

Abstract
Objectives:
Cisplatin is commonly used in the treatment of head and neck squamous cell carcinoma (HNSCC), and the repair of cisplatin-induced DNA damage involves activation of the DNA damage response protein ataxia telangiectasia and Rad3-related (ATR). Resistance to cisplatin therapy exacerbates adverse toxicities and is associated with poor outcomes. Since repair of cisplatin-induced DNA damage contributes to resistance, we hypothesized that inhibition of ATR using AZD6738, a well-tolerated and orally-bioavailable inhibitor, would enhance the sensitivity of HNSCC cells and tumors to cisplatin.
Materials and methods:
A panel of human papilloma virus-negative (HPV−) and HPV+ HNSCC cell lines were treated with cisplatin in the absence or presence of AZD6738, and effects on cell viability, colony formation, apoptosis signaling, and DNA damage were assessed. The impact of co-treatment with cisplatin plus AZD6738 on the growth of HPV− and HPV+ cell line- and patient-derived xenograft tumors was also examined.
Results:
Inhibition of ATR with AZD6738 enhanced cisplatin-induced growth inhibition of HNSCC cell lines and tumors, in association with increased apoptosis signaling and DNA damage. Both HPV− and HPV+ models were sensitized to cisplatin by ATR inhibition.
Conclusion:
Inhibition of ATR promotes sensitization to cisplatin in preclinical in vitro and in vivo models of HPV− and HVP+ HNSCC, supporting clinical evaluation of this strategy in this disease.
Keywords: ATR, cisplatin, AZD6738, HNSCC
Introduction
Exposure to tobacco products and an increasing incidence of oropharyngeal infection with human papilloma virus (HPV) have contributed to making head and neck squamous cell carcinoma (HNSCC) one of the leading causes of cancer mortality worldwide (1,2). Historically, surgery, radiation and chemotherapy have served as the primary treatment options for HNSCC, although these approaches are commonly associated with multiple adverse toxicities and effects on speech and swallowing. Three molecular targeting agents have been approved by the US Food and Drug Administration (FDA) for the treatment of HNSCC. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, was approved in 2006 (3, 4), while pembrolizumab and nivolumab, antibodies targeting the checkpoint receptor PD-1 on effector T cells, were approved in 2016 (5-7). Although the application of these new agents represents a significant advance in the treatment of HNSCC, in each case, only a minority subset of patients respond (3-7). Hence, the use of conventional chemotherapy remains a mainstay of treatment.
The most commonly used chemotherapy drug for HNSCC is cisplatin (8, 9). Cisplatin acts by forming covalent adducts with DNA bases, primarily guanine, promoting the formation of intra-strand DNA crosslinks (and less frequently, inter-strand crosslink) (10-12). Failure to repair these crosslinks blocks the progression of DNA and RNA polymerases, leading to induction of cell death (13). The repair of cisplatin-induced DNA intra-strand crosslinks occurs via nucleotide excision repair and involves activation of the kinase activity of ataxia telangiectasia and Rad3-related (ATR) and the ATR substrate CHK1 (14-18). The development of small molecule inhibitors of ATR, including M6620 (formerly called VX-970 and VE-822) (19, 20) and AZD6738 (21,22), has confirmed the critical role of ATR in the repair of cisplatin-induced damage. Moreover, treatment with ATR inhibitors has been shown to enhance cell death induced by chemotherapy drugs and radiation in cancer cell lines in vitro (19-21,23-30). In vivo studies have shown that M6620 and AZD6738 sensitize lung cancer and pancreatic ductal adenocarcinoma tumors to cisplatin and gemcitabine, respectively (20, 21, 31). The ability of ATR inhibitors to enhance responsiveness to chemotherapy is amplified in cells and tumors deficient in ataxia telangiectasia mutated (ATM) (21,23, 28, 32). The impact of ATR inhibitors on HNSCC has not been reported.
The profile of HNSCC in the United States is characterized by a dramatically increasing incidence of HPV-positive (HPV+) oropharyngeal carcinoma (33, 34). Current estimates indicate that roughly 75% of oropharyngeal HNSCC cases are associated with HPV infection (35, 36). Recent investigations implicate a complex interaction between HPV and DNA damage response proteins, including ATR and ATM (37-39). In particular, HPV recruits and exploits DNA damage response proteins to facilitate viral replication (37-39). However, the impact of HPV on ATR-mediated repair of chemotherapy-induced DNA damage is incompletely understood. HNSCC offers a unique opportunity for investigation of chemosensitization by ATR inhibitors as both HPV-negative (HPV−) and HPV+ HNSCC preclinical models exist. In this report we utilized a broad panel of HPV− and HPV+ HNSCC cell lines to determine the effects of ATR inhibition on cellular growth and survival following cisplatin treatment. Inhibition of ATR signaling was achieved using AZD6738, an orally bioavailable inhibitor currently being evaluated in clinical trials (40), or by downregulation of ATR mRNA and protein. We further evaluated the in vivo anti-tumor activities of AZD6738 and cisplatin, alone and in combination, against both HPV− and HPV+ cell line-derived and patient-derived xenograft tumors. Our findings support clinical evaluation of AZD6738/cisplatin co-treatment in patients with either HPV− or HPV+ HNSCC.
Materials and methods
Cell lines and cell culture
All cell lines were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin. The authenticity of each cell line was validated by Short Tandem Repeat (STR) profiling prior to use and every 6 months thereafter.
Crystal violet assays
Cells were plated in 96-well plates (500-5,000 cells/well depending on growth rate) and incubated with drugs for 48 hours. Treated cells were washed with 50 μL PBS then incubated in 50 μL of 0.5% crystal violet in 25% methanol. After 30 minutes the staining solution was removed and the plates washed with water 5 times. After overnight air-drying, crystal violet stain was dissolved using a 1:1 mixture of 200 mM sodium citrate (pH 6.0) and 100% ethanol. Plates were then briefly placed on a shaker and the resulting absorbance of the solution was read at 590 nm. IC50 values were calculated using GraphPad Prism (GraphPad Software).
Annexin V/PI flow cytometry
Annexin V/PI staining was performed using FITC Annexin V Apoptosis Detection kits (BD Biosciences, cat# 556547). Cells were removed from plates by trypsinization, washed twice with PBS, then resuspended in 1x Binding Buffer. The cells (1×105 cells in 100 μL) were incubated with 2 μL FITC-annexin V and 2 μL propidium iodide (PI) for 15 minutes at room temperature. Binding buffer (400 μL) was then added to each tube prior to analysis on a FACS Calibur DxP8 (BD Biosciences). Data was analyzed using FlowJo software (FlowJo, LLC).
Immunoblotting
Immunoblotting was performed as previously described (41). The following primary antibodies were used in this study: rabbit anti-pATR Ser428 (CST, cat# 2853), rabbit anti-ATR (CST, cat# 13934), rabbit anti-ATM (CST, cat# 2873), rabbit anti-PARP-1 (CST, cat# 9542), rabbit anti-pCHK1 Ser345 (CST, cat# 2348), mouse anti-CHK1 (CST, cat# 2360), rabbit anti-pH2AX Ser139 (CST, cat# 9718), mouse anti-TP53 (Santa Cruz, cat# sc-126), and rabbit anti-GAPDH (CST, cat# 5174). Goat anti-mouse HRP (Bio-rad, cat# 1706516) and goat anti-rabbit HRP (Bio-rad, cat# 1706515) antibodies were used as secondary antibodies.
Colony formation assays
Cells were plated in 6-well plates (1.7-2.0×105 cells/well) and incubated with drugs for 24 hours. Afterwards, cells were trypsinized, plated (400-800 cells/well depending on growth rate) in triplicate in new 6-well plates, then allowed to grow for at least 14 days. Colonies were then washed with 1 mL of PBS and incubated in 650 μL of 0.5% crystal violet. Colonies were counted using the ImageJ software and later dissolved with a 1:1 mixture of 200 mM sodium citrate and 100% ethanol. The absorbance (590 nm) of the resulting solution was then measured.
siRNA knockdown
ON-TARGETplus SMARTpool siRNAs were obtained from Dharmacon for ATR (cat# L-003202-00) and ATM (cat# L-003201-00). ON-TARGETplus non-targeting pool siRNA (siNT; cat# D-001810-10) was used as a control. Cells (1×106 per 6 cm dish) were transfected with 60 nmol of siRNA using Lipofectamine RNAiMAX transfection reagent (cat# 13778150), followed by incubation for 48 hours prior to analysis.
In vivo xenograft models
To generate xenografts tumors from human HNSCC cell lines, 1×106 cells were injected into each flank of nude mice. Patient-derived xenograft (PDX) tumor models were propagated bilaterally in NOD/SCID gamma (NSG) mice (NOD.Cg-Prkdcscid ll2rgtm1WJl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ) as previously described (42). Tumors were grown to an average volume of 100 mm3 before randomization and treatment with the indicated dosing regimens. Tumors were subsequently measured by calipers twice per week. This work was performed in accordance with UCSF IACUC protocol # AN109582.
Statistical analysis
For in vitro studies, statistical differences between treatment groups were determined using two-tailed Student’s t-test. Figures for in vitro studies are representative of at least three independent experiments, except where indicated. For in vivo studies, Mann-Whitney test was used to determine statistical differences between treatment groups. Error bars in all figures indicate standard deviations.
Results
ATR inhibition sensitizes HPV− and HPV+ HNSCC cells to growth inhibition by cisplatin
We first determined the sensitivity of a panel of 21 human HNSCC cells lines toward cisplatin (Figure 1A and Supplemental Table 1). Cells were treated with a range of cisplatin concentrations for 48 hours, followed by assessment of growth in crystal violet assays. A relatively narrow range of IC50 values was observed, with CAL-33 cells being the most sensitive line (IC50 = 2.0 μM) and SCC-9 being the most resistant (IC50 = 21.7 μM). Somewhat surprisingly, the 4 HPV+ cell lines (UM-SCC-47, UPCI-SCC-090, 93VU147T, UD-SCC-2) in the panel were among the most resistant (IC50’s from 8.9-18.0 μM).
Fig. 1.
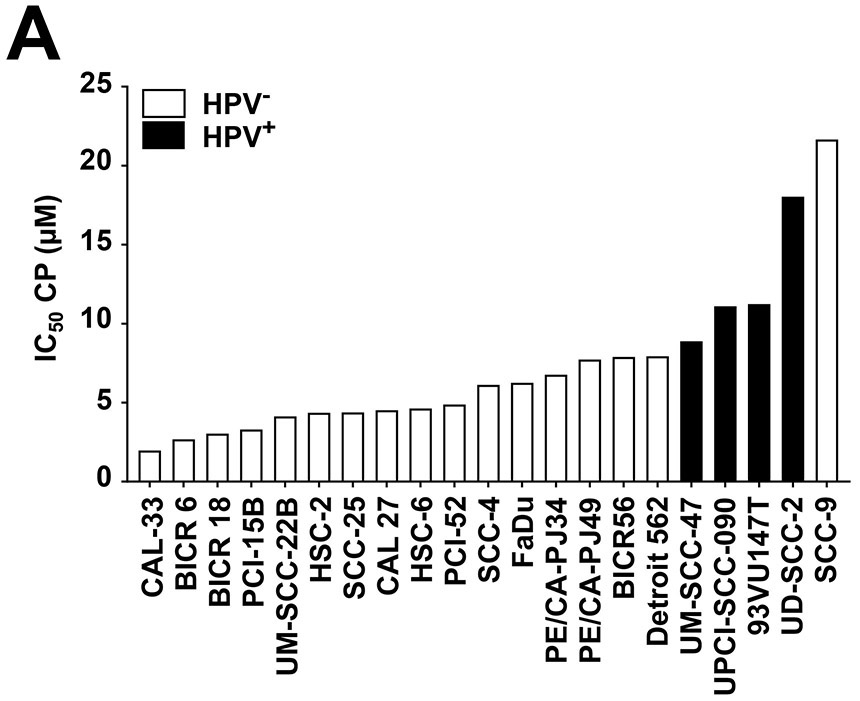
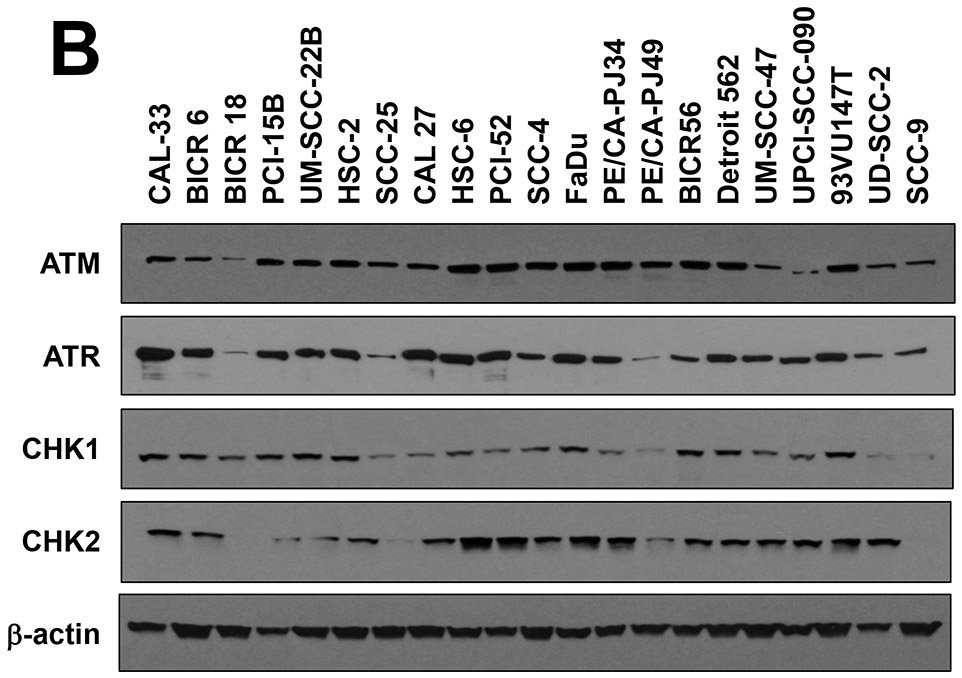
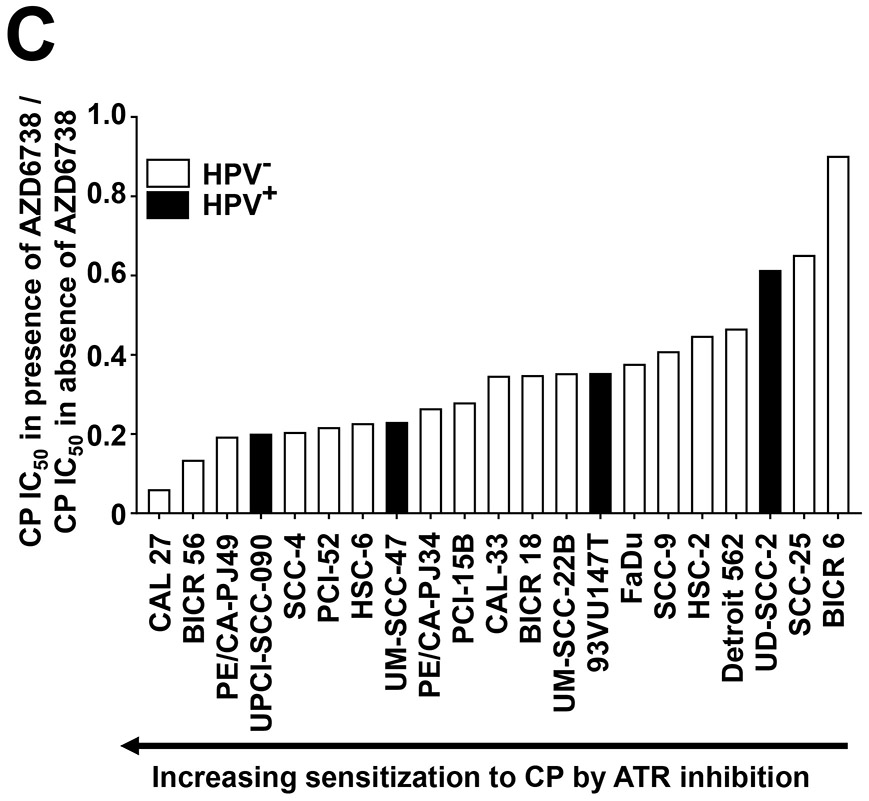
ATR inhibition by AZD6738 sensitizes HNSCC cell lines to cisplatin (CP). A, IC50 values for CP are shown for HPV− (open bars) and HPV+ (solid bars) HNSCC cell lines. Cells were incubated for 48 hours with varying concentrations of CP, followed by performance of crystal violet assays. IC50 values were extrapolated from titration treatment curves performed in triplicate. B, Whole cell lysates of HNSCC cell lines were subjected to immunoblotting for ATM, ATR, CHK1, or CHK2, with β-actin used as a control for protein loading. C, CP IC50 values determined in the absence (vehicle treatment) or presence of 500 nM AZD6738. The plotted values represent the ratio of CP IC50s in the presence of AZD6738 to CP IC50s in the absence of AZD6738. Solid bars again indicate HPV+ cell lines.
To determine whether potential relationships exist between sensitivity to cisplatin and the expression levels of total ATR, ATM, CHK1, and CHK2 proteins we performed immunoblotting, with β-actin serving as the loading control (Figure 1B). Varying levels of the four proteins were detected among the different HNSCC cell lines, with no absolute correlation with cisplatin sensitivity.
Despite the fact that no clear correlation was established between ATR levels and cisplatin sensitivity, prior studies have indicated that targeted inhibition of ATR may provide a means for enhancing the in vitro and in vivo sensitivity to cisplatin in lung cancer models (20, 21). To determine the impact of inhibiting ATR on cisplatin-induced growth inhibition, we assessed IC50’s for cisplatin in the absence or presence of 500 nM AZD6738, a highly selective ATR inhibitor (21, 22). At the 500 nM concentration, AZD6738 alone did not impact the growth of the cells in 48-hour assays. Figure 1C (and Supplemental Table 2) depicts the ratio of the IC50 obtained in the presence versus absence of AZD6738. As shown, AZD6738 enhanced sensitivity to cisplatin in all 21 HNSCC cell lines, although a range of sensitization was observed. The greatest sensitization was observed in CAL 27 cells, where AZD6738 caused a roughly 16.7-fold decrease in IC50 for cisplatin. By contrast, BICR 6 cells, the least sensitized cell line, exhibited only a 1.1-fold decrease in IC50 for cisplatin in the presence of the ATR inhibitor. Notably, AZD6738 caused a greater than 2-fold decrease in cisplatin IC50’s in 3 out of 4 HPV+ cell lines (UPCI-SCC-090, UM-SCC-47, 93VU147T), but a less than 2-fold decrease in HPV+ UD-SCC-2 cells.
To confirm the impact of AZD6738 on cisplatin-induced growth inhibition in HNSCC cells we performed colony formation assays. CAL 27 (highly sensitized in crystal violet assays) and Detroit 562 (weakly sensitized in crystal violet assays) were treated with vehicle, 500 nM AZD6738 alone, 2 μM cisplatin alone, or the combination of AZD6738 and cisplatin. Following 24 hours of treatment, cells were replated and grown for at least 14 days, followed by determination of colony formation (Figure 2A and Supplemental Figure 1). As shown, the combination of AZD6738 and cisplatin resulted in enhanced loss of colony formation in both cell line models, relative to treatment with either agent alone. The combination also resulted in modestly enhanced loss of colony formation, relative to single agents, in the HPV+ cell line UM-SCC-47 (Supplemental Figure 1). By contrast, co-treatment with AZD6738 and paclitaxel, which exerts anticancer activity via effects on microtubules, did not result in enhanced loss of colony formation (Figure 2B).
Fig. 2.
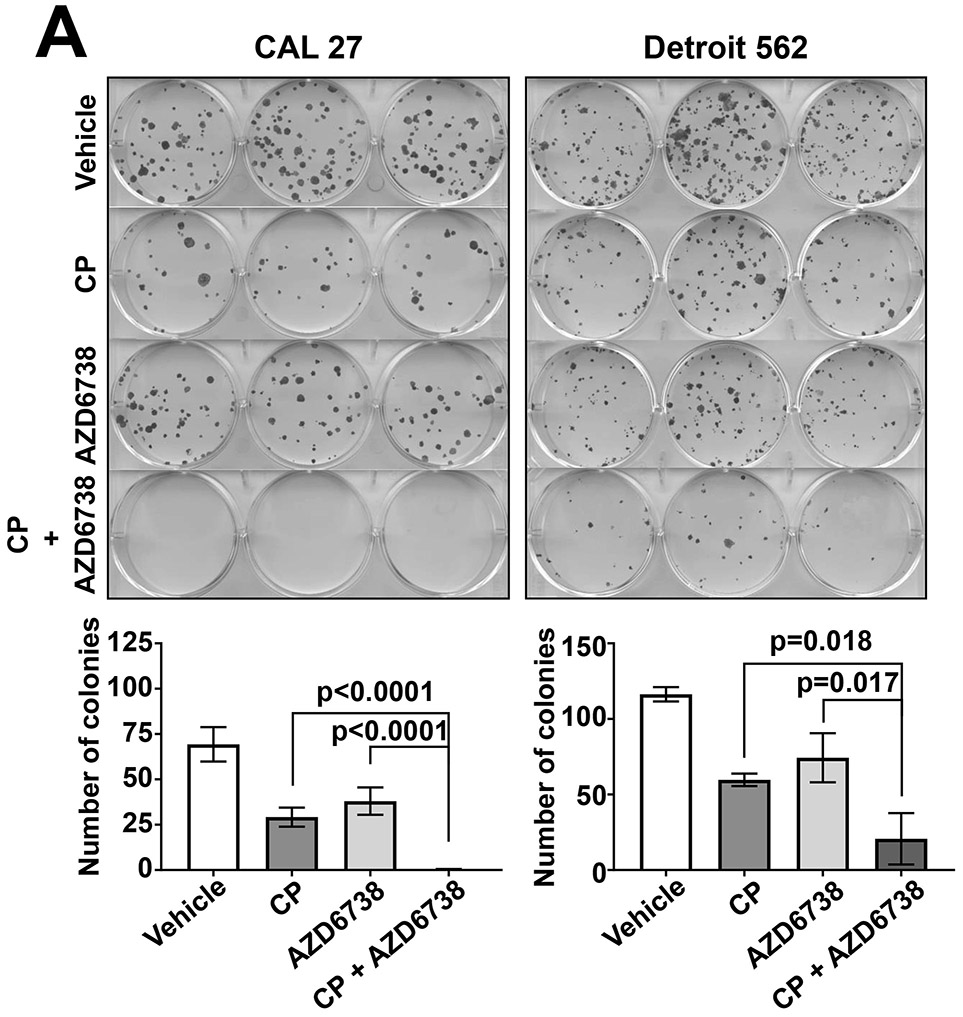
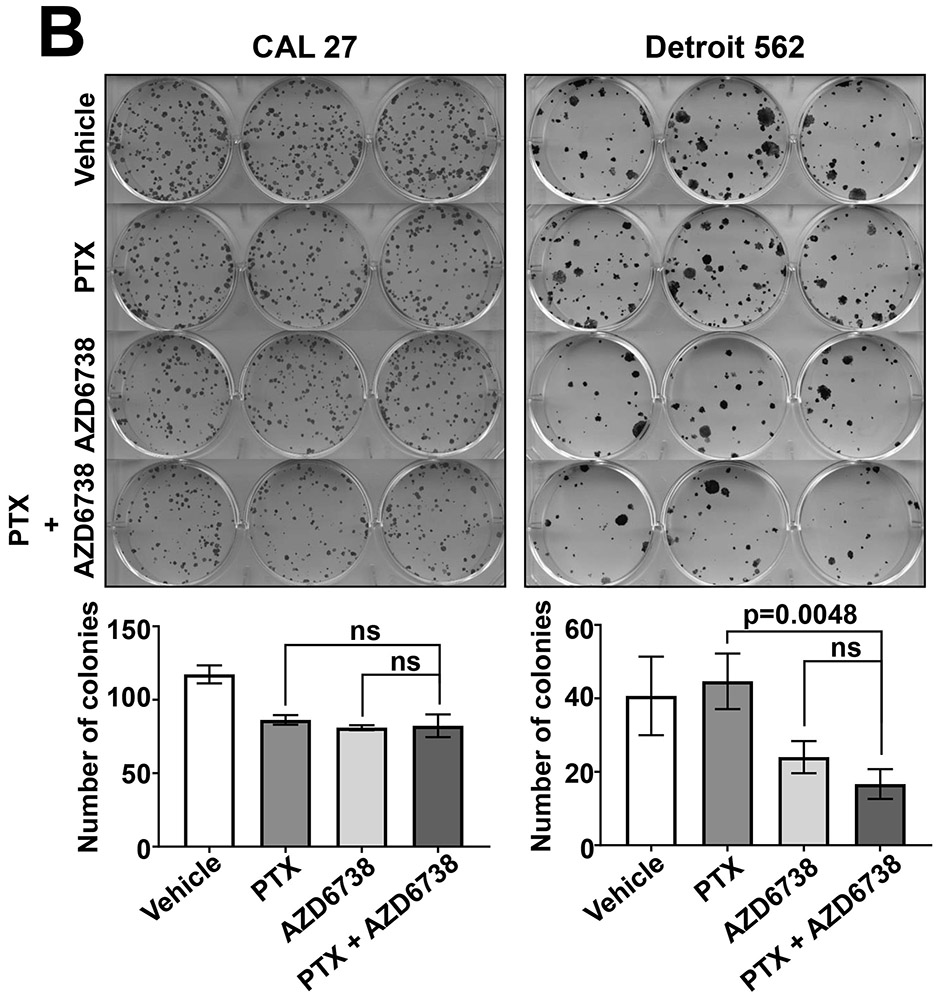
AZD6738 enhances loss of colony formation by cisplatin (CP). A, CAL 27 and Detroit 562 cells were treated for 24 hours with vehicle, 2 μM CP alone, 500 nM AZD6738 alone, or CP plus AZD6738. The treated cells were then replated in 6-well plates, then incubated for an additional 14 days before staining with crystal violet and counting of colonies. Colonies were defined as consisting of 50 cells or more. Bar graphs represent the average number of colonies from triplicate wells and error bars represent standard deviations. B, CAL 27 and Detroit 562 were treated with vehicle, 0.15 nM paclitaxel (PTX) alone, 500 nM AZD6738 alone, or PTX plus AZD6738, then analyzed in colony formation assays as in panel A.
ATR inhibition enhances cisplatin-induced DNA damage and cell death induction in HNSCC cells
To investigate the mechanism whereby AZD6738 enhanced cisplatin-induced growth inhibition of HNSCC cells, we first evaluated cell death induction. For these experiments we utilized the 3 HPV− cell lines that were sensitized to cisplatin to the greatest degree by AZD6738 (CAL 27, BICR 56, PE/CA-PJ49; see Figure 1C) and the 3 HPV− cell lines that were sensitized to the least degree (Detroit 562, SCC-25, BICR 6). Cells were treated for 24 hours with vehicle, 500 nM AZD6738 alone, 2 μM cisplatin alone, or the combination, followed by flow cytometric analysis of annexin V and PI staining as an indicator of cell death (Figure 3A). Consistent with the cell growth assays in Figure 1C, ATR inhibition with AZD6738 markedly enhanced cisplatin-induced cell death in CAL 27, BICR 56, and PE/CA-PJ49. Similarly, AZD6738 enhanced cisplatin-induced death in HPV+ UM-SCC-47 cells (Figure 3A). By contrast, Detroit 562, SCC-25, and BICR 6 failed to show substantially enhanced cell death induction following treatment with the combination.
Fig. 3.

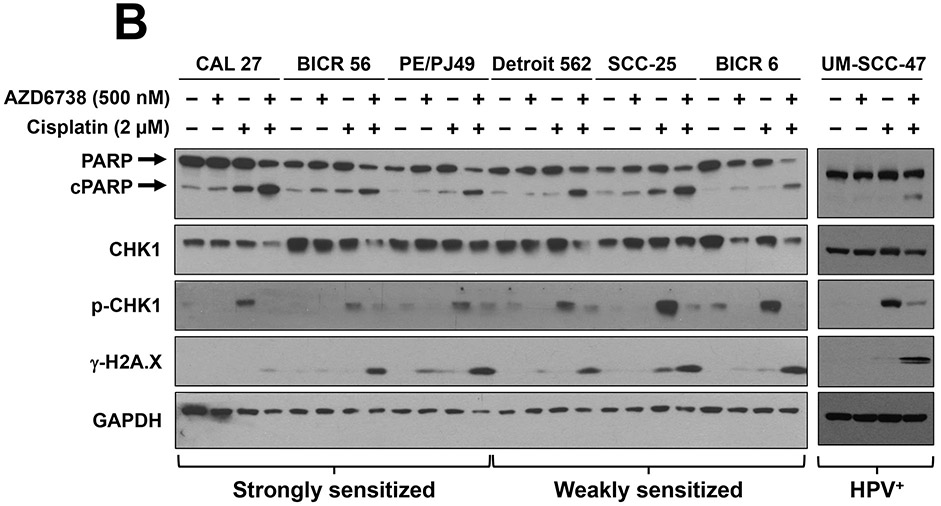
ATR inhibition enhances cisplatin-induced apoptosis and DNA damage. The indicated cell lines were treated for 24 hours with either vehicle, 500 nM AZD6738, 2 μM cisplatin or 500 nM AZD6738 plus 2 μM cisplatin (A, B). A, Annexin V and PI staining was performed and the percentage of Annexin V-positive and/or PI-positive cells determined by flow cytometry. The figure represents one experiment. B, Immunoblots were performed for common indicators of apoptosis signaling (cleaved PARP (cPARP)), DNA damage (γ-H2A.X), and DNA damage/repair (p-CHK1). PE/CA-PJ49 cells are abbreviated as PE/PJ49.
We next examined the impact of AZD6738, cisplatin, and the combination on apoptosis signaling, DNA damage, and ATR activity in HNSCC cells. Immunoblot analyses revealed increased cleavage of poly(ADP-ribose) polymerase (PARP), a marker of apoptosis signal transduction, in all 6 cell lines (as well as HPV+ UM-SCC-47) following treatment with the combination of AZD6738 and cisplatin, relative to cells treated with either agent alone (Figure 3B). Similarly, co-treatment with AZD6738 plus cisplatin resulted in markedly elevated levels of phospho-H2A.X (γ-H2A.X), a marker of DNA damage. Protein lysates were also analyzed for expression of phospho-CHK1 (p-CHK1), the downstream product of ATR kinase activity. Treatment with AZD6738 reduced the baseline levels of p-CHK1 to undetectable levels, consistent with inhibition of ATR activity. As expected, treatment with cisplatin alone led to induction of p-CHK1 in all 6 cell lines. Interestingly, however, p-CHK1 upregulation by cisplatin was markedly greater in the cell lines that were more weakly sensitized by AZD6738 compared to the cell lines that were more strongly sensitized by the ATR inhibitor. Expression of p-CHK1 returned to baseline levels in all 6 HNSCC cell lines following co-treatment with cisplatin and AZD6738.
Inhibition of expression of ATR, but not ATM, potentiates cisplatin-induced growth inhibition
To confirm that abrogation of ATR signaling enhances growth inhibition by cisplatin in HNSCC cells, we used siRNA to prevent ATR expression. Experiments were performed using 2 HNSCC cell lines that were strongly sensitized to cisplatin by AZD6738 (BICR 56, PE/CA-PJ49) and 2 that were weakly sensitized (Detroit 562, BICR 6). Immunoblotting confirmed efficient downregulation of ATR protein by the ATR siRNA (siATR) relative to transfection with nontargeting siRNA (siNT) (Figure 4A). Following transfection of siRNAs, cells were treated with cisplatin and cell growth analyzed by crystal violet assays. Consistent with findings using AZD6738, siATR-mediated downregulation of ATR resulted in strong sensitization to cisplatin in BICR 56 and PE/CA-PJ49 cells, and only weak sensitization in Detroit 562 and BICR 6 cells (Figure 4B).
Fig. 4.

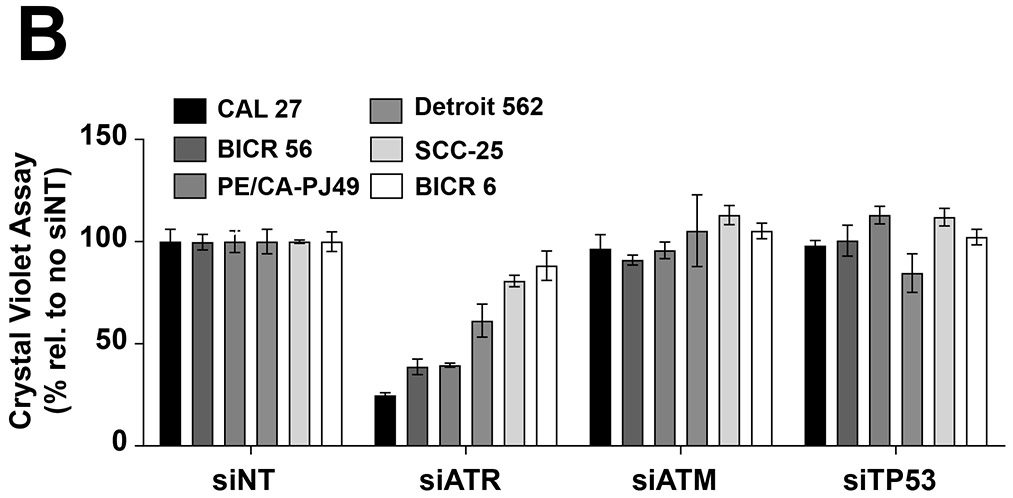
Inhibition of ATR expression, but not ATM or p53 expression, sensitizes HNSCC cell lines to cisplatin. A, HNSCC cell lines were transfected with nontargeting siRNA (siNT) or siRNAs directed against ATR, ATM, or p53 mRNAs, as described in Materials and Methods. After 48 hours, whole cell lysates were prepared and subjected to immunoblotting to confirm effective knockdown of the intended targets. B) Cells transfected for 48 hours with the indicated siRNAs were treated with 10 μM cisplatin for an additional 48 hours, followed by performance of crystal violet assays. Data points were normalized to treatment with siNT for each cell line. Columns and error bars represent the average and standard deviation of triplicate wells, respectively.
We also assessed the impact of ATM downregulation, using siRNA directed against ATM mRNA (siATM) (Figure 4). In contrast to ATR downregulation, inhibition of ATM expression had little impact on cisplatin sensitivity in any of the 4 cell lines tested. Similarly, siRNA-mediated downregulation of p53 (data not shown), a key substrate of ATR signaling, had negligible effect on cisplatin sensitivity in the 3 cell lines that expressed basal levels of p53 protein (BICR 6 did not express detectable p53). These findings related to p53 are perhaps not surprising, as p53 has been reported to be mutated, and likely nonfunctional, in BICR 56 cells.
AZD6738 enhances the in vivo efficacy of cisplatin against HNSCC cell line-derived and patient-derived xenograft tumors
We next assessed the impact of AZD6738 on the in vivo activity of cisplatin against HNSCC xenograft tumors. Nude mice were inoculated subcutaneously with HPV− CAL 27 cells or HPV+ UM-SCC-47 cells. Following the development of palpable tumors, mice were randomized, then treated with vehicle alone, cisplatin alone, AZD6738 alone, or the combination of cisplatin and AZD6738 (Figure 5A and 5B). For CAL 27-derived tumors, treatment with the combination resulted in greater inhibition of tumor growth than treatment with either agent alone (cisplatin vs. combination p=0.0082; AZD6738 vs. combination p=0.0041). Similarly, statistically significant greater inhibition of tumor growth was observed with the combination in mice harboring UM-SCC-47-derived tumors (cisplatin vs. combination p=0.0026; AZD6738 vs. combination p=0.0030).
Fig. 5.
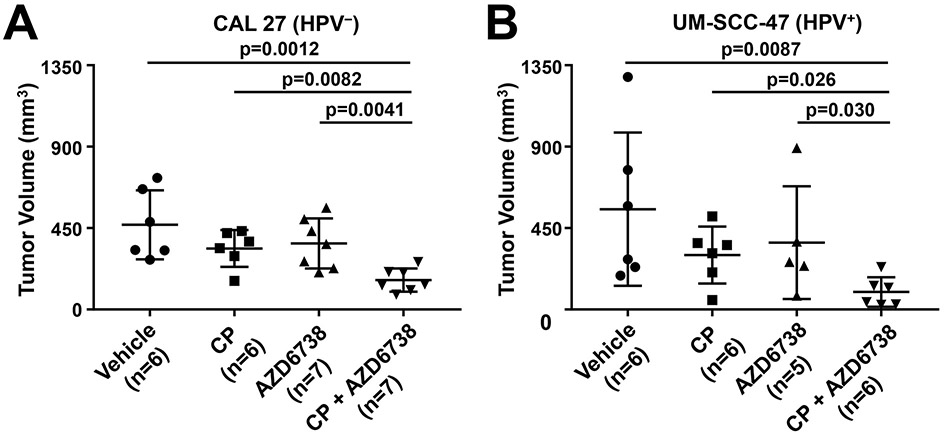
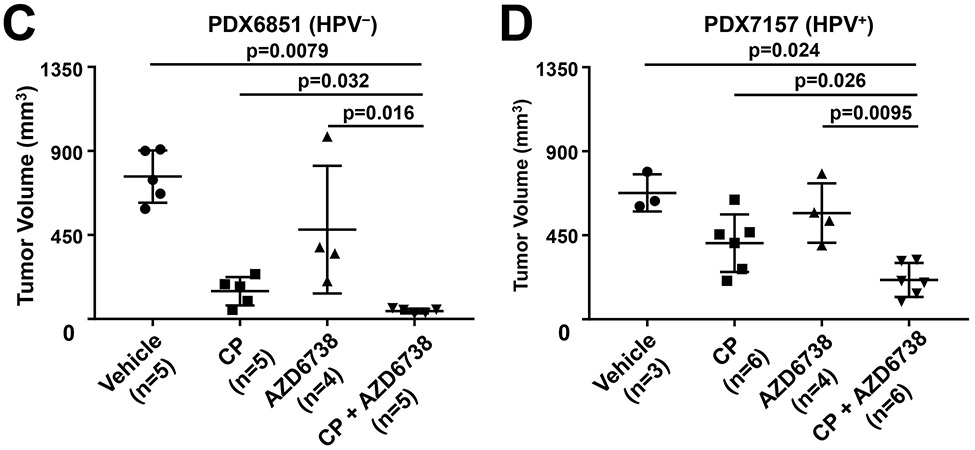
Co-treatment with AZD6738 and cisplatin enhances growth inhibition of HPV− and HPV+ HNSCC cell line and PDX tumors. A-D, Mice harboring tumors derived from HPV− (CAL 27) or HPV+ (UM-SCC-47) cell lines, or HPV− (PDX6851) or HPV+ (PDX7157) PDX models were treated with vehicle, AZD6738 (25 mg/kg, PO, 5 days on, 2 days off), cisplatin (5 mg/kg, IP, once per week), or the combination. Tumor volumes for CAL 27 (A), UM-SCC-47 (B), PDX6851 (C), and PDX7157 (D) are reported upon experiment completion on days 29, 16, 24, and 25 post-initial treatment, respectively. The number of tumors in each group is indicated by “n”. P values were calculated using Mann-Whitney t tests.
To confirm our findings in more clinically relevant in vivo models, we evaluated AZD6738 activity against 2 PDX models. NSG mice were implanted with tumor tissue derived from an HPV− PDX (PDX6851) or an HPV+ PDX (PDX7157), followed by randomization and treatment with vehicle, cisplatin, AZD6738, or the combination (Figure 5C and 5D). As was seen with the cell line-derived xenografts, treatment with cisplatin plus AZD6738 resulted in greater tumor growth inhibition than treatment with either agent alone. The enhanced anti-tumor activity of the combination was observed in both the HPV− and HPV+ models.
Discussion
Despite substantial adverse toxicities associated with its use, cisplatin remains a mainstay treatment for head and neck cancer. Continuing efforts are needed to identify agents that can be used in combination to enhance cisplatin activity against HNSCC tumors, and thereby allow lower doses of cisplatin to be used. Such agents also might be useful for overcoming the inherent or acquired resistance to cisplatin that is commonly seen in HNSCC tumors and markedly impacts the success of treatment. Since cisplatin exerts its killing effects against cancer cells by causing DNA damage, it is reasonable to hypothesize that inhibition of the processes that repair this damage are likely to enhance cisplatin potency. The repair of cisplatin-induced DNA damage is known to occur, in large part, via activation of ATR (43). Small molecule inhibitors of ATR, including orally bioavailable AZD6738, have recently been developed and shown to enhance cisplatin activity in preclinical models of lung cancer and gynecologic cancers (20-22, 30). The impact of AZD6738 on cisplatin sensitivity of HNSCC cell lines and tumors, however, has not been reported. We find that a nontoxic dose of AZD6738 sensitized all 21 HNSCC cell lines tested to cisplatin, although variation in the degree of sensitization was observed. The combination of AZD6738 and cisplatin also produced greater inhibition of HNSCC tumor growth, relative to either agent alone, in in vivo experiments. AZD6738 is currently being evaluated in early phase clinical trials; our studies support clinical evaluation of AZD6738 plus cisplatin in HNSCC.
The prevalence of HPV+ HNSCC is increasing dramatically, particularly among younger adults (33, 44-48). Greater than 75% of oropharyngeal cancers in the developed world test positive for HPV. HNSCC patients with HPV+ disease typically have better prognoses and respond better to therapy than patients with HPV− disease (49-55). However, recurrent and metastatic HPV+ HNSCC remains difficult to treat and is often lethal. Moreover, recent findings indicate that cetuximab therapy is largely ineffective for HPV+ HNSCC (56, 57). Thus, improved therapeutic options are needed for both HPV− and HPV+ HNSCC. In our studies, the four HPV+ HNSCC cell lines studied were moderately more resistant to cisplatin alone compared to most of the 17 HPV− cell lines. None-the-less, AZD6738 sensitized both HPV+ and HPV− cell lines to cisplatin. Indeed, 3 of the 4 HPV+ cell lines exhibited a greater than two-fold enhancement of sensitivity to cisplatin in the presence of AZD6738. In our in vivo studies, combined treatment with cisplatin and AZD6738 exhibited greater anti-tumor effects, relative to either agent alone, against both HPV+ and HPV− xenograft models, including PDX models. These findings suggest that inhibition of ATR may be a useful strategy for enhancing response to cisplatin in both HPV+ and HPV− HNSCC patients.
Our studies utilized simultaneous treatment with cisplatin and AZD6738. Using this approach, we found that the impact of ATR inhibition varied amongst the 21 cell lines. Although AZD6738 enhanced sensitivity to cisplatin in all 21 cell lines, varying degrees of sensitization were observed. While this variability may be reflective of differing gene expression patterns, the timing of ATR inhibition also may be a factor. As AZD6738 and other inhibitors of ATR advance to clinical testing in combination with cisplatin, it will be important to evaluate the impact of dosing and schedule of administration. Identification of baseline predictive biomarkers, such as loss of ATM or other DNA damage response proteins, will also help to optimize combination regimens incorporating ATR inhibitors and cisplatin. Collectively, these studies may provide an effective therapeutic strategy for enhancing anti-tumor activity and reducing the adverse toxicities of cisplatin in patients with HNSCC.
Supplementary Material
Highlights.
ATR inhibition with AZD6738 sensitized a panel of 21 HNSCC cell line to cisplatin.
AZD6738 enhanced tumor growth inhibition by cisplatin in vivo.
AZD6738 enhancement of cisplatin effects was observed in both HPV− and HPV+ models.
Acknowledgments
Financial support
This work was supported by National Institutes of Health grants R01 DE024728 (DEJ), R35 CA231998 (JRG), and R01 CA204173 (CJB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
DEJ and JRG are co-inventors of cyclic STAT3 decoy and have financial interests in STAT3 Therapeutics. STAT3 Therapeutics holds an interest in cyclic STAT3 decoy, which is a not a focus of the studies in this manuscript. The remaining authors declare no conflicts.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. PubMed PMID: 26742998. [DOI] [PubMed] [Google Scholar]
- 2.Global Burden of Disease Cancer C, Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, Dandona L, Fleming T, Forouzanfar MH, Hancock J, Hay RJ, Hunter-Merrill R, Huynh C, Hosgood HD, Johnson CO, Jonas JB, Khubchandani J, Kumar GA, Kutz M, Lan Q, Larson HJ, Liang X, Lim SS, Lopez AD, MacIntyre MF, Marczak L, Marquez N, Mokdad AH, Pinho C, Pourmalek F, Salomon JA, Sanabria JR, Sandar L, Sartorius B, Schwartz SM, Shackelford KA, Shibuya K, Stanaway J, Steiner C, Sun J, Takahashi K, Vollset SE, Vos T, Wagner JA, Wang H, Westerman R, Zeeb H, Zoeckler L, Abd-Allah F, Ahmed MB, Alabed S, Alam NK, Aldhahri SF, Alem G, Alemayohu MA, Ali R, Al-Raddadi R, Amare A, Amoako Y, Artaman A, Asayesh H, Atnafu N, Awasthi A, Saleem HB, Barac A, Bedi N, Bensenor I, Berhane A, Bernabe E, Betsu B, Binagwaho A, Boneya D, Campos-Nonato I, Castaneda-Orjuela C, Catala-Lopez F, Chiang P, Chibueze C, Chitheer A, Choi JY, Cowie B, Damtew S, das Neves J, Dey S, Dharmaratne S, Dhillon P, Ding E, Driscoll T, Ekwueme D, Endries AY, Farvid M, Farzadfar F, Fernandes J, Fischer F, TT GH, Gebru A, Gopalani S, Hailu A, Horino M, Horita N, Husseini A, Huybrechts I, Inoue M, Islami F, Jakovljevic M, James S, Javanbakht M, Jee SH, Kasaeian A, Kedir MS, Khader YS, Khang YH, Kim D, Leigh J, Linn S, Lunevicius R, El Razek HMA, Malekzadeh R, Malta DC, Marcenes W, Markos D, Melaku YA, Meles KG, Mendoza W, Mengiste DT, Meretoja TJ, Miller TR, Mohammad KA, Mohammadi A, Mohammed S, Moradi-Lakeh M, Nagel G, Nand D, Le Nguyen Q, Nolte S, Ogbo FA, Oladimeji KE, Oren E, Pa M, Park EK, Pereira DM, Plass D, Qorbani M, Radfar A, Rafay A, Rahman M, Rana SM, Soreide K, Satpathy M, Sawhney M, Sepanlou SG, Shaikh MA, She J, Shiue I, Shore HR, Shrime MG, So S, Soneji S, Stathopoulou V, Stroumpoulis K, Sufiyan MB, Sykes BL, Tabares-Seisdedos R, Tadese F, Tedla BA, Tessema GA, Thakur JS, Tran BX, Ukwaja KN, Uzochukwu BSC, Vlassov VV, Weiderpass E, Wubshet Terefe M, Yebyo HG, Yimam HH, Yonemoto N, Younis MZ, Yu C, Zaidi Z, Zaki MES, Zenebe ZM, Murray CJL, Naghavi M. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017;3(4):524–48. doi: 10.1001/jamaoncol.2016.5688. PubMed PMID: 27918777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, Youssoufian H, Amellal N, Rowinsky EK, Ang KK. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567–78. PubMed PMID: 16467544. [DOI] [PubMed] [Google Scholar]
- 4.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, Peyrade F, Benasso M, Vynnychenko I, De Raucourt D, Bokemeyer C, Schueler A, Amellal N, Hitt R. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116–27. Epub 2008/09/12. doi: 359/11/1116 [pii] 10.1056/NEJMoa0802656. PubMed PMID: 18784101. [DOI] [PubMed] [Google Scholar]
- 5.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, Cheng JD, Chow LQ. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17(7):956–65. doi: 10.1016/S1470-2045(16)30066-3. PubMed PMID: 27247226. [DOI] [PubMed] [Google Scholar]
- 6.Chow LQ, Haddad R, Gupta S, Mahipal A, Mehra R, Tahara M, Berger R, Eder JP, Burtness B, Lee SH, Keam B, Kang H, Muro K, Weiss J, Geva R, Lin CC, Chung HC, Meister A, Dolled-Filhart M, Pathiraja K, Cheng JD, Seiwert TY. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. J Clin Oncol. 2016. doi: 10.1200/JCO.2016.68.1478. PubMed PMID: 27646946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, Worden F, Saba NF, Iglesias Docampo LC, Haddad R, Rordorf T, Kiyota N, Tahara M, Monga M, Lynch M, Geese WJ, Kopit J, Shaw JW, Gillison ML. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375(19):1856–67. doi: 10.1056/NEJMoa1602252. PubMed PMID: 27718784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szturz P, Wouters K, Kiyota N, Tahara M, Prabhash K, Noronha V, Adelstein D, Vermorken JB. Altered fractionation radiotherapy combined with concurrent low-dose or high-dose cisplatin in head and neck cancer: A systematic review of literature and meta-analysis. Oral Oncol. 2018;76:52–60. doi: 10.1016/j.oraloncology.2017.11.025. PubMed PMID: 29290286. [DOI] [PubMed] [Google Scholar]
- 9.Szturz P, Wouters K, Kiyota N, Tahara M, Prabhash K, Noronha V, Adelstein D, Van Gestel D, Vermorken JB. Low-Dose vs. High-Dose Cisplatin: Lessons Learned From 59 Chemoradiotherapy Trials in Head and Neck Cancer. Front Oncol. 2019;9:86. doi: 10.3389/fonc.2019.00086. PubMed PMID: 30847300; PMCID: PMC6394212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamieson ER, Lippard SJ. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem Rev. 1999;99(9):2467–98. PubMed PMID: 11749487. [DOI] [PubMed] [Google Scholar]
- 11.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–20. doi: 10.1038/nrd1691. PubMed PMID: 15789122. [DOI] [PubMed] [Google Scholar]
- 12.Hu J, Lieb JD, Sancar A, Adar S. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc Natl Acad Sci U S A. 2016;113(41):11507–12. doi: 10.1073/pnas.1614430113. PubMed PMID: 27688757; PMCID: PMC5068337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelland L The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–84. doi: 10.1038/nrc2167. PubMed PMID: 17625587. [DOI] [PubMed] [Google Scholar]
- 14.Zamble DB, Mu D, Reardon JT, Sancar A, Lippard SJ. Repair of cisplatin--DNA adducts by the mammalian excision nuclease. Biochemistry. 1996;35(31):10004–13. doi: 10.1021/bi960453+. PubMed PMID: 8756462. [DOI] [PubMed] [Google Scholar]
- 15.Reardon JT, Vaisman A, Chaney SG, Sancar A. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 1999;59(16):3968–71. PubMed PMID: 10463593. [PubMed] [Google Scholar]
- 16.Wang D, Hara R, Singh G, Sancar A, Lippard SJ. Nucleotide excision repair from site-specifically platinum-modified nucleosomes. Biochemistry. 2003;42(22):6747–53. doi: 10.1021/bi034264k. PubMed PMID: 12779329. [DOI] [PubMed] [Google Scholar]
- 17.Lee TH, Park JM, Leem SH, Kang TH. Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair. Oncogene. 2014;33(1):19–25. doi: 10.1038/onc.2012.539. PubMed PMID: 23178497. [DOI] [PubMed] [Google Scholar]
- 18.Jarrett SG, Wolf Horrell EM, Christian PA, Vanover JC, Boulanger MC, Zou Y, D'Orazio JA. PKA-mediated phosphorylation of ATR promotes recruitment of XPA to UV-induced DNA damage. Mol Cell. 2014;54(6):999–1011. doi: 10.1016/j.molcel.2014.05.030. PubMed PMID: 24950377; PMCID: PMC4076709. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, Vallis KA, Hammond EM, Olcina MM, Gillies McKenna W, Muschel RJ, Brunner TB. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. doi: 10.1038/cddis.2012.181. PubMed PMID: 23222511; PMCID: PMC3542617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y, Hare B, Johnson MA, Milton S, Murphy CE, Takemoto D, Tolman C, Wood M, Charlton P, Charrier JD, Furey B, Golec J, Reaper PM, Pollard JR. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. 2014;5(14):5674–85. doi: 10.18632/oncotarget.2158. PubMed PMID: 25010037; PMCID: PMC4170644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vendetti FP, Lau A, Schamus S, Conrads TP, O'Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015;6(42):44289–305. doi: 10.18632/oncotarget.6247. PubMed PMID: 26517239; PMCID: PMC4792557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foote KM, Nissink JWM, McGuire T, Turner P, Guichard S, Yates JWT, Lau A, Blades K, Heathcote D, Odedra R, Wilkinson G, Wilson Z, Wood CM, Jewsbury PJ. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J Med Chem. 2018;61(22):9889–907. doi: 10.1021/acs.jmedchem.8b01187. PubMed PMID: 30346772. [DOI] [PubMed] [Google Scholar]
- 23.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM, Pollard JR. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7(7):428–30. doi: 10.1038/nchembio.573. PubMed PMID: 21490603. [DOI] [PubMed] [Google Scholar]
- 24.Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18(6):721–7. doi: 10.1038/nsmb.2076. PubMed PMID: 21552262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, Muschel RJ, Brunner TB. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13(11):1072–81. doi: 10.4161/cbt.21093. PubMed PMID: 22825331; PMCID: PMC3461814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huntoon CJ, Flatten KS, Wahner Hendrickson AE, Huehls AM, Sutor SL, Kaufmann SH, Karnitz LM. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013;73(12):3683–91. doi: 10.1158/0008-5472.CAN-13-0110. PubMed PMID: 23548269; PMCID: PMC3687010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohni KN, Thompson PS, Luzwick JW, Glick GG, Pendleton CS, Lehmann BD, Pietenpol JA, Cortez D. A Synthetic Lethal Screen Identifies DNA Repair Pathways that Sensitize Cancer Cells to Combined ATR Inhibition and Cisplatin Treatments. PLoS One. 2015;10(5):e0125482. doi: 10.1371/journal.pone.0125482. PubMed PMID: 25965342; PMCID: PMC4428765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, Parry H, Taylor M, Moss P, Hillmen P, Stankovic T. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016;127(5):582–95. doi: 10.1182/blood-2015-05-644872. PubMed PMID: 26563132. [DOI] [PubMed] [Google Scholar]
- 29.Ma J, Li X, Su Y, Zhao J, Luedtke DA, Epshteyn V, Edwards H, Wang G, Wang Z, Chu R, Taub JW, Lin H, Wang Y, Ge Y. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci Rep. 2017;7:41950. doi: 10.1038/srep41950. PubMed PMID: 28176818; PMCID: PMC5296912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teng PN, Bateman NW, Darcy KM, Hamilton CA, Maxwell GL, Bakkenist CJ, Conrads TP. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol Oncol. 2015;136(3):554–61. doi: 10.1016/j.ygyno.2014.12.035. PubMed PMID: 25560806; PMCID: PMC4382918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, Bernaldo de Quiros Fernandez S, Lau A, Richards FM, Jodrell DI. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther. 2018;17(8):1670–82. doi: 10.1158/1535-7163.MCT-18-0010. PubMed PMID: 29891488; PMCID: PMC6076438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, Yang Y, Kim HJ, Lee KH, Kim JW, Kim TY, Oh DY, Brown J, Lau A, O’Connor MJ, Bang YJ. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol Cancer Ther. 2017;16(4):566–77. doi: 10.1158/1535-7163.MCT-16-0378. PubMed PMID: 28138034. [DOI] [PubMed] [Google Scholar]
- 33.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, Jiang B, Goodman MT, Sibug-Saber M, Cozen W, Liu L, Lynch CF, Wentzensen N, Jordan RC, Altekruse S, Anderson WF, Rosenberg PS, Gillison ML. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29(32):4294–301. doi: 10.1200/JCO.2011.36.4596. PubMed PMID: 21969503; PMCID: 3221528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jemal A, Simard EP, Dorell C, Noone AM, Markowitz LE, Kohler B, Eheman C, Saraiya M, Bandi P, Saslow D, Cronin KA, Watson M, Schiffman M, Henley SJ, Schymura MJ, Anderson RN, Yankey D, Edwards BK. Annual Report to the Nation on the Status of Cancer, 1975-2009, featuring the burden and trends in human papillomavirus(HPV)-associated cancers and HPV vaccination coverage levels. J Natl Cancer Inst. 2013;105(3):175–201. doi: 10.1093/jnci/djs491. PubMed PMID: 23297039; PMCID: PMC3565628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C, Kim H, Axelrod R, Silverman CC, Redmond KP, Gillison ML. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. PubMed PMID: 20530316; PMCID: PMC2943767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang H, Kiess A, Chung CH. Emerging biomarkers in head and neck cancer in the era of genomics. Nat Rev Clin Oncol. 2015;12(1):11–26. doi: 10.1038/nrclinonc.2014.192. PubMed PMID: 25403939. [DOI] [PubMed] [Google Scholar]
- 37.Wallace NA, Galloway DA. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin Cancer Biol. 2014;26:30–42. doi: 10.1016/j.semcancer.2013.12.003. PubMed PMID: 24412279; PMCID: PMC4050178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gautam D, Moody CA. Impact of the DNA Damage Response on Human Papillomavirus Chromatin. PLoS Pathog. 2016;12(6):e1005613. doi: 10.1371/journal.ppat.1005613. PubMed PMID: 27310012; PMCID: PMC4910971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anacker DC, Moody CA. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2017;231:41–9. doi: 10.1016/j.virusres.2016.11.006. PubMed PMID: 27836727; PMCID: PMC5325762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dillon MT, Boylan Z, Smith D, Guevara J, Mohammed K, Peckitt C, Saunders M, Banerji U, Clack G, Smith SA, Spicer JF, Forster MD, Harrington KJ. PATRIOT: A phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid tumours. Clin Transl Radiat Oncol. 2018;12:16–20. doi: 10.1016/j.ctro.2018.06.001. PubMed PMID: 30073210; PMCID: PMC6068075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zang Y, Kirk CJ, Johnson DE. Carfilzomib and oprozomib synergize with histone deacetylase inhibitors in head and neck squamous cell carcinoma models of acquired resistance to proteasome inhibitors. Cancer Biol Ther. 2014;15(9):1142–52. doi: 10.4161/cbt.29452. PubMed PMID: 24915039; PMCID: 4128857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Wheeler S, Park Y, Ju Z, Thomas SM, Fichera M, Egloff AM, Lui VW, Duvvuri U, Bauman JE, Mills GB, Grandis JR. Proteomic Characterization of Head and Neck Cancer Patient-Derived Xenografts. Mol Cancer Res. 2016;14(3):278–86. doi: 10.1158/1541-7786.MCR-15-0354. PubMed PMID: 26685214; PMCID: PMC4794346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang TH, Leem SH. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. 2014;42(7):4427–34. doi: 10.1093/nar/gku094. PubMed PMID: 24489120; PMCID: PMC3985666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shiboski CH, Schmidt BL, Jordan RC. Tongue and tonsil carcinoma: increasing trends in the U.S. population ages 20-44 years. Cancer. 2005;103(9):1843–9. Epub 2005/03/18. doi: 10.1002/cncr.20998. PubMed PMID: 15772957. [DOI] [PubMed] [Google Scholar]
- 45.Nasman A, Attner P, Hammarstedt L, Du J, Eriksson M, Giraud G, Ahrlund-Richter S, Marklund L, Romanitan M, Lindquist D, Ramqvist T, Lindholm J, Sparen P, Ye W, Dahlstrand H, Munck-Wikland E, Dalianis T. Incidence of human papillomavirus (HPV) positive tonsillar carcinoma in Stockholm, Sweden: an epidemic of viral-induced carcinoma? Int J Cancer. 2009;125(2):362–6. doi: 10.1002/ijc.24339. PubMed PMID: 19330833. [DOI] [PubMed] [Google Scholar]
- 46.Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J Clin Oncol. 2008;26(4):612–9. PubMed PMID: 18235120. [DOI] [PubMed] [Google Scholar]
- 47.Maxwell JH, Grandis JR, Ferris RL. HPV-Associated Head and Neck Cancer: Unique Features of Epidemiology and Clinical Management. Annu Rev Med. 2016;67:91–101. doi: 10.1146/annurev-med-051914-021907. PubMed PMID: 26332002; PMCID: PMC5242186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sonawane K, Suk R, Chiao EY, Chhatwal J, Qiu P, Wilkin T, Nyitray AG, Sikora AG, Deshmukh AA. Oral Human Papillomavirus Infection: Differences in Prevalence Between Sexes and Concordance With Genital Human Papillomavirus Infection, NHANES 2011 to 2014. Ann Intern Med. 2017;167(10):714–24. doi: 10.7326/M17-1363. PubMed PMID: 29049523; PMCID: PMC6203692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindel K, Beer KT, Laissue J, Greiner RH, Aebersold DM. Human papillomavirus positive squamous cell carcinoma of the oropharynx: a radiosensitive subgroup of head and neck carcinoma. Cancer. 2001;92(4):805–13. PubMed PMID: 11550151. [DOI] [PubMed] [Google Scholar]
- 50.Li W, Thompson CH, O'Brien CJ, McNeil EB, Scolyer RA, Cossart YE, Veness MJ, Walker DM, Morgan GJ, Rose BR. Human papillomavirus positivity predicts favourable outcome for squamous carcinoma of the tonsil. Int J Cancer. 2003;106(4):553–8. doi: 10.1002/ijc.11261. PubMed PMID: 12845651. [DOI] [PubMed] [Google Scholar]
- 51.Weinberger PM, Yu Z, Haffty BG, Kowalski D, Harigopal M, Brandsma J, Sasaki C, Joe J, Camp RL, Rimm DL, Psyrri A. Molecular classification identifies a subset of human papillomavirus--associated oropharyngeal cancers with favorable prognosis. J Clin Oncol. 2006;24(5):736–47. doi: 10.1200/JCO.2004.00.3335. PubMed PMID: 16401683. [DOI] [PubMed] [Google Scholar]
- 52.Licitra L, Perrone F, Bossi P, Suardi S, Mariani L, Artusi R, Oggionni M, Rossini C, Cantu G, Squadrelli M, Quattrone P, Locati LD, Bergamini C, Olmi P, Pierotti MA, Pilotti S. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J Clin Oncol. 2006;24(36):5630–6. Epub 2006/12/21. doi: 24/36/5630 [pii] 10.1200/JCO.2005.04.6136. PubMed PMID: 17179101. [DOI] [PubMed] [Google Scholar]
- 53.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, Forastiere A, Gillison ML. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100(4):261–9. Epub 2008/02/14. doi: djn011 [pii] 10.1093/jnci/djn011. PubMed PMID: 18270337. [DOI] [PubMed] [Google Scholar]
- 54.Gillison ML, D'Souza G, Westra W, Sugar E, Xiao W, Begum S, Viscidi R. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. 2008;100(6):407–20. Epub 2008/03/13. doi: djn025 [pii] 10.1093/jnci/djn025. PubMed PMID: 18334711. [DOI] [PubMed] [Google Scholar]
- 55.Psyrri A, Gouveris P, Vermorken JB. Human papillomavirus-related head and neck tumors: clinical and research implication. Curr Opin Oncol. 2009;21(3):201–5. PubMed PMID: 19370803. [DOI] [PubMed] [Google Scholar]
- 56.Gillison ML, Trotti AM, Harris J, Eisbruch A, Harari PM, Adelstein DJ, Sturgis EM, Burtness B, Ridge JA, Ringash J, Galvin J, Yao M, Koyfman SA, Blakaj DM, Razaq MA, Colevas AD, Beitler JJ, Jones CU, Dunlap NE, Seaward SA, Spencer S, Galloway TJ, Phan J, Dignam JJ, Le QT. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): a randomised, multicentre, non-inferiority trial. Lancet. 2018. doi: 10.1016/S0140-6736(18)32779-X. PubMed PMID: 30449625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mehanna H, Robinson M, Hartley A, Kong A, Foran B, Fulton-Lieuw T, Dalby M, Mistry P, Sen M, O'Toole L, Al Booz H, Dyker K, Moleron R, Whitaker S, Brennan S, Cook A, Griffin M, Aynsley E, Rolles M, De Winton E, Chan A, Srinivasan D, Nixon I, Grumett J, Leemans CR, Buter J, Henderson J, Harrington K, McConkey C, Gray A, Dunn J, De EHPVTG. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): an open-label randomised controlled phase 3 trial. Lancet. 2018. doi: 10.1016/S0140-6736(18)32752-1. PubMed PMID: 30449623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.