

A method is presented for efficient co-crystal structure determination for G protein-coupled receptors taking advantage of serial femtosecond crystallography.
Keywords: drug discovery, G protein-coupled receptors, serial femtosecond crystallography, X-ray free-electron lasers, structure determination, membrane proteins, protein structure, molecular recognition
Abstract
Rational structure-based drug design (SBDD) relies on the availability of a large number of co-crystal structures to map the ligand-binding pocket of the target protein and use this information for lead-compound optimization via an iterative process. While SBDD has proven successful for many drug-discovery projects, its application to G protein-coupled receptors (GPCRs) has been limited owing to extreme difficulties with their crystallization. Here, a method is presented for the rapid determination of multiple co-crystal structures for a target GPCR in complex with various ligands, taking advantage of the serial femtosecond crystallography approach, which obviates the need for large crystals and requires only submilligram quantities of purified protein. The method was applied to the human β2-adrenergic receptor, resulting in eight room-temperature co-crystal structures with six different ligands, including previously unreported structures with carvedilol and propranolol. The generality of the proposed method was tested with three other receptors. This approach has the potential to enable SBDD for GPCRs and other difficult-to-crystallize membrane proteins.
1. Introduction
Structure-based drug design (SBDD) is a powerful approach that can substantially accelerate the process of drug discovery and optimization as well as guide medicinal chemists towards the selection of the most promising lead candidates for subsequent clinical trials (Jazayeri et al., 2015 ▸). While even a single structure of the target protein can be very helpful for the discovery of new compounds, the most successful SBDD programs exploit a large number of co-crystal structures to comprehensively map the ligand-binding pocket and ligand-binding modes in an iterative manner. For example, a recent study on inhibitors of the aspartic protease endothiapepsin has shown that even minor chemical modifications in the ligand can cause dramatic and unexpected changes in the ligand-binding mode (Kuhnert et al., 2015 ▸).
G protein-coupled receptors (GPCRs) are ubiquitous cellular gatekeepers in eukaryotic organisms. They mediate sensory stimuli and cell signaling, regulating all major physiological processes, and therefore have historically been primary targets in the pharmaceutical industry (Allen & Roth, 2011 ▸). GPCRs represent the largest protein superfamily in humans, comprising over 800 members (Lagerström & Schiöth, 2008 ▸) that are commonly grouped into five classes according to sequence similarity. The largest of the classes, class A, accounts for nearly 85% of the GPCR superfamily and includes, among others, aminergic receptors targeted by about a quarter of current prescription drugs (Rask-Andersen et al., 2011 ▸).
Since their discovery several decades ago, GPCRs have become some of the most valuable targets for structural studies. Their high-resolution structure determination has been enabled by a number of breakthroughs in several fields, such as protein engineering (Rosenbaum et al., 2007 ▸; Chun et al., 2012 ▸; Serrano-Vega et al., 2008 ▸), crystallization in the native-membrane-mimicking environment of the lipidic cubic phase (LCP; Landau & Rosenbusch, 1996 ▸; Cherezov, 2011 ▸) and microcrystallography (Cherezov et al., 2009 ▸; Smith et al., 2012 ▸). Combined efforts from several laboratories during the last decade have produced over 300 structures of ∼60 unique GPCRs captured in different signaling states (Pándy-Szekeres et al., 2018 ▸). These structures shed light on ligand-recognition and signal-transduction mechanisms. Despite this impressive progress in structural studies of GPCRs, their crystallization remains extremely challenging and constitutes one of the largest hurdles for SBDD applications.
Most commonly in SBDD, multiple co-crystal structures are obtained by soaking various ligands into crystals of unliganded (apo) proteins. Unfortunately, owing to their highly dynamic nature, a routine approach to the crystallization of apo GPCRs has not yet been established, and the use of ligands remains critical for successful receptor solubilization, purification and crystallization. Even after the structure of the target receptor in complex with one of the ligands has been solved, co-crystal structure determination with other ligands still represents a major challenge, as it often requires an extensive optimization of the receptor purification and crystallization conditions in order to obtain sufficiently large crystals for crystallographic data collection at synchrotron sources. Fully optimized GPCR crystals grown in LCP often tend to be well ordered, but owing to their small size and the rapid onset of radiation damage the acquisition of a complete data set requires screening hundreds of crystals and combining the data from dozens of them. Each of these crystals has to be manually harvested, cryocooled and aligned with the X-ray beam using rastering approaches, making the entire process exceedingly tedious and resource-consuming (Cherezov et al., 2009 ▸).
Most of these problems, however, can be essentially overcome by using the serial femtosecond crystallography (SFX) data-collection approach (Chapman et al., 2011 ▸; Liu et al., 2013 ▸; Mishin et al., 2019 ▸). SFX utilizes extremely bright and short pulses (tens of femtoseconds) produced by an X-ray free-electron laser (XFEL) to enable room-temperature data collection from micrometre-sized crystals, outrunning structure-altering radiation damage in a ‘diffraction-before-destruction’ manner (Neutze et al., 2000 ▸). The requirement of small crystal size in SFX experiments simplifies the crystallization optimization process. Furthermore, SFX offers additional advantages, such as a typically higher resolution, owing to higher order and a lower occurrence of defects in small crystals, in comparison to conventional synchrotron data collection, as well as room-temperature data collection that can potentially reveal important features related to protein dynamics (Fraser et al., 2009 ▸, 2011 ▸). Recent progress in SFX data-processing software has considerably lowered the amount of data that is required for structure determination (Kabsch, 2014 ▸; Uervirojnangkoorn et al., 2015 ▸; White et al., 2016 ▸).
Here, we present a simple and efficient method, dubbed Complex-LCP (Crystallization of membrane proteins using transient ligand exchange in LCP), for structure determination of a target GPCR in complex with a panel of different ligands by taking advantage of the SFX method. This represents a major step towards high-throughput GPCR–ligand co-crystal structure determination. We validated our approach using the human β2-adrenergic receptor (β2AR) that has been extensively studied over the last few decades and structures of which are available in both active and inactive states (Cherezov et al., 2007 ▸; Rasmussen et al., 2011 ▸; Wacker et al., 2010 ▸; Ring et al., 2013 ▸). Our method requires at least three components: a transient ligand, a protein and a ligand of interest. Using timolol or alprenolol as a transient ligand for receptor solubilization and purification, we were able to successfully exchange these ligands during crystallization for six β2AR ligands with diverse modes of action (MoAs) ranging from inverse agonism and antagonism to arrestin-biased agonism. Overall, eight β2AR structures with resolutions of between 2.4 and 3.4 Å were determined in this work. The exchanged ligands were unambiguously identified in the difference electron-density maps obtained with SFX data. The structures of two of these ligands, carvedilol and propranolol, have not previously been reported in complex with β2AR. The general applicability of the method was then demonstrated with adenosine A2A (A2AAR), serotonin 1B (5-HT1B) and 2B (5-HT2B) and MT1 melatonin receptors.
2. Materials and methods
2.1. Receptor constructs
The gene sequences of the human receptors β2AR, A2AAR, 5-HT1B, 5-HT2B and MT1 were modified to increase protein expression and stability as described previously (Hanson et al., 2008 ▸; Liu et al., 2012 ▸; Wacker et al., 2013 ▸; Wang et al., 2013 ▸; Stauch et al., 2019 ▸). Briefly, the gene for human β2AR (UniProt ID P07550) was altered as follows: (i) ICL3 residues 231–262 were replaced with cysteine-less T4 lysozyme to improve protein stability and increase the polar surface area for crystallization, (ii) the C-terminus was truncated at residue 348 and (iii) a point mutation E1223.41W was introduced to improve the protein yield and stability (Roth et al., 2008 ▸).
The A2AAR (UniProt ID P29274) construct was prepared by replacing ICL3 residues Lys209–Gly218 with a thermostabilized apocytochrome b 562 from Escherichia coli (BRIL) and truncating the C-terminal residues 317–412. The 5-HT2B (UniProt ID P41595) construct was generated by replacing ICL3 residues Tyr249–Val313 with BRIL and truncating the N-terminal residues 1–35 and the C-terminal residues 406–481 from the original sequence. Similarly to β2AR, a thermostabilizing M1443.41W mutation (Roth et al., 2008 ▸) was introduced into 5-HT2B. The wild-type human 5-HT1B (UniProt ID P28222) was fused to BRIL by replacing ICL3 residues Leu240–Lys303. The N-terminus of 5-HT1B was truncated at Asn32 to remove all glycosylation sites. In addition, a single point mutation L1383.41W (Roth et al., 2008 ▸) was introduced to increase the thermostability.
The MT1 (UniProt ID P48039) crystallization construct was obtained by the truncation of 11 N-terminal and 25 C-terminal residues, the replacement of intracellular receptor amino-acid residues 219–227 with the catalytic domain of Pyrococcus abyssi glycogen synthase (PGS; UniProt ID Q9V2J8) and the introduction of nine essential, stabilizing point mutations (D732.50N, L95ECL1F, G1043.29A, F1163.41W, N1243.49D, C1273.52L, W2516.48F, A2927.50P and N2998.47D) (Stauch et al., 2019 ▸).
2.2. Expression and purification
All GPCR constructs were cloned into a modified pFastBac1 expression vector bearing N-terminal HA- and FLAG-tags and expressed using the baculovirus expression system. In brief, the recombinant baculovirus was obtained according to the standard protocols in the Bac-to-Bac system (Invitrogen) and used to infect Spodoptera frugiperda (Sf9) insect cells at a multiplicity of infection of 5. The cells were harvested after incubation for 48 h. Insect-cell membranes were disrupted by osmotic shock in a hypotonic buffer consisting of 10 mM HEPES pH 7.5, 20 mM KCl, 10 mM MgCl2 and EDTA-free protease-inhibitor cocktail (Roche). The hypotonic wash was repeated once and was followed by a high-salt wash in buffer consisting of 1.0 M NaCl, 10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl and EDTA-free protease-inhibitor cocktail (Roche). The high-salt wash was repeated 1–2 times. Extensive washing of the membranes was performed by repeated centrifugation and Dounce homogenization to strip the membranes of soluble and membrane-associated proteins. The nembranes were flash-frozen and stored at −80°C until further use. Prior to solubilization, the prepared membranes were thawed on ice in the presence of 20 µM of the corresponding transient ligand (timolol or alprenolol), 2 mg ml−1 iodoacetamide and protease inhibitors. The membranes were then solubilized by incubation in the presence of 0.5%(w/v) n-dodecyl-β-d-maltopyranoside (DDM; Avanti Polar Lipids) and 0.1% cholesteryl hemisuccinate (CHS; Sigma) for 3 h at 4°C. After solubilization, the solution was clarified at 100 000g and the resulting supernatant was incubated with TALON IMAC resin overnight at 4°C. The resin was washed with ten column volumes (CV) of wash buffer I (50 mM HEPES pH 7.5, 300 mM NaCl, 20 mM imidazole, 0.1/0.02% DDM/CHS) and 5 CV of wash buffer II (50 mM HEPES pH 7.5, 300 mM NaCl, 40 mM imidazole, 0.05/0.01% DDM/CHS) to remove impurities, followed by elution of the receptor with 25 mM HEPES pH 7.5, 300 mM NaCl, 200 mM imidazole, 10% glycerol, 0.015/0.003% DDM/CHS. The transient ligand (timolol or alprenolol) was maintained at a concentration of 50 µM throughout solubilization, washing and elution. MT1–agomelatine was purified as previously described (Stauch et al., 2019 ▸).
2.3. Lipidic cubic phase crystallization
Prior to crystallization, all proteins were concentrated to 20–30 mg ml−1. LCP was made by mixing two parts (by volume) of protein solution with three parts of molten lipid (monoolein supplemented with 10% cholesterol by weight). Initial LCP crystallization screening of β2AR was performed using an NT8-LCP robot (Formulatrix) in 96-well glass sandwich plates (Marienfield) by dispensing 40 nl LCP drops and covering them with 800 nl precipitant solution. In this way, hit conditions were identified that yielded high concentrations of small crystals (∼5 µm) suitable for SFX data collection. The standard 48-salt screens (Xu, Liu et al., 2011 ▸) supplemented with 2 mM of the target ligand (from a 100 mM stock in DMSO) in each well were used for the setup in sandwich plates. Control Plate A supplemented with a matching concentration of DMSO (no ligand) in the screen was set up in parallel to identify conditions in which the concentration of the transient ligand is not high enough to generate crystals without introduction of the secondary ligand, and Control Plate B was set up supplemented with 2 mM of the transient ligand as a positive control of protein quality and crystallogenesis.
Crystallizations of 5-HT1B and 5-HT2B in plates were performed similarly to that of β2AR. In the case of 5-HT1B, washing buffers were supplemented with 20 µM ergotamine (ERG) and the crystallization buffer contained 2 mM of one of the exchange ligands (methylergometrine, oxymetazoline, sumatripan or RU-24969). In the case of 5-HT2B the washing buffers were supplemented with 50 µM serotonin, and the precipitant (400 mM ammonium chloride, 30% PEG 400, 100 mM Tris pH 8) contained one of the exchange ligands (ERG or dihydroergotamine) at 2 mM.
Crystals for XFEL data collection were obtained in Hamilton gas-tight syringes using the previously reported procedure (Liu et al., 2014 ▸). Purified β2AR in complex with an intermediate ligand (timolol or alprenolol) at a concentration of 25 mg ml−1 was reconstituted in LCP as described above. Approximately 5 µl of protein-laden LCP was carefully injected as a continuous filament of ∼400 µm in diameter into a 100 µl syringe filled with 60 µl precipitant solution [0.1 M HEPES pH 7.0, 0.1 M ammonium sulfate, 30%(v/v) PEG 400, 2 mM ligand] and incubated for 24 h at 20°C.
For the ligand-exchange experiments with A2AAR, the protein was purified following the previously published protocols (Liu et al., 2012 ▸) using 50 µM LUF5834 as a transient ligand. Microcrystals in syringes were obtained using 50 mM sodium thiocyanate, 100 mM sodium citrate pH 4.8, 28%(v/v) PEG 400 supplemented with 2 mM ZM241385. Showers of small (∼5 µm) crystals appeared overnight and were used for data collection at the Linac Coherent Light Source (LCLS).
MT1 was crystallized as described previously (Stauch et al., 2019 ▸) but using the ligand agomelatine during purification and a precipitant solution consisting of 60–100 mM potassium phosphate monobasic, 100 mM HEPES pH 7.0, 32–35%(v/v) PEG 400, the target ligand 2-phenylmelatonin (2-PMT) at 1 mM, 2.5%(v/v) DMSO, 1.5%(v/v) propan-2-ol.
After crystals had formed, excess precipitant solution was carefully removed, followed by the addition of ∼3 µl 7.9 MAG (Misquitta et al., 2004 ▸) to absorb the residual precipitant solution. The microcrystal samples were characterized on-site at LCLS using a zoom stereomicroscope (Leica) equipped with linear rotating polarizers.
2.4. XFEL data collection
LCP-SFX data collection for β2AR with carazolol, timolol, alprenolol and ICI-118,551, for A2AAR with ZM241385 and for MT1 with 2-PMT was performed using the CXI instrument (Boutet & Williams, 2010 ▸) at LCLS at SLAC National Accelerator Laboratory, Menlo Park, California, USA. LCLS operated at a wavelength of 1.33 Å (9.5 keV), delivering individual X-ray pulses of 42 fs duration with 1012 photons per pulse focused into a spot size of approximately 1.5 µm in diameter using a pair of Kirkpatrick–Baez mirrors. Protein microcrystals in LCP medium were injected into the focus region using the LCP injector (Weierstall et al., 2014 ▸) with a 50 µm diameter nozzle at a flow rate of 0.2 µl min−1. Microcrystals ranged in size from 1 to ∼10 µm, with an average size of 5 × 2 × 2 µm. Single-shot diffraction patterns of randomly oriented crystals were recorded at a rate of 7200 patterns per minute (120 Hz) with the 2.3 megapixel Cornell–SLAC Pixel Array Detector (CSPAD; Hart et al., 2012 ▸). The beam was attenuated to ∼10% (9 × 1010 photons per pulse) of the full intensity to avoid detector saturation.
SFX data for β2AR in complex with propranolol and carvedilol were collected on the BL3 beamline at the SPring-8 Angstrom Compact free-electron LAser (SACLA) in Japan using a multiport charge-coupled device (MPCCD) detector (Tono et al., 2015 ▸). The instrument operated at a wavelength of 1.76 Å (7 keV) with a pulse duration of <10 fs and a repetition rate of 30 Hz. The XFEL pulse (471 µJ per pulse) was focused into a spot size of approximately 1.5 µm in diameter. Data collection at SACLA was guided by a real-time data-processing pipeline (Nakane et al., 2016 ▸) based on Cheetah (Barty et al., 2014 ▸) and CrystFEL (White et al., 2016 ▸).
The overall time of data collection from eight β2AR samples (six ligands and two controls) with a total volume of ∼180 µl was about ∼14.8 h and yielded ∼230 000 indexed patterns (Table 1 ▸). Potential single-crystal diffraction patterns were identified using Cheetah with a threshold of 15 potential Bragg peaks (Barty et al., 2014 ▸). Indexing, integration and merging of the crystal diffraction data was performed using CrystFEL (White et al., 2016 ▸), which involved application of the indexing algorithms in MOSFLM (Leslie, 2006 ▸), XDS (Kabsch, 2010 ▸) and DIRAX (Duisenberg, 1992 ▸) followed by averaging and integration of Bragg peaks using a Monte Carlo integration algorithm (Kirian et al., 2011 ▸). The data-collection statistics are summarized in Supplementary Table S1. Data for MT1–Ago-2-PMT were processed as described previously (Stauch et al., 2019 ▸), solving the indexing ambiguity resulting from space group P4212 and the very similar lengths of the axes: c ≃ a = b.
Table 1. XFEL data-collection statistics.
The names of the data sets contain the transient ligand followed by the ligand of interest. Alp, alprenolol; Tim, timolol; Cara, carazolol; Carv, carvedilol; ICI, ICI-118,551; Prop, propanolol; LUF, LUF5834; ZM, ZM241385; Ago, agomelatine; 2-PMT, 2-phenylmelatonin.
| Data set | XFEL | Data-collection time (min) | Total No. of images | No. of hits | Hit rate (%) | No. of indexed images | Indexing rate (%) | Resolution at CC* = 0.5† (Å) |
|---|---|---|---|---|---|---|---|---|
| β2AR–Alp-Alp | LCLS | 195 | 1405887 | 149024 | 10.6 | 39599 | 26.6 | 2.8 |
| β2AR–Alp-Tim | LCLS | 209 | 1503057 | 238986 | 15.9 | 59814 | 25.0 | 3.2 |
| β2AR–Alp-Cara | LCLS | 147 | 1061949 | 41416 | 3.9 | 9493 | 22.9 | 3.4 |
| β2AR–Tim-Alp | LCLS | 108 | 775070 | 99984 | 12.9 | 60694 | 60.7 | 2.4 |
| β2AR–Tim-Carv | SACLA | 33 | 59426 | 28465 | 47.9 | 14579 | 51.2 | 2.5 |
| β2AR–Tim-ICI | LCLS | 103 | 739335 | 116815 | 15.8 | 43660 | 37.4 | 2.6 |
| β2AR–Tim-Tim | LCLS | 128 | 918091 | 80792 | 8.8 | 17952 | 22.2 | 2.7 |
| β2AR–Tim-Prop | SACLA | 65 | 117972 | 8494 | 7.2 | 5201 | 61.2 | 2.9 |
| A2AAR–LUF-ZM | LCLS | 46 | 241932 | 68623 | 28.4 | 39281 | 57.2 | 1.85 |
| MT1–Ago-2-PMT | LCLS | 136 | 977748 | 87453 | 8.9 | 65260 | 74.6 | 3.3 |
The reported resolution may depend on the number of indexed patterns used for each data set.
2.5. Structure determination
All structures were solved using molecular replacement. The β2AR and A2AAR data sets were phased using the models of the previously solved structure of β2AR bound to timolol (Hanson et al., 2008 ▸; PDB entry 3d4s), the previously solved structure of A2AAR bound to ZM241385 (Liu et al., 2012 ▸; PDB entry 4eiy) and the previously solved structure of MT1 bound to agomelatine (Stauch et al., 2019 ▸; PDB entry 6me5), respectively, which had all heteroatoms removed from the search model. The structures were further optimized by iterative cycles of rebuilding in Coot (Emsley & Cowtan, 2004 ▸) and refinement in phenix.refine (Adams et al., 2010 ▸) or REFMAC5 (Murshudov et al., 2011 ▸) in the case of MT1–Ago-2-PMT. After the receptor refinement had converged, the respective ligand was inserted into the electron density inside the ligand-binding pocket. The final refinement runs were performed with BUSTER v.2.10.2 (Smart et al., 2012 ▸) for the β2AR structures, phenix.refine for A2AAR–LUF-ZM and REFMAC5 in the case of MT1–Ago-2-PMT (Supplementary Tables S1 and S3).
The atomic coordinates and structure factors have been deposited in the Protein Data Bank under the following accession codes (see Table 1 ▸ for an explanation of the data-set names): 6prz (β2AR–Alp-Alp), 6ps0 (β2AR–Alp-Cara), 6ps1 (β2AR–Alp-Tim), 6ps2 (β2AR–Tim-Alp), 6ps3 (β2AR–Tim-Carv), 6ps4 (β2AR–Tim-ICI), 6ps5 (β2AR–Tim-Prop), 6ps6 (β2AR–Tim-Tim), 6ps7 (A2AAR–LUF-ZM) and 6ps8 (MT1–Ago-2-PMT).
3. Results
3.1. Development of the Complex-LCP method
The typical GPCR structure-determination process consists of several major steps, including construct design, expression of the target receptor in a heterologous system, purification of the protein in the presence of a stabilizing ligand and crystallization in LCP (Stevens et al., 2013 ▸). The ligand of interest is usually added before receptor solubilization and is continuously supplied throughout purification and crystallization. Following this traditional structure-determination approach, each receptor–ligand combination requires individual screening and optimization of purification and, more importantly, crystallization conditions, which may take weeks to months. Thus, SBDD studies demand substantial efforts proportional to the number of compounds being investigated.
Recently, a more efficient method for multiple GPCR co-crystal structure determination was introduced (Rucktooa et al., 2018 ▸) by crystallizing the target receptor in complex with a low-affinity ‘carrier’ ligand and subsequent soaking of crystals in solutions containing the desired higher affinity compounds. This approach, however, has several limitations. Firstly, it relies on the availability of relatively large high-quality crystals of the receptor in a complex with a low-affinity ligand, which is often challenging as such ligands typically do not sufficiently stabilize the receptors. Secondly, inhomogeneous ligand exchange in a large crystal could potentially disrupt its structure and integrity, leading to a loss of diffraction quality and dissolution of the entire crystal. These difficulties can be overcome by using a receptor thermally stabilized in a specific pre-defined conformation, such as, for example, the adenosine A2A receptor stabilized in an inactive state by nine mutations using the STaR technology (Doré et al., 2011 ▸).
We approached these challenges from a different angle, taking advantage of the LCP-SFX approach (Liu et al., 2013 ▸) that has proven to be highly successful for GPCR structure determination using micrometre-sized crystals grown in LCP (Stauch & Cherezov, 2018 ▸). As a model system, we have selected the human β2-adrenergic receptor (β2AR), which is one of the most extensively studied GPCRs to date and has a large set of pharmacologically and structurally diverse ligands, including approved drugs such as beta blockers (β2AR antagonists) and anti-asthmatic medicines (β2AR agonists).
We modified our XFEL sample-preparation protocol (Liu et al., 2014 ▸) by introducing a transient ligand. The function of this ligand is to stabilize the receptor during purification and to enable its crystallization. A suitable transient ligand should have an off-rate that is fast compared with the crystallization timescale in order to facilitate its exchange to the ligand of interest. Therefore, in our Complex-LCP approach the target receptor is first purified in the presence of a minimal amount of the transient ligand and reconstituted in LCP. Concurrent ligand exchange and crystallization is then initiated by overlaying a protein-laden LCP bolus with precipitant solution containing a large excess of the ligand of interest. Dozens of different ligands can be tested in parallel using a single batch (∼1 mg) of the purified receptor reconstituted in LCP. The transient ligand is replaced during the process, leading to co-crystals of the receptor in complex with the desired ligands. Finally, SFX data sets are collected for each ligand of interest and structures are solved using molecular replacement.
3.2. Implementation of the protocol
An overall scheme for the Complex-LCP method is shown in Fig. 1 ▸. In a proof-of-concept study, we used β2AR fused to T4 lysozyme (β2AR-T4L) and aimed at obtaining structures in complex with eight ligands with different MoAs ranging from inverse agonism to agonism (Table 2 ▸). Two of these ligands (alprenolol and timolol) were selected to play the role of a transient ligand owing to their favorable kinetic properties (Supplementary Table S1). Initial high-throughput crystallization trials were performed in 96-well glass sandwich LCP plates to identify suitable crystallization conditions that would produce showers of small crystals of the receptor purified with each of the two transient ligands. Both ligands were used at a 50 µM concentration in the purified protein sample. In the case of timolol no supplementation of ligand to the crystallization screen was necessary, whereas in the case of alprenolol the screens were supplemented with 100 µM ligand to maintain crystal growth. Following these trials, a condition based on ammonium sulfate as a precipitant salt was chosen for further steps (100 mM ammonium sulfate, 100 mM HEPES pH 7.0, 30% PEG 400, 1 mM ligand).
Figure 1.

Schematic diagram of the Complex-LCP method. After crystallization conditions have been identified and optimized, the target receptor is purified in complex with a transient ligand and screened against a panel of N ligands using nanolitre-volume high-throughput robotic crystallization in 96-well glass sandwich plates. Those ligands that support crystallization are then used to prepare samples for XFEL data collection in syringes.
Table 2. β2AR ligands used in the exchange experiments with their MoAs, molecular weights (MW) and affinity (K i) values.
Data are from the ChEMBL database (ChEMBL_23; Gaulton et al., 2012 ▸). Chemical structures of the ligands used for the other receptors in this study are shown in Supplementary Fig. S4.
| Ligand | Chemical structure | MoA | ChEMBL ID | MW (Da) | K i (nM) | Reference |
|---|---|---|---|---|---|---|
| ICI-118,551 |
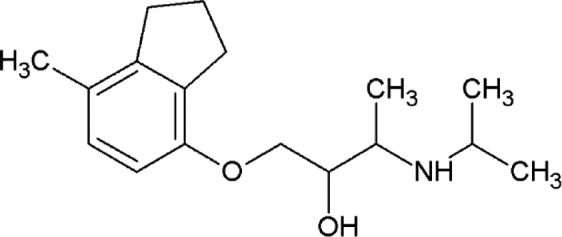
|
Inverse agonist | CHEMBL513389 | 277.4 | 0.13 | Dolušić et al. (2011 ▸) |
| Carazolol |
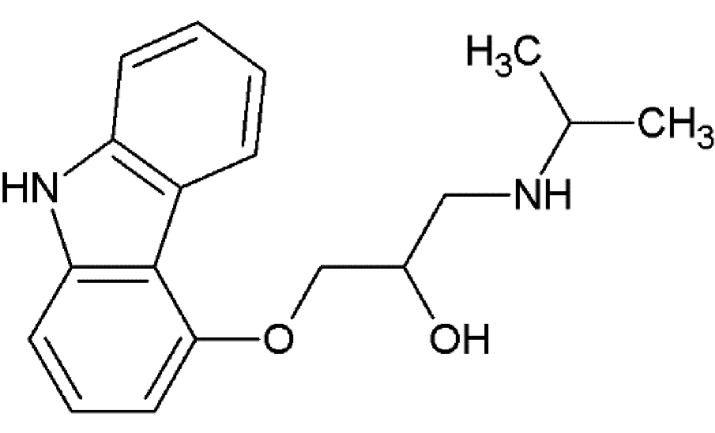
|
Inverse agonist | CHEMBL324665 | 298.4 | 0.114 | Sabio et al. (2008 ▸) |
| Timolol |
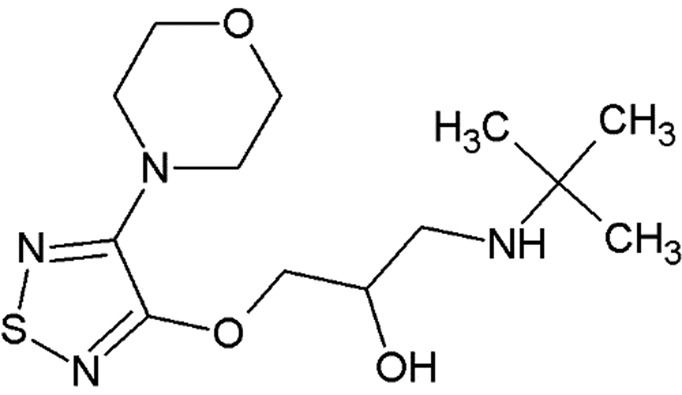
|
Inverse agonist | CHEMBL499 | 316.4 | 0.201† | |
| Propranolol |
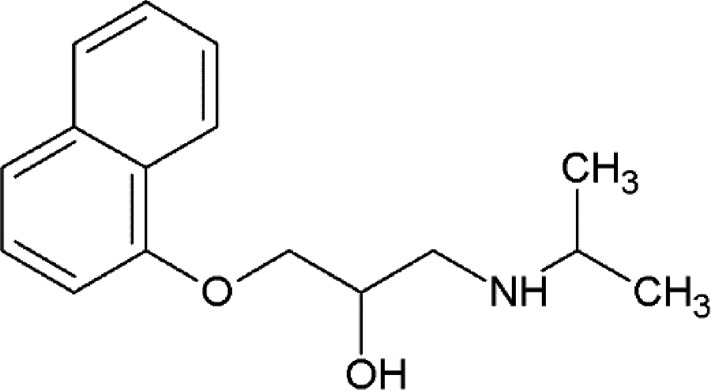
|
Inverse agonist | CHEMBL27 | 259.3 | 3.69 | Plazinska et al. (2014 ▸) |
| Alprenolol |
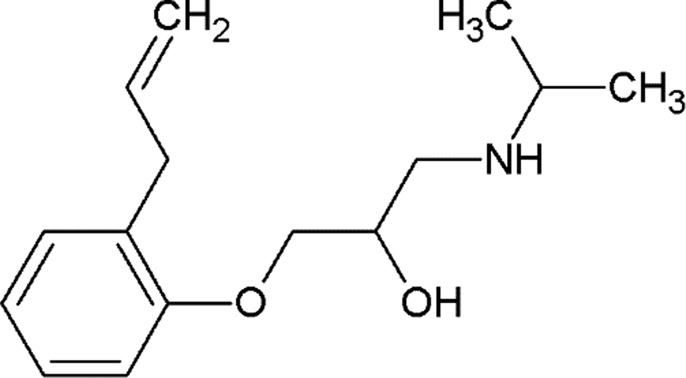
|
Antagonist | CHEMBL266195 | 249.4 | 1 | Aristotelous et al. (2013 ▸) |
| Carvedilol |

|
β-Arrestin-biased agonist | CHEMBL723 | 406.5 | 0.166† | |
| Procaterol |

|
Agonist | CHEMBL160519 | 290.4 | 78 | Baker (2010 ▸) |
| Formoterol |
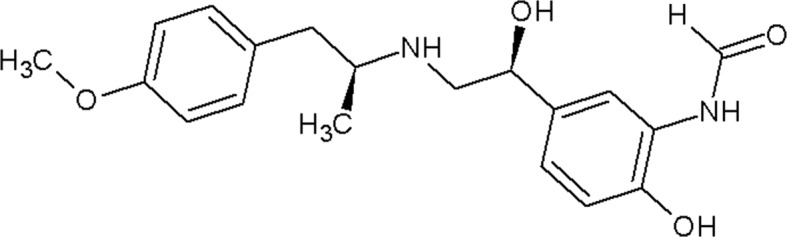
|
Agonist | CHEMBL3989798 | 344.4 | 23 | Baker (2010 ▸) |
Values are from the DrugMatrix Database (https://ntp.niehs.nih.gov/drugmatrix/index.html).
Next, the receptor was purified in the presence of 50 µM of the transient ligand (alprenolol or timolol), and LCP crystallization trials were set up in 96-well glass sandwich plates using the precipitant solution supplemented with 1 mM of the target ligand, with one plate per ligand. Two additional control plates were also set up: Control Plate A without any ligand in the precipitant solutions (negative control) and Control Plate B with the same transient ligand as that used during purification added to the precipitant solutions (positive control). Crystallization plates were stored at 20°C in a RockImager 1000 (Formulatrix) and were inspected at 12 h intervals.
On the first day after setup, showers of small crystals appeared in most wells of all plates with ligands added to the precipitant solutions (Supplementary Fig. S1). On the second day, however, crystals in plates with the agonists formoterol and procaterol started to dissolve, and they completely disappeared on day 3 [Supplementary Fig. S1(b)]. We interpreted this phenomenon as an indication that agonist binding induces a substantially different receptor conformation that is incompatible with pre-formed crystal contacts. Crystals in plates with the remaining six ligands reached a maximal size of up to 5–10 µm and remained stable until the end of the observation period of one month. As expected, the same behavior was observed in Control Plate B, while no crystals or only very small (∼1 µm) and sparse crystals were found in Control Plate A.
After confirming crystal formation in plates, the crystallization volume was scaled up ∼100 times in Hamilton gas-tight syringes for LCP-SFX data collection, following our previously developed protocol (Liu et al., 2014 ▸). Owing to the large excess of precipitant solution over LCP used in the crystallization setup, the concentration of the transient ligand decreases 50 times upon equilibration, resulting in a ∼1000:1 molar excess of each of the target ligands over the transient ligand to ensure efficient ligand exchange.
3.3. LCP-SFX data collection
LCP-SFX data were collected as described previously (Liu et al., 2013 ▸), with microcrystals delivered to the XFEL beam within their crystal-growth medium by an LCP injector (Weierstall et al., 2014 ▸). Except for two data sets (Table 1 ▸), the data were acquired at the CXI experimental station (Boutet & Williams, 2010 ▸) of LCLS using a vacuum sample chamber with the XFEL beam focused to about 1.5 µm diameter and the CSPAD (Hart et al., 2012 ▸), operating at an XFEL pulse repetition rate of 120 Hz. Data for carvedilol and propranolol were collected at SACLA (Ishikawa et al., 2012 ▸) using a helium-filled sample chamber (Kameshima et al., 2014 ▸), a 1.5 µm diameter beam size and an MPCCD detector (Tono et al., 2015 ▸), operating at an XFEL pulse repetition rate of 30 Hz. Crystals were delivered using an LCP injector with a 50 µm inner diameter (ID) nozzle and a flow rate of 150–200 nl min−1 for experiments at LCLS and with a 100 µm ID nozzle and a flow rate of 250 nl min−1 at SACLA. All acquired LCP-SFX data were processed using a Monte Carlo integration approach implemented in CrystFEL (White et al., 2016 ▸), which requires a large number of indexed patterns for the accurate determination of intensities. In our experience, 20 000–30 000 indexed images are sufficient to assemble a high-quality data set. However, when the overall receptor structure is known and only the binding pose of the ligand is being determined, fewer indexed images, of the order of 5000–10 000, may be sufficient (Table 1 ▸). With the parameters described above and a crystal hit/indexing rate of >1%, it is currently possible to collect a complete data set at LCLS within less than 2 h using about 25 µl of crystal-laden LCP.
3.4. Ligand electron-density maps and structure validation
Successful molecular replacement using the known β2AR structure immediately revealed strong mF o − DF c electron densities in the ligand-binding pocket resembling the shapes of the target ligands (Fig. 2 ▸). To further validate the ligand exchange, we refined the coordinates after placing the corresponding transient ligand in the density. The resulting mF o − DF c maps showed substantial positive and negative electron density around the ligand (Fig. 2 ▸ and Supplementary Fig. S2), indicating that the presence of the transient ligand in the complex is not supported by the experimental data and thus confirming that the transient ligand has successfully exchanged during the process of crystallization. Conversely, the refinement of structures against the experimental data after placing the corresponding target ligands in the density produced well defined maps fully consistent with the chemical structures of the ligands used. The final data-processing and refinement statistics are shown in Supplementary Table S2.
Figure 2.

Electron densities for the β2AR ligands obtained using the Complex-LCP method. 2mF o − DF c electron densities in the ligand-binding pocket (purple circle) right after molecular replacement are shown as superimposed meshes contoured at 0.7σ (timolol, green; alprenolol, red; carvedilol, blue; propranolol, cyan). The mF o − DF c polder ligand OMIT maps (Liebschner et al., 2017 ▸) contoured at 3σ are shown for each ligand inside blue (transient ligand alprenolol) and green (transient ligand timolol) boxes. The inset in the black box demonstrates cross-validation of the ligand exchange using crystallographic data. When alprenolol-to-timolol exchange data are refined with alprenolol, the residual mF o − DF c electron density (green) contoured at 3σ clearly indicates that the transient ligand has been successfully exchanged.
3.5. Comparison of synchrotron and XFEL structures
The XFEL β2AR structures in complex with carazolol, timolol, alprenolol and ICI-118,551 obtained in this work are almost identical to those previously determined using synchrotron data collected from cryocooled crystals (Supplementary Table S3; Cherezov et al., 2007 ▸; Wacker et al., 2010 ▸; Hanson et al., 2008 ▸), validating our approach. The resolution cutoff values of the data sets range from 2.4 Å for alprenolol to 3.4 Å for carazolol. The variation in data quality most likely arises from a combination of factors such as crystal quality for the particular ligand and the number of indexed images for a given data set. Crystal quality also appears to depend on the identity of the transient ligand used for the exchange: the resolution of the structures based on alprenolol as a transient ligand is consistently lower compared with those based on timolol. This observation can be explained by potentially stronger crystal contacts in the initial seed crystals in the complex with timolol; however, it requires further investigation. The mean B factor of the obtained structures correlates with resolution (Supplementary Fig. S3) and on average is ∼30 Å2 higher than the mean B factor of the corresponding synchrotron structures, reflecting the differences in data-collection temperature and data processing.
3.6. Structures of β2AR bound to carvedilol and propranolol
In addition to the four ligands that have previously been co-crystallized with β2AR (Wacker et al., 2010 ▸; Cherezov et al., 2007 ▸; Hanson et al., 2008 ▸), we used the Complex-LCP method to determine two new β2AR structures in complex with carvedilol and propranolol. Both of these ligands belong to the class of beta blockers; however, carvedilol acts as a β-arrestin-biased agonist, while concomitantly antagonizing G protein activity (Drake et al., 2008 ▸). This characteristic of carvedilol has important pharmacological implications owing to its improved cardioprotective effects compared with the majority of current beta blockers (Leonetti & Egan, 2012 ▸).
The propranolol-bound β2AR structure reveals a canonical ligand-binding pose with the ethanolamine moiety occupying the same position and engaging in hydrogen bonds to Asp1133.32 and Asn3127.39 (where the superscripts refer to the Ballesteros–Weinstein Class A GPCR numbering scheme; Ballesteros & Weinstein, 1995 ▸), as in the structures with other antagonists (for example alprenolol), and with the naphthalene ring reinforcing the hydrophobic interactions [Fig. 3 ▸(a)].
Figure 3.

Binding of propranolol and carvedilol to β2AR. (a) Superposition of propranolol-bound (cyan) and alprenolol-bound (pink) β2AR structures. (b) Chemical structure of carvedilol with contacting β2AR residues within 4 Å of the ligand. (c) Superposition of carvedilol-bound β2AR (green) and β1AR (orange; PDB entry 4amj; two molecules from one asymmetric unit; Warne et al., 2012) structures. (d) Superposition of carvedilol-bound (green) and carazolol-bound (yellow) β2AR structures. Hydrogen bonds are shown as dashed lines for the propranolol- and carvedilol-bound β2AR structures only. Helices are labeled with Roman numerals.
Carvedilol also binds β2AR in a similar pose, anchored by Asp1133.32 and Asn3127.39, with the terminal methoxybenzene group of its tail, which is believed to be responsible for the β-arrestin-biased activity of carvedilol, participating in hydrophobic interactions with His932.64, Ile942.65, Trp1093.28 and Trp3137.40 [Fig. 3 ▸(b)]. Superposition of our human β2AR–carvedilol structure with the structure of turkey β1AR–carvedilol reported previously using the receptor stabilized by eight point mutations (Warne et al., 2012 ▸) reveals several notable differences [Fig. 3 ▸(c)]. While the ligands overlap well in the carbazole head and the oxypropanolamine tail (r.m.s.d. of 0.45 Å) common to many β2AR ligands (Hanson et al., 2008 ▸; Cherezov et al., 2007 ▸; Wacker et al., 2010 ▸), the terminal methoxybenzene group adopts slightly different orientations in these two receptors. Comparing the structures of the receptors, we observe a 1–2 Å outward tilt of the extracellular tips of helices II and VII in β1AR. The extra space in the β1AR pocket created by these displacements potentially allows a more dynamic conformation of the carvedilol tail (different orientations in the two molecules in the asymmetric unit) compared with β2AR. Additionally, the extracellular tip of helix I that does not interact with the ligand is also shifted ∼3 Å between these structures, which may reflect differences between these receptors and/or differences in their crystallization environment and crystal packing.
A comparison of carvedilol- and carazalol-bound β2AR structures shows displacements of several residues interacting with the methoxybenzene group of carvedilol, leading to an ∼0.6 Å outward shift of helix II and extracellular loop 2 (ECL2) and expanding the volume of the ligand-binding pocket [Fig. 3 ▸(d)]. These small structural differences may be responsible for the differences in the reported ligand MoA.
3.7. Application of the Complex-LCP method to other receptors
To demonstrate the general applicability of the described Complex-LCP method, we applied it to four other GPCRs: serotonin receptors 1B (5-HT1B) and 2B (5-HT2B), adenosine A2A receptor (A2AAR) and melatonin receptor type 1A (MT1). An overview of all of the compounds used as transient or target ligands in this study can be found in Supplementary Fig. S4. At the time these experiments were performed, only two agonists, ergotamine (ERG) and dihydroergotamine (DHE), had been co-crystallized with serotonin receptors (Wacker et al., 2013 ▸; Wang et al., 2013 ▸); therefore, one of them, ERG, was initially chosen as a transient ligand. Microcrystals of 5-HT1B were generated following the same procedure as for β2AR with the goal of exchanging ERG for the antagonist methylergometrine or for the agonists oxymetazoline, sumatripan and RU-24969 (Supplementary Fig. S4). However, after collecting diffraction data and solving the structures it was observed that the ligand exchange had been unsuccessful and that the receptor was still bound to the transient ligand ERG. This result emphasizes the importance of proper transient ligand selection. ERG is a relatively large molecule with multiple interactions within the binding pocket of 5-HT1B, leading to a slow off-rate (k off = 0.0125 min−1; Unett et al., 2013 ▸), which is likely to explain its low exchange efficiency. After realizing the need for a transient ligand with a high off-rate, we switched to serotonin (k off = 0.1216 min−1; Unett et al., 2013 ▸) and performed crystallization experiments with 5-HT2B, exchanging serotonin for ERG or DHE. Microcrystals were obtained in both cases within one day [Figs. 4 ▸(a) and 4 ▸(b)], while no crystals were observed in the control experiment without any ligand supplemented in the precipitant.
Figure 4.

Ligand exchange in 5-HT2B and A2AAR. (a, b) Ligand-exchange experiment in LCP crystallization plates with 5-HT2B. The transient ligand serotonin was exchanged for ERG (a) and DHE (b). The images were taken under cross-polarized light. No crystals appeared in control experiments (insets) set up without adding ligands to the precipitant solution. (c, d) Ligand-exchange experiment in glass syringes with A2AAR. The transient ligand LUF5834 is exchanged for ZM241385. (c) LCP string immersed in precipitant solution in a glass syringe. (d) LCP sample titrated with 7.9 MAG before loading into an LCP injector.
A2AAR is another prototypical GPCR, which similarly to β2AR has a large number of available ligands. A2AAR has been crystallized in complex with many different ligands, including the relatively large antagonists ZM241385 (Liu et al., 2012 ▸) and comp-1 (Sun et al., 2017 ▸), the agonist UK-432097 (Xu, Wu et al., 2011 ▸) and some smaller, high-off-rate antagonists such as XAC, caffeine, PSB36 and theophylline (Cheng et al., 2017 ▸; Doré et al., 2011 ▸). The latter structures with xanthines could only be obtained using a thermostabilized construct of A2AAR. We applied our approach using the antagonist LUF5834 (Supplementary Fig. S4) as a transient ligand, successfully exchanging it for the higher affinity antagonist ZM241385 [Figs. 4 ▸(c) and 4 ▸(d)], and solved the co-crystal structure by SFX (Supplementary Table S4 and Supplementary Fig. S5). As expected, the data quality and the overall A2AAR–LUF-ZM structure derived via the Complex-LCP method are nearly identical (the Cα r.m.s.d. with PDB entry 5k2d is 0.21 Å) to published A2AAR–ZM241385 structures obtained using conventional LCP crystallization (Batyuk et al., 2016 ▸; Liu et al., 2012 ▸).
Melatonin receptor MT1 has recently been co-crystallized with several agonists, including two melatonin analogs as well as two drugs: the sleeping aid ramelteon and the atypical antidepressant agomelatine (Stauch et al., 2019 ▸). Since addition of the ligand to the precipitant solution was essential for crystallization in all cases, indicating that ligand-exchange events happen on a timescale comparable to crystallization, we expected MT1 to be a suitable target for our Complex-LCP method. We used the antidepressant agomelatine (Ago) as a transient ligand during crystallization setups and successfully exchanged it for the larger 2-phenylmelatonin (2-PMT), as unambiguously identified in the resulting electron densities (Supplementary Fig. S4 and Fig. 5 ▸). The size and diffraction quality of the resulting MT1–Ago-2-PMT crystals were comparable with those of published crystals of MT1–Ago (PDB entry 6me5; Stauch et al., 2019 ▸), suggesting that the transient ligand (in this case Ago) was the limiting factor to the final resolution. Again, as in the case with A2AAR, our MT1–Ago-2-PMT structure is nearly identical to the published structure of MT1–2-PMT (the Cα r.m.s.d. with PDB entry 6me3 is 0.49 Å), microcrystals of which were obtained using conventional LCP crystallization (Stauch et al., 2019 ▸).
Figure 5.

Electron density in the MT1 ligand-binding pocket. (a) Ligand electron density for MT1 (white cartoon) after ligand exchange of agomelatine (Ago) to 2-phenylmelatonin (2-PMT; orange). (b) Ligand electron density after incorrect placement of the transient ligand Ago (cyan) and refinement starting from the same molecular-replacement structure (see Section 2). Both structural models are of comparable stereochemical quality, but the structure refined with Ago shows slightly worse refinement statistics (R and R free of 0.257 and 0.314 compared with 0.257 and 0.308, respectively). 2mF o − DF c maps (blue mesh) are contoured at 1σ. mF o − DF c electron-density difference maps contoured at ±3.5σ (green and red for positive and negative peaks, respectively) show strong (∼6.7σ) positive difference density for the missing phenyl ring in the structure refined with Ago.
4. Discussion
We have introduced a new method, Complex-LCP, for facilitating the structure determination of multiple GPCR–ligand complexes. The method allows the rapid identification of ligand-binding poses and interactions for a panel of about ten ligands in a single experiment. The two most critical aspects of this method are the use of a transient ligand to increase the stability and conformational homogeneity of the target receptor and the application of an XFEL source for crystallographic data collection from micrometre-sized crystals. Several considerations were deemed to be essential for the selection of the transient ligand, such as a fast ligand off-rate to ensure efficient ligand exchange and a relatively low dissociation constant, K d, compared with the ligands of interest to maintain a high target ligand/transient ligand concentration ratio during ligand exchange. As conceived, ligand exchange can occur at different stages of the process: before, during or after crystal nucleation. For example, in the case of A2AAR the transient ligand LUF5834 is likely to be exchanged with ZM241385 before crystallization, as no crystals were obtained when using LUF5834 alone. On the other hand, in the case of β2AR the transient ligand is apparently involved in crystal nucleation and is replaced after the crystals have already formed. This conclusion is supported by the observation of crystal dissolution in the presence of the agonists procaterol and formoterol and by the prominent effect of the transient ligand on the resolution of the obtained structures, so that using timolol rather than alprenolol as the transient ligand resulted in higher resolution structures (Table 1 ▸). Most strikingly, exchanging timolol for alprenolol substantially improved the resolution (β2AR–Tim-Alp data set, 2.4 Å resolution) compared with the control sample using alprenolol alone (β2AR–Alp-Alp data set, 2.8 Å resolution) and with the previously reported β2AR–alprenolol synchrotron structure (PDB entry 3nya, 3.16 Å resolution; Wacker et al., 2010 ▸). It is evident that such improvements in resolution are highly desirable for SBDD applications.
The availability of a protein construct with an adequate conformational stability to generate crystals is a prerequisite for the Complex-LCP method. The method has not been devised to provide an alternative to thermostabilization by point mutations, the introduction of which might still be necessary for crystallogenesis. Indeed, most of the GPCR constructs used for successful crystallization in this study and elsewhere (Xiang et al., 2016 ▸) contained one or more point mutations (see Section 2 for details). Compounds with fast off-rates that are potential candidates for the role of transient ligands are often less stabilizing than super-high-affinity ligands and may require additional receptor engineering for crystallization.
The Complex-LCP method relies on the ability to collect high-resolution crystallographic data from micrometre-sized crystals, which is enabled by the SFX approach at XFEL sources. The small size of the microcrystals facilitates ligand exchange without affecting the crystal quality and integrity. Additionally, the lower mosaicity and the fewer growth defects in microcrystals compared with their larger counterparts used for data collection at synchrotron sources, as well as bypassing crystal harvesting and potential artifacts from cryocooling, often lead to higher quality diffraction, as demonstrated by several examples (Fenalti et al., 2015 ▸; Zhang et al., 2015 ▸; Kang et al., 2015 ▸). Recent advances in SFX data-processing algorithms and XFEL sample-delivery instrumentation have reduced the requirements for the amount of crystalline sample per data set. Further optimizations and improvements are imminent, such as fixed-target-based crystal delivery (Roedig et al., 2017 ▸; Mueller et al., 2015 ▸; Hunter et al., 2015 ▸) combined with higher repetition-rate XFEL sources, which could substantially increase the throughput of this method.
While this approach and its variations have a strong potential to accelerate drug-discovery applications using difficult-to-crystallize membrane proteins, their broader acceptance by academic and industrial laboratories may be limited by the shortage of available XFEL beamtime. Recent demonstrations of serial crystallography at synchrotron sources (Martin-Garcia et al., 2017 ▸; Meents et al., 2017 ▸; Weinert et al., 2017 ▸) indicate that new-generation diffraction-limited storage rings and other related developments (Eberhardt, 2015 ▸; Yabashi & Tanaka, 2017 ▸) promise to deliver crystallographic data from micrometre-sized crystals that are comparable in quality to those from XFELs. We believe that these advancements will produce a strong impact on the development of more efficient and safe therapies.
5. Related literature
The following reference is cited in the supporting information for this article: Sykes et al. (2014 ▸).
Supplementary Material
PDB reference: β2-adrenergic receptor, complex with alprenolol, 6prz
PDB reference: 6ps2
PDB reference: complex with carazolol, 6ps0
PDB reference: complex with timolol, 6ps1
PDB reference: 6ps6
PDB reference: complex with carvedilol, 6ps3
PDB reference: complex with ICI-118,551, 6ps4
PDB reference: complex with propranolol, 6ps5
PDB reference: adenosine A2A receptor, complex with ZM241385, 6ps7
PDB reference: melatonin receptor MT1, complex with 2-phenylmelatonin, 6ps8
Supplementary Tables and Figures. DOI: 10.1107/S2052252519013137/cw5024sup1.pdf
Acknowledgments
Parts of this research were carried out at the LCLS, a National User Facility operated by Stanford University on behalf of the US Department of Energy and supported by the US Department of Energy Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The XFEL data sets from SACLA (BL3) were collected with the approval of the Japan Synchrotron Radiation Research Institute (Proposal 2015A8025). CG kindly thanks SLAC National Accelerator Laboratory and the Department of Energy for financial support through the Panofsky fellowship. IM thanks the Membrane Protein Laboratory at Diamond Light Source (Wellcome Trust grant No. 099165/Z/12/Z). NZ acknowledges the support of the Australian Research Council through the Centre of Excellence in Advanced Molecular Imaging (CE140100011).
Funding Statement
This work was funded by National Institutes of Health, National Institute of General Medical Sciences grants R35 GM127086 and R01 GM124152. National Institutes of Health, National Institute on Drug Abuse grant R21 DA042298 to Wei Liu. National Science Foundation grants 1231306 and 1565180. European Molecular Biology Organization grant ALTF 677-2014 to Benjamin Stauch. Ministry of Education, Culture, Sports, Science and Technology grant . GPCR Consortium grant .
References
- Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L.-W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. & Zwart, P. H. (2010). Acta Cryst. D66, 213–221. [DOI] [PMC free article] [PubMed]
- Allen, J. A. & Roth, B. L. (2011). Annu. Rev. Pharmacol. Toxicol. 51, 117–144. [DOI] [PubMed]
- Aristotelous, T., Ahn, S., Shukla, A. K., Gawron, S., Sassano, M. F., Kahsai, A. W., Wingler, L. M., Zhu, X., Tripathi-Shukla, P., Huang, X.-P., Riley, J., Besnard, J., Read, K. D., Roth, B. L., Gilbert, I. H., Hopkins, A. L., Lefkowitz, R. J. & Navratilova, I. (2013). ACS Med. Chem. Lett. 4, 1005–1010. [DOI] [PMC free article] [PubMed]
- Baker, J. G. (2010). Br. J. Pharmacol. 160, 1048–1061. [DOI] [PMC free article] [PubMed]
- Ballesteros, J. A. & Weinstein, H. (1995). Methods Neurosci. 25, 366–428.
- Barty, A., Kirian, R. A., Maia, F. R. N. C., Hantke, M., Yoon, C. H., White, T. A. & Chapman, H. (2014). J. Appl. Cryst. 47, 1118–1131. [DOI] [PMC free article] [PubMed]
- Batyuk, A., Galli, L., Ishchenko, A., Han, G. W., Gati, C., Popov, P. A., Lee, M.-Y., Stauch, B., White, T. A., Barty, A., Aquila, A., Hunter, M. S., Liang, M., Boutet, S., Pu, M., Liu, Z., Nelson, G., James, D., Li, C., Zhao, Y., Spence, J. C. H., Liu, W., Fromme, P., Katritch, V., Weierstall, U., Stevens, R. C. & Cherezov, V. (2016). Sci. Adv. 2, e1600292. [DOI] [PMC free article] [PubMed]
- Boutet, S. & Williams, G. J. (2010). New J. Phys. 12, 035024.
- Chapman, H. N., Fromme, P., Barty, A., White, T. A., Kirian, R. A., Aquila, A., Hunter, M. S., Schulz, J., DePonte, D. P., Weierstall, U., Doak, R. B., Maia, F. R. N. C., Martin, A. V., Schlichting, I., Lomb, L., Coppola, N., Shoeman, R. L., Epp, S. W., Hartmann, R., Rolles, D., Rudenko, A., Foucar, L., Kimmel, N., Weidenspointner, G., Holl, P., Liang, M., Barthelmess, M., Caleman, C., Boutet, S., Bogan, M. J., Krzywinski, J., Bostedt, C., Bajt, S., Gumprecht, L., Rudek, B., Erk, B., Schmidt, C., Hömke, A., Reich, C., Pietschner, D., Strüder, L., Hauser, G., Gorke, H., Ullrich, J., Herrmann, S., Schaller, G., Schopper, F., Soltau, H., Kühnel, K.-U., Messerschmidt, M., Bozek, J. D., Hau-Riege, S. P., Frank, M., Hampton, C. Y., Sierra, R. G., Starodub, D., Williams, G. J., Hajdu, J., Timneanu, N., Seibert, M. M., Andreasson, J., Rocker, A., Jönsson, O., Svenda, M., Stern, S., Nass, K., Andritschke, R., Schröter, C.-D., Krasniqi, F., Bott, M., Schmidt, K. E., Wang, X., Grotjohann, I., Holton, J. M., Barends, T. R. M., Neutze, R., Marchesini, S., Fromme, R., Schorb, S., Rupp, D., Adolph, M., Gorkhover, T., Andersson, I., Hirsemann, H., Potdevin, G., Graafsma, H., Nilsson, B. & Spence, J. C. H. (2011). Nature (London), 470, 73–77.
- Cheng, R. K. Y., Segala, E., Robertson, N., Deflorian, F., Doré, A. S., Errey, J. C., Fiez-Vandal, C., Marshall, F. H. & Cooke, R. M. (2017). Structure, 25, 1275–1285.e4. [DOI] [PubMed]
- Cherezov, V. (2011). Curr. Opin. Struct. Biol. 21, 559–566. [DOI] [PMC free article] [PubMed]
- Cherezov, V., Hanson, M. A., Griffith, M. T., Hilgart, M. C., Sanishvili, R., Nagarajan, V., Stepanov, S., Fischetti, R. F., Kuhn, P. & Stevens, R. C. (2009). J. R. Soc. Interface, 6, s587. [DOI] [PMC free article] [PubMed]
- Cherezov, V., Rosenbaum, D. M., Hanson, M. A., Rasmussen, S. G. F., Thian, F. S., Kobilka, T. S., Choi, H.-J., Kuhn, P., Weis, W. I., Kobilka, B. K. & Stevens, R. C. (2007). Science, 318, 1258–1265. [DOI] [PMC free article] [PubMed]
- Chun, E., Thompson, A. A., Liu, W., Roth, C. B., Griffith, M. T., Katritch, V., Kunken, J., Xu, F., Cherezov, V., Hanson, M. A. & Stevens, R. C. (2012). Structure, 20, 967–976. [DOI] [PMC free article] [PubMed]
- Dolušić, E., Larrieu, P., Moineaux, L., Stroobant, V., Pilotte, L., Colau, D., Pochet, L., Van den Eynde, B., Masereel, B., Wouters, J. & Frédérick, R. (2011). J. Med. Chem. 54, 5320–5334. [DOI] [PubMed]
- Doré, A. S., Robertson, N., Errey, J. C., Ng, I., Hollenstein, K., Tehan, B., Hurrell, E., Bennett, K., Congreve, M., Magnani, F., Tate, C. G., Weir, M. & Marshall, F. H. (2011). Structure, 19, 1283–1293. [DOI] [PMC free article] [PubMed]
- Drake, M. T., Violin, J. D., Whalen, E. J., Wisler, J. W., Shenoy, S. K. & Lefkowitz, R. J. (2008). J. Biol. Chem. 283, 5669–5676. [DOI] [PubMed]
- Duisenberg, A. J. M. (1992). J. Appl. Cryst. 25, 92–96.
- Eberhardt, W. (2015). J. Electron Spectrosc. Relat. Phenom. 200, 31–39.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fenalti, G., Zatsepin, N. A., Betti, C., Giguere, P., Han, G. W., Ishchenko, A., Liu, W., Guillemyn, K., Zhang, H., James, D., Wang, D., Weierstall, U., Spence, J. C. H., Boutet, S., Messerschmidt, M., Williams, G. J., Gati, C., Yefanov, O. M., White, T. A., Oberthuer, D., Metz, M., Yoon, C. H., Barty, A., Chapman, H. N., Basu, S., Coe, J., Conrad, C. E., Fromme, R., Fromme, P., Tourwé, D., Schiller, P. W., Roth, B. L., Ballet, S., Katritch, V., Stevens, R. C. & Cherezov, V. (2015). Nat. Struct. Mol. Biol. 22, 265–268. [DOI] [PMC free article] [PubMed]
- Fraser, J. S., Clarkson, M. W., Degnan, S. C., Erion, R., Kern, D. & Alber, T. (2009). Nature (London), 462, 669–673. [DOI] [PMC free article] [PubMed]
- Fraser, J. S., van den Bedem, H., Samelson, A. J., Lang, P. T., Holton, J. M., Echols, N. & Alber, T. (2011). Proc. Natl Acad. Sci. USA, 108, 16247–16252. [DOI] [PMC free article] [PubMed]
- Gaulton, A., Bellis, L. J., Bento, A. P., Chambers, J., Davies, M., Hersey, A., Light, Y., McGlinchey, S., Michalovich, D., Al-Lazikani, B. & Overington, J. P. (2012). Nucleic Acids Res. 40, D1100–D1107. [DOI] [PMC free article] [PubMed]
- Hanson, M. A., Cherezov, V., Griffith, M. T., Roth, C. B., Jaakola, V.-P., Chien, E. Y. T., Velasquez, J., Kuhn, P. & Stevens, R. C. (2008). Structure, 16, 897–905. [DOI] [PMC free article] [PubMed]
- Hart, P., Boutet, S., Carini, G., Dubrovin, M., Duda, B., Fritz, D., Haller, G., Herbst, R., Herrmann, S., Kenney, C., Kurita, N., Lemke, H., Messerschmidt, M., Nordby, M., Pines, J., Schafer, D., Swift, M., Weaver, M., Williams, G., Zhu, D., Van Bakel, N. & Morse, J. (2012). Proc. SPIE, 8504, 85040C.
- Hunter, M. S., Segelke, B., Messerschmidt, M., Williams, G. J., Zatsepin, N. A., Barty, A., Benner, W. H., Carlson, D. B., Coleman, M., Graf, A., Hau-Riege, S. P., Pardini, T., Seibert, M. M., Evans, J., Boutet, S. & Frank, M. (2015). Sci. Rep. 4, 6026. [DOI] [PMC free article] [PubMed]
- Ishikawa, T., Aoyagi, H., Asaka, T., Asano, Y., Azumi, N., Bizen, T., Ego, H., Fukami, K., Fukui, T., Furukawa, Y., Goto, S., Hanaki, H., Hara, T., Hasegawa, T., Hatsui, T., Higashiya, A., Hirono, T., Hosoda, N., Ishii, M., Inagaki, T., Inubushi, Y., Itoga, T., Joti, Y., Kago, M., Kameshima, T., Kimura, H., Kirihara, Y., Kiyomichi, A., Kobayashi, T., Kondo, C., Kudo, T., Maesaka, H., Maréchal, X. M., Masuda, T., Matsubara, S., Matsumoto, T., Matsushita, T., Matsui, S., Nagasono, M., Nariyama, N., Ohashi, H., Ohata, T., Ohshima, T., Ono, S., Otake, Y., Saji, C., Sakurai, T., Sato, T., Sawada, K., Seike, T., Shirasawa, K., Sugimoto, T., Suzuki, S., Takahashi, S., Takebe, H., Takeshita, K., Tamasaku, K., Tanaka, H., Tanaka, R., Tanaka, T., Togashi, T., Togawa, K., Tokuhisa, A., Tomizawa, H., Tono, K., Wu, S., Yabashi, M., Yamaga, M., Yamashita, A., Yanagida, K., Zhang, C., Shintake, T., Kitamura, H. & Kumagai, N. (2012). Nat. Photon. 6, 540–544.
- Jazayeri, A., Dias, J. M. & Marshall, F. H. (2015). J. Biol. Chem. 290, 19489–19495. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2014). Acta Cryst. D70, 2204–2216. [DOI] [PMC free article] [PubMed]
- Kameshima, T., Ono, S., Kudo, T., Ozaki, K., Kirihara, Y., Kobayashi, K., Inubushi, Y., Yabashi, M., Horigome, T., Holland, A., Holland, K., Burt, D., Murao, H. & Hatsui, T. (2014). Rev. Sci. Instrum. 85, 033110. [DOI] [PubMed]
- Kang, Y., Zhou, X. E., Gao, X., He, Y., Liu, W., Ishchenko, A., Barty, A., White, T. A., Yefanov, O., Han, G. W., Xu, Q., de Waal, P. W., Ke, J., Tan, M. H. E., Zhang, C., Moeller, A., West, G. M., Pascal, B. D., Van Eps, N., Caro, L. N., Vishnivetskiy, S. A., Lee, R. J., Suino-Powell, K. M., Gu, X., Pal, K., Ma, J., Zhi, X., Boutet, S., Williams, G. J., Messerschmidt, M., Gati, C., Zatsepin, N. A., Wang, D., James, D., Basu, S., Roy-Chowdhury, S., Conrad, C. E., Coe, J., Liu, H., Lisova, S., Kupitz, C., Grotjohann, I., Fromme, R., Jiang, Y., Tan, M., Yang, H., Li, J., Wang, M., Zheng, Z., Li, D., Howe, N., Zhao, Y., Standfuss, J., Diederichs, K., Dong, Y., Potter, C. S., Carragher, B., Caffrey, M., Jiang, H., Chapman, H. N., Spence, J. C. H., Fromme, P., Weierstall, U., Ernst, O. P., Katritch, V., Gurevich, V. V., Griffin, P. R., Hubbell, W. L., Stevens, R. C., Cherezov, V., Melcher, K. & Xu, H. E. (2015). Nature (London), 523, 561–567.
- Kirian, R. A., White, T. A., Holton, J. M., Chapman, H. N., Fromme, P., Barty, A., Lomb, L., Aquila, A., Maia, F. R. N. C., Martin, A. V., Fromme, R., Wang, X., Hunter, M. S., Schmidt, K. E. & Spence, J. C. H. (2011). Acta Cryst. A67, 131–140. [DOI] [PMC free article] [PubMed]
- Kuhnert, M., Köster, H., Bartholomäus, R., Park, A. Y., Shahim, A., Heine, A., Steuber, H., Klebe, G. & Diederich, W. E. (2015). Angew. Chem. Int. Ed. 54, 2849–2853. [DOI] [PubMed]
- Lagerström, M. C. & Schiöth, H. B. (2008). Nat. Rev. Drug Discov. 7, 339–357. [DOI] [PubMed]
- Landau, E. M. & Rosenbusch, J. P. (1996). Proc. Natl Acad. Sci. USA, 93, 14532–14535. [DOI] [PMC free article] [PubMed]
- Leonetti, G. & Egan, C. G. (2012). Vasc. Health Risk Manag. 8, 307–322. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Liebschner, D., Afonine, P. V., Moriarty, N. W., Poon, B. K., Sobolev, O. V., Terwilliger, T. C. & Adams, P. D. (2017). Acta Cryst. D73, 148–157. [DOI] [PMC free article] [PubMed]
- Liu, W., Chun, E., Thompson, A. A., Chubukov, P., Xu, F., Katritch, V., Han, G. W., Roth, C. B., Heitman, L. H., IJzerman, A. P., Cherezov, V. & Stevens, R. C. (2012). Science, 337, 232–236. [DOI] [PMC free article] [PubMed]
- Liu, W., Ishchenko, A. & Cherezov, V. (2014). Nat. Protoc. 9, 2123–2134. [DOI] [PMC free article] [PubMed]
- Liu, W., Wacker, D., Gati, C., Han, G. W., James, D., Wang, D., Nelson, G., Weierstall, U., Katritch, V., Barty, A., Zatsepin, N. A., Li, D., Messerschmidt, M., Boutet, S., Williams, G. J., Koglin, J. E., Seibert, M. M., Wang, C., Shah, S. T. A., Basu, S., Fromme, R., Kupitz, C., Rendek, K. N., Grotjohann, I., Fromme, P., Kirian, R. A., Beyerlein, K. R., White, T. A., Chapman, H. N., Caffrey, M., Spence, J. C. H., Stevens, R. C. & Cherezov, V. (2013). Science, 342, 1521–1524. [DOI] [PMC free article] [PubMed]
- Martin-Garcia, J. M., Conrad, C. E., Nelson, G., Stander, N., Zatsepin, N. A., Zook, J., Zhu, L., Geiger, J., Chun, E., Kissick, D., Hilgart, M. C., Ogata, C., Ishchenko, A., Nagaratnam, N., Roy-Chowdhury, S., Coe, J., Subramanian, G., Schaffer, A., James, D., Ketwala, G., Venugopalan, N., Xu, S., Corcoran, S., Ferguson, D., Weierstall, U., Spence, J. C. H., Cherezov, V., Fromme, P., Fischetti, R. F. & Liu, W. (2017). IUCrJ, 4, 439–454. [DOI] [PMC free article] [PubMed]
- Meents, A., Wiedorn, M. O., Srajer, V., Henning, R., Sarrou, I., Bergtholdt, J., Barthelmess, M., Reinke, P. Y. A., Dierksmeyer, D., Tolstikova, A., Schaible, S., Messerschmidt, M., Ogata, C. M., Kissick, D. J., Taft, M. H., Manstein, D. J., Lieske, J., Oberthuer, D., Fischetti, R. F. & Chapman, H. N. (2017). Nat. Commun. 8, 1281. [DOI] [PMC free article] [PubMed]
- Mishin, A., Gusach, A., Luginina, A., Marin, E., Borshchevskiy, V. & Cherezov, V. (2019). Exp. Opin. Drug. Discov. 14, 933–945. [DOI] [PMC free article] [PubMed]
- Misquitta, L. V., Misquitta, Y., Cherezov, V., Slattery, O., Mohan, J. M., Hart, D., Zhalnina, M., Cramer, W. A. & Caffrey, M. (2004). Structure, 12, 2113–2124. [DOI] [PubMed]
- Mueller, C., Marx, A., Epp, S. W., Zhong, Y., Kuo, A., Balo, A. R., Soman, J., Schotte, F., Lemke, H. T., Owen, R. L., Pai, E. F., Pearson, A. R., Olson, J. S., Anfinrud, P. A., Ernst, O. P. & Dwayne Miller, R. J. (2015). Struct. Dyn. 2, 054302. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nakane, T., Joti, Y., Tono, K., Yabashi, M., Nango, E., Iwata, S., Ishitani, R. & Nureki, O. (2016). J. Appl. Cryst. 49, 1035–1041. [DOI] [PMC free article] [PubMed]
- Neutze, R., Wouts, R., van der Spoel, D., Weckert, E. & Hajdu, J. (2000). Nature (London), 406, 752–757. [DOI] [PubMed]
- Pándy-Szekeres, G., Munk, C., Tsonkov, T. M., Mordalski, S., Harpsøe, K., Hauser, A. S., Bojarski, A. J. & Gloriam, D. E. (2018). Nucleic Acids Res. 46, D440–D446. [DOI] [PMC free article] [PubMed]
- Plazinska, A., Pajak, K., Rutkowska, E., Jimenez, L., Kozocas, J., Koolpe, G., Tanga, M., Toll, L., Wainer, I. W. & Jozwiak, K. (2014). Bioorg. Med. Chem. 22, 234–246. [DOI] [PMC free article] [PubMed]
- Rask-Andersen, M., Almén, M. S. & Schiöth, H. B. (2011). Nat. Rev. Drug Discov. 10, 579–590. [DOI] [PubMed]
- Rasmussen, S. G. F., DeVree, B. T., Zou, Y., Kruse, A. C., Chung, K. Y., Kobilka, T. S., Thian, F. S., Chae, P. S., Pardon, E., Calinski, D., Mathiesen, J. M., Shah, S. T. A., Lyons, J. A., Caffrey, M., Gellman, S. H., Steyaert, J., Skiniotis, G., Weis, W. I., Sunahara, R. K. & Kobilka, B. K. (2011). Nature (London), 477, 549–555. [DOI] [PMC free article] [PubMed]
- Ring, A. M., Manglik, A., Kruse, A. C., Enos, M. D., Weis, W. I., Garcia, K. C. & Kobilka, B. K. (2013). Nature (London), 502, 575–579. [DOI] [PMC free article] [PubMed]
- Roedig, P., Ginn, H. M., Pakendorf, T., Sutton, G., Harlos, K., Walter, T. S., Meyer, J., Fischer, P., Duman, R., Vartiainen, I., Reime, B., Warmer, M., Brewster, A. S., Young, I. D., Michels-Clark, T., Sauter, N. K., Kotecha, A., Kelly, J., Rowlands, D. J., Sikorsky, M., Nelson, S., Damiani, D. S., Alonso-Mori, R., Ren, J., Fry, E. E., David, C., Stuart, D. I., Wagner, A. & Meents, A. (2017). Nat. Methods, 14, 805–810. [DOI] [PMC free article] [PubMed]
- Rosenbaum, D. M., Cherezov, V., Hanson, M. A., Rasmussen, S. G. F., Thian, F. S., Kobilka, T. S., Choi, H.-J., Yao, X.-J., Weis, W. I., Stevens, R. C. & Kobilka, B. K. (2007). Science, 318, 1266–1273. [DOI] [PubMed]
- Roth, C. B., Hanson, M. A. & Stevens, R. C. (2008). J. Mol. Biol. 376, 1305–1319. [DOI] [PMC free article] [PubMed]
- Rucktooa, P., Cheng, R. K. Y., Segala, E., Geng, T., Errey, J. C., Brown, G. A., Cooke, R. M., Marshall, F. H. & Doré, A. S. (2018). Sci. Rep. 8, 41. [DOI] [PMC free article] [PubMed]
- Sabio, M., Jones, K. & Topiol, S. (2008). Bioorg. Med. Chem. Lett. 18, 5391–5395. [DOI] [PubMed]
- Serrano-Vega, M. J., Magnani, F., Shibata, Y. & Tate, C. G. (2008). Proc. Natl Acad. Sci. USA, 105, 877–882. [DOI] [PMC free article] [PubMed]
- Smart, O. S., Womack, T. O., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., Vonrhein, C. & Bricogne, G. (2012). Acta Cryst. D68, 368–380. [DOI] [PMC free article] [PubMed]
- Smith, J., Fischetti, R. & Yamamoto, M. (2012). Curr. Opin. Struct. Biol. 22, 602–612. [DOI] [PMC free article] [PubMed]
- Stauch, B. & Cherezov, V. (2018). Annu. Rev. Biophys. 47, 377–397. [DOI] [PMC free article] [PubMed]
- Stauch, B., Johansson, L. C., McCorvy, J. D., Patel, N., Han, G. W., Huang, X.-P., Gati, C., Batyuk, A., Slocum, S. T., Ishchenko, A., Brehm, W., White, T. A., Michaelian, N., Madsen, C., Zhu, L., Grant, T. D., Grandner, J. M., Shiriaeva, A., Olsen, R. H. J., Tribo, A. R., Yous, S., Stevens, R. C., Weierstall, U., Katritch, V., Roth, B. L., Liu, W. & Cherezov, V. (2019). Nature (London), 569, 284–288.
- Stevens, R. C., Cherezov, V., Katritch, V., Abagyan, R., Kuhn, P., Rosen, H. & Wüthrich, K. (2013). Nat. Rev. Drug Discov. 12, 25–34. [DOI] [PMC free article] [PubMed]
- Sun, B., Bachhawat, P., Chu, M. L.-H., Wood, M., Ceska, T., Sands, Z. A., Mercier, J., Lebon, F., Kobilka, T. S. & Kobilka, B. K. (2017). Proc. Natl Acad. Sci. USA, 114, 2066–2071. [DOI] [PMC free article] [PubMed]
- Sykes, D. A., Parry, C., Reilly, J., Wright, P., Fairhurst, R. A. & Charlton, S. J. (2014). Mol. Pharmacol. 85, 608–617. [DOI] [PubMed]
- Tono, K., Nango, E., Sugahara, M., Song, C., Park, J., Tanaka, T., Tanaka, R., Joti, Y., Kameshima, T., Ono, S., Hatsui, T., Mizohata, E., Suzuki, M., Shimamura, T., Tanaka, Y., Iwata, S. & Yabashi, M. (2015). J. Synchrotron Rad. 22, 532–537. [DOI] [PMC free article] [PubMed]
- Uervirojnangkoorn, M., Zeldin, O. B., Lyubimov, A. Y., Hattne, J., Brewster, A. S., Sauter, N. K., Brunger, A. T. & Weis, W. I. (2015). Elife, 4, e05421. [DOI] [PMC free article] [PubMed]
- Unett, D. J., Gatlin, J., Anthony, T. L., Buzard, D. J., Chang, S., Chen, C., Chen, X., Dang, H. T.-M., Frazer, J., Le, M. K., Sadeque, A. J. M., Xing, C. & Gaidarov, I. (2013). J. Pharmacol. Exp. Ther. 347, 645–659. [DOI] [PubMed]
- Wacker, D., Fenalti, G., Brown, M. A., Katritch, V., Abagyan, R., Cherezov, V. & Stevens, R. C. (2010). J. Am. Chem. Soc. 132, 11443–11445. [DOI] [PMC free article] [PubMed]
- Wacker, D., Wang, C., Katritch, V., Han, G. W., Huang, X.-P., Vardy, E., McCorvy, J. D., Jiang, Y., Chu, M., Siu, F. Y., Liu, W., Xu, H. E., Cherezov, V., Roth, B. L. & Stevens, R. C. (2013). Science, 340, 615–619. [DOI] [PMC free article] [PubMed]
- Wang, C., Jiang, Y., Ma, J., Wu, H., Wacker, D., Katritch, V., Han, G. W., Liu, W., Huang, X.-P., Vardy, E., McCorvy, J. D., Gao, X., Zhou, X. E., Melcher, K., Zhang, C., Bai, F., Yang, H., Yang, L., Jiang, H., Roth, B. L., Cherezov, V., Stevens, R. C. & Xu, H. E. (2013). Science, 340, 610–614. [DOI] [PMC free article] [PubMed]
- Warne, T., Edwards, P. C., Leslie, A. G. W. & Tate, C. G. (2012). Structure, 20, 841–849. [DOI] [PMC free article] [PubMed]
- Weierstall, U., James, D., Wang, C., White, T. A., Wang, D., Liu, W., Spence, J. C. H., Doak, R. B., Nelson, G., Fromme, P., Fromme, R., Grotjohann, I., Kupitz, C., Zatsepin, N. A., Liu, H., Basu, S., Wacker, D., Han, G. W., Katritch, V., Boutet, S., Messerschmidt, M., Williams, G. J., Koglin, J. E., Seibert, M. M., Klinker, M., Gati, C., Shoeman, R. L., Barty, A., Chapman, H. N., Kirian, R. A., Beyerlein, K. R., Stevens, R. C., Li, D., Shah, S. T. A., Howe, N., Caffrey, M. & Cherezov, V. (2014). Nat. Commun. 5, 3309. [DOI] [PMC free article] [PubMed]
- Weinert, T., Olieric, N., Cheng, R., Brünle, S., James, D., Ozerov, D., Gashi, D., Vera, L., Marsh, M., Jaeger, K., Dworkowski, F., Panepucci, E., Basu, S., Skopintsev, P., Doré, A. S., Geng, T., Cooke, R. M., Liang, M., Prota, A. E., Panneels, V., Nogly, P., Ermler, U., Schertler, G., Hennig, M., Steinmetz, M. O., Wang, M. & Standfuss, J. (2017). Nat. Commun. 8, 542. [DOI] [PMC free article] [PubMed]
- White, T. A., Mariani, V., Brehm, W., Yefanov, O., Barty, A., Beyerlein, K. R., Chervinskii, F., Galli, L., Gati, C., Nakane, T., Tolstikova, A., Yamashita, K., Yoon, C. H., Diederichs, K. & Chapman, H. N. (2016). J. Appl. Cryst. 49, 680–689. [DOI] [PMC free article] [PubMed]
- Xiang, J., Chun, E., Liu, C., Jing, L., Al-Sahouri, Z., Zhu, L. & Liu, W. (2016). Trends Pharmacol. Sci. 37, 1055–1069. [DOI] [PubMed]
- Xu, F., Liu, W., Hanson, M. A., Stevens, R. C. & Cherezov, V. (2011). Cryst. Growth Des. 11, 1193–1201. [DOI] [PMC free article] [PubMed]
- Xu, F., Wu, H., Katritch, V., Han, G. W., Jacobson, K. A., Gao, Z.-G., Cherezov, V. & Stevens, R. C. (2011). Science, 332, 322–327. [DOI] [PMC free article] [PubMed]
- Yabashi, M. & Tanaka, H. (2017). Nat. Photonics, 11, 12–14.
- Zhang, H., Unal, H., Gati, C., Han, G. W., Liu, W., Zatsepin, N. A., James, D., Wang, D., Nelson, G., Weierstall, U., Sawaya, M. R., Xu, Q., Messerschmidt, M., Williams, G. J., Boutet, S., Yefanov, O. M., White, T. A., Wang, C., Ishchenko, A., Tirupula, K. C., Desnoyer, R., Coe, J., Conrad, C. E., Fromme, P., Stevens, R. C., Katritch, V., Karnik, S. S. & Cherezov, V. (2015). Cell, 161, 833–844. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: β2-adrenergic receptor, complex with alprenolol, 6prz
PDB reference: 6ps2
PDB reference: complex with carazolol, 6ps0
PDB reference: complex with timolol, 6ps1
PDB reference: 6ps6
PDB reference: complex with carvedilol, 6ps3
PDB reference: complex with ICI-118,551, 6ps4
PDB reference: complex with propranolol, 6ps5
PDB reference: adenosine A2A receptor, complex with ZM241385, 6ps7
PDB reference: melatonin receptor MT1, complex with 2-phenylmelatonin, 6ps8
Supplementary Tables and Figures. DOI: 10.1107/S2052252519013137/cw5024sup1.pdf