessed by laser Doppler flowmetry and showed no significant difference among the groups (Supplementary Figure 1). I/R strongly induced the expression of LOX-1 mRNA in MCAs from tMCAO animals, as compared to sham-operated rodents (P < 0.05, Figure 1(a)). Furthermore, circulating oxLDL plasma levels were reduced in mice undergoing tMCAO compared to sham-operated ones (P < 0.05, Figure 1(b)); meanwhile, oxLDL levels were increased in ipsilateral MCAs compared to contralateral ones, suggesting a physiological relevance of increased LOX-1 expression following stroke (P < 0.05, Figure 1(c)).
Figure 1.
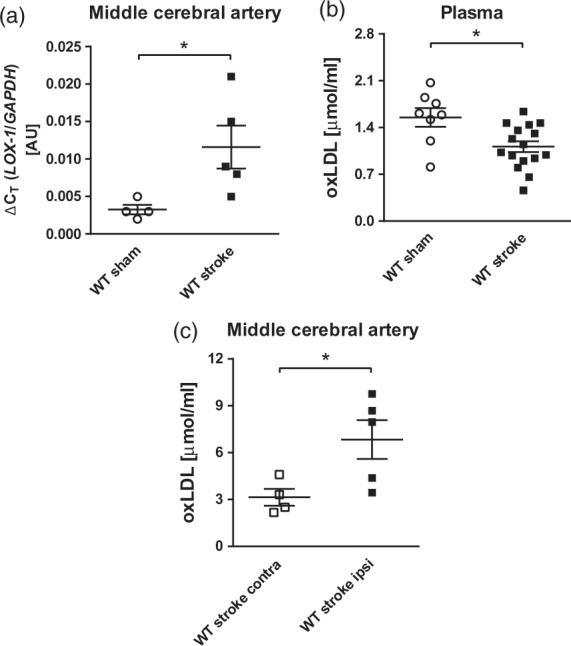
LOX-1 mRNA expression and oxLDL uptake in middle cerebral arteries after tMCAO. (a) Vascular LOX-1 gene expression increased in MCAs of WT mice undergoing tMCAO for 45 min followed by 24 h reperfusion, as compared with sham-operated WT animals (WT sham, n = 4; WT stroke, n = 5). (b) Plasma levels of oxidized LDL were decreased in WT animals that underwent tMCAO (WT stroke, n = 16), compared with sham-operated WT animals (WT sham, n = 8). (c) Further, oxidized LDL uptake in ipsilateral MCAs, which underwent ischaemia reperfusion (WT stroke ipsi, n = 5), was increased, compared with contralateral MCAs (WT stroke contra, n = 4). *P < 0.05. AU: arbitrary unit; MCA: middle cerebral artery; oxLDL: oxidized low-density lipoprotein receptor; LOX-1: lectin-like oxidized low-density lipoprotein receptor-1; WT: wild type.
Endothelial LOX-1 overexpression increases ischaemia/reperfusion-induced cerebral damage and neurologic deficit in mice by activating caspase-3
To assess the relevance of endothelial LOX-1 in cerebral I/R injury, eLOX-1TG mice underwent tMCAO. Following 45 min of ischaemia and 24 h of reperfusion, eLOX-1TG mice revealed increased infarct volumes, as assessed by TTC staining (P < 0.05, Figure 2(a)). Neurological deficits after tMCAO were quantified by two different tests. At baseline, no differences in neurological- and motor function were noticeable between eLOX-1TG mice and WT littermates as assessed by Bederson-based neurological scale and RotaRod test, respectively (Figure 2(b) and (c)); 24 h after stroke, however, eLOX-1TG mice displayed a significantly shorter latency to fall from the rotating rod compared to WT animals, indicating a greater impairment of motor function (P < 0.05, Figure 2(b)). In line with this, also the neurological deficits according to the Bederson-based scale were significantly greater in eLOX-1TG mice than in WT rodents (P < 0.05, Figure 2(c)). All sham-operated animals showed consistently normal neurologic performances during the experimental period (Figure 2(b) and (c)).
Figure 2.

Impact of endothelial LOX-1 overexpression on cerebral lesion and neurological deficit after tMCAO in mice. (a) Endothelial LOX-1 overexpressing mice (eLOX-1TG) revealed increased stroke volumes (n = 7) and (b) decreased neurological functions as assessed by RotaRod (n = 7) or (c) Bederson-based neurological score (n = 7), as compared to WT littermates (n = 7); sham-operated animals (WT sham and LOX1 sham, respectively) remained unaffected (n = 4). (d) After tMCAO, ipsilateral hemispheres from eLOX-1TG mice showed a higher increase in cleaved caspase-3 content from baseline (contralateral side), as compared to WT animals. *P < 0.05. AU: arbitrary unit; MCA: middle cerebral artery; tMCAO: transient middle cerebral artery occlusion; LOX-1: lectin-like oxidized low-density lipoprotein receptor-1; WT: wild-type.
Caspase-3 is part of the caspase-dependent apoptotic cascade22 and is a known downstream target of LOX-1.14 After tMCAO, ipsilateral hemispheres from eLOX-1TG mice showed a higher increase in cleaved caspase-3 content as compared to WT animals (P < 0.05, Figure 2(d)).
In vivo LOX-1 silencing reduces ischaemia/reperfusion-induced cerebral damage
In order to assess the potential of LOX-1 as a pharmacological target, thus increasing the clinical relevance of our above-described findings, LOX-1 was knocked-down in vivo in WT mice, before performing tMCAO for 45 min followed by 48 h of reperfusion. Intravenous injection of siRNA against LOX-1 was the method of choice because previously showed to primarily target endothelium in the brain.15 Stroke sizes as well as neurological deficits were then compared to siScr rodents. Infarct volume was decreased by 30% in siLOX-1 mice as compared to siScr control animals (P < 0.05, Figure 3(a)); accordingly, neuromotor deficit as assessed by the RotaRod test 24 h post-tMCAO was decreased by LOX-1 silencing (P < 0.05; Figure 3(b)). Meanwhile, Bederson-based neurological test was unable to detect any difference in post stroke neurological impairment between siLOX-1 and siScr animals 24 h after stroke (Figure 3(c)).
Figure 3.
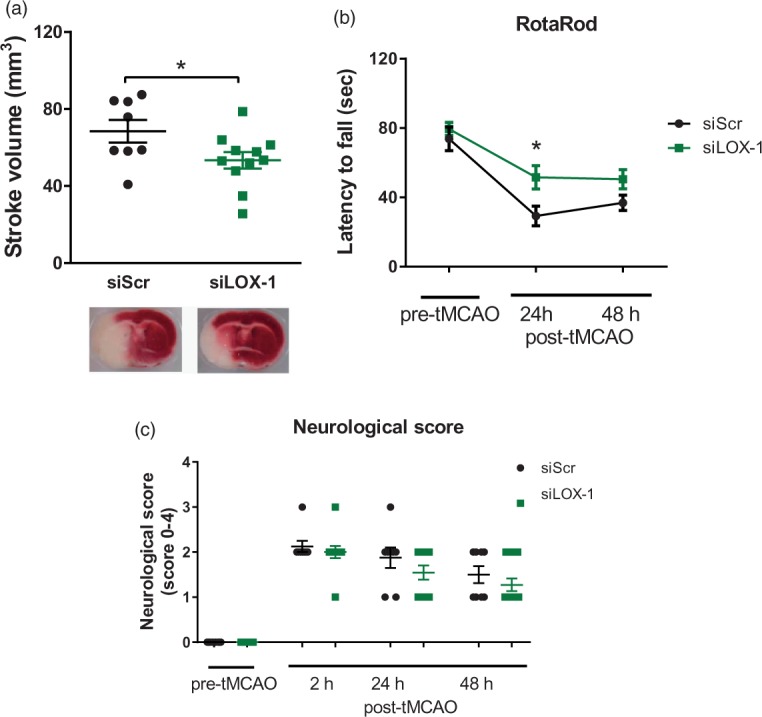
Effects of LOX-1 silencing on cerebral damage and neurological deficit after tMCAO. (a) LOX-1-silenced mice (siLOX-1) showed decreased stroke volumes (n = 11) and (b) decreased neurological function as assessed by RotaRod (n = 11) 24 h after tMCAO, compared to animals treated with control scrambled siRNA (siScr) (n = 8). (c) Bederson-based neurological score test was comparable between the groups (n = 11/8). *P < 0.05. AU: arbitrary unit; tMCAO: transient middle cerebral artery occlusion; LOX-1: lectin-like oxidized low-density lipoprotein receptor-1; WT: wild-type.
Increased cell death and caspase-3 activation in LOX-1-transfected primary HBMVECs after hypoxia/reoxygenation
To translate our findings in mice to human cerebral endothelial cells, primary HBMVECs were exposed to hypoxia for 4 h followed by 4 h of reoxygenation. LOX-1 protein induction in HBMVECs was achieved by transfection with a human LOX-1 expression construct as compared to EV-transfected control cells (P < 0.05, Figure 4(b)). In line with the observed upregulation of vascular LOX-1 following I/R in mice, we observed increased LOX-1 expression in primary HBMVECs after exposure to hypoxia/reoxygenation (P < 0.05, Figure 4(a)). LOX-1 overexpressing cells continue to exhibit increased LOX-1 protein levels compared to EV-treated ones even after hypoxia/reoxygenation (data not shown). LOX-1 overexpression did not affect cell death rate under normoxic conditions (Figure 4(c)). In line with the increased brain injury observed in eLOX-1TG mice, increased cell death rate was also confirmed in LOX-1-transfected HBMVECs following hypoxia/reoxygenation (P < 0.05, Figure 4(d)). Furthermore, we also confirmed an enhanced activation of the pro-apoptotic caspase-3 after hypoxia/reoxygenation in LOX-1 overexpressing cells (P < 0.05, Figure 4(e)) similarly to what observed in eLOX-1TG mice.
Figure 4.

LOX-1 responsiveness and effects of endothelial LOX-1 overexpression on apoptosis and cell death in primary HBMVECs after hypoxia/reoxygenation. (a) LOX-1 expression increased in primary HBMVECs after exposure to hypoxia/reoxygenation (4 h/4 h), as compared with normoxia (8 h) (n = 10). (b) LOX-1 protein expression (n = 4) increased significantly (c) without affecting cell death (n = 4) after LOX-1 transfection of HBMVECs. (d) Endothelial cell death in LOX-1-transfected HBMVECs was increased, compared with EV-transfected cells after exposure to hypoxia/reoxygenation (n = 9). (e) Activated (cleaved) caspase-3 protein expression was upregulated in LOX-1-transfected HBMVECs, compared with EV-transfected cells after exposure to hypoxia/reoxygenation (n = 9). *P < 0.05. EV: empty vector; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; H/R: hypoxia/reoxygenation; HBMVEC: human brain microvascular endothelial cells; kDA: kilodalton; LDH: lactate dehydrogenase; LOX-1: lectin-like oxidized low-density lipoprotein receptor-1; Nx: normoxia.
LOX-1 gene expression is increased in monocytes of patients with ischaemic stroke and correlates with disease severity
To increase the translational relevance of our data, we analysed LOX-1 expression levels in monocytes of patients suffering from ischaemic stroke. In light of the fact that endothelial cells could not be easily obtained, we chose monocytes as a surrogate cell type. A total of 22 ischaemic stroke patients and 14 age- and sex-matched control subjects were included in the analysis. Clinical characteristics between the groups did not differ significantly (Table 1). In ischaemic stroke patients, LOX-1 mRNA levels significantly increased 6 h after initial stroke symptoms and returned to baseline after 24 h (Figure 5(a)) confirming LOX-1 responsiveness to ischaemic stroke also in humans. When analysing patients according to thrombolysis treatment, LOX-1 levels significantly increased after 6 h and returned to control levels after 24 h in both subgroups (n = 11 each, Figure 5(b) and (c)). Interestingly, LOX-1 transcript levels of all study subjects positively correlated with the neurological deficit at admission, assessed according to NIHSS (n = 36, Figure 5(d)).
Table 1.
Characteristics of clinical study population.
| Study population | Controls (n = 14) | Stroke patients (n=22) | p |
|---|---|---|---|
| Age, years (range) | 70.6 (63–82) | 74.0 (52–90) | 0.2732 |
| Female, n (%) | 5 (35.7) | 11 (50.0) | 0.5007 |
| Smoking, n (%) | 0 (0) | 6 (27.3) | 0.0625 |
| Hypertension, n (%) | 6 (42.9) | 11 (50.0) | 0.7419 |
| Dyslipidaemia, n (%) | 1 (7.1) | 4 (18.2) | 0.6283 |
| Atrial fibrillation, n (%) | 0 (0) | 6 (27.3) | 0.0672 |
| Coronary artery disease, n (%) | 0 (0) | 5 (22.7) | 0.1336 |
| Previous TIA/stroke, n (%) | 0 (0) | 3 (13.6) | 0.2667 |
| Peripheral artery disease, n (%) | 0 (0) | 3 (13.6) | 0.2667 |
| Thrombolysis (%) | 0 (0) | 11 (50.0) | 0.0020 |
Note: Clinical characteristics of controls and stroke patients are comparable for age, sex and cerebrovascular risk factors.
TIA: transient ischaemic attack.
Figure 5.

Transiently increased LOX-1 gene expression in patients with ischaemic stroke. (a) LOX-1 mRNA expression in monocytes of patients who have suffered from ischaemic stroke was increased 6 h after initial symptom onset (n = 22), as compared with age- and sex-matched control subjects (n = 14). After 24 h, LOX-1 mRNA expression (n = 11) returned to levels of control subjects. (b) In patients that did undergo thrombolysis (n = 11) as well as (c) in patients without thrombolysis (n = 11), LOX-1 mRNA increased significantly, compared with control subjects, before returning to basal levels after 24 h of symptom onset. (d) LOX-1 transcript levels positively correlated with the neurological deficit assessed by the National Institute of Health Stroke Scale (NIHSS) (n = 36). *P < 0.05. LOX-1: lectin-like oxidized low-density lipoprotein receptor-1; TBP: TATA-binding protein.
Discussion
In recent years, LOX-1 has been increasingly recognised as a crucial regulator of cardiovascular disease by promoting inflammation, oxidative stress and apoptosis.7 Here, we investigated the role of endothelial LOX-1 in stroke making use of eLOX-1TG as well as WT animals treated with LOX-1 silencing RNA (siRNA), exposed to I/R-induced brain injury. Additionally, the translational relevance of this study was assessed by investigating LOX-1 in primary HBMVECs exposed to hypoxia/reoxygenation and monocytes isolated from stroke patients.
We demonstrated for the first time, that functionally active endothelial LOX-1 receptors are expressed in murine cerebral arteries after I/R brain injury as well as in HBMVECs exposed to hypoxia/reoxygenation and monocytes of ischaemic stroke patients. Indeed, the LOX-1 receptors expressed in the setting of an experimental stroke were functionally active as reflected by the increased oxLDL levels in the MCA of eLOX-1TG mice. Importantly, we also showed that specific endothelial LOX-1 overexpression increases stroke size and neurological deficits in mice undergoing tMCAO alongside augmented caspase-3 activation. On the contrary, in vivo LOX-1 silencing reduced post-stroke cerebral damage and improved short-term neuromotor function. Similarly, primary HBMVECs transfected with LOX-1 construct displayed increased cell death following hypoxia/reoxygenation and further investigation of potential mechanisms identified, as observed in vivo, an increased activation of caspase-3 – a known LOX-1 downstream target involved in apoptosis regulation.14 Lastly, given the fact that endothelial cells could not be easily obtained from patients, we chose monocytes as a surrogate cell to investigate LOX-1 in stroke patients. In this setting, we showed a transient increase in LOX-1 expression in monocytes of ischaemic stroke patients within 6 h of symptom-onset, as compared to control subjects. Interestingly, we found a positive correlation between LOX-1 transcript levels and the disease severity.
A large body of evidence links LOX-1 to increased cardio- and cerebrovascular risk.7 Although constitutive levels of this receptor are low, several mediators of cardiovascular diseases, such as pro-inflammatory cytokines and chemokines, shear stress, oxidative stress and angiotensin II can induce its expression.7 Interestingly, some of them also have been recently described as alternative ligands (e.g. advanced glycation end products23 and C-reactive protein24), opening the field to new, oxLDL-independent LOX-1 functions. Vascular LOX-1 expression is increased in different pathological conditions including atherosclerosis,25 diabetes,26 hypertension27 and dyslipidaemia.28 In this sense, our data add novel information showing LOX-1 induction in response to cerebral I/R injury in mice, human primary endothelial cells upon hypoxia/reoxygenation and monocytes isolated from ischaemic stroke patients thus, suggesting a potential novel role for LOX-1 in the pathogenesis of stroke. Although the exact mechanisms mediating the increased expression and activation of LOX-1 in our experimental settings are not fully understood, we speculate that hypoxia-inducible factor (HIF)-1α may be the primary activator of hypoxia-dependent LOX-1 increase. Indeed, our group previously showed that HIF-1α knockdown attenuates hypoxic induction of LOX-1 in macrophages.29
In our experiments, endothelial LOX-1 mediates cerebral damage and neurological impairment after cerebral I/R, complementing what has been already shown in cardiomyocytes by different research groups.10 The solidity of our findings is elegantly confirmed by the fact that LOX-1 silencing reduced stroke size and neuromotor deficit. Interestingly, by using the same eLOX-1TG line, we previously showed endothelial LOX-1 to play a significant role in both atherogenesis through increased vascular inflammation11 and arterial thrombosis via oxLDL-dependent activation of the ERK1/2 pathway.12 Thus, because of its deleterious effects on the cerebrovasculature, endothelial LOX-1 not only increases the risk of major cerebrovascular events by acting on atherosclerosis and arterial thrombus formation but also contributes to the extent of brain damage and resulting neurological deficit. Accordingly, LOX-1 could be envisaged as a potential target for both stroke prevention and treatment. Indeed, tMCAO as a model of stroke well represents the clinical situation of patients presenting within an accepted time frame for pharmacological or mechanical recanalization of an occluded cerebral artery to limit brain damage.
TTC staining of viable brain in vivo and LDH assay of HBMVECs’ supernatant in vitro suggest LOX-1 overexpression to increase I/R- and hypoxia/reoxygenation-mediated cell death rate. To this end, we identified activation of pro-apoptotic caspase-3 as a potential mechanism confirmed in both in brain homogenates of eLOX-1TG mice and in cell lysates of LOX-1 overexpressing primary HBMVECs. Indeed, caspase-3 is a known downstream target of LOX-1 activation and our findings are in line with the results from Chen et al. showing increased cleaved caspase-3 in human coronary artery endothelial cells exposed to oxLDL.30 On the other hand, we could not observe increased levels of oxidative stress (data not shown) in response to oxLDL-mediated LOX-1 activation as previously reported in endothelial cells31 as well as murine vessels.11 This suggests endothelial LOX-1 activation per se to be sufficient to increase caspase-3 cleavage and subsequent cell death after I/R, independently of ROS. On the other hand, increased LOX-1 expression in murine MCAs following I/R was associated with increased uptake of oxLDL, providing confirmation of its functionality.
In order to increase the translational relevance of our preclinical findings, we investigated LOX-1 expression also in ischaemic stroke patients. Since isolation of cerebrovascular endothelial cells from ischaemic stroke patients was not feasible, monocytes from whole blood were selected as a surrogate cell type to investigate LOX-1 gene expression changes in response to stroke as previously described.15,32 Here, we found that LOX-1 mRNA levels were transiently increased 6 h after onset of stroke symptoms and returned to baseline at 24 h, irrespectively of thrombolytic treatment. Also, LOX-1 expression positively correlated with the disease severity at admission. These data confirmed what was also observed in mice and HBMVECs, thus supporting a potential role for LOX-1 in determining outcome after stroke.
Some limitations should be taken into consideration when interpreting the herein reported data. First, although we already demonstrated that i.v. injection of siRNA primarily targets the brain’s endothelium,15 we cannot exclude that other cell types minimally concur to determine the results showed in LOX-1 knockdown experiments. Second, the exact molecular mechanisms leading to increased LOX-1 expression following I/R remain unclear. However, as we previously reported,29 HIF-1α is a likely candidate. Finally, given the difficulty of obtaining cerebrovascular specimens from stroke patients, we chose peripheral blood monocytes as a surrogate cell for proof of principle experiments to confirm our findings in patients; this, however, will require additional confirmatory follow-up investigations.
In conclusion, LOX-1 was found to be upregulated in cerebral vessels from mice undergoing cerebral I/R, in HBMVECs exposed to hypoxia/reperfusion and in monocytes from ischaemic stroke patients. In particular, we report that LOX-1 plays a detrimental role in determining infarct size and neurological impairment in mice after tMCAO. Accordingly, in vivo LOX-1 targeting by siRNA could ameliorate morphological and functional stroke outcome. Furthermore, human brain endothelial cells exposed to hypoxia/reoxygenation showed increased death rate when overexpressing LOX-1 and, in our clinical cohort, LOX-1 expression in monocytes positively correlated with the disease severity. Interestingly, increased activation of LOX-1 downstream target caspase-3 was observed both in vivo and in vitro likely representing a key mediator of the observed effect. Our data implicate, for the first time, LOX-1 as a potential therapeutic target, not only for the prevention of ischaemic cerebrovascular disease but also for its treatment.
Supplemental Material
Supplemental material for Deleterious role of endothelial lectin-like oxidized low-density lipoprotein receptor-1 in ischaemia/reperfusion cerebral injury by Alexander Akhmedov, Nicole R Bonetti, Martin F Reiner, Remo D Spescha, Heidi Amstalden, Mario Merlini, Daniel S Gaul, Candela Diaz-Cañestro, Rebecca S Spescha, Aurora Semerano, Giacomo Giacalone, Gianluigi Savarese, Fabrizio Montecucco, Luka Kulic, Roger M Nitsch, Christian M Matter, Gerd A Kullak-Ublick, Maria Sessa, Thomas F Lüscher, Jürg H Beer, Luca Liberale and Giovanni G Camici in Journal of Cerebral Blood Flow & Metabolism
Acknowledgements
We would like to thank Christian Hiller from the Department of Clinical Pharmacology and Toxicology, University Hospital Zurich, Zurich, Switzerland for his support in hypoxic chamber experiments.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The present work was supported by the Swiss National Science Foundation (Drs. Camici [310030_175546] and Lüscher [310030_166576]), the Alfred and Annemarie von Sick Grants for Translational and Clinical Research Cardiology and Oncology to Dr. Camici and the Foundation for Cardiovascular Research–Zurich Heart House. G.G. Camici is the recipient of a Sheikh Khalifa's Foundation Assistant Professorship at the Faculty of Medicine, University of Zurich.
Declaration conflicting interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
AA, NRB, LL, and GGC contributed equally to this work. This study was conceived and planned by AA, NRB, MFR, RDS, TFL, CMM, JHB, LL and GGC. Experiments were performed by AA, NRB, MFR, RDS, HA, MM, DSG, CDC, RSS, GS and LL. AA, MFR, LL and GGC analysed the data and drafted the article. AS, GG, MS enrolled the clinical cohort, processed human samples and gave important insights to this work. FM, LK, RMN and GAKU contributed to the interpretation of data. All authors revised the article for important intellectual content, reviewed the data and their analysis and approved this article.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Atlas Writing G, Timmis A, Townsend N, et al. European Society of Cardiology: Cardiovascular Disease Statistics 2017. Eur Heart J 2017; 39: 508–579. [DOI] [PubMed] [Google Scholar]
- 2.Camici GG, Liberale L. Aging: the next cardiovascular disease? Eur Heart J 2017; 38: 1621–1623. [DOI] [PubMed] [Google Scholar]
- 3.Ramos-Cabrer P, Campos F, Sobrino, et al. Targeting the ischemic penumbra. Stroke 2011; 42: S7–S11. [DOI] [PubMed] [Google Scholar]
- 4.Bonaventura A, Liberale L, Vecchie A, et al. Update on inflammatory biomarkers and treatments in ischemic stroke. Int J Mol Sci 2016; 17: 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol 2015; 6: 524–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawamura T, Kume N, Aoyama T, et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997; 386: 73–77. [DOI] [PubMed] [Google Scholar]
- 7.Pothineni NVK, Karathanasis SK, Ding Z, et al. LOX-1 in atherosclerosis and myocardial ischemia: biology, genetics, and modulation. J Am Coll Cardiol 2017; 69: 2759–2768. [DOI] [PubMed] [Google Scholar]
- 8.Paneni F, Diaz Canestro C, Libby P, et al. The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol 2017; 69: 1952–1967. [DOI] [PubMed] [Google Scholar]
- 9.Camici GG, Savarese G, Akhmedov A, et al. Molecular mechanism of endothelial and vascular aging: implications for cardiovascular disease. Eur Heart J 2015; 36: 3392–3403. [DOI] [PubMed] [Google Scholar]
- 10.Kataoka K, Hasegawa K, Sawamura T, et al. LOX-1 pathway affects the extent of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun 2003; 300: 656–660. [DOI] [PubMed] [Google Scholar]
- 11.Akhmedov A, Rozenberg I, Paneni F, et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur Heart J 2014; 35: 2839–2848. [DOI] [PubMed] [Google Scholar]
- 12.Akhmedov A, Camici GG, Reiner MF, et al. Endothelial LOX-1 activation differentially regulates arterial thrombus formation depending on oxLDL levels: role of the Oct-1/SIRT1 and ERK1/2 pathways. Cardiovasc Res 2017; 113: 498–507. [DOI] [PubMed] [Google Scholar]
- 13.Lu J, Mitra S, Wang X, et al. Oxidative stress and lectin-like ox-LDL-receptor LOX-1 in atherogenesis and tumorigenesis. Antioxid Redox Signal 2011; 15: 2301–2333. [DOI] [PubMed] [Google Scholar]
- 14.Lubrano V, Balzan S. LOX-1 and ROS, inseparable factors in the process of endothelial damage. Free Radic Res 2014; 48: 841–848. [DOI] [PubMed] [Google Scholar]
- 15.Spescha RD, Klohs J, Semerano A, et al. Post-ischaemic silencing of p66Shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. Eur Heart J 2015; 36: 1590–1600. [DOI] [PubMed] [Google Scholar]
- 16.Spescha RD, Shi Y, Wegener S, et al. Deletion of the ageing gene p66(Shc) reduces early stroke size following ischaemia/reperfusion brain injury. Eur Heart J 2013; 34: 96–103. [DOI] [PubMed] [Google Scholar]
- 17.Liberale L, Diaz-Canestro C, Bonetti NR, et al. Post-ischaemic administration of the murine Canakinumab-surrogate antibody improves outcome in experimental stroke. Eur Heart J. Epub ahead of print 16 May 2018. DOI: 10.1093/eurheartj/ehy286. [DOI] [PubMed] [Google Scholar]
- 18.Bederson JB, Pitts LH, Tsuji M, et al. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 1986; 17: 472–476. [DOI] [PubMed] [Google Scholar]
- 19.Bamford J, Sandercock P, Dennis M, et al. Classification and natural history of clinically identifiable subtypes of cerebral infarction. Lancet 1991; 337: 1521–1526. [DOI] [PubMed] [Google Scholar]
- 20.Adams HP, Jr, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993; 24: 35–41. [DOI] [PubMed] [Google Scholar]
- 21.Franzeck FC, Hof D, Spescha RD, et al. Expression of the aging gene p66Shc is increased in peripheral blood monocytes of patients with acute coronary syndrome but not with stable coronary artery disease. Atherosclerosis 2012; 220: 282–286. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene 2008; 27: 6194–6206. [DOI] [PubMed] [Google Scholar]
- 23.Jono T, Miyazaki A, Nagai R, et al. Lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) serves as an endothelial receptor for advanced glycation end products (AGE). FEBS Lett 2002; 511: 170–174. [DOI] [PubMed] [Google Scholar]
- 24.Shih HH, Zhang S, Cao W, et al. CRP is a novel ligand for the oxidized LDL receptor LOX-1. Am J Physiol Heart Circ Physiol 2009; 296: H1643–H1650. [DOI] [PubMed] [Google Scholar]
- 25.Kataoka H, Kume N, Miyamoto S, et al. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation 1999; 99: 3110–3117. [DOI] [PubMed] [Google Scholar]
- 26.Chen M, Nagase M, Fujita T, et al. Diabetes enhances lectin-like oxidized LDL receptor-1 (LOX-1) expression in the vascular endothelium: possible role of LOX-1 ligand and AGE. Biochem Biophys Res Commun 2001; 287: 962–968. [DOI] [PubMed] [Google Scholar]
- 27.Nagase M, Hirose S, Sawamura T, et al. Enhanced expression of endothelial oxidized low-density lipoprotein receptor (LOX-1) in hypertensive rats. Biochem Biophys Res Commun 1997; 237: 496–498. [DOI] [PubMed] [Google Scholar]
- 28.Chen H, Li D, Sawamura T, Inoue K, et al. Upregulation of LOX-1 expression in aorta of hypercholesterolemic rabbits: modulation by losartan. Biochem Biophys Res Commun 2000; 276: 1100–1104. [DOI] [PubMed] [Google Scholar]
- 29.Crucet M, Wust SJA, Spielmann P, et al. Hypoxia enhances lipid uptake in macrophages: role of the scavenger receptors Lox1, SRA, and CD36. Atherosclerosis 2013; 229: 110–117. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Mehta JL, Haider N, et al. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ Res 2004; 94: 370–376. [DOI] [PubMed] [Google Scholar]
- 31.Cominacini L, Pasini AF, Garbin U, et al. Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL receptor-1 in endothelial cells induces the activation of NF-kappaB through an increased production of intracellular reactive oxygen species. J Biol Chem 2000; 275: 12633–12638. [DOI] [PubMed] [Google Scholar]
- 32.Diaz-Canestro C, Merlini M, Bonetti NR, et al. Sirtuin 5 as a novel target to blunt blood-brain barrier damage induced by cerebral ischemia/reperfusion injury. Int J Cardiol 2018; 260: 148–155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material for Deleterious role of endothelial lectin-like oxidized low-density lipoprotein receptor-1 in ischaemia/reperfusion cerebral injury by Alexander Akhmedov, Nicole R Bonetti, Martin F Reiner, Remo D Spescha, Heidi Amstalden, Mario Merlini, Daniel S Gaul, Candela Diaz-Cañestro, Rebecca S Spescha, Aurora Semerano, Giacomo Giacalone, Gianluigi Savarese, Fabrizio Montecucco, Luka Kulic, Roger M Nitsch, Christian M Matter, Gerd A Kullak-Ublick, Maria Sessa, Thomas F Lüscher, Jürg H Beer, Luca Liberale and Giovanni G Camici in Journal of Cerebral Blood Flow & Metabolism