

Abstract
The regenerative potential of bone marrow cells could be harnessed for tissue engineering applications. Bone marrow can be easily collected from patients, providing a valuable autologous source of therapeutic cells. However, years of delivery of bone marrow cells have highlighted the need for their genetic manipulation to overcome heterogeneity and to confer specificity to the regenerative process. In this study, we optimized the use of condensed mRNA as a non-viral alternative. As a proof of concept, we used mRNA encoding for reporter proteins such as EGFP or Firefly luciferase, which was condensed by complexing agents and delivered to human bone marrow cells using mineral-coated microparticles. We demonstrated that human bone marrow cells could be transfected with complexed mRNA, and that this approach was more efficient than the delivery of complexed plasmid DNA. In addition, human bone marrow cells were vulnerable to the toxicity of mRNA complexing agents, but these deleterious effects were mitigated by using mineral-coated microparticles as a carrier of complexed mRNA. Microparticle-mediated delivery of complexed mRNA also enabled higher cell metabolic activity and higher transfection in multiple in vitro culture conditions, including suspension culture and three-dimensional culture.
Keywords: mRNA delivery, microparticles, non-viral vectors, gene therapy, bone marrow, T cells, transfection, bone marrow aspirates, Jurkat, mineral coating
Graphical Abstract
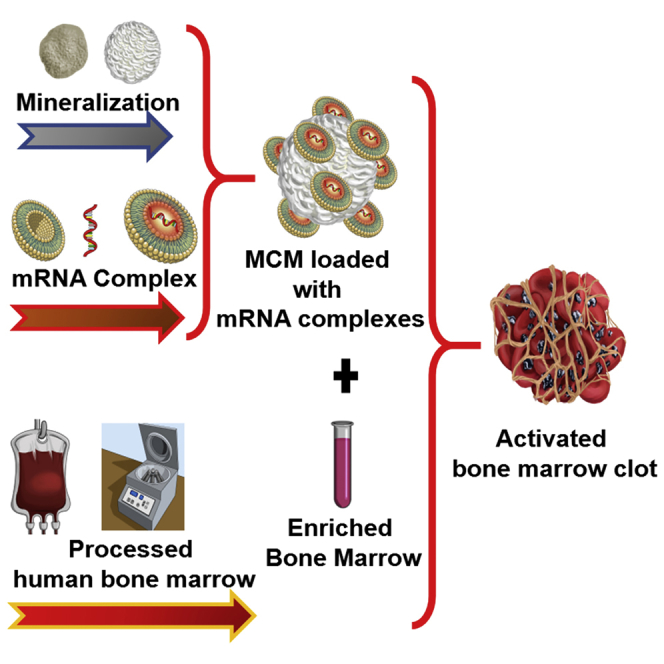
Introduction
The goal of tissue engineering is to restore function to damaged tissues. To be successful, three key elements should be present: (1) a scaffold with the adequate structural features to support tissue growth, (2) cell populations that can deposit the desired extracellular matrix, and (3) an environment bearing the adequate biochemical cues to direct tissue growth and evoke the right cell phenotype.1 The design of systems that can embrace all of these three elements presents several challenges from technical and regulatory standpoints. For this reason, there is still a limited number of clinical trials about scaffold-based tissue engineering.1
To address these issues, autologous tissues have been used as scaffolds in clinical practices, because they can provide structural support and a cellular component already immersed in biological cues that can guide tissue regeneration.1 The use of autografts has been shown to lead to positive outcomes, especially in musculoskeletal applications.2, 3, 4 In particular, human bone marrow (hBM) cells have been used in the clinic for several years because of their therapeutic potential.5 Although bone marrow is heralded as a source of marrow stromal cells, they comprise only a small fraction of all the cells that populate bone marrow tissue.5 The other 99% of bone marrow cells5 are likely to play an important role in tissue regeneration.6, 7 For instance, delivery of bone marrow cells was found to be conducive to angiogenesis because of the presence of vascular cells,8, 9 and the high concentration of white blood cells in marrow can impart anti-inflammatory properties because they secrete high levels of interleukin-1 receptor antagonist (IL-1ra).5, 10 However, it remains challenging to control the effects of such a vast array of different cell types and to achieve consistency in therapeutic outcomes. Recent studies showed that by genetically engineering bone marrow aspirates it is possible to increase their therapeutic potential while directing their regenerative capabilities toward a specific tissue.11, 12, 13 Nevertheless, most studies on engineering bone marrow reported the use of viral vectors,11, 14 which may be difficult to translate into clinical application because of cost and safety concerns.15 In the last decade, several studies assessed the non-viral delivery of plasmid DNA (pDNA) as an alternative to viral vectors, but this approach showed limited efficacy.16 One of the main limitations is that pDNA needs to be delivered inside the nucleus to be expressed, and this is challenging to achieve in non-proliferative cells.16 In contrast, mRNA can be translated directly in the cytoplasm, and thus mRNA delivery can be used to transfect cells in a non-proliferative state.17 The potential of mRNA delivery for the expression of therapeutic proteins was known already from the 1970s,18, 19 but for decades its use was limited to the development of vaccines because of its inherent immunogenicity.20, 21
In 2008, Karikó et al.17 showed that mRNA synthesized in vitro using modified nucleosides had significantly lower immunogenicity and had a higher translation rate because of increased stability. Nonetheless, modified mRNA has been found to be vulnerable to enzymatic degradation,17 and the ubiquitous presence of RNases in various tissues may shorten considerably the half-life of the therapeutic mRNA.22, 23, 24 On the other hand, the prompt degradation of mRNA can be seen as a safety feature,22 which when combined with mRNA’s lack of integration in the host’s genome and the ability to transfect non-proliferating cells could encourage clinical adoption.
One obstacle to the therapeutic use of mRNA is its intracellular delivery. The endocytosis of mRNA is significantly limited by the negative charge and high molecular weight. This issue can be bypassed with the use of cationic complexing agents. These can be either polymeric or lipidic, and can bind and condense mRNA through electrostatic interactions, forming mRNA complexes that can be internalized by the cells.25 Cationic lipids have shown great efficacy for the intracellular delivery of DNA and mRNA.25, 26 However, this increased delivery efficacy comes at a cost: delivery of high concentrations of complexed mRNA was associated with increased toxicity in vivo.27 It has been shown that by controlling the delivery of complexed nucleic acids using nanoparticles and microparticles as reservoir system, it is possible to mitigate their cytotoxicity.28 In this study, we proposed to use mineral-coated microparticles (MCM) as a reservoir system for the delivery of complexed mRNA. The mineral coating was applied generally to a bioresorbable core material. The core materials are immersed in a solution of modified simulated body fluid (mSBF) containing twice as many calcium and phosphate ions than in the human blood plasma. This allows the growth of a mineral layer on the core materials that has controllable properties such as porosity, charge, and dissolution rate.29, 30 We demonstrated in previous studies that by altering the mSBF composition it is possible to obtain mineral coatings with faster or slower dissolution rates that were used to fine-tune the release of therapeutic proteins with different timing.29 Moreover, the nanostructural features of the mineral coatings were shown to stabilize proteins31 and may exert the same stabilizing effect on several other biologics. We hypothesized that MCM could mitigate the toxicity of mRNA complexes while increasing transfection of hBM cells. Our previous studies have demonstrated that the mineral coatings allow high-affinity binding of biologics and sustained release of the biologics over time.29, 32, 33, 34, 35 Moreover, we showed that MCM could be used to bind and deliver pDNA-lipid complexes, thereby decreasing cytotoxicity34 and improving non-viral transfection in vitro.34, 36, 37 The focus of this study was on optimizing conditions for complexed mRNA delivery using MCM. We compared regular MCM with fluoride-doped mineral coated microparticles (FMCM) in their ability to deliver complexed mRNA to T cells and hBM cells. The optimal formulation was then selected to transfect hBM in suspension and in three-dimensional (3D) cultures. This study demonstrates that MCM decreased the toxicity of mRNA complexes while significantly improving their transfection capability in hBM cultures.
Results
hBM Cells Can Be Transfected Using mRNA Complexes
When mRNA is condensed with cationic lipids it can be effectively delivered to hBM cells as shown by the co-localization of the acridine orange stain with hBM cells (Figure 1B). The condensation of pDNA and mRNA using similar cationic lipids enabled the transfection of Jurkat cells (Figure 1C) and hBM cells (Figure 1D). However, the delivery of complexed mRNA allowed superior transfection compared with pDNA; this was observed in Jurkat (Figure 1E) and hBM cells (Figure 1F).
Figure 1.

mRNA Delivery Can Overcome Current Limitations in Gene Therapy of Bone Marrow Cells
(A) Schematic representation of the hBM processing. (B) hBM stained using acridine orange dye; the images show control cells and cells exposed to complexed mRNA, respectively. The cells in red have higher amounts of mRNA. (C and D) Jurkat cells (C) and hBM cells (D) incubated with pDNA or mRNA, respectively, encoding for EGFP and complexed with lipidic complexing agents. (E and F) Quantification of EGFP-positive Jurkat cells (E) and hBM (F) shows that the delivery of complexed mRNA is more effective than pDNA. Asterisk represents statistically significant differences using paired Student’s t test (n = 4), p < 0.05.
MCMs Enable Higher Transfection of hBM Cells Than FMCMs When Using Complexed mRNA
MCM-mediated delivery of complexed mRNA (Figure 2) enabled transfection of Jurkat cells with mRNA encoding for luciferase, even with low amounts of complexed mRNA (83 ng) (Figure 3A). The ratio of MCM to mRNA did not affect luciferase activity, but higher ratios shielded the cells from the toxicity of mRNA complexes. The highest metabolic activity was obtained when using 60 microparticles per nanogram of complexed mRNA (Figure 3A). Delivery of complexed mRNA via FMCM required a higher amount of mRNA (about 333 ng) to achieve comparable transfection. The optimal ratio of FMCM to complexed mRNA was found to be 15 particles per nanogram of mRNA (Figure 3B). When comparing the most promising conditions to deliver complexed mRNA via MCM or via FMCM, we found that FMCMs had the highest protective effect of Jurkat cell’s metabolic activity, but the highest transfection was recorded when MCMs were used as a delivery system (Figure 3C).
Figure 2.

MCM as a Reservoir System for the Delivery of Complexed mRNA
Schematic representation of the procedure used to manufacture MCM and their loading with mRNA complexes prior to exposure to hBM cells. The MCM facilitates the interaction between cells and complexed mRNA. The rate of release of complexed mRNA can be controlled by tuning the dissolution rate of the mineral coatings.
Figure 3.

Optimization of mRNA Delivery via MCM
(A and B) Contour plots displaying the results of the screening design performed on Jurkat cells. (A) Complexed mRNA encoding for Gaussia luciferase delivered via MCM. (B) Complexed mRNA encoding for Gaussia luciferase delivered via FMCM. (C) Direct comparison of complexed mRNA delivery to Jurkat cells using MCM or FMCM. Asterisk represents statistically significant differences using paired Student’s t test (n = 4), *p < 0.05. (D and E) Contour plots displaying the results of the screening design performed on hBM cells for the delivery of complexed mRNA encoding for Gaussia luciferase via MCM (D) or FMCM (E). (F) Direct comparison of complexed mRNA delivery to hBM cells using MCM or FMCM. Asterisks represent statistically significant differences using paired Student’s t test (n = 4), ****p < 0.05.
In the screening design conducted on hBM, we observed higher transfection with lower ratios of MCM to mRNA, but this also coincided with a lower cell metabolic activity, highlighting the trade-off between transfection efficiency and cytotoxicity of mRNA complexes (Figure 3D). The FMCM enabled delivery of higher amounts of complexed mRNA while maintaining high metabolic activity in hBM (Figure 3E). However, higher transfection efficiency was obtained when complexed mRNA was delivered via MCM (Figure 3F). Therefore, we selected MCM over FMCM as a delivery vehicle for complexed mRNA in the remaining parts of the study.
MCM-Mediated Delivery of mRNA Complexes Improves hBM Transfection and Decreases Cytotoxicity
The delivery of complexed mRNA in either soluble form or mediated by MCM was effective in transfecting Jurkat cells in suspension culture (Figure 4A). However, MCM-mediated delivery significantly decreased the toxicity associated with the lipidic complexing agents (Figure 4E). The culture conditions were also critical in determining the outcome of transfection. Delivery of complexed mRNA to Jurkat cells cultured in serum-supplemented RPMI media resulted in high luciferase expression, whereas no transfection was observed when the cells were cultured in bone marrow aspirates (Figure 4E). A similar behavior was also observed with hBM cells. hBM cells in suspension could be transfected either with soluble or MCM-mediated delivery of complexed mRNA (Figure 4B). However, hBM cells were highly vulnerable to the toxicity of lipidic complexing agents, because only MCM-mediated delivery maintained 100% of the metabolic activity. In contrast, hBM incubated directly with mRNA complexes showed a poor metabolic activity, below 50% (Figure 4F). The culture conditions also affected the outcomes of hBM transfection. In particular, suspension culture in bone marrow aspirates severely decreased the expression of luciferase-encoding mRNA, whereas culture in cell culture medium allowed for effective expression. MCM-mediated delivery of complexed mRNA allowed three times higher expression relative to delivery without MCMs (Figure 4F). Interestingly, the presence of MCMs was shown to increase the production of endosomes in Jurkat (Figure 4C) and in hBM cells (Figure 4D), confirming similar findings reported in a previous study.34 This may indicate a higher likelihood for the internalization of mRNA complexes in the presence of MCMs.
Figure 4.

Transfection of hBM Is More Effective When mRNA Is Delivered via MCM
(A and B) Transfection of Jurkat cells (A) and hBM cells (B) using complexed mRNA encoding for EGFP. (C and D) Exposure of Jurkat cells (C) or hBM cells (D) to MCM causes an increase of endosomal activity as shown by an increased uptake of labeled dextran. (E and F) Jurkat cells (E) or hBM cells (F) exposed to complexed mRNA encoding for Firefly luciferase. The extracellular environment had a considerable impact on transfection efficiency; when cells were cultured in whole bone marrow the transfection was significantly lower than cells cultured in media. The delivery of complexed mRNA via MCM enables significantly higher cell metabolic activity. Asterisk represents statistically significant differences using paired Student’s t test (n = 4), p < 0.05.
MCM-Mediated mRNA Delivery Allows for Transfection of hBM Cells in a Clinically Relevant Setting
We harnessed the ability of fresh bone marrow aspirates to form 3D clots, and we determined that they can be diluted with up to 1 equivalent volume of MCM solution without compromising the ability to clot (Figure 5B). We also showed that the hurdle of forming clots with heparinized bone marrow sources can be circumvented by washing the cells and by the addition of clotting agents such as fibrinogen and thrombin (Figures 5C and 5D) as described in Figure 5A. The transfection of hBM with complexed mRNA was rapid. Interestingly, we found that incubations of less than 30 s led to higher transfection of hBM compared with 24 h of exposure (Figure 6B). The rapid delivery of complexed mRNA to hBM allowed to transfect and encapsulate hBM cells in less than 30 min (Figure 5E). Therefore, this is a procedure that can be applicable in clinically relevant settings. In 3D clot mimics, both Jurkat and hBM showed significantly higher metabolic activity when mRNA complexes were delivered via MCM (Figure 6C and 6D), and as a result, hBM showed also higher expression of the delivered mRNA (Figure 6D). However, similar to what was observed in suspension cultures, also in 3D clot mimics the culture in bone marrow aspirates was found to suppress the expression of the delivered mRNA (Figures 6C and 6D).
Figure 5.

Cells Can Be Transfected Also in 3D Clot Mimic Hydrogels
(A) Schematic representation of the bone marrow clots formation. Fresh bone marrow aspirates can be mixed with a solution of MCM at a 1:1 volume ratio and then let clot at 37°C. Alternatively, heparinized bone marrow can be concentrated by centrifugation, mixed with MCM solution and then enriched with clotting agents such as fibrinogen and thrombin, and let clot at 37°C. (B–D) Color-enhanced scanning electron micrographs showing, respectively, (B) a spontaneous clot formed using fresh bone marrow aspirates mixed with MCM and clots formed using heparinized bone marrow washed and then mixed with clotting agents, without MCM (C) or with MCM (D). (E) Delivery of complexed mRNA encoding for EGFP to 3D-clot mimic obtained by mixing hBM with clotting agents. mRNA complexes can transfect hBM cells also in 3D.
Figure 6.

hBM Cells Can Be Enriched, Transfected, and Encapsulated within 30 min
(A) Effect of incubation time on transfection of Jurkat cells. Complexed mRNA encoding for Firefly luciferase was incubated with Jurkat cells from a few seconds up to 24 h. The incubation time did not seem to have significative impact on transfection of Jurkat cells, but there was a trend indicating that longer incubation time could lead to lower transfection. (B) Incubation time had a significant impact on the transfection of hBM; the highest transfection was observed with the shortest incubation time. (C and D) Transfection of Jurkat cells (C) or hBM cells (D) encapsulated in 3D clot-mimic hydrogels. The delivery of mRNA complexes via MCM coincided with a higher preservation of metabolic activity. The extracellular environment had a significant effect on the transfection efficiency; in fact, higher transfection was observed when cells were cultured in media compared with culture in whole bone marrow. Asterisk represents statistically significant differences using paired Student’s t test (n = 4), p < 0.05.
Discussion
In this study we developed a non-viral approach to transfect hBM cells. Although the use of complexed pDNA is a standard common alternative to viral gene therapy,38 we showed that pDNA-based transfection is not effective in transfecting hBM cells (Figure 1D). The complexed pDNA must overcome several biological barriers before being expressed, including the most limiting step, which is the need for nuclear localization.38 The likelihood of this event is higher in dividing cells because of the temporary breakdown of the nuclear envelope, but it is very unlikely to occur in non-proliferative cells such as hBM-borne cells.39 mRNA, on the other hand, can be readily translated within minutes upon entry in the cell’s cytoplasm.17 However, most cell types do not spontaneously uptake naked mRNA, and its intracellular delivery can be greatly improved by condensation with complexing agents.40 We found that mRNA complexed with cationic lipids is effective in transfecting hBM cells (Figure 1D) showing significantly higher transfection than complexed pDNA (Figure 1F). A number of studies have reported that complexed mRNA is effective in transfecting cells in vivo,27 but this has also led to increased cell apoptosis in the area of interest.27 These prior studies highlight the trade-off between transfection efficiency and toxicity caused by the transfecting agent. We hypothesized that the toxic effects of mRNA complexes could be mitigated by using MCM to locally deliver mRNA complexes to hBM cells.
We manufactured these microparticles using β-TCP as a core material because it can be resorbed in the body.41 Moreover, in our previous in vivo studies using the MCM, we did not observe any side effects when these were injected to the medial collateral ligament42 or in critical-sized bone defects.43 There is also ample literature supporting the claim that calcium phosphate biomaterials are generally safe in vivo.44 The mineral coatings used in this study were previously found to be highly adaptable,29 because their dissolution rate can be controlled by altering the composition of mSBF used to grow the mineral coating.29 For example, the addition of inorganic dopants such as fluoride were shown to decrease the dissolution rate of the coating, thus influencing the binding and release of DNA complexes32, 33, 34 and enhancing pDNA-based non-viral transfection.34, 36, 37 In the current study, we compared the performance of two formulations of mineral coatings: MCM obtained using regular mSBF and fluoride-doped mineral coated microparticles (FMCM) obtained with the addition of fluoride as inorganic dopant. We found that MCM-mediated delivery of complexed mRNA enables higher levels of transfection than FMCM (Figure 3F). This was possibly due to the higher dissolution rate of the MCM formulation when compared with the FMCM formulation.
When we compared the direct exposure of cells to complexed mRNA versus the delivery mediated by MCM, we found that MCM-mediated delivery allowed significantly higher cell metabolic activity in Jurkat cells (Figure 4E) and hBM cells (Figure 4F). This cytoprotective effect on cells was more pronounced in hBM (the MCM group had 60% higher metabolic activity than the soluble approach; Figure 4F) compared with Jurkat cells (the MCM group had 10% higher metabolic activity than the soluble approach; Figure 4E). This could be due to the fact that MCM-mediated delivery gradually releases the mRNA complexes, therefore mitigating their disruptive effect on the cell’s membrane, responsible for cell death.25 This allows to transfect cells without compromising their viability. We found that Jurkat cells can tolerate mRNA complexes better than hBM. Therefore, the presence of MCM is more beneficial for hBM cells (Figure 4).
Interestingly, we also observed an increase in endosomal activity in Jurkat cells (Figure 4C) and hBM cells (Figure 4D) cultured in the presence of MCMs, which was consistent with our previous findings.34 It is highly unlikely that the MCMs themselves could be internalized because of their large size (∼8 μm),34 but the higher endosome production could be triggered by high extracellular concentrations of soluble mineral ions such as Ca2+ and PO43− derived from the dissolution of the mineral coating.36 This increase in endosome production in the presence of MCMs suggests that mRNA complexes may have a higher likelihood of being internalized in the presence of MCMs. Therefore, the role of MCMs is that of a reservoir system that can increase the interactions between cells and mRNA complexed with lipidic transfecting agents.
We also observed that briefly washing the hBM cells in media before mRNA delivery increased significantly the transfection efficiency (Figure 4F). This is likely due to the removal of the abundant RNases known to be present in the bone marrow environment.22, 45 We found that lipidic complexing agents can partially shield mRNA from degradation when cultured in the presence of RNase A (Figure S6). However, the complexes were vulnerable to changes in osmolarity (Figure S7), which may limit their efficacy in tissues such as cartilage that are characterized by high osmolarity.46 These are limitations of the current lipidic transfecting agents that will need to be overcome to enable higher transfection efficiency in vivo.
Interestingly, we found that a 30-s exposure to complexed mRNA was sufficient to effectively transfect Jurkat cells (Figure 6A) and hBM cells (Figure 6B). The swift transfection is probably the result of electrostatic interactions that enable the loading/release of mRNA complexes from the MCMs. We believe that mRNA complexes may be constantly released/sequestered by the MCM, allowing the mRNA to transfect only cells neighboring the MCM. This was demonstrated in a previous study where MCMs were loaded with pDNA complexed with cationic lipids34 and also by using mineral nanoparticles to locally deliver mRNA/lipid complexes.47 This rapid process may enable transfection of hBM cells also in clinically relevant settings, where the ability of bone marrow to spontaneously clot can be harnessed to manufacture 3D clots (Figure 5B). Toward that end, we showed here that hBM cells can be transfected with mRNA complexes prior to encapsulation in 3D clots, using either fresh bone marrow aspirates or clotting agents (Figure 5E). We demonstrated that the entire procedure of transfection and clotting takes less than 30 min to complete; thus, it could potentially be performed during a surgical procedure without requiring additional ex vivo cell culture manipulation. The results obtained in 3D cultures corroborated what was observed in suspension culture: MCM-mediated delivery of complexed mRNA enabled higher transfection of hBM cells while maintaining high metabolic activity, consistently outperforming direct delivery of complexed mRNA (Figure 6D).
The results of this study correlate with recent reports endorsing mRNA delivery as a promising alternative to viral gene therapy.17, 27, 48 The limitations such as high immunogenicity and low stability that prevented mRNA’s adoption for several years can now be circumvented. For instance, immunogenicity of synthetic mRNA can be decreased by nucleoside modification17 or by sequence engineering,49 and the current complexing agents available can at least partially shield mRNA from enzymatic degradation. Moreover, in this study we show that there are several advantages in using MCM to mediate the delivery of mRNA complexes, because they enable higher transfection with lower toxicity.
We found that in vitro, the expression of complexed mRNA peaks between 6 and 10 hours and slowly decreases over time. We observed that modified mRNA can be expressed up to 4 days in Jurkat and hBM cells (Figure S8), and this is consistent with what has been reported in the literature in other cell types.50 However, different cell types, delivery environment, use of modified mRNA, different 3′ UTR sequences, and length of poly A tail are likely to dictate the duration of the mRNA expression. These are all factors that can be customized, allowing further control over the expression. Nevertheless, some applications may require the expression of therapeutic mRNA for weeks. It will be a focus of our future studies to harness the stabilizing effect of our mineral coatings to protect mRNA complexes, enabling higher control over duration of expression. One consideration that must be made is that the short-term expression of mRNA is not necessarily a disadvantage. In fact, the fast degradation of mRNA could be seen as a safety feature that could ease the path to clinical adoption.
One limitation of this study is that we used mainly mRNA encoding for reporter proteins. As a proof of concept we have also synthesized mRNA encoding for human TGF-β1, and we assessed that it can be used to increase the expression of therapeutic proteins in bone marrow cells (Figure S9). However, future studies will assess whether the expression of therapeutic mRNA can increase the regenerative potential of the target cells. It will be critical to study mRNA delivery in vivo, to assess whether the delivery of therapeutic mRNA can increase tissue regeneration.
Conclusions
The combination of safety and efficacy is pivotal for the success of gene therapy strategies and their clinical adoption. The delivery of mRNA is emerging as a promising alternative to viral approaches. In this study, we showed that MCMs can be a valuable tool that enables higher transfection with lower toxicity. Moreover, the use of bone marrow aspirates to generate autologous biological scaffolds not only offers high regenerative potential for several tissues, but it may also be a clinically achievable approach.
Materials and Methods
Bone Marrow Cell Enrichment
hBM cells were isolated from discarded bone marrow filters and leftover bags from unidentified normal healthy bone marrow donors in University of Wisconsin Hospital, based on an institutional review board (IRB)-approved protocol, as described previously.51 The key steps of the procedure are depicted in Figure 1A. This protocol was approved by the University of Wisconsin (UW)-Madison IRB. The marrow bag was rinsed with 1× PBS + 1,000 U/mL penicillin and 1,000 μg/mL streptomycin (P/S), and filtered out through a 100-μm cell strainer. The marrow solution was centrifuged for 5 min at 2,000 rpm. The supernatant was removed and the cells were diluted to 10 mL with PBS + P/S. The cell suspension was washed twice and treated with 1× RBC Lysis Buffer (Fisher Scientific) for 5 min to eliminate red blood cells. Enriched bone marrow cells were then resuspended in RPMI media (Corning) containing 10% fetal bovine serum (FBS).
Fabrication of MCM
The biomineralization of microparticles was performed in mSBF as previously described34 using beta-tricalcium phosphate powder (Cam Bioceramics) as a core material. We compared two formulations: (1) particles coated with regular mSBF (MCM) and (2) particles coated with mSBF supplemented with fluoride (FMCM). The powder was suspended at a concentration of 1 mg/mL in mSBF prepared as shown in Table S1 to obtain MCM or FMCM and rotated at 37°C for 3 days. Each day, the microparticles were centrifuged and the supernatant was replaced with fresh mSBF to ensure uniformity of the coating. The microparticles were then collected by centrifugation, washed in Deionized water, filtered through a cell strainer, and suspended in 5 mL DI water. Finally, they were frozen using liquid nitrogen and lyophilized. The resultant coated microparticles were sterilized using UV light for 30 min, suspended in Opti-MEM (Thermo Fisher Scientific), and stored at −20°C until use.
Acridine Orange Stain
The acridine orange dye has a different emission when bound to DNA (green) or associated to mRNA (red), and it was used to visualize the mRNA present with the cells after incubation with mRNA complexes. The staining solution was obtained by resuspending 6 μg/mL of acridine orange (Sigma-Aldrich) in 0.1 M citric acid and Na2HPO4 0.2M (pH 2.6). The cells were washed in PBS and then fixed in 1% paraformaldehyde for 15 min. After fixation, the cells were washed again in PBS and then incubated in the staining solution for 20 min at room temperature. Before imaging, the cells were washed one more time in PBS.
Screening Design
We used the software JMP Pro version 14.0 to generate a screening design using the factors: (1) ratio of complexing agent to mRNA, (2) number of particles, and (3) number of particles per nanogram of mRNA. For the screening design we delivered complexed mRNA encoding for Gaussia luciferase, and the outcomes measured were Gaussia luciferase activity and cell metabolic activity. We decided to use T cells as a model cell line because they constitute 8% of all the nucleated bone marrow cells;52 in particular we used Jurkat cells, an immortalized T cell line, for our protocol optimization and initial screening design. The results were then validated on hBM cells.
Formation of mRNA and pDNA Complexes
EGFP-encoding mRNA (EGFP, TriLink L-7201), Gaussia luciferase-encoding mRNA (TriLink L-7205), and Firefly luciferase-encoding mRNA (TriLink L-7202) were used as reporter genes. The mRNA was polyadenylated, optimized for mammalian systems, and modified with 5-methoxyuridine. GFP-encoding pDNA (Clontech) was also used in one experiment. We used Lipofectamine MessengerMAX (Life Technologies) as a complexing agent for mRNA and Lipofectamine 2000 (Life Technologies) to complex pDNA. The lipidic transfecting agents were resuspended in Opti-MEM, and mRNA was added at a final concentration of 15 μg/mL. The solution was incubated for 10 min at room temperature to allow the formation of mRNA-lipid complexes (Figure 2).
Transfection of Cells in Suspension or in 3D Cultures
hBM and Jurkat (ATCC) cells were cultured in RPMI medium supplemented with 10% FBS (v/v) and 1,000 U/mL P/S. For the complexed mRNA-only groups, the solution of complexed mRNA was added directly to the cells of interest for incubation. For MCM or FMCM-mediated groups, the sterile microparticles were added to the complex solution and incubated at room temperature for 1 h, at which point complex-bound MCM or FMCM was centrifuged and resuspended in fresh Opti-MEM to remove any unbound complexes. The MCMs were then added to the cells. Cells were dispensed directly onto a 96-well plate. For the transfection in 3D clot mimics, the cell suspension was quickly mixed with fibrinogen (0.2 mg/mL media) and thrombin (0.2 U/mL media) to form a hydrogel and plated at 100 μL/well. After about 30 min at 37°C, 100 μL of fresh media was added to each gel. The cells were cultured at 37°C overnight. The varying reagent ratios and concentrations, as well as the incubation protocols, are specified for each experiment in the Results.
Measurement of Transfection
Transfection was assessed at 24 h after mRNA delivery (or 48 h for pDNA experiments). Cells incubated with EGFP mRNA or pDNA were centrifuged and resuspended in Opti-MEM for better visualization. EGFP-expressing cells were imaged using a Nikon Eclipse Ti Microscope, and positive cells were counted using ImageJ software. Cells transfected with Gaussia or Firefly luciferase mRNA were centrifuged, and media were removed from each well. The activity of Gaussia luciferase was determined from the supernatant and using a commercially available kit (Biolux, New England Biolabs) and a plate reader. Firefly luciferase activity was measured by adding directly the substrate luciferin (150 μg/mL in 1× PBS) (Biosynth) to cells, and luminescence was measured immediately using a plate reader.
Measurement of Metabolic Activity
After microscopic imaging of GFP-transfected cells, metabolic activity was measured using a CellTiter-Blue (Promega) assay. A total of 10 μL dye per 200 μL culture media was added to each well and incubated at 37°C for about 4 h. Absorbance was measured at 570 and 600 nm using a plate reader, and the difference between absorbance at these two wavelengths was used as a measure of metabolic activity. Experimental well absorbance was normalized to control wells with untreated cells and expressed as a percentage.
Visualization of Endocytosis Using Labeled Dextran
Enriched bone marrow or Jurkat cells were incubated with MCMs and 250 ng dextran (Alexa Fluor 594; Thermo Fisher) per milliliter culture media for 2 h. After incubation, cells were washed in sterile PBS, fixed in neutral-buffered formalin for 15 min, and washed again. A total of 100 mM HCl was added to the cells and incubated for 15 min at 37°C to remove extracellular dextran and dissolve MCMs. Intracellular fluorescence was visualized with fluorescence microscopy and compared with control cells without MCMs added to assess endocytosis.
Scanning Electron Micrographs of MCMs in 3D Clots
Samples were fixed in 2% paraformaldehyde for 15 min at the time of the collection and then washed in 1× PBS. Samples were further fixed in 1.5% glutaraldehyde resuspended in freshly prepared 70 mM sodium cacodylate buffer (pH 7.4) for 2 h at room temperature. The samples were then rinsed in 70 mM sodium cacodylate buffer supplemented with 2.5% sucrose and dehydrated by immersion in a graduated series of ethanol in H2O and hexamethyldisilazane (HDMS) in ethanol baths of, respectively, 30%, 50%, 80%, and 95%. The samples were air-dried and then gold sputter coated before being imaged.
Statistical Analysis
The results are presented as mean ± SD. The statistical comparisons were made using paired Student’s t test or one-way ANOVA with Tukey’s post hoc test, and the statistical significance was determined to be p < 0.05. The statistical analysis was performed using GraphPad Prism version 6.
Author Contributions
G.F. wrote the paper, designed and conducted most of the experiments, and characterized the clots using the scanning electron microscope; H.L.M. conducted some of the transfection experiments and helped with imaging; J.S.L. helped with imaging and provided scientific support; K.S. helped with the synthesis and characterization of mRNA; P.H. helped with the bone marrow collection and provided scientific support; and W.L.M. supervised the study.
Acknowledgments
This project was funded by a generous philanthropic gift from The Shannon Family in support of the Musculoskeletal Regeneration Partnership, by NIH Training Grant 5T35OD011078-08 for the support of H.L.M., and by University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520. We also want to thank Paolo Bianchini for drawing the schematics.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2019.09.004.
Supplemental Information
References
- 1.Smith B.D., Grande D.A. The current state of scaffolds for musculoskeletal regenerative applications. Nat. Rev. Rheumatol. 2015;11:213–222. doi: 10.1038/nrrheum.2015.27. [DOI] [PubMed] [Google Scholar]
- 2.Gamie Z., Tran G.T., Vyzas G., Korres N., Heliotis M., Mantalaris A., Tsiridis E. Stem cells combined with bone graft substitutes in skeletal tissue engineering. Expert Opin. Biol. Ther. 2012;12:713–729. doi: 10.1517/14712598.2012.679652. [DOI] [PubMed] [Google Scholar]
- 3.Li M.T.A., Willett N.J., Uhrig B.A., Guldberg R.E., Warren G.L. Functional analysis of limb recovery following autograft treatment of volumetric muscle loss in the quadriceps femoris. J. Biomech. 2014;47:2013–2021. doi: 10.1016/j.jbiomech.2013.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mariscalco M.W., Magnussen R.A., Mehta D., Hewett T.E., Flanigan D.C., Kaeding C.C. Autograft versus nonirradiated allograft tissue for anterior cruciate ligament reconstruction: a systematic review. Am. J. Sports Med. 2014;42:492–499. doi: 10.1177/0363546513497566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King W., Toler K., Woodell-May J. Role of White Blood Cells in Blood- and Bone Marrow-Based Autologous Therapies. BioMed Res. Int. 2018;2018:6510842. doi: 10.1155/2018/6510842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pang W.W., Price E.A., Sahoo D., Beerman I., Maloney W.J., Rossi D.J., Schrier S.L., Weissman I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhatnagar A., Bolli R., Johnstone B.H., Traverse J.H., Henry T.D., Pepine C.J., Willerson J.T., Perin E.C., Ellis S.G., Zhao D.X., Cardiovascular Cell Therapy Research Network (CCTRN) Bone marrow cell characteristics associated with patient profile and cardiac performance outcomes in the LateTIME-Cardiovascular Cell Therapy Research Network (CCTRN) trial. Am. Heart J. 2016;179:142–150. doi: 10.1016/j.ahj.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guerrero J., Oliveira H., Catros S., Siadous R., Derkaoui S.M., Bareille R., Letourneur D., Amédée J. The use of total human bone marrow fraction in a direct three-dimensional expansion approach for bone tissue engineering applications: focus on angiogenesis and osteogenesis. Tissue Eng. Part A. 2015;21:861–874. doi: 10.1089/ten.tea.2014.0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heo S.-H., Park Y.S., Kang E.S., Park K.B., Do Y.S., Kang K.S., Kim D.I. Early Results of Clinical Application of Autologous Whole Bone Marrow Stem Cell Transplantation for Critical Limb Ischemia with Buerger’s Disease. Sci. Rep. 2016;6:19690. doi: 10.1038/srep19690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cassano J.M., Kennedy J.G., Ross K.A., Fraser E.J., Goodale M.B., Fortier L.A. Bone marrow concentrate and platelet-rich plasma differ in cell distribution and interleukin 1 receptor antagonist protein concentration. Knee Surg. Sports Traumatol. Arthrosc. 2018;26:333–342. doi: 10.1007/s00167-016-3981-9. [DOI] [PubMed] [Google Scholar]
- 11.Qi X., Pay S.L., Yan Y., Thomas J., Jr., Lewin A.S., Chang L.J., Grant M.B., Boulton M.E. Systemic Injection of RPE65-Programmed Bone Marrow-Derived Cells Prevents Progression of Chronic Retinal Degeneration. Mol. Ther. 2017;25:917–927. doi: 10.1016/j.ymthe.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frisch J., Rey-Rico A., Venkatesan J.K., Schmitt G., Madry H., Cucchiarini M. Chondrogenic Differentiation Processes in Human Bone Marrow Aspirates upon rAAV-Mediated Gene Transfer and Overexpression of the Insulin-Like Growth Factor I. Tissue Eng. Part A. 2015;21:2460–2471. doi: 10.1089/ten.tea.2014.0679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venkatesan J.K., Moutos F.T., Rey-Rico A., Estes B.T., Frisch J., Schmitt G., Madry H., Guilak F., Cucchiarini M. Chondrogenic differentiation processes in human bone-marrow aspirates seeded in three-dimensional woven poly (ε-caprolactone) scaffolds enhanced by recombinant adeno-associated virus-mediated SOX9 gene transfer. Hum. Gene Ther. 2018;29:1277–1286. doi: 10.1089/hum.2017.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pascher A., Palmer G.D., Steinert A., Oligino T., Gouze E., Gouze J.N., Betz O., Spector M., Robbins P.D., Evans C.H., Ghivizzani S.C. Gene delivery to cartilage defects using coagulated bone marrow aspirate. Gene Ther. 2004;11:133–141. doi: 10.1038/sj.gt.3302155. [DOI] [PubMed] [Google Scholar]
- 15.Thomas C.E., Ehrhardt A., Kay M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 16.Ramamoorth M., Narvekar A. Non viral vectors in gene therapy- an overview. J. Clin. Diagn. Res. 2015;9:GE01–GE06. doi: 10.7860/JCDR/2015/10443.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karikó K., Muramatsu H., Welsh F.A., Ludwig J., Kato H., Akira S., Weissman D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008;16:1833–1840. doi: 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurdon J.B., Lane C.D., Woodland H.R., Marbaix G. Use of frog eggs and oocytes for the study of messenger RNA and its translation in living cells. Nature. 1971;233:177–182. doi: 10.1038/233177a0. [DOI] [PubMed] [Google Scholar]
- 19.Laskey R.A., Gurdon J.B., Crawford L.V. Translation of encephalomyocarditis viral RNA in oocytes of Xenopus laevis. Proc. Natl. Acad. Sci. USA. 1972;69:3665–3669. doi: 10.1073/pnas.69.12.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kübler H., Scheel B., Gnad-Vogt U., Miller K., Schultze-Seemann W., Vom Dorp F., Parmiani G., Hampel C., Wedel S., Trojan L. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: a first-in-man phase I/IIa study. J. Immunother. Cancer. 2015;3:26. doi: 10.1186/s40425-015-0068-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kallen K.-J., Theß A. A development that may evolve into a revolution in medicine: mRNA as the basis for novel, nucleotide-based vaccines and drugs. Ther. Adv. Vaccines. 2014;2:10–31. doi: 10.1177/2051013613508729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Probst J., Brechtel S., Scheel B., Hoerr I., Jung G., Rammensee H.G., Pascolo S. Characterization of the ribonuclease activity on the skin surface. Genet. Vaccines Ther. 2006;4:4. doi: 10.1186/1479-0556-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harder J., Schröder J.-M. RNase 7, a novel innate immune defense antimicrobial protein of healthy human skin. J. Biol. Chem. 2002;277:46779–46784. doi: 10.1074/jbc.M207587200. [DOI] [PubMed] [Google Scholar]
- 24.Hooper L.V., Stappenbeck T.S., Hong C.V., Gordon J.I. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat. Immunol. 2003;4:269–273. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 25.Wasungu L., Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J. Control. Release. 2006;116:255–264. doi: 10.1016/j.jconrel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 26.Bally M.B., Harvie P., Wong F.M., Kong S., Wasan E.K., Reimer D.L. Biological barriers to cellular delivery of lipid-based DNA carriers. Adv. Drug Deliv. Rev. 1999;38:291–315. doi: 10.1016/s0169-409x(99)00034-4. [DOI] [PubMed] [Google Scholar]
- 27.Sultana N., Magadum A., Hadas Y., Kondrat J., Singh N., Youssef E., Calderon D., Chepurko E., Dubois N., Hajjar R.J., Zangi L. Optimizing Cardiac Delivery of Modified mRNA. Mol. Ther. 2017;25:1306–1315. doi: 10.1016/j.ymthe.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Browne S., Fontana G., Rodriguez B.J., Pandit A. A protective extracellular matrix-based gene delivery reservoir fabricated by electrostatic charge manipulation. Mol. Pharm. 2012;9:3099–3106. doi: 10.1021/mp300231d. [DOI] [PubMed] [Google Scholar]
- 29.Yu X., Khalil A., Dang P.N., Alsberg E., Murphy W.L. Multilayered inorganic microparticles for tunable dual growth factor delivery. Adv. Funct. Mater. 2014;24:3082–3093. doi: 10.1002/adfm.201302859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suárez-González D., Barnhart K., Migneco F., Flanagan C., Hollister S.J., Murphy W.L. Controllable mineral coatings on PCL scaffolds as carriers for growth factor release. Biomaterials. 2012;33:713–721. doi: 10.1016/j.biomaterials.2011.09.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X., Biedrzycki A.H., Khalil A.S., Hess D., Umhoefer J.M., Markel M.D., Murphy W.L. Nanostructured Mineral Coatings Stabilize Proteins for Therapeutic Delivery. Adv. Mater. 2017;29:1701255. doi: 10.1002/adma.201701255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suárez-González D., Lee J.S., Diggs A., Lu Y., Nemke B., Markel M., Hollister S.J., Murphy W.L. Controlled multiple growth factor delivery from bone tissue engineering scaffolds via designed affinity. Tissue Eng. Part A. 2014;20:2077–2087. doi: 10.1089/ten.tea.2013.0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suárez-González D., Lee J.S., Lan Levengood S.K., Vanderby R., Jr., Murphy W.L. Mineral coatings modulate β-TCP stability and enable growth factor binding and release. Acta Biomater. 2012;8:1117–1124. doi: 10.1016/j.actbio.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khalil A.S., Yu X., Xie A.W., Fontana G., Umhoefer J.M., Johnson H.J., Hookway T.A., McDevitt T.C., Murphy W.L. Functionalization of microparticles with mineral coatings enhances non-viral transfection of primary human cells. Sci. Rep. 2017;7:14211. doi: 10.1038/s41598-017-14153-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J.S., Suarez-Gonzalez D., Murphy W.L. Mineral coatings for temporally controlled delivery of multiple proteins. Adv. Mater. 2011;23:4279–4284. doi: 10.1002/adma.201100060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi S., Yu X., Jongpaiboonkit L., Hollister S.J., Murphy W.L. Inorganic coatings for optimized non-viral transfection of stem cells. Sci. Rep. 2013;3:1567. doi: 10.1038/srep01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi S., Murphy W.L. Sustained plasmid DNA release from dissolving mineral coatings. Acta Biomater. 2010;6:3426–3435. doi: 10.1016/j.actbio.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lächelt U., Wagner E. Nucleic Acid Therapeutics Using Polyplexes: A Journey of 50 Years (and Beyond) Chem. Rev. 2015;115:11043–11078. doi: 10.1021/cr5006793. [DOI] [PubMed] [Google Scholar]
- 39.Remaut K., Symens N., Lucas B., Demeester J., De Smedt S.C.C. Cell division responsive peptides for optimized plasmid DNA delivery: the mitotic window of opportunity? J. Control. Release. 2014;179:1–9. doi: 10.1016/j.jconrel.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 40.Sahin U., Karikó K., Türeci Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 41.Koepp H.E., Schorlemmer S., Kessler S., Brenner R.E., Claes L., Günther K.P., Ignatius A.A. Biocompatibility and osseointegration of β-TCP: histomorphological and biomechanical studies in a weight-bearing sheep model. J. Biomed. Mater. Res. B Appl. Biomater. 2004;70:209–217. doi: 10.1002/jbm.b.30034. [DOI] [PubMed] [Google Scholar]
- 42.Clements A.E.B., Leiferman E.M., Chamberlain C.S., Vanderby R., Murphy W.L. Addition of Mineral-Coated Microparticles to Soluble Interleukin-1 Receptor Antagonist Injected Subcutaneously Improves and Extends Systemic Interleukin-1 Inhibition. Adv. Ther. 2018;1:1800048. [Google Scholar]
- 43.Orth M., Kruse N.J., Braun B.J., Scheuer C., Holstein J.H., Khalil A., Yu X., Murphy W.L., Pohlemann T., Laschke M.W., Menger M.D. BMP-2-coated mineral coated microparticles improve bone repair in atrophic non-unions. Eur. Cell. Mater. 2017;33:1–12. doi: 10.22203/eCM.v033a01. [DOI] [PubMed] [Google Scholar]
- 44.Sheikh Z., Abdallah M.N., Hanafi A.A., Misbahuddin S., Rashid H., Glogauer M. Mechanisms of in Vivo Degradation and Resorption of Calcium Phosphate Based Biomaterials. Materials (Basel) 2015;8:7913–7925. doi: 10.3390/ma8115430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nitto T., Dyer K.D., Czapiga M., Rosenberg H.F. Evolution and function of leukocyte RNase A ribonucleases of the avian species, Gallus gallus. J. Biol. Chem. 2006;281:25622–25634. doi: 10.1074/jbc.M604313200. [DOI] [PubMed] [Google Scholar]
- 46.Urban J.P., Hall A.C., Gehl K.A. Regulation of matrix synthesis rates by the ionic and osmotic environment of articular chondrocytes. J. Cell. Physiol. 1993;154:262–270. doi: 10.1002/jcp.1041540208. [DOI] [PubMed] [Google Scholar]
- 47.Zohra F.T., Chowdhury E.H., Nagaoka M., Akaike T. Drastic effect of nanoapatite particles on liposome-mediated mRNA delivery to mammalian cells. Anal. Biochem. 2005;345:164–166. doi: 10.1016/j.ab.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y., Miao L., Satterlee A., Huang L. Delivery of oligonucleotides with lipid nanoparticles. Adv. Drug Deliv. Rev. 2015;87:68–80. doi: 10.1016/j.addr.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thess A., Grund S., Mui B.L., Hope M.J., Baumhof P., Fotin-Mleczek M., Schlake T. Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015;23:1456–1464. doi: 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phua K.K.L.L., Leong K.W., Nair S.K. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J. Control. Release. 2013;166:227–233. doi: 10.1016/j.jconrel.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bouchlaka M.N., Moffitt A.B., Kim J., Kink J.A., Bloom D.D., Love C., Dave S., Hematti P., Capitini C.M. Human Mesenchymal Stem Cell-Educated Macrophages Are a Distinct High IL-6-Producing Subset that Confer Protection in Graft-versus-Host-Disease and Radiation Injury Models. Biol. Blood Marrow Transplant. 2017;23:897–905. doi: 10.1016/j.bbmt.2017.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Rosa F., Pabst R. The bone marrow: a nest for migratory memory T cells. Trends Immunol. 2005;26:360–366. doi: 10.1016/j.it.2005.04.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.