

Abstract
Autoimmune diseases, such as celiac disease, multiple sclerosis, and type 1 diabetes, are leading causes of morbidity and mortality in the United States. In these disease states, immune regulatory mechanisms fail that result in T and B cell-mediated destruction of self-tissues. The known role of T cells in mediating autoimmune diseases has led to the emergence of numerous therapies aimed at inactivating T cells, however successful ‘tolerance-inducing’ strategies have not yet emerged for approved standard-of-care clinical use. In this review, we describe relevant examples of antigen-specific tolerance approaches that have been applied in clinical trials for human diseases. Furthermore, we describe the evolution of biomaterial approaches from cell-based therapies to induce immune tolerance with a focus on the Tolerogenic Immune-Modifying nanoParticle (TIMP) platform. The TIMP platform can be designed to treat various autoimmune conditions and is currently in clinical trials testing its ability to reverse celiac disease.
Keywords: Nanoparticle, Immune tolerance, Allergy, Autoimmune disease, Drug delivery, Clinical trial
An important goal of current research is to develop new therapies for autoimmune diseases that specifically inhibit and/or tolerize antigen-specific (Ag-specific) CD4+ and CD8+ T cells. The requirement of naïve T cells to receive two signals to become activated was first proposed by Lafferty and Cunningham.1 Subsequently, this model has been revised to incorporate a third signal for T cell activation where modulation of these signals has become the basis for many potential immune modulatory biologic therapies currently under development.2 Ligation of the T cell receptor (TCR) by its cognate peptide-loaded major histocompatibility (MHC) complex expressed on the surface of an Ag presenting cell (APC), serves as signal one for naïve CD4+ T cells. The second set of signals that a CD4+ T cell receives is the co-signaling molecules (i.e., B7-1 (CD80) and B7-2 (CD86)) which interact with the co-receptor CD28 that is constitutively expressed on the surface of CD4+ T cells.3,4 The third signal is provided by cytokines that act on APCs or T cells. Importantly, it should be noted that the modulation of these signals using either small molecules or biologics ultimately results in systemic and non-specific immunosuppression that carries significant risks to patients that are already immunocompromised. Therefore, the development of technologies that act to modulate T and B cell responses in an Ag-specific manner is highly desirable.
The complexity of autoimmune disease pathogenesis necessitates the appropriate choice of model to evaluate novel approaches to induce tolerance. During the various stages of autoimmune disease, the number and specificity of Ag-specific CD4+ T cells expand via a process known as epitope spreading and T cell proliferation.5,6 Research has focused on characterizing the molecular and cellular basis of epitope spreading in various autoimmune-mediated diseases and disease in the hope of uncovering new Ag-specific treatments. This has been complicated in humans due to the difficulty in identifying and verifying the underlying pathological Ag(s) responsible for causing disease. Conversely, animal models have the advantage that the initiating Ag in many cases is known, genetically identical animals are used, and the immune response is analyzed over a specified time course. Clinical studies that have attempted to induce immune tolerance in humans have been done by high zone tolerance, which involves the administration of high concentrations of the autoantigen to patients. While this method is highly effective in rodents, high zone tolerance protocols have not successfully translated to humans. Instead, years of research have provided evidence that the delivery of Ag in the context of a carrier, either an apoptotic cell or by using a synthetic apoptotic surrogate, such as a nanoparticle, may yield improved results and be more clinically translatable. In this review, we describe several relevant clinical examples that have had mixed success for reversing autoimmune disease (celiac disease (CD) and multiple sclerosis (MS)), the evolution of cell-based Ag carriers to the development of Tolerogenic Immune-Modifying nanoParticles (TIMP) for tolerance induction, and the safety of administered nanoparticles. We believe that the development of the TIMP platform represents a significant step forward for the treatment of immune-mediated pathologies and that they have great promise due to their biomimetic properties.
Mechanism for antigen-specific tolerance induction
Autoimmune diseases develop due to a breakdown of central tolerance mechanisms. In rodents, peripheral immune tolerance can be induced in vivo using a variety of technologies.7 To induce tolerance, Ag must be delivered to the appropriate APC subsets and initiate a cascade of tolerogenic signaling pathways.7 Specifically, the antigenic peptide needs to be processed and presented in the context of MHC II by antigen presenting cells (APCs) in the presence of low levels of co-stimulatory molecule expression, and in the absence of other activating stimuli (i.e. inflammation, infection, or other pathologies).8,9 Ag-specific effector CD4+ T cells recognize peptide-MHC complexes on the surface of APCs through cognate T cell receptors (TCR). The context of this interaction between APCs and T cells is critical to drive effector T cell phenotypes towards an unreactive state (anergy or deletion) or induce differentiation of regulatory T cells (Tregs).10 Through the noted mechanisms, aberrant CD4+ T cell responses can be controlled in an Ag-specific manner to induce immune tolerance.
Celiac disease: a rational starting point to test immune tolerance therapies
CD results from the breakdown of peripheral immune tolerance to gliadins, a component of gluten and the major glycoproteins present in wheat, oat, barely, and rye. The ideal treatment of CD would involve the restoration or induction of Ag-specific immune tolerance to gliadin-specific CD4+ T cells. Correspondingly, a therapy that induces T cell tolerance would limit immune-mediated damage in the small bowel. To date there is no available therapy capable of inducing immune tolerance to gliadin, or any other Ag, in humans. Unlike other autoimmune conditions where the underlying Ags that are responsible for disease initiation and progression remain unknown or controversial, in CD, many of the immune dominant epitopes found within the gliadin proteins have been identified.11,12 Furthermore, the accessibility to raw antigen-containing materials and the ability to isolate and manufacture gliadin proteins using good manufacturing practices (GMP) support the development of a tolerance-inducing therapeutic for human use. One other unique aspect of CD is the ability to control exposure to the underlying disease-causing proteins, which is not possible in other autoimmune conditions such as MS and Type 1 Diabetes (T1D). Therefore, an Ag-specific treatment capable of inactivating gliadin-specific CD4+ T cells will not only address a significant unmet medical need, but also transform the future of therapies for multiple autoimmune diseases.
Our team, in collaboration with Northwestern University, University of Sydney, and University of Michigan, has developed a novel approach to induce immune tolerance using nanoparticles that encapsulate gliadin (TIMP-GLIA) for the treatment of CD. This therapy has resulted from the culmination of multiple decades of research into immune tolerance, CD, and polymer chemistry to address this specific need.
Clinical experience in administering autoantigen in humans
Clinical trials have been carried out in autoimmune diseases including CD, MS, rheumatoid arthritis (RA), and T1D employing various Ag-specific strategies that were based on promising results obtained from early rodent studies (Table 1). Most critically, in the context of the TIMP platform, none of the strategies tested to date have specifically delivered diseaserelevant Ag in a coordinated and targeted fashion to tolerogenic APC populations in the liver and spleen. Additionally, lessons learned from previous Ag-specific approaches support the ability to safely administer antigenic proteins and peptides to humans that are known to have activated CD4+ T cell-specific responses.
Table 1.
Human antigen-specific tolerance-based clinical trials for autoimmune disease.
| Citation | Disease | Antigen Specific Treatment | Results and Safety Observations |
|---|---|---|---|
| 16 | CD | Nexvax2 mixture of DQ2.5-glia-α1, -α2 DQ2.5-glia-ω1, and ω2 DQ2.5-hor3 immunodominant epitopes Ascending dose study testing 3 intradermal injections of 60 μg, 90 μg, or 150 μg given over 15 days (total exposure max exposure 450 μg) |
No efficacy was determined. Biopsy showed no worsening of intestinal pathology in patients receiving Nexvax2. Most common side effects included vomiting, nausea, and headache and were the only treatment-emergent adverse events that occurred in at least 5% of participants. Study showed that administration of immunodominant epitopes to celiac patients shown to be reactive to these epitopes was associated with self-limited acute gastrointestinal symptoms. |
| 16 | CD | Nexvax 2 mixture of DQ2.5-glia-α1, -α2 DQ2.5-glia-ω1, and ω2 DQ2.5-hor3 immunodominant epitopes 16 intradermal injections given over 53 days of either 150 μg, or 300 μg (total 4800 μg) |
No efficacy was determined. Biopsy showed no worsening of intestinal pathology in patients receiving Nexvax2. Most common side effects included vomiting, nausea, and headache and were the only treatment-emergent adverse events that occurred in at least 5% of participants in either study. Study showed that administration of immunodominant epitopes to celiac patients shown to be reactive to these epitopes was associated with self-limited acute gastrointestinal symptoms. |
| 44 | MS | Myelin basic peptide altered peptide ligand 50 mg weekly subcutaneous injections up to 9 months (total exposure 1800 mg). |
2 out of 8 patients showed increased brain lesions; 1 showed hypersensitivity; 3 others had non-specific side effects; 1 patient dose lowered to 5 mg and still developed increased MS lesions; Study suspended. |
| 44 | MS | Myelin basic peptide altered peptide ligand Randomized, double-blind clinical trial in 142 patients; 5 mg, 20 mg, or 50 mg weekly subcutaneous injections for 4 months and then decreased to 5 mg |
No significant difference in relapse rate of treated and placebo groups but volume of new brain lesions was reduced in some of the patients that received 5 mg throughout; 9% developed hypersensitivity to the therapy and the trial was suspended. |
| 45 | MS | Glatiramer acetate injection (Trade name Copaxone) Randomized, double-blind trl; dose-comparison of 20 mg and 40 mg subcutaneous injection in relapsing-remitting MS |
Higher dose was well tolerated and a decrease in relapse rate observed. Drug is approved for treating MS. |
| 17 | MS | Myelin Basic Peptide Intravenous Infusion Randomized, double-blind trail; Phase 2 dose-infusing 500 mg of MBP peptide |
Drug was well tolerated but had questionable efficacy. Showed intravenous infusion of 500 mg of immunodominant T cell epitope was safe in MS patients |
| 20 | MS | Peptide coupled leukocytes (PBL) Intravenous administration of ECDI-fixed, autologous PBLs coupled with 7 immunodominant myelin peptides; a total estimated dose of 0.5 mg to 3.5 mg of total peptide This is further discussed in Section 3.1.2 |
Phase 1 clinical trial data show a good safety profile; patients were dosed with 1 × 103 up to 3 × 109 peptide-coupled PBLs. Patients receiving over 1 × 109 peptide-coupled PBLs showed a decrease in the level of peptide-specific CD4+ T cell responses in vitro. Showed that the delivery of immunodominant epitopes attached to the surface of ECDI-fxed cells could be safely administered via intravenous infusion. |
CD = Celiac Disease; MS = Multiple Sclerosis.
In CD, oral exposure to gluten has been used to examine disease pathogenesis and for diagnostic purposes for over two decades (experience from oral gluten challenge is well documented and reviewed by13). These studies have been useful in delineating the temporal and kinetic profile of symptoms, intestinal pathology and peripheral CD4+ T cell responses.12,14,15 After several gluten challenge studies, Anderson and colleagues defined the primary immunodominant CD4+ T cell epitopes associated with CD. Using this information, Nexvax2 was developed. Nexvax2 is an adjuvant-free mixture of DQ2.5-glia-α1,-α2, DQ2.5-glia-ω1, and ω2 DQ2.5-hor3 immunodominant epitopes for gluten-specific CD4+ T cells. Intradermal administration of Nexvax2 to CD patients was tested to render gluten-specific CD4+ T cells unresponsive to further antigenic stimulation. To test this hypothesis, two Phase 1 clinical trials were conducted.16 In these studies, patients that responded to the immunodominant CD4+ T cell epitopes were enrolled into one of two Phase 1 studies. In one ascending dose study, patients received 3 doses of 60 μg, 90 μg, or 150 μg of Nexvax2 over 15 days (total patient exposure was 180-450 μg). In the second study, patients received either 150 μg or 300 μg of Nexvax2 twice weekly for 53 days (total patient exposure was 2400-7200 μg).16
In these studies, the maximum tolerated dose (MTD) was determined as 150 μg due to the development of transient CD like symptoms including, acute gastrointestinal adverse events with onset 2-5 h after initial doses of the vaccine.16 In a three dose escalation study, 55% of placebo recipients had at least one drug associated adverse event, as did all the placebo recipients in the biopsy study. In the treatment arms 56%, 78%, and 63% of patients receiving 60 μg, 90 μg, and 150 μg respectively, developed Nexvax2-associated adverse events. In a follow up study, examining ascending doses in a 16-dose regimen, 71% of placebo recipients, 75% of the 150 μg Nexvax2 cohort, and 100% of patients that received 300 μg Nexvax2 had at least one treatment-emergent adverse event. 16 The most common effects, affecting greater than 5% of patients were vomiting, nausea, and headache.16 Interestingly, Nexvax2 did not alter the median villous height to crypt depth ratio in distal duodenal biopsies when compared to the placebo group.16 In conclusion, the intradermal injection of pure CD4+ T cell epitopes in CD patients was associated with acute gastrointestinal symptoms, there were no noted sequelae into more severe life-threatening conditions, and treatment was not associated with pathologic change on duodenal biopsy. In other words, the treatment was argued to be relatively safe, and larger Phase 2 studies are currently being planned by the developing company, ImmusanT.16
The administration of immunodominant peptides as well as Altered Peptide Ligands (APLs), has also been shown to be safe in MS patients (Table 1). The induction of tolerance to myelin basic protein (MBP) peptide was examined in Phase 1 and 2 clinical trials in patients with chronic-progressive MS using an immunodominant peptide for MBP-specific CD4+ T cells and B cells. In this study, 500 mg of MBP85-96 was infused every 6 months and the induction of tolerance was monitored by quantification of MBP-specific autoantibodies in cerebrospinal fluid (CSF). While this study showed that intravenous treatment with MBP85-96 peptide decreased the level of autoantibodies present in the CSF for 3-4 months post-treatment and a correlation between the decreased levels of MBP85-96-specific autoantibodies and a decrease in disease severity, the direct indication of tolerance was not shown. Subsequently, the study and the drug development program were terminated. 17
To date, the clinical efficacy of Ag/peptide-specific immunotherapies for the treatment of pre-existing autoimmune disease has not recapitulated the promising results often observed in rodent studies. Multiple factors need to be considered when moving from animal models to humans including, but not limited to: the route of Ag administration, the dosage and frequency Ag administration, development of robust immunological assays to measure outcomes and monitor follow-up, and the time of administration (prevention versus intervention). It is proposed that the primary underlying causes for the failures of these Ag-specific treatments have been the inability to safely deliver disease-associated proteins to the appropriate subsets of cells in the body (i.e. tolerogenic APCs in the spleen and liver) in a fashion that would restore peripheral immune tolerance.
Evolution of tolerance therapies from cell-based to synthetic antigen carriers
The development of Ag-specific immune tolerance strategies has been studied for nearly four decades and the experimental findings have been published extensively. A critical discovery was the recognition that intravenous delivery of Ag in the context of an apoptotic-non-inflammatory milieu was a highly efficient method for inducing immune tolerance to the Ag. The finding was first observed by the Miller lab using Ag-coupled splenocytes (Ag-SP).18 Mechanistic studies subsequently showed that the process used to covalently cross-link Ag to splenocytes results in cellular apoptosis.8,19 Intravenous infusion of Ag-SP resulted in their distribution to the marginal zone of the spleen where macrophages phagocytose and process these cells and the conjugated Ag.19 Subsequently, the macrophages represent the Ag coupled to the apoptotic cells on MHC I and II molecules, as well as upregulate negative co-stimulatory molecules (i.e. programmed death-ligand 1 (PD-L1)).19 Together, this culminates in the induction of Ag-specific immune tolerance. From 2008 to 2013, this approach was tested in humans suffering MS, where autologous peripheral blood leukocytes (PBLs) were chemically linked to a cocktail of seven myelin T cell epitopes and the autologous PBLs were then reinfused to the patients. This process was shown to be safe and effective in reducing T cell responses of patients to four of the seven myelin epitopes three months post treatment.20 However, the manufacturing process was found to be laborious and extremely expensive. During this time, it was discovered that 500 nm, highly negatively-charged nanoparticles could be substituted for apoptotic cells to deliver Ag in a tolerogenic fashion.21 Over the last decade, subsequent modification and optimization of the platform have been performed, resulting in the final TIMP platform (Figure 1).
Figure 1.
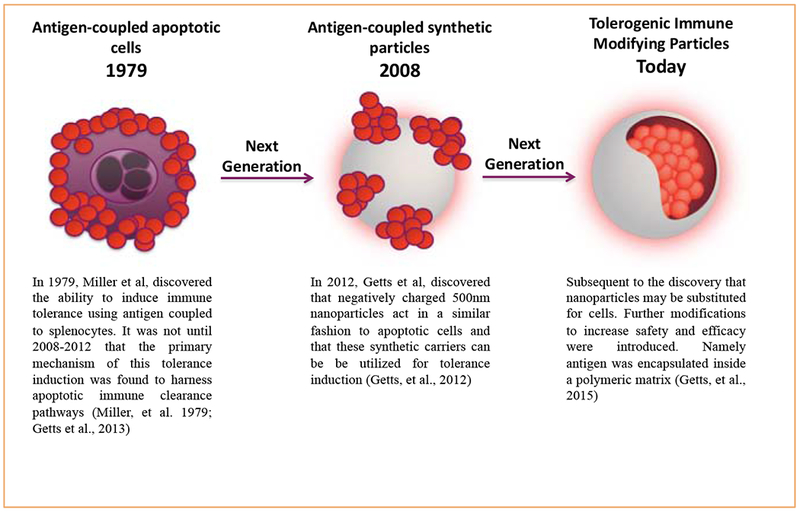
The evolution of the TIMP platform from antigen-coupled apoptotic cells.
Apoptotic Cells and the induction of immune tolerance
Murine studies showing antigen-coupled cell immune tolerance induction
As noted above, the first iteration of the TIMP technology utilized cells to deliver Ag in a tolerogenic manner. In these studies, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (ECDI) was used to conjugate Ag to the syngeneic donor leukocytes. As noted above, the use of the chemical crosslinker ECDI led to the induction of apoptosis in the donor cell population (splenocytes). The non-specific cross-linking of Ag to the cell surface while inducing apoptosis resulted in recognition by the recipient immune system via a non-inflammatory (non-immunogenic) pathway. When infused, Ag-SP are cleared by marginal zone macrophages, in a similar fashion to dead/dying red and white blood cells.19,22 Using this strategy, our team has demonstrated the efficacy of Ag-SP in numerous models of immune disease (Table 2), which included Experimental Autoimmune Encephalomyelitis (EAE), a murine model of MS,9,19 T1D,6 and allogeneic islet cell transplantation.23
Table 2.
Experience of using antigen-coupled cell based tolerance in rodent models of disease.
| Citation | Disease Model | Antigen-Specific Therapy | Treatment Regimen | Estimated Antigen per Dose | Results |
|---|---|---|---|---|---|
| 18,22,46,47 | EAE | Myelin peptide/proteins | 50 × 106 antigen-coupled splenocytes (coupled with 0.5 mg/mL peptide in the presence of ECDI). | 24-55 μg/dose | Treatment prevented or reversed disease depending on when treatment was initiated. |
| 6 | TID | Full length bovine insulin | NOD mice were injected i.v. with 5 × 107 insulin coupled splenocytes at numerous time points. (coupled with 0.5 mg/mL peptide in the presence of ECDI). | Not defined | Demonstration of disease prevention or remission, inhibition of pathogenic T cell proliferation, decreased cytokine production, and induction of anergy. |
| 48 | TID | 1040-p31 peptide | NOD.BDC2.5.Thy1.1 TCR Tg+ cells transfer into NOD/scid mice; mice received 50 × 106 antigen-coupled splenocytes (coupled with 0.5 mg/mL peptide in the presence of ECDI). | Not defined | Demonstration of disease prevention or remission, inhibition of pathogenic T cell proliferation, decreased cytokine production, and induction of anergy. |
| 23 | Islet Transplant | ECDI fixed allogeneic splenocytes | 1 × 108 ECDI-treated allogeneic splenocytes were transferred into streptozotocin-treated mice that received allogeneic beta-islet cells under the kidney capsule. | Not defined | Demonstration of indefinite survival of allogeneic islet grafts in the absence of immunosuppression |
| 49 | EAT | Thyroglobulin-coupled spleen cells | Suppression of experimental autoimmune thyroiditis in guinea pigs. | 133 μg/dose | Prevention of Thyroiditis in all pre-treated animals |
| 50 | Uveitis | S-antigen (S-Ag) and interphotoreceptor retinoid binding protein (IRBP) | Injection of S-Ag or IRBP coupled to spleen cells 5 days prior to immunization with native S-Ag or IRBP, respectively. | Protein homogenate, unknown concentration of antigens | Prevention of Uveitis in 6/8 rats |
| 51 | Neuritis | Myelin P253-78 | Lewis rats receive 100 μg of SP-26/CFA; treated with 20 μg/1 × 107 splenocytes and each rat received 50 × 106 coupled cells. | Not defined | Prevention of neuritis |
Translation of apoptotic cells combined with antigen into human MS patients
To translate the animal studies using apoptotic Ag coupled cells, a Phase 1 clinical trial () in MS patients was conducted.20 The aim of this first-in-man trial was to assess the feasibility, safety, and tolerability of a tolerization regimen in MS patients that uses a single infusion of autologous PBLs chemically coupled with seven myelin peptides derived from myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MBP), and proteolipid protein (PLP) (i.e. MOG1-20, MOG35-55, MBP13-32, MBP83-99, MBP111-129, MBP146-170, and PLP139-154).20 An open-label, single-center, dose-escalation study was performed in seven relapsing-remitting and two secondary progressive MS patients that were off-treatment for standard therapies. All patients had to show T cell reactivity against at least one of the myelin peptides used in the trial. Neurological, magnetic resonance imaging, laboratory, and immunological examinations were performed to assess the safety, tolerability, and in vivo mechanisms of action of this regimen.20 The data generated from the aforementioned Phase 1 study show that the administration of Ag-coupled cells was feasible, had a favorable safety profile, and was well tolerated in MS patients. Additionally, the highest dose of cells used in this study (3 × 109) delivered an estimated 2.6 mg of total peptides and led to post-treatment diminution of T cell responses to four of seven myelin epitopes without affecting the T cell response to tetanus toxoid.20
Evolution to synthetic biodegradable nanoparticles for tolerance induction
The translational challenges associated with the cell-based approach and findings that polymeric biodegradable nanoparticles could be substituted for apoptotic cells motivated the development of the TIMP platform. TIMPs are highly-negatively charged, Ag-associated PLGA nanoparticles that are administered by intravenous injection, which target MARCO+ macrophages in the liver and spleen.24 TIMPs have been applied to models of autoimmunity, allergy, and allogeneic cell transplantation (Table 3). Relevant studies highlighting the efficacy of the TIMP platform are described below. Furthermore, readers are directed towards several review articles, which describe the development of TIMPs from Ag-coupled splenocytes.7,10,24–26
Table 3.
Literature supporting tolerogenic immune-modifying nanoparticles in rodent models.
| Citation | Disease Model (Tolerogenic Nanopeptide Used) | Size (mean) | (mv) | Ag μg/mg Polymer | Dose (mg/polymer) | Antigen (μg)/dose | Total Ag Dose (μg) | Result |
|---|---|---|---|---|---|---|---|---|
| 21 | Experimental Autoimmune Encephalomyelitis (Myelin antigen PLP coupled to surface of PLGA nanoparticles) |
500 | ND | 16 | 1 infusion of 62 mg/kg | 20 | 20 | The induction of specific tolerance to TIMP-Ag associated peptide, as determined by decreases in disease severity and decreases in peptide-specific CD4+ T cell responses. |
| 31 | Experimental Autoimmune Encephalomyelitis (Myelin antigen PLP coupled to surface of PLGA nanoparticles) |
500 | −67 | 8.4 | 1 infusion of 62 mg/kg | 10.5 | 10.5 | |
| 36 | Experimental Autoimmune Encephalomyelitis TIMP-PLP139-151 |
621 | −43 | 2.4 | 1 infusion of 125 mg/kg | 6 | 6 | |
| 34 | Experimental Autoimmune Encephalomyelitis TIMP-PLP139-151 |
398 | −43 | 2.6 | 2 infusions of 125 mg/kg | 6.5 | 13 | |
| 33 | Experimental airway hypersensitivity TIMP-OVA |
543 | −41 | 14.2 | 2 infusions of 125 mg/kg | 35.5 | 71 | Abrogation of allergic airway inflammation |
| 38 | Bone Marrow Transplantation | 500 | −45 | 12.1 | 2 infusions 62 mg-62 mg/kg | 0.15-15.13 | 0.3-33.26 | 40-60% engraftment/Transplant acceptance |
| Dby-IMP1 | 500 | −45 | 9.1 | |||||
| Uty-IMP1 | ||||||||
| Bone Marrow Transplantation | 500 | −45 | 12 | 1 infusion of 62 mg/kg | 15 | 15 | 0% (0.15 dose) −60% engraftment/Transplant acceptance | |
| Dby-IMP1 | 1.2 | 1 infusion of 6.2 mg/kg | 1.5 | 1.5 | ||||
| 0.12 | 1 infusion of 0.062 mg/kg | 0.15 | 0.15 | |||||
| Bone Marrow Transplantation | 403 | −53 | 2.53 | 2 infusion of 125 mg/kg | 6.3 | 12.6 | 20-week graft acceptance/Transplant acceptance | |
| TIMP-Dby | 497 | −46 | 3.21 | 2 infusion of 125 mg/kg | 8.0 | 16 | ||
| TIMP-Uty | ||||||||
| 37 | Islet Transplantation TIMP-Lysate |
460 | −73 | 100-150 | 2 infusions of 150 mg/kg | 300-450 | 600-900 | 20% graft acceptance and 60% graft acceptance with short course of rapamycin/ Transplant acceptance |
Originally, intravenous injection of 500 nm ‘non-biodegradable’ carboxylated polystyrene (PS) particles coupled with peptides (Ag-PS) was shown to prevent the onset of disease in the mouse model of multiple sclerosis (EAE).21 Interestingly, these studies using polystyrene (PS) nanoparticles identified that particles with diameters ranging from 500 to 1000 nm were most effective, and tolerance was dependent on the presence of scavenger receptors, including the macrophage receptor with collagenous structures (MARCO).27,28 MARCO has been shown to be responsible for uptake of PS beads that have anionic surfaces.29
Given these promising results using non-biodegradable Ag-PS particles, nanoparticles developed from poly(lactide-co-glycolide) (PLGA) were tested. The high negative charge was provided to the PLGA nanoparticles by using poly(ethylene-alt-maleic anhydride) (PEMA) as the stabilizing surfactant during the emulsification procedure for TIMP formation.30 Myelin Ags were coupled to the carboxylated surface of PLGA nanoparticles and were shown to induce Ag-specific tolerance in EAE.21,31 Administration of Ag-coupled PLGA nanoparticles resulted in significantly reduced CNS infiltration of encephalitogenic Th1 (IFN-γ) and Th17 (IL-17a, GM-CSF) cells as well as inflammatory monocytes/macrophages. Tolerance was most effectively induced by intravenous infusion, with intraperitoneal and subcutaneous delivery having significantly reduced efficacy.31 The intravenous route has greater efficacy due to the direct delivery of Ag to the tolerogenic APC populations in the spleen and liver.19 It was later described that nanoparticle administration to APCs promoted an anti-inflammatory response, which resulted in downregulation of co-stimulatory molecule expression (CD80, CD86, and CD40) and increased levels of expression of the co-inhibitory molecule PD-L1.32 For a more detailed description of the proposed mechanism of action for TIMPs, please see Ref [24].
The previous examples demonstrated that tolerance could be induced by particles with surface-coupled Ag. However, the covalent modification of the particle surface with Ag has potential to induce adverse reactions upon injection such as anaphylaxis due to immune recognition of the Ag on the nanoparticle surface as well as issues with impacting physico-chemical properties of particles such as their size, charge and solution stability.7,31,33,34 Therefore, encapsulation of Ag within particles represents a more advantageous method to deliver Ag in vivo (Table 3). Additionally, we have recently demonstrated that other bioactive molecules such as transforming growth factor beta-1 could be conjugated to the surface of PLGA particles, while maintaining its bioactivity.35 McCarthy et al recently described the encapsulation of peptide Ag (OVA323-339, PLP139-151, and PLP178-191) into PLGA particles (TIMP) to treat EAE.36 In these studies, a double emulsion process was used to generate TIMPs with encapsulated myelin Ag. Treatment significantly abrogated EAE induction in vivo using prophylactic and therapeutic disease models, while inhibiting Th1/17 antigen recall responses (proliferation, IFN-γ, and IL-17a) in vitro.
TIMPs have also been applied for use in allogeneic cell transplantation. The induction of donor-specific tolerance to transplanted cells and organs remains of utmost importance to mitigate allograft rejection. Donor-specific tolerance was induced using TIMPs coupled with donor cell lysates that resulted in long-term acceptance of full MHC-mismatched allografts using an islet transplant model (BALB/c to C57BL/6). Delivery of PLGA particles with surface-coupled donor Ags to transplanted C57BL/6 mice led to tolerance in 20% of recipients. In combination with a short course of low-dose rapamycin at the time of transplant, tolerance was greatly improved to 60%.37 In another study, PLGA particles were used to induce tolerance in a minor histocompatibility Ag sex-mismatched C57BL/6 model of bone marrow transplantation. Peptide Ags Dby and Uty are respective CD4+ and CD8+ T cell Ags that mediate male bone marrow transplant rejection in females. Interestingly, delivery of Dby peptide either by conjugation to the surface or encapsulation promoted transplant tolerance. However, delivery of Uty peptide using particles did not induce tolerance. In this model, depletion of Tregs using anti-CD25 antibody did not alter tolerance induction, suggesting other potential tolerance induction mechanisms.38 TIMPs have also been applied in the setting of allergy. TIMPs were loaded with ovalbumin (OVA) protein to tolerize against OVA in a Th2-mediated allergic airway inflammation model both pre- and post-sensitization.33 In a prophylactic model, TIMPs reduced Th2 cytokine production (IL-4, IL-5, IL-13) and inhibited the generation of anti-OVA IgE antibodies. In a therapeutic disease model (i.e. pre-sensitization to OVA), TIMPs reduced Ag-specific T cell proliferation and reduced Th2 cytokine production.
Considering the results obtained from these studies, distinct physicochemical properties of PLGA TIMPs that are important for the induction of Ag-specific tolerance in a number of unrelated models of autoimmunity, allergy, and allogeneic cell transplantation were identified: (1) particle sizes (400 to 800 nm); (2) zeta potentials less than −30 mV; and (3) antigen loading (approximately 10 μg/mg polymer). Using these parameters, PLGA particles encapsulating gliadin (TIMP-GLIA) were then developed for application as a CD therapy and a Phase 1 clinical trial () began in 2018.
Rationale for using PLGA nanoparticles for human studies
The notable success of the TIMP platform for inducing Ag-specific immune tolerance to a wide variety of peptide and protein Ags provided rationale to pursue the application of these particles in humans. PLGA is a biodegradable and biocompatible aliphatic polymer that is approved by the Food and Drug Administration for internal use in humans.39,40 PLGA is a random copolymer comprised of lactic acid and glycolic acid subunits that is synthesized by ring opening polymerization of lactide and glycolide. 39,40 By varying the ratio of lactide and glycolide at the start of the polymerization, the degradation of the polymer can be controlled. The presence of the methyl group in lactic acid makes PLGA more hydrophobic than glycolic acid and this means that PLGA polymers that contain higher proportions of lactic acid to glycolic acid will degrade slower than polymers with higher glycolic acid contents.39,40 The one exception is that a 50:50 lactide/glycolide ratio will degrade the fastest. The mechanism by which degradation proceeds is either through enzymatic or non-enzymatic pathways. The by-products (lactic and glycolic acids) that result from the degradation of PLGA are non-toxic and eliminated through metabolic pathways. Both molecules are endogenous are readily metabolized by the body through the Krebs cycle into water and carbon dioxide. Therefore, minimal toxicity is usually observed for PLGA-based materials and it represents an ideal starting material to produce drug carriers for intravenous administration.
Toxicological studies and one human clinical trial have been carried out using nanoparticles that share some similarities to TIMPs with regard to polymer composition and size. The results obtained from these studies are discussed below and summarized in Table 4. In one study, poly(ethylene glycol)-poly(lactide-co-glycolide) (PEG-PLGA) microcapsules (1.5 μm in diameter) that encapsulated perfluorodecalin (PFD) were intravenously administered to Wistar rats at a dose of 1247 mg/kg (i.e., 202 mg/kg, human equivalent dose; HED).41 The purpose of this study was to evaluate PFD-filled PEG-PLGA microcapsules as artificial oxygen/drug carriers. Microcapsules were administered by syringe pump (30 min, 20 mL/kg body weight × hour). Notably, all rats survived the microcapsule administration, however at this extremely high dose, marked toxicity was observed (increased liver enzyme activities, increased pro-inflammatory cytokines and complement factors, and mild metabolic acidosis). Immediately after the start of dosing of the PFD-filled PEG-PLGA microcapsules, hypotension was observed but it was transient and returned to baseline after 70 min. Importantly, the infusion of similarly prepared microcapsules without PFD did not result in hypotension indicating that the change in mean arterial pressure was caused by the PFD and less likely due to the microcapsule itself. Infusion of microcapsules increased ALAT (3.6-fold), ASAT (6-fold), CK (5-fold), and LDH (24-fold). Histological analysis of the spleen, liver, and lung indicated some abnormal findings in these organs. The results obtained in this study are important as they have identified that microcapsules could be administered safely to rats when the dose is controlled for appropriately.
Table 4.
Literature supporting safety of PLGA nanoparticles.
| Citation | Nanoparticle Formulation | Dosing Regimen | Follow-Up | Observations/Results |
|---|---|---|---|---|
| 41 | Perfluorodecalin (PFD)-filled PLGA microcapsules (92% PLGA, 8% PEG-PLGA) | Single dose i.v. (Rat) (1247 mg/kg) | 0 to 4.5 h | All animals survived dosing. Marked toxicity (increased enzyme activity, increased pro-inflammatory cytokines and complement factors). Transient hypotension in PFD-filled microcapsule group. |
| 42 | PLA/Cholate | Single dose i.v. (Rat) 1. 440 mg/kg 2. 220 mg/kg 3. 75 mg/kg |
0 to 9 days | 1. Reduced motor activity D1, dyspnea D1, 100% mortality D1. 2. Reduced motor activity D1, dyspnea D1, 40% mortality D4 D5. 3. No clinical signs detected. 0% mortality. |
| PEG-PLA | Single dose i.v. (Rat) 1. 440 mg/kg 2. 220 mg/kg 3. 75 mg/kg |
0 to 9 days | 1. No clinical signs detected. 0% mortality. 2. No clinical signs detected. 0% mortality. 3. No clinical signs detected. 0% mortality. |
|
| 43 | Docetaxel-encapsulated PEG-PLA | Single dose i.v. (Rat) 1. 2000 mg/kg 2. 1500 mg/kg 3. 1000 mg/kg |
0 to 6 days | 1. No changes in blood chemistry or body weight. 0% mortality. 2. No changes in blood chemistry or body weight. 0% mortality. 3. No changes in blood chemistry or body weight. 0% mortality. |
| Docetaxel-encapsulated PEG-PLA | Single dose for 3 weeks i.v. (Cynomolgus monkey) 5 to 50 mg/m2 | 0 to 15 days | Increased circulation half-life. No change in pharmacokinetic profile following repeat dosing. 0% mortality. | |
| Docetaxel-encapsulated PEG-PLA | Single dose every 3 weeks i.v. (Human) 3.5 to 75 mg/m2 | - | Increased circulation half-life. No change in pharmacokinetic profile following repeat dosing. 0% mortality. |
In another study, the safety profiles of Methoxy-PEG-PLA50 (Me.PEG-PLA50) and PLA50 nanoparticles (140 nm and 100 nm, respectively) after single dose intravenous administration to rat were evaluated.42 The two types of nanoparticles were prepared from PLA50 and Me.PEG-PLA50 by emulsion/solvent evaporation using cholate as tensioactive agent. Me.PEG-PLA50 nanoparticles displayed a hydrophobic core of PLA and a hydrophilic surface of PEG, whereas PLA50 nanoparticles were hydrophobic and prepared from a single component. With PLA50/Cholate nanoparticles, no signs of toxicity were noted at 75 mg/kg. At higher doses, 220 and 440 mg/kg, marked toxicity (mortality and marked clinical signs) was observed with dose-related hematological changes (decrease of thrombocytes count, increase of Prothrombin Time and Activated Partial Thromboplastin Time) and biochemical changes (increase of aminotransferases activities). For the PLA50/Chol. suspension, doses of 220 and 440 mg/kg resulted in mortality in 2/5 rats and 5/5 rats, respectively. Furthermore, marked clinical signs (dypsnea, reduced motor activity) prior to death were observed Day 1 (3 to 4 h after treatment at 440 mg/kg) to Day 5 (220 mg/kg). A mild decrease in white blood cells was also observed for the three animals examined on Day 1 treated with PLA50/Chol. nanoparticles at 440 mg/kg. In the surviving animals, all changes were transient and disappeared by Day 9. No toxicity (no deaths and no clinical signs) was observed for both PLA50/Chol. and Me.PEG-PLA50/Chol. suspensions when administered at 75 mg/kg. The results obtained in this study demonstrate that delivery of PLA50 nanoparticle was safe and resulted in no measurable toxicity at doses up to 75 mg/kg in rats. Although distinct physicochemical differences exist between the nanoparticles utilized in this study, it can be concluded that when the appropriate dose levels are chosen nanoparticles can be delivered through the intravenous route of administration in a safe manner.
The tolerability of PEG-polylactide (PEG-PLA) nanoparticles that encapsulated docetaxel (approximately 100 nm) was evaluated in rats.43 The doses evaluated were 1000, 1500, and 2000 mg/kg by a single intravenous injection. Rats were monitored for clinical observations, changes in body weight, and blood chemistry for 6 days (study termination). Importantly, no abnormal clinical observations or appreciable body weight loss was observed at all doses and no changes in blood chemistry parameters were observed. These results indicated that the PEG-PLA nanoparticles were well tolerated and no apparent hypersensitivity or anaphylactic responses were observed, which have been observed with high dose administration of nanoparticles.
In the same study,43 the docetaxel-encapsulated PEG-PLA nanoparticles were administered to cynomolgus monkeys by 30 min infusion at doses ranging from 5 to 50 mg/m2 and humans with advanced solid tumors at doses ranging between 3.5 and 75 mg/m2. The objective of these studies was to evaluate the pharmacokinetic profile of the nanoparticles. It was determined that the nanoparticle formulation increased the plasma concentrations of docetaxel by at least an order of magnitude over 24 h.
The few studies available demonstrate that intravenous administration of polymer-based particles, including PLGA, can be performed safely and with minimal toxicological effects when the appropriate dosing regimen is chosen.
Conclusion
Immune tolerance remains the holy grail for treating autoimmunity. Currently, tolerance in humans has been elusive. This is obviously due to the complexity and multifactorial nature of most autoimmune diseases. The relatively recent description of CD-associated T cell epitopes and the ability to control Ag exposure in patients may hold the key for testing novel immune tolerance inducing therapies.
It is clear from human experience that the administration of T cell epitopes without careful targeting or modification has had significant setbacks. One potential avenue to address this failure may include TIMPs. TIMPs have emerged following four decades of research and development in the field of immune tolerance that have progressed from cell-based therapies to the polymeric particle-based platform that is being used today in the clinic. The careful design of future in human studies will be necessary if tolerance therapies are to reach wide-spread implementation. For example, one of the most promising avenues for development is in the area of allergy, where the Ags of interest are well known. However, to be successful, the design of new TIMP platforms will need to be carefully designed to mitigate any potential risks of adverse reactions upon injection like those that have been seen when specific immunotherapies (SITs) have been used. In the context of CD, we believe that the unique combination of gliadin proteins with the TIMP platform will enable the precise delivery of Ags to tolerogenic APCs in the spleen and liver, subsequently resulting in regulation of gliadin-specific CD4+ T cell responses. As additional data are generated for the TIMP platform through the current clinical trial, we will move one step closer towards the development of a curative therapy for autoimmune diseases.
Acknowledgments
Funding Disclosure: Supported in part by National Institutes of Health Grants EB-013198 (L.D.S. and S.D.M.), NS-026543 (S.D.M.).
Footnotes
Conflict of interest disclosure: All authors are inventors of immune-modifying nanoparticles and Tolerogenic Immune-modifying Nanoparticles. All receive research support and hold stock and/or stock-options in Cour Pharmaceuticals Development Company.
References
- 1.Lafferty KJ, Cunningham AJ. A new analysis of allogeneic interactions. Aust J Exp Biol Med Sci 1975;53:27–42. [DOI] [PubMed] [Google Scholar]
- 2.Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol 2010;22:333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damle NK, Klussman K, Linsley PS, Aruffo A. Differential costimulatory effects of adhesion molecules B7, ICAM-1, LFA-3, and VCAM-1 on resting and antigen-primed CD4+ T lymphocytes. J Immunol 1992;148:1985–92. [PubMed] [Google Scholar]
- 4.Gross JA, Callas E, Allison JP. Identification and distribution of the costimulatory receptor CD28 in the mouse. J Immunol 1992;149:380–8. [PubMed] [Google Scholar]
- 5.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med 2005;11:335–9. [DOI] [PubMed] [Google Scholar]
- 6.Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9–23 epitope and involves functional epitope spreading. J Autoimmun 2012;39:347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearson RM, Casey LM, Hughes KR, Miller SD, Shea LD. In vivo reprogramming of immune cells: technologies for induction of antigen-specific tolerance. Adv Drug Deliv Rev 2017;114:240–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turley DM, Miller SD. Peripheral tolerance induction using ethylene-carbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol 2007;178:2212–20. [DOI] [PubMed] [Google Scholar]
- 9.Podojil JR, Miller SD. Molecular mechanisms of T-cell receptor and costimulatory molecule ligation/blockade in autoimmune disease therapy. Immunol Rev 2009;229:337–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo X, Miller SD, Shea LD. Immune tolerance for autoimmune disease and cell transplantation. Annu Rev Biomed Eng 2016;18:181–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang DW, Li D, Wang J, Zhao Y, Wang Z, Yue G, et al. Genome-wide analysis of complex wheat gliadins, the dominant carriers of celiac disease epitopes. Sci Rep 2017;744609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tye-Din JA, Stewart JA, Dromey JA, Beissbarth T, van Heel DA, Tatham A, et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci Transl Med 2010;2:41ra51. [DOI] [PubMed] [Google Scholar]
- 13.Bruins MJ. The clinical response to gluten challenge: a review of the literature. Nutrients 2013;5:4614–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cook L, Munier CML, Seddiki N, van Bockel D, Ontiveros N, Hardy MY, et al. Circulating gluten-specific FOXP3+CD39+ regulatory T cells have impaired suppressive function in patients with celiac disease. J Allergy Clin Immunol 2017;140(6):1592–603. [DOI] [PubMed] [Google Scholar]
- 15.Hardy MY, Girardin A, Pizzey C, Cameron DJ, Watson KA, Picascia S, et al. Consistency in polyclonal T-cell responses to gluten between children and adults with celiac disease. Gastroenterology 2015;149:1541–52 [ e1542]. [DOI] [PubMed] [Google Scholar]
- 16.Goel G, King T, Daveson AJ, Andrews JM, Krishnarajah J, Krause R, et al. Epitope-specific immunotherapy targeting CD4-positive T cells in coeliac disease: two randomised, double-blind, placebo-controlled phase 1 studies. Lancet Gastroenterol Hepatol 2017;2:479–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warren KG, Catz I, Ferenczi LZ, Krantz MJ. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA class II-defined cohort ofpatients with progressive multiple sclerosis: results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur J Neurol 2006;13:887–95. [DOI] [PubMed] [Google Scholar]
- 18.Miller SD, Wetzig RP, Claman HN. The induction of cell-mediated immunity and tolerance with protein antigens coupled to syngeneic lymphoid cells. J Exp Med 1979;149:758–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, et al. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol 2011;187:2405–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lutterotti A, Yusef S, Sputtek A, Stumer K, Stellmann J-P, Breiden P, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med 2013;5:188ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZH, Yap WT, et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol 2012;30:1217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Getts DR, McCarthy DP, Miller SD. Exploiting apoptosis for therapeutic tolerance induction. J Immunol 2013;191:5341–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo X, Pothoven KL, McCarthy D, DeGutes M, Martin A, Getts DR, et al. ECDI-fixed allogeneic splenocytes induce donor-specific tolerance for long-term survival of islet transplants via two distinct mechanisms. Proc Natl Acad Sci U S A 2008;105:14527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Getts DR, Shea LD, Miller SD, King NJ. Harnessing nanoparticles for immune modulation. Trends Immunol 2015;36:419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hlavaty KA, Luo X, Shea LD, Miller SD. Cellular and molecular targeting for nanotherapeutics in transplantation tolerance. Clin Immunol 2015;160:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarthy DP, Hunter ZN, Chackerian B, Shea LD, Miller SD. Targeted immunomodulation using antigen-conjugated nanoparticles. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2014;6:298–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol 2013;13:621–34. [DOI] [PubMed] [Google Scholar]
- 28.Jing J, Yang IV, Hui L, Patel JA, Evans CM, Prikeris R, et al. Role of macrophage receptor with collagenous structure in innate immune tolerance. J Immunol 2013;190:6360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanno S, Furuyama A, Hirano S. A murine scavenger receptor MARCO recognizes polystyrene nanoparticles. Toxicol Sci 2007;97:398–406. [DOI] [PubMed] [Google Scholar]
- 30.Keegan ME, Falcone JL, Leung TC, Saltzman WM. Biodegradable microspheres with enhanced capacity for covalently bound surface ligands. Macromolecules 2004;37:9779–84. [Google Scholar]
- 31.Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, et al. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano 2014;8:2148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuo R, Saito E, Miller SD, Shea LD. Peptide-conjugated nanoparticles reduce positive co-stimulatory expression and T cell activity to induce tolerance. Mol Ther 2017;25:1676–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smarr CB, Yap WT, Neef TP, Pearson RM, Hunter ZN, Ifergan I, et al. Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre- and postsensitization. Proc Natl Acad Sci U S A 2016;113:5059–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pearson RM, Casey LM, Hughes KR, Wang LZ, North MG, Getts DR, et al. Controlled delivery of single or multiple antigens in tolerogenic nanoparticles using peptide-polymer bioconjugates. Mol Ther 2017;25:1655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casey LM, Pearson RM, Hughes KR, Liu JMH, Rose JA, North MG, et al. Conjugation of transforming growth factor beta to antigen-loaded poly(lactide-co-glycolide) nanoparticles enhances efficiency of antigen-specific tolerance. Bioconjug Chem 2017, 10.1021/acs.bioconjchem.7b00624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, et al. An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy. Nanomedicine 2017;13:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bryant J, Hlavaty KA, Zhang X, Yap WT, Zhang L, Shea LD, et al. Nanoparticle delivery of donor antigens for transplant tolerance in allogeneic islet transplantation. Biomaterials 2014;35:8887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hlavaty KA, McCarthy DP, Saito E, Yap WT, Miller SD, Shea LD. Tolerance induction using nanoparticles bearing HY peptides in bone marrow transplantation. Biomaterials 2016;76:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 2011;3:1377–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y FDA’s regulatory science program for generic PLA/PLGA-based drug products. Am Pharm Rev 2016. [Google Scholar]
- 41.Ferenz KB, Waack IN, Laudien J, Mayer C, Broecker-Preuss M, Groot H, et al. Safety of poly (ethylene glycol)-coated perfluorodecalin-filled poly (lactide-co-glycolide) microcapsules following intravenous administration of high amounts in rats. ResultsPharma Sci 2014;4:8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plard JP, Bazile D. Comparison of the safety profiles of PLA50 and me.PED-PLA50 nanoparticles after single dose intravenous administration to rat. Colloids Surf B Biointerfaces 1999;16:173–83. [Google Scholar]
- 43.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med 2012;4:128ra139. [DOI] [PubMed] [Google Scholar]
- 44.Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med 2000;6:1167–75. [DOI] [PubMed] [Google Scholar]
- 45.Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 1995;45:1268–76. [DOI] [PubMed] [Google Scholar]
- 46.Kennedy MK, Dal Canto MC, Trotter JL, Miller SD. Specific immune regulation of chronic-relapsing experimental allergic encephalomyelitis in mice. J Immunol 1988;141:2986–93. [PubMed] [Google Scholar]
- 47.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol 2002;2:85–95. [DOI] [PubMed] [Google Scholar]
- 48.Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med 2006;203:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Braley-Mullen H, Tompson JG, Sharp GC, Kyriakos M. Suppression of experimental autoimmune thyroiditis in guinea pigs by pretreatment with thyroglobulin-coupled spleen cells. Cell Immunol 1980;51:408–13. [DOI] [PubMed] [Google Scholar]
- 50.Dua HS, Gregerson DS, Donoso LA. Inhibition of experimental autoimmune uveitis by retinal photoreceptor antigens coupled to spleen cells. Cell Immunol 1992;139:292–305. [DOI] [PubMed] [Google Scholar]
- 51.Gregorian SK, Clark L, Heber-Katz E, Amento EP, Rostami A. Induction of peripheral tolerance with peptide-specific anergy in experimental autoimmune neuritis. Cell Immunol 1993;150:298–310. [DOI] [PubMed] [Google Scholar]