

Temperate bacteriophages can integrate their genomes into the bacterial host chromosome and exist as prophages whose gene products play key roles in bacterial fitness and interactions with eukaryotic host organisms. Most bacterial chromosomes contain “cryptic” prophages that have lost genes required for production of phage progeny but retain genes of unknown function that may be important for regulating bacterial host physiology. This study provides such an example, where a cryptic-prophage-encoded product can perform multiple roles in the bacterial host and influence processes, including metabolism, cell division, and susceptibility to phage infection. Further functional characterization of cryptic-prophage-encoded functions will shed new light on host-phage interactions and their cellular physiological implications.
KEYWORDS: Hfq, cell division, prophages, small RNA, small protein, sugars
ABSTRACT
Bacterial genomes harbor cryptic prophages that have lost genes required for induction, excision from host chromosomes, or production of phage progeny. Escherichia coli K-12 strains contain a cryptic prophage, Qin, that encodes a small RNA, DicF, and a small protein, DicB, that have been implicated in control of bacterial metabolism and cell division. Since DicB and DicF are encoded in the Qin immunity region, we tested whether these gene products could protect the E. coli host from bacteriophage infection. Transient expression of the dicBF operon yielded cells that were ∼100-fold more resistant to infection by λ phage than control cells, and the phenotype was DicB dependent. DicB specifically inhibited infection by λ and other phages that use ManYZ membrane proteins for cytoplasmic entry of phage DNA. In addition to blocking ManYZ-dependent phage infection, DicB also inhibited the canonical sugar transport activity of ManYZ. Previous studies demonstrated that DicB interacts with MinC, an FtsZ polymerization inhibitor, causing MinC localization to midcell and preventing Z ring formation and cell division. In strains producing mutant MinC proteins that do not interact with DicB, both DicB-dependent phenotypes involving ManYZ were lost. These results suggest that DicB is a pleiotropic regulator of bacterial physiology and cell division and that these effects are mediated by a key molecular interaction with the cell division protein MinC.
IMPORTANCE Temperate bacteriophages can integrate their genomes into the bacterial host chromosome and exist as prophages whose gene products play key roles in bacterial fitness and interactions with eukaryotic host organisms. Most bacterial chromosomes contain “cryptic” prophages that have lost genes required for production of phage progeny but retain genes of unknown function that may be important for regulating bacterial host physiology. This study provides such an example, where a cryptic-prophage-encoded product can perform multiple roles in the bacterial host and influence processes, including metabolism, cell division, and susceptibility to phage infection. Further functional characterization of cryptic-prophage-encoded functions will shed new light on host-phage interactions and their cellular physiological implications.
INTRODUCTION
Bacteriophages are abundant in the environment, with an estimated 1031 bacteriophage (phage) particles, and outnumber their bacterial hosts by a factor of 10 to 1 (1, 2). They are found in all ecosystems that harbor bacteria and play a vital role in driving bacterial evolution (3). Based on their life cycles, phages can be broadly classified as virulent or temperate. Virulent phages use a lytic life cycle, wherein they infect bacterial hosts, use the host cell’s resources to make more phage particles, and ultimately lyse the cell to release progeny virions into the environment. Temperate phages can grow using a lytic life cycle or, alternatively, can undergo lysogeny, integrating their genomes at a specific attachment site in the host chromosome and remaining stably associated with the host. A bacterium with an integrated phage genome (prophage) is called a lysogen. Changes in host metabolic conditions or external environmental triggers can induce the prophage, which then excises out of the host chromosome and resumes a lytic life cycle (4, 5).
Nearly half of all sequenced bacterial genomes have been found to contain at least one prophage, with many genomes containing multiple prophages (6). Lysogeny comes at a cost to the bacterial host due to the extra burden of replication of prophage DNA and the threat of lysogen induction, which is lethal to the host cell. On the other hand, there are many well-documented examples of lysogenic conversion, where prophage-encoded products confer new and advantageous characteristics on the host (7, 8). Many prophages carry virulence genes that contribute to the pathogenicity of a bacterial host, e.g., phage-encoded Shiga toxin in Escherichia coli O157 strains (9), phage-encoded diphtheria toxin in Corynebacterium diphtheriae (10), and neurotoxin in Clostridium botulinum (11). Prophage-encoded toxins, host cell invasion factors, and serum resistance proteins promote various aspects of the infection processes carried out by bacterial pathogens (7). Another well-documented benefit of prophages is superinfection immunity. In a mixed population of lysogens and other bacteria, if a prophage becomes induced and lyses a host cell, the active phage particles released infect and lyse only the nonlysogens, while the lysogens are protected by the prophage-encoded immunity functions (5). Less well characterized at a mechanistic level are examples of prophage genes that increase the host’s ability to grow under different environmental or stress conditions (12–14).
Growing evidence suggests that in many genomes, most of the resident prophages are cryptic (defective), having suffered mutations that leave them unable to excise from the host chromosome, lyse host cells, or produce infectious phage particles (15–18). A recent study identified and characterized orthologous prophages that were integrated into an ancestral host genome and subsequently passed down vertically with the host chromosome in E. coli and Salmonella (16). Most of these prophages showed evidence of loss of large portions of the original prophage genome, but the remaining genes were under purifying selection (16). These results suggest that certain prophage genes are selected for during host evolution because they encode products that are advantageous to the host under some condition. The cryptic prophages of E. coli K-12 have been associated with several host phenotypes, including biofilm formation, stress sensitivity, and antibiotic resistance (19). To understand the molecular basis of cryptic-prophage-associated phenotypes, functional characterization of prophage genes is essential.
In E. coli K-12, the cryptic prophage Qin carries an operon encoding a small protein, DicB, and a small RNA (sRNA), DicF, that both function as cell division inhibitors (20–25). The sRNA DicF represses ftsZ translation by directly base pairing with the ftsZ mRNA near the Shine-Dalgarno sequence (24, 25). DicF also regulates other mRNAs that encode a variety of regulatory and metabolic functions (25). The 62-amino-acid protein DicB inhibits cell division by directly interacting with MinC and recruiting it to the septum via interactions with the septal protein ZipA, where MinC stimulates depolymerization of the Z ring, resulting in cell filamentation (23, 26–28). The region immediately upstream of the dicBF operon includes dicA and dicC and is similar in sequence and structural arrangement to the lambdoid phage immunity locus. DicA is analogous to the P22 phage C2 repressor and DicC to the P22 Cro repressor (29). DicA represses the dicBF operon promoter (which is similar to the λ phage PL promoter), and the natural conditions leading to induction of the operon are unknown (29). DicB and DicF are conserved in many strains of E. coli, and, interestingly, many pathogenic strains of E. coli possess multiple cryptic prophages including dicBF operons (25, 30, 31).
In this study, we identified a role for the E. coli dicBF operon in resistance to bacteriophage infection. Short-term expression of the dicBF operon promotes E. coli resistance to λ phage infection. The resistance phenotype is primarily attributable to DicB. DicB does not affect λ phage adsorption to host cells. Instead, our results suggest that DicB inhibits injection of λ DNA into the cytoplasm through the inner membrane proteins ManYZ, which are components of the mannose phosphotransferase system. Consistent with an effect of DicB on ManYZ activity, we found that growth of dicB-expressing cells on minimal medium with mannose as the sole carbon source was strongly inhibited. Our results suggest that products encoded by the dicBF operon, found in cryptic prophages in many E. coli and Shigella strains, can impact bacterial physiology, including by altering the cells’ susceptibility to bacteriophage infection. We postulate that this may be a common reason why certain cryptic-prophage genes are retained in host chromosomes.
RESULTS
Transient induction of the dicBF operon protects against λ phage infection.
The region of the Qin prophage containing the dicBF operon (Fig. 1A) resembles the immunity regions of P22 and other lambdoid phages (29). While functions have not been identified for most of the products of the dicBF operon, DicB (a small protein) and DicF (a small RNA) have been shown to inhibit cell division (20, 22–25). We showed previously that DicF posttranscriptionally regulates a variety of genes involved in cell division, growth, and metabolism (25). Given their positions in the immunity region of the prophage genome and the fact that the characterized gene products impact cell physiology, we hypothesized that products of the dicBF operon could cause changes in the host cell that promote resistance to phage infection. We tested this by comparing phage infections of control and dicBF-expressing cells. Though some recent studies have found higher expression of the sRNA DicF under microaerobic or anaerobic conditions (31, 32), we have not yet found a condition that stimulates production of detectable levels of DicF or the polycistronic dicBF mRNA from the chromosomal locus in our strain. Thus, to test our hypothesis, we used an inducible expression system we described previously (25), where the dicBF operon promoter was replaced with a Plac promoter at the native locus. In addition to the Plac-dicBF strain, we used strains with deletions of different genes in the operon (Fig. 1B).
FIG 1.

Transient induction of the dicBF operon protects against λvir infection. (A) dicBF locus on the Qin prophage of E. coli K-12. The red arrow indicates where Plac is inserted on the chromosome, replacing the native dicBp promoter. (B) EOP is defined as the λvir titer on the test strain divided by the λvir titer on the control strain. The strains used in the experiment were DJ480 (control), DB240 (Plac-dicBF), PR165 (Plac-dicBF ΔdicB), DB247 (Plac-dicBF ΔdicF), PR164 (Plac-dicBF ΔydfD::kan), and DB248 (Plac-dicBF ΔdicF ΔdicB). All the strains were grown to the same state of growth with the dicBF operon induced with 0.5 mM IPTG for 60 min. The inducer was washed off, and the strains were resuspended in TM buffer, infected with λvir, and plated to calculate the titer. The error bars were calculated as standard deviations of values from three biological replicates. (C) ECOI is calculated as follows: (number of infectious centers per milliliter from the test strain) × 100/(number of infectious centers per milliliter from the control strain). The strains used in the experiment were DJ480 (control), DB240 (Plac-dicBF), DB243 (Plac-dicBF ΔdicB), DB247 (Plac-dicBF ΔdicF), and DB248 (Plac-dicBF ΔdicF ΔdicB). The cells were grown with induction of the dicBF operon with 0.5 mM IPTG and infected with λvir at an MOI of 0.1. The unadsorbed phages were removed, and the phage-host complex was added to phage-sensitive cells (DJ480) and plated onto LB agar for plaque counting (infectious centers). The error bars were calculated as standard deviations of values from three biological replicates.
We measured phage infection of the strains by efficiency-of-plaquing (EOP) assays, initially using phage λ. In this assay, the titer of the phage is determined in all bacterial strains, and the titer (in PFU per milliliter) is calculated for each strain. The EOP is defined as the phage titer on the test strain divided by the phage titer on the control strain. The control strain lacked the Plac promoter. Strains with the Plac promoter driving dicBF expression were exposed to isopropyl-β-d-thiogalactopyranoside (IPTG) for 60 min. Then, the strains were infected with λvir (a λ mutant that grows only via the lytic (and not the lysogenic) cycle during infection of host cells) and plated to determine its titer as described in Materials and Methods. The EOP for the Plac-dicBF strain was 0.04 (Fig. 1B), meaning that the rate of infection of the dicBF-expressing strain was only 4% relative to the control strain. This result suggested that transient expression of the dicBF operon conferred resistance to infection by λvir. To further characterize the basis for this phenotype, we deleted dicF, dicB, and ydfD, singly and in combination, because previous studies identified growth or cell division phenotypes associated with these genes (23, 25, 33). The phenotypes of deletion mutants demonstrated that dicB played the most prominent role in the resistance phenotype (Fig. 1B). Deletion of dicB alone or dicB in combination with dicF restored the EOP of λvir to nearly that of the control. In contrast, deletion of dicF or ydfD alone had a minimal effect on the resistance phenotype (Fig. 1B).
We observed previously that insertion of the Plac promoter upstream of the dicBF locus leads to low-level expression of the operon even in the absence of the inducer (25). We checked the EOP of λvir on the Plac-dicBF strain with and without induction. In the absence of IPTG, the EOP was 58% compared with 4% in the presence of inducer (see Fig. S1 in the supplemental material). This result is consistent with leaky expression from the Plac promoter. We also carried out infections using the same host strains with wild-type λ phage and saw similar results for EOP on control, dicBF-expressing, and deletion mutant strains (see Fig. S2 in the supplemental material).
Because previous studies showed that ectopic expression of the dicBF operon impairs growth of the host strain, we reasoned that poor growth of test strains could influence the results of EOP assays. To more accurately assess the outcome of a phage infection on cells expressing the dicBF operon, we conducted center of infection (COI) assays. For this assay, strains (Fig. 1C) were induced with 0.5 mM IPTG, and λvir infection was carried out at a multiplicity of infection (MOI) of 0.1. After adsorption of the phage to the test strains, the unadsorbed phages were removed by washing, and the infected test cells were diluted and mixed with the phage-sensitive control strain. Productive infections of the test strain were detected as plaques (centers of infection) on the phage-sensitive control strain. The efficiency of λvir forming centers of infection (ECOI) was calculated as follows: (number of centers of infection per milliliter from the test strain) × 100/(number of centers of infection per milliliter from the control strain). The ECOI for λvir on Plac-dicBF cells was 3% (Fig. 1C). This result is similar to the results of EOP assays (Fig. 1B), suggesting that the growth characteristics of the Plac-dicBF test strain did not impact the experimental outcome. Deletion of dicB, alone or in combination with dicF, restored the ECOI to ∼80%. The ΔdicF strain gave an ECOI of 5% (Fig. 1C). These results are again consistent with our EOP experiments (Fig. 1B), implicating DicB as the major player in the phage resistance phenotype.
The dicBF operon promotes resistance to λ, but not other phages.
To determine if transient expression of dicBF conferred resistance to other phages, we conducted infection experiments using control and Plac-dicBF strains with nine different lytic and temperate phages (Fig. 2). In this experiment, the EOP of λvir on the Plac-dicBF strain was 0.016, or 1.6%, compared to the control strain, which was the lowest of the nine phages tested (Fig. 2). Partial resistance was observed for the T3 phage, which had an EOP of 0.14 on Plac-dicBF cells. However, the EOPs for the remaining seven phages, including phages ϕ80 and HK97, which are closely related to λ, were similar to that for the control cells (Fig. 2). These results suggest that DicB does not provide a broad spectrum of resistance against bacteriophages.
FIG 2.
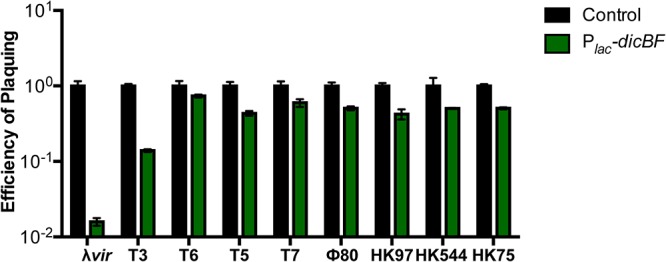
The dicBF operon confers resistance against λ phage but not other phages. For each of the nine phages, the titer (in PFU per milliliter) was calculated by infection of DJ480 (control) and DB240 (Plac-dicBF) cells. The cells were prepared for infection, and the EOP was calculated for each phage as described for Fig. 1B. The error bars were calculated as standard deviations of values from three biological replicates.
Effect of dicBF expression on λ phage growth.
The classical experiment to study the growth cycle of phages in bacteria is the one-step growth curve, as described by Ellis and Delbrück (34). They observed a latent period, when the numbers of phages recovered from infected cells remained low as new phage particles were being synthesized inside the host cell. After the latent period is the “burst,” when the numbers of infectious phage particles increase rapidly as the phage life cycle is completed and cells are lysed to release mature progeny. We conducted one-step growth curves for λvir on control and dicBF-expressing strains, essentially as described above for ECOI experiments over a time course following infection. λvir was added at an MOI of 0.1 to control and Plac-dicBF cells resuspended in TM buffer (10 mM Tris-HCl and 10 mM MgSO4). After phage adsorption, the cells were washed to remove unadsorbed phages, and the phage-host complexes were diluted in fresh medium with IPTG (see Materials and Methods). At each time point, the number of infectious phage particles in each culture was calculated by removing samples and plating for PFU on a phage-sensitive control strain.
As expected based on previous results (Fig. 1B and C), the ECOI for λvir on Plac-dicBF cells was reduced by almost 2 log units compared with the control strain at the early time points, and the reduced numbers of phages produced by Plac-dicBF cells persisted across the phage growth curve (Fig. 3). The latent period for Plac-dicBF cells (75 min) was ∼10 min longer than for control cells (65 min) (Fig. 3). The calculated burst sizes were 343 phages/Plac-dicBF cell compared to 169 phages/control cell. This increase in burst size in dicBF-expressing cells is likely due to filamentation of cells caused by DicB and DicF. It has been shown previously that filamenting cells produce more phages than normal-size cells (35, 36). Importantly, we observed that the ∼3% of phages that escaped DicB-mediated resistance followed a growth curve similar to that of phages growing on control cells. Collectively, these data led us to hypothesize that DicB affects an early step of the phage life cycle, like adsorption or DNA injection, since phages that escape this DicB effect complete a relatively normal life cycle.
FIG 3.
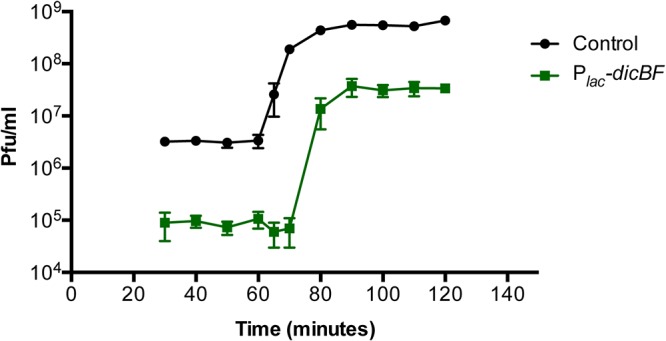
One-step growth curve of λvir on control and Plac-dicBF cells. The cells were grown with induction of the dicBF operon, and infection was carried out at an MOI of 0.1, similar to the center of infection assay. After removing unadsorbed phages, the cells were diluted in LB medium (with IPTG to induce the dicBF operon) and incubated at 37°C for the entire duration of the growth curve. At each time point, starting at 30 min from the start of infection, samples were removed and added to the phage-sensitive strain (DJ480) and plated to count plaques. The burst size was calculated as follows: (phage titer at 100 min − initial titer at 30 min)/initial titer at 30 min. The latent period was calculated as the time at the midpoint of the exponential phase of the curve. The error bars were calculated as standard deviations of values from three biological replicates.
The dicBF operon does not affect phage adsorption to host cells.
To test if expression of the dicBF operon affects the first step of phage infection, we tested the ability of λvir to adsorb to host cells expressing the operon. During the ECOI experiment, once phage infection was carried out with cells in TM buffer at an MOI of 0.1 and incubated at 37°C for 10 min, the phage-cell mixture was centrifuged and the supernatant containing the unadsorbed phages was removed. The titer of the supernatant was determined on phage-sensitive control cells by standard plaque assay (residual titer). The control titer was calculated using the same procedure described above, with phages added to TM buffer instead of bacterial cells in the first step of the experiment. The percent adsorption was calculated as follows: (control titer − residual titer) × 100/control titer.
The adsorption of λvir to strains expressing the dicBF operon was the same as adsorption to the control strain (Table 1). Notably, while λvir and HK97 both adsorb to the same outer membrane receptor, LamB (37, 38), the effects of dicBF expression on the EOP of these two phages are significantly different, with a reduced EOP only for λvir (Fig. 2). These observations strongly suggest that DicB does not affect the phage life cycle at the step of adsorption to host cells.
TABLE 1.
The dicBF operon does not affect phage adsorption to host cellsa
| Strain descriptionb | % adsorption | ±SE |
|---|---|---|
| Control | 99 | 0.22 |
| Plac-dicBF | 99 | 0.25 |
| Plac-dicBF ΔdicB | 99 | 0.69 |
| Plac-dicBF ΔdicF | 99 | 0.23 |
| Plac-dicBF ΔdicF ΔdicB | 99 | 0.15 |
The percent adsorption was calculated as follows: (control titer − residual titer) × 100/control titer. The standard error was calculated as the standard deviation of values from three biological replicates.
The strains used in this experiment were DJ480 (control), DB240 (Plac-dicBF), DB243 (Plac-dicBF ΔdicB), DB247 (Plac-dicBF ΔdicF), and DB248 (Plac-dicBF ΔdicF ΔdicB). The cells were infected at an MOI of 0.1 with λvir, which was allowed to adsorb for 10 min at 37°C. After adsorption, the samples were centrifuged, and the supernatant containing the unadsorbed phages was measured on phage-sensitive cells to quantify the titer (residual titer). The control titer was calculated by carrying out the assay with TM buffer without bacterial cells.
Recombinant λ phages with the host range region of ϕ80 are not affected by DicB.
The genomes of λ phage and ϕ80 (a lambdoid phage) have strikingly similar organizations, allowing easy construction of recombinant phages (39, 40). One prominent difference between λ and ϕ80 is their use of different outer and inner membrane receptors. λ uses LamB (outer membrane) and ManYZ (inner membrane) for adsorption and DNA injection, respectively (41–45), while ϕ80 uses FhuA (outer membrane) and the TonB complex (inner membrane) (39, 46, 47). The phage genes encoding determinants for utilization of host outer and inner membrane receptors are located in the host range region of lambdoid phage genomes. Our results so far suggest that the DicB-dependent phage resistance phenotype is not due to an effect on adsorption to the outer membrane receptor but might be mediated at another early step of infection, such as injection of the phage genome through the inner membrane receptor. To test this idea, we measured phenotypes of control and dicBF-expressing cells challenged with recombinant λ phage containing the host range region of ϕ80 (λh80). The λh80 phages carry most of the wild-type λ genome but have an altered host range region specifying use of the ϕ80 outer and inner membrane receptors. To confirm this, we tested the plaquing ability of λh80 phages on wild-type, ΔfhuA, ΔtonB, and ΔmanXYZ E. coli strains. As expected, the λh80 phages, like ϕ80, did not plaque on ΔfhuA and ΔtonB strains but formed normal plaques on the ΔmanXYZ strain (see Table S3 in the supplemental material). Next, we carried out EOP assays using λvir and a panel of recombinant phages with the host range of λ or ϕ80 (see Table S1 in the supplemental material) on control and Plac-dicBF cells (Fig. 4). We hypothesized that if DicB mediates resistance to λ phage by impairing injection of phage DNA across the cytoplasmic membrane, then phages with the host range of λ would remain inhibited by DicB, whereas λh80 phages with altered inner membrane receptor specificity would not be impacted by DicB. The results of the EOP assays demonstrate that phages with the λ host range remained sensitive to DicB-mediated inhibition while λh80 phages had similar EOP on Plac-dicBF and control cells (Fig. 4). Together with our previous results, this observation suggests that DicB-mediated resistance acts at the level of the inner membrane receptor ManYZ used for λ phage DNA injection into the cytoplasm of E. coli. We note that the panel of phages that we tested in this experiment had other genetic differences, aside from the different host ranges (see Table S1). Only the host range was correlated with susceptibility to DicB-mediated resistance.
FIG 4.
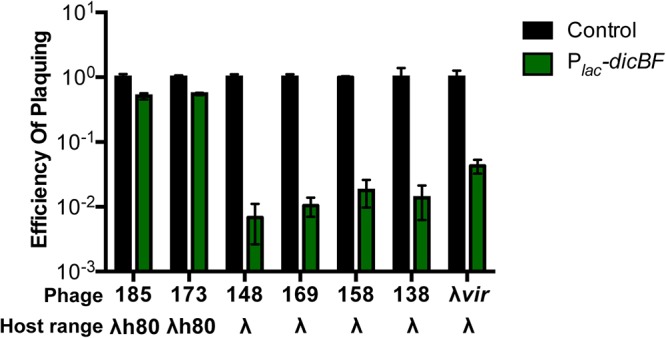
λ phage with the host range of ϕ80 is not affected by DicB. Recombinant λ phages with either the λ or ϕ80 host range were plaqued on DJ480 (control) or DB240 (Plac-dicBF) cells (see Table S1 for phage genotypes). The cells were prepared for infection, and the EOP was calculated for each phage as described for Fig. 1B. The error bars were calculated as standard deviations of values from three biological replicates.
Phage 434 (44) is another phage that uses ManYZ for injection of DNA through the cytoplasmic membrane (Fig. 5A). Previous studies have shown that manXYZ deletion mutants (also known as pel mutants) were resistant to infection by λ and phage 434, but not ϕ80 (44). To further test our hypothesis that DicB inhibits phage infection at the level of DNA entry through ManYZ, we tested the abilities of λvir, phage 434, and ϕ80 to infect control and dicBF-expressing cells in manXYZ+ and ΔmanXYZ backgrounds. We verified that λ, phage 434, and ϕ80 plaqued as expected on the wild-type strain and strains with mutations in specific receptors (see Table S4 in the supplemental material). As shown in Fig. 2, ϕ80 plaquing efficiency is not impacted by expression of the dicBF operon in a manXYZ+ background. The ΔmanXYZ mutant host also supported wild-type EOP for ϕ80 plaquing regardless of whether dicBF was expressed (Fig. 5B). For λvir, the EOP on Plac-dicBF cells was 4% relative to control in manXYZ+ cells, whereas the ΔmanXYZ host did not support λvir growth (Fig. 5B). The pattern of growth for phage 434 was very similar to that of λvir, with a reduced EOP of ∼10% on Plac-dicBF cells in the manXYZ+ host and no plaques on the ΔmanXYZ host (Fig. 5B). The results of this experiment are consistent with the hypothesis that DicB inhibits the use of the mannose phosphotransferase system (PTS) proteins ManYZ as an inner membrane receptor for productive phage infection.
FIG 5.

Phage 434 plaquing on ManYZ+ strains is inhibited by the dicBF operon. (A) Outer and inner membrane receptor specificities of phages λvir, 434, and ϕ80. (B) An EOP assay was carried out by preparing cells and calculating the titers of phages on the different strains as described in the legend to Fig. 1B. The strains used in the experiment were DJ624 (control), DB240 (Plac-dicBF), PR187 (control ΔmanXYZ::kan), and PR191 (Plac-dicBF ΔmanXYZ::kan). The EOP of phages for each strain was calculated with respect to the control strain in the same background. The error bars were calculated as standard deviations of values from three biological replicates. The stars represent strains for which plaques could not be counted accurately, since the plaques were smaller than pinpoints. This has been observed before for λ plaques on ΔmanXYZ mutant strains (44).
Growth of dicBF-expressing cells is inhibited on plates with mannose as the C source.
Our previous results pointed to the small protein DicB inhibiting the activity of the mannose transporter ManYZ proteins with regard to DNA uptake during phage infection. To test whether DicB inhibits the functions of these proteins more broadly, we checked the growth of dicBF-expressing cells on mannose as the sole C source. For this experiment, we used control, Plac-dicBF, and Plac-dicBF ΔdicB strains. The strains were streaked on M63 minimal plates with different sugars with or without 0.025 mM IPTG (to induce dicBF expression) and incubated for 44 h at 37°C. Growth was scored using the parent strain’s growth as a reference (see the legends to Table 2 and to Fig. S3 in the supplemental material and examples of growth phenotypes in Fig. S3). In the absence of inducer, all the strains had nearly normal growth on the different sugars used. When dicBF expression was induced using 0.025 mM IPTG, we observed growth inhibition of Plac-dicBF cells on mannose and glucosamine, but not on glucose (see Fig. S3), fructose, or N-acetylglucosamine (Table 2). On glucosamine plates, we observed apparent suppressors of the Plac-dicBF strain on the plate with IPTG, which resulted in uneven growth of dense and light streaks on that plate. Comparing the single-colony sizes on the glucose-plus-IPTG and glucosamine-plus-IPTG plates demonstrates clearly that growth of the Plac-dicBF strain is inhibited on glucosamine (see Fig. S3). Deletion of dicB relieved the growth inhibition on mannose and glucosamine (Table 2; see Fig. S3). Both mannose and glucosamine sugars are transported via the ManXYZ transporter in E. coli (48–50). These results demonstrate that DicB affects growth specifically on substrates of ManYZ. Growth on sugars that are transported by other PTS proteins was unaffected. These data suggest that DicB impacts at least two different functions of ManYZ: uptake of phage DNA during infection and transport of sugar substrates.
TABLE 2.
Growth of dicBF-expressing cells is inhibited on plates with mannose as the C source
| C source | Growtha |
|||||
|---|---|---|---|---|---|---|
| Control |
Plac-dicBF |
Plac-dicBF ΔdicB |
||||
| Uninduced | Induced | Uninducedb | Induced | Uninducedb | Induced | |
| Mannose | +++ | +++ | ++ | + | +++ | +++ |
| Glucose | +++ | +++ | +++ | +++ | +++ | +++ |
| Glucosamine | +++ | +++ | ++ | + | +++ | +++ |
| Fructose | +++ | +++ | +++ | +++ | +++ | +++ |
| N-Acetylglucosamine | +++ | +++ | +++ | +++ | +++ | +++ |
The strains were streaked on M63 minimal medium with 0.2% sugars as the C source with or without 0.025 mM IPTG and incubated for 44 h at 37°C. The strains used were DJ624 (control), DB240 (Plac-dicBF), and PR165 (Plac-dicBF ΔdicB). +++ indicates growth of the control strain on the respective sugars; ++ and + indicate decremental growth based on the sizes of the single colonies on the plate compared to the control strain on that sugar (for examples of growth phenotypes corresponding to each score, see Fig. S3).
The Plac promoter is leaky, and we suspect low-level expression of the dicBF operon even at 0 mM IPTG.
MinC mutants that do not interact with DicB lose the phage resistance and sugar phenotypes.
The only characterized activity of DicB is inhibition of cell division (20, 51). The mechanism by which DicB impacts cell division requires a protein-protein interaction with MinC, one of the proteins involved in controlling septal ring placement in E. coli. MinC is an inhibitor of FtsZ polymerization, and normally MinC concentrations are highest at cell poles, so that septum formation is inhibited at polar sites and directed instead to midcell (52). Previous work demonstrated that DicB interacts with MinC and brings it to midcell via an interaction with ZipA, a septal protein (28). DicB-mediated localization of MinC to the cell center inhibits FtsZ polymerization and promotes filamentation (27, 28). To determine if the DicB-MinC interaction is necessary for the DicB-dependent phenotypes we found in this study, we constructed mutant strains that produce MinC proteins that are defective for interaction with DicB. We used two different MinC mutants: MinC R172A, which interacts weakly with DicB, and MinC E156A, which does not interact with DicB (53). In strains expressing these minC alleles, DicB had a modest (MinC R172A) or no (MinC E156A) impact on cell division, consistent with their reduced binding to DicB. We used MinC E156A and R172A mutant hosts to test whether the DicB-mediated phage resistance or sugar growth phenotype required the DicB-MinC interaction.
As observed previously, in the wild-type minC+ background, dicBF-expressing cells showed reduced EOP for λvir compared to control cells (Fig. 6). However, in the MinC R172A (reduced binding to DicB) strain, the resistance phenotype was diminished; dicBF expression in this host gave an EOP of 12% compared to the control strain. In the MinC E156A (abrogated binding to DicB) background, the EOP of λvir on dicBF-expressing cells was very similar to that of the control strain (Fig. 6). These results suggested that the DicB-MinC interaction is required for DicB-mediated resistance to λ phage infection. The same strains were grown on M63 minimal plates with different sugars with or without 0.025 mM IPTG to induce the dicBF operon. As shown above, in the wild-type minC+ background, expression of dicBF inhibited growth on plates with mannose and glucosamine, but not on plates with glucose (Table 3). In contrast, dicBF expression in minC mutant strains (E156A and R172A) did not inhibit growth on any of the sugars tested (Table 3). Collectively, these data indicate that the new DicB-associated phenotypes we have identified—phage resistance and inhibition of growth on sugars that are transported by ManYZ—require the previously defined molecular mechanism of DicB interaction with the host protein MinC.
FIG 6.
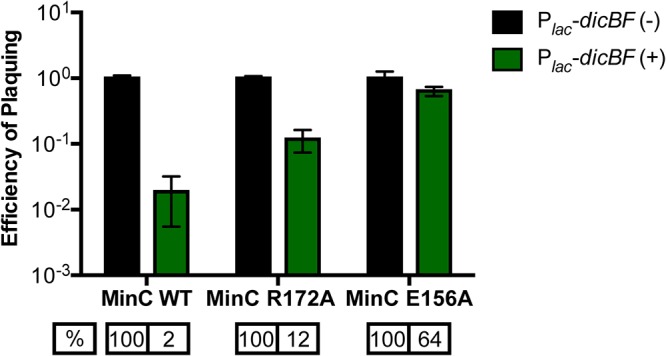
MinC mutants that do not interact with DicB lose the phage resistance effect. The cells were grown with induction of the dicBF operon with 0.5 mM IPTG and infected with λvir, and the EOP was calculated as described in the legend to Fig. 1B. The EOP of λvir for each strain was calculated with respect to the control strain in the same background. The strains used in the experiment were DJ624 (control), DB240 (Plac-dicBF), PR181 (control minC R172A), PR183 (Plac-dicBF minC R172A), PR180 (control minC E156A), and PR182 (Plac-dicBF minC E156A). The error bars were calculated as standard deviations of values from three biological replicates. WT, wild type.
TABLE 3.
MinC mutants that do not interact with DicB regain the ability to grow on mannose and glucosamine
| Strain description | Growtha |
|||||
|---|---|---|---|---|---|---|
| Glucose |
Mannose |
Glucosamine |
||||
| Uninduced | Induced | Uninduced | Induced | Uninduced | Induced | |
| Control | +++ | +++ | +++ | +++ | +++ | +++ |
| Plac-dicBF | +++ | +++ | ++ | + | ++ | + |
| Control minC R172A | +++ | +++ | +++ | +++ | +++ | +++ |
| Plac-dicBF minC R172A | +++ | +++ | +++ | +++ | +++ | +++ |
| Control minC E156A | +++ | +++ | +++ | +++ | +++ | +++ |
| Plac-dicBF minC E156A | +++ | +++ | +++ | +++ | +++ | +++ |
The strains were streaked on M63 minimal medium plates with 0.2% sugars and 0.025 mM IPTG to induce the dicBF operon. The plates were incubated for 44 h at 37°C. The strains used in the experiment were DJ624 (control), DB240 (Plac-dicBF), PR181 (control minC R172A), PR183 (Plac-dicBF minC R172A), PR180 (control minC E156A), and PR182 (Plac-dicBF minC E156A). Growth on the plates was scored as described for Table 2. The Plac promoter is leaky, and we suspect low-level expression of the dicBF operon even at 0 mM IPTG.
DISCUSSION
The existence of cryptic or defective prophages on bacterial chromosomes was discovered long ago (4), but their potential beneficial functions for host cells are still coming to light. In part, this is because we do not know the functions of the majority of genes carried on these prophages. In this study, we have identified a new functional role for the cryptic-prophage-encoded protein DicB in E. coli K-12. We showed that induction of the dicBF operon makes cells resistant to infection by phages that use the ManYZ PTS proteins as inner membrane receptors for DNA injection (Fig. 1 and 5). DicB, a 62-amino-acid protein encoded by the dicBF operon, plays the primary role in conferring this phage resistance phenotype (Fig. 1). Our results are consistent with the model that DicB inhibits phage DNA injection through the mannose transporter proteins ManYZ (Fig. 4, 5, and 7). The DicB effect on ManYZ also inhibits ManYZ-dependent transport of sugar substrates (Table 2), suggesting that DicB affects the general structure or function of these transport proteins. Previous work demonstrated that DicB inhibits cell division by interacting with and affecting the localization and activity of the cell division proteins MinC and FtsZ (23, 26–28). In this study, we found that the DicB-associated phage resistance and sugar utilization phenotypes are dependent on DicB-MinC interactions (Fig. 6 and Table 3).
FIG 7.

Working model for regulation of host cell physiology by DicF and DicB. The small protein DicB and the sRNA DicF of the dicBF operon are encoded on the Qin cryptic prophage of E. coli K-12. The physiological conditions that induce expression of the operon are not yet known. However, when ectopically expressed, DicB and DicF regulate multiple processes in the cell. DicF inhibits cell division and metabolism genes posttranscriptionally, including manXYZ, encoding the mannose PTS. DicB interacts with MinC, which is a negative regulator of FtsZ polymerization. The DicB-MinC complex interacts with the septal ring component ZipA and causes depolymerization of FtsZ at the septum, which leads to filamentation of cells. In this study, we showed that DicB confers on the host cell resistance to phages that use the inner membrane proteins ManYZ to inject their DNA into the cell. DicB also affects growth on mannose and glucosamine sugars, which are transported into the cell via ManXYZ. Interaction with MinC is necessary for DicB to promote the phage resistance and sugar phenotypes.
Prior to this work, the only known function of DicB was inhibition of cell division. DicB directly interacts with MinC of the Min system, which consists of the proteins MinC, MinD, and MinE, which play roles in the spatial positioning of the FtsZ ring at midcell for cell division. MinC is the only component of the Min system required for DicB-dependent cell division inhibition (23). MinC is a negative regulator of FtsZ polymerization, and in E. coli, MinC oscillates between the two cell poles (driven by MinD and MinE) in order to inhibit Z ring assembly at the poles (54, 55). However, when DicB is expressed, a DicB-MinC complex is formed which interacts with the septal ring component ZipA and stimulates Z ring depolymerization at midcell, leading to cell filamentation (28). Both activities of DicB, cell division inhibition and the ManYZ inhibition phenotypes reported in this study, involve interaction with MinC. These results imply a previously unsuspected link between the Min system or other components of the cell division machinery and the mannose PTS. A few studies have examined localization of various PTS proteins. The general PTS proteins, EI and HPr, were found to localize primarily to cell poles (56, 57). Localization of EII sugar permeases has been less studied, but there is recent evidence that these proteins cluster together around the cell membrane (58). It will be interesting in future work to explore the subcellular localization of ManYZ and to examine if or how it is impacted by MinC and DicB.
It is clear that active prophages can protect their hosts from superinfection by other phages (59, 60), a phenotype called superinfection exclusion. One mechanism of superinfection exclusion is mediated by prophage-encoded proteins that block entry of a superinfecting phage’s DNA by mechanisms that are not well defined but may involve modifying the activity or function of inner membrane receptors (38, 59, 61–64). Our results suggest that in at least some cases defective prophages can play similar roles in protecting their hosts from phage infection. It has been speculated that another beneficial role of defective prophages could be encoding functions that are important for host cell adaptation to stress conditions. In a study by Wang et al. (19), defective prophages of E. coli K-12 were shown to increase resistance to environmental stresses, like oxidative stress and osmotic stress, and to certain antibiotics, like quinolones and beta-lactams. The study reported that Δqin derivatives of the parent strain were more sensitive to beta-lactam antibiotics and that ΔdicB strains showed greater sensitivity to azlocillin and nalidixic acid (19). We constructed Δqin and ΔdicB strains and examined their sensitivities to nalidixic acid and ampicillin, and we found no differences in sensitivity between parent strains and mutants (data not shown). It is possible that phenotypes vary with strain background—our strains are MG1655 derivatives, and Wang et al. used BW25113 (19).
Identifying the signals or conditions that induce prophage genes will be key to understanding their physiological roles in host cells. We have exposed our strains to various conditions that are known to induce prophage gene expression, including DNA damage, starvation, and exposure to antibiotics, and have not yet identified conditions that substantially induce transcription from the native dicBF promoter (data not shown). Another study (32) reported that E. coli K-12 MG1655 cells undergo DicF-dependent filamentation under anaerobic conditions (growth in large-volume anaerobic fermentors). The authors of the study suggest that the stability of DicF is differentially regulated so that it is more stable under anaerobic growth conditions and degrades faster under aerobic conditions. A very recent study found that four DicF orthologs encoded by different prophages in E. coli O157:H7 are produced under microaerobic growth conditions (31). These DicF sRNAs promote low-oxygen-responsive virulence gene expression via base pairing-mediated regulation of a key virulence transcription factor. These studies suggest that in at least some E. coli strain backgrounds, oxygen is an important signal for modulation of dicBF operon transcription or DicF mRNA stability. However, we have not observed any DicF- or DicB-mediated filamentation of MG1655 cells grown in small-volume LB liquid cultures in an anaerobic chamber (data not shown), so we speculate that additional signals or conditions might contribute to dicBF operon expression in our host strain background.
Previous studies from our laboratory characterized the mRNA target regulon of DicF (25). In addition to the previously discovered DicF target ftsZ mRNA, we found that DicF base pairs with and represses translation of xylR, pykA, and manXYZ mRNAs, encoding the xylose repressor, pyruvate kinase, and mannose PTS components, respectively (25, 65). Thus, the dicBF operon encodes a base pairing-dependent sRNA regulator (DicF) and a small protein (DicB) that act at different levels to inhibit the synthesis and activity of a PTS sugar transporter (ManXYZ) (Fig. 7). This is strikingly similar to the regulation of the glucose PTS (ptsG; enzyme IICBGlc) by the dual-function sRNA SgrS and the small protein it encodes, SgrT. SgrS base pairs with and represses translation of ptsG mRNA (66, 67), while SgrT inhibits PtsG activity at a posttranslational level (68–70). Perhaps regulation of PTS enzyme synthesis and activity by sRNAs and small proteins is a common mechanism for posttranscriptional control of these systems. Future studies on the multitude of sRNAs and small proteins encoded on prophages and bacterial chromosomes promise to reveal more surprising connections between phages and their hosts.
MATERIALS AND METHODS
Strain construction and media.
All the strains and phages used in this study are summarized in Table S1 in the supplemental material, and the oligonucleotides (from Integrated DNA Technologies) are listed in Table S2. The strains used in the study are derivatives of E. coli K-12 strains MG1655 and BW25113. Chromosomal mutations were constructed using the λ red recombination method as described previously (71–73) or were moved into the required strain background using P1 transduction (74).
Construction of strain DB240, which has a Plac promoter inserted upstream of ydfA replacing the native dicBF operon promoter, was described previously (25). Oligonucleotides O-PR185 and O-PR186 were used to amplify the kanamycin resistance gene from pKD13, and the PCR product was recombined into the chromosome of DB240 using λ red functions produced by pSIM6 (73). The resulting ΔdicB::kan strain was called PR163. The kanamycin cassette was removed and replaced with an “FRT scar” using pCP20 (72) to create the ΔdicB::scar strain PR165. Similarly, a ΔydfD::kan strain called PR164 was constructed by amplifying the kanamycin cassette of pKD13 using oligonucleotides O-PR189 and O-PR190 and recombining it into DB240.
A ΔmanXYZ::kan deletion was moved into DJ624 and DB240 by P1 transduction from YS208 (75) to create PR187 and PR191, respectively. MinC mutants with single amino acid changes E156A and R172A (53) were constructed by first inserting a kan-araC-PBAD-ccdB PCR product into the minC gene in strain DJ624. Oligonucleotides O-PR209/O-PR210 (for E156A) and O-PR205/O-PR206 (for R172A) were used to amplify the kan-ccdB region of strain YS243, and the PCR product was recombined into DJ624(pSIM6) to generate strains PR178 and PR179, respectively. Oligonucleotides O-PR211 and O-PR212 (containing the E156A mutation) were used to amplify a segment of DNA from the control strain DJ480 to generate a PCR product with the desired mutations for minC E156A and recombined into PR178 pSIM6 to generate strain PR180. Oligonucleotides O-PR213 and O-PR214 (containing the R172A mutation) were used similarly to generate a PCR product with mutations for minC R172A and recombined into PR179 pSIM6 to create strain PR181. A Plac promoter replacing the promoter of the dicBF operon was introduced in PR180 and PR181 to generate strains PR182 and PR183, respectively.
E. coli K-12 strains were grown in LB medium at 37°C on a rotary shaker. All phage dilutions were made in TM buffer containing 10 mM Tris-HCl and 10 mM MgSO4, and phage infections were carried out using the same buffer. For phage infections, the top agar was made with equal parts LB agar and TM buffer, unless otherwise specified (76). Top agar was added to the infection mixture and plated on LB agar plates.
Phage propagation.
New stocks of each phage were prepared as described by Rotman et al. (76). Plating cultures were prepared by growing DJ480 in tryptone broth (TB) medium with 5 mM MgSO4 (and 0.2% maltose exclusively for λ stock preparation) until late log phase, after which an equal amount of TM buffer was added and the mixture was vortexed vigorously. The titers of old phage stocks were determined by combining phage stocks with prepared plating cultures, mixing in top agar (made of equal parts TB agar and TM buffer), and plating onto TB agar plates that were subsequently incubated overnight at 37°C. The next day, a single individual plaque was punched out and incubated in TM buffer at room temperature for 1 to 2 h with occasional vortexing. Between 10 and 30 μl of the single-plaque eluate was mixed with 300 μl of DJ480 plating culture and incubated at 37°C for 15 min; 3 ml TB-TM top agar was added and plated onto TB plates for incubation at 37°C. After 3 to 7 h, when the lysis was confluent, the plate was overlaid with 5 ml TM buffer overnight at room temperature. The TM buffer containing phages was collected in the morning, and 4 ml fresh TM buffer was added to the plate and kept at room temperature. After 8 h, the remaining TM buffer containing phages was collected, and the combined eluate was centrifuged to pellet the agar and cells down. The supernatant was transferred into a fresh tube, 50 μl chloroform was added, and the fresh phage lysate was stored at 4°C.
EOP assay.
The strains used in the EOP experiment were precultured from overnight cultures in LB and subcultured in LB medium to ensure all the strains were in the same state of growth when phage infection was carried out. After 1 h of subculturing (when the optical density at 600 nm [OD600] was ∼0.1 to 0.2), IPTG was added to a final concentration of 0.5 mM to induce the Plac promoter. After 1 h with IPTG induction, the cells were washed and resuspended in LB medium. The final OD600 was measured, and 1 ml of the culture was centrifuged and resuspended in 1 ml TM buffer; 100 μl of the phage dilution was added to 100 μl of bacteria from the previous step and incubated for 10 min at 37°C. After 10 min, 3 ml prewarmed LB top agar was added to the mixture and plated onto LB agar plates. The plates were incubated overnight at 37°C, and the plaques were counted. The EOP was calculated as the phage titer on the test strain (in PFU per milliliter) divided by the phage titer on the control strain (in PFU per milliliter) (77, 78).
ECOI assay.
The strains were prepared for infection as described for the EOP assay. The only difference was in the last step of sample preparation, when the cells were resuspended in TM buffer with 0.5 mM IPTG to induce Plac-dicBF during phage infection. The procedure followed for the ECOI assay was based on that described by Moineau et al. (79). λvir lysates were added to 500 μl of prepared strains at an MOI of 0.1 or less and incubated at 37°C for 10 min. The infection mixture was washed with TM buffer containing 0.5 mM IPTG to remove unadsorbed phages and resuspended in 500 μl of fresh buffer. The infected cells were diluted in TM-IPTG buffer, and 100 μl of each dilution of strains was added to 100 μl of DJ480 cells in TM buffer, LB-TM top agar was added to this mixture and plated onto LB agar plates. The plates were incubated overnight at 37°C, and the plaques arising from each individual infection were observed and counted. The ECOI was calculated as follows: (number of centers of infection per milliliter from the test strain) × 100/(number of centers of infection per milliliter from the control strain) (79).
One-step growth curve.
The samples were prepared for infection as described for the ECOI assays. The one-step growth curve experiment was designed based on previously published methods (79). After resuspending the cells, λvir was added at an MOI of 0.1 or less to 500 μl of cells and incubated for 10 min at 37°C. The infection mixture was washed to remove unadsorbed phages and resuspended in 500 μl of TM buffer with 0.5 mM IPTG. The strains were diluted 1:10,000 for DJ480 and 1:1,000 for DB240 (Plac-dicBF) to a final volume of 20 ml in LB with 10 mM MgSO4 and 0.5 mM IPTG in flasks and incubated in a 37°C water bath. Immediately, 100 μl was withdrawn from the flask and added to 100 μl phage-sensitive DJ480 cells in TM buffer (for lawn formation); prewarmed top agar was added to this mixture, and it was plated onto LB agar plates. The first time point was 30 min after the start of infection. The same procedure was repeated for each time point. The burst size was calculated as follows: (phage titer at 100 min − initial titer at 30 min)/initial titer at 30 min. The latent period was calculated as the midpoint of the exponential phase of the growth curve (79).
Adsorption assay.
The procedure described above for ECOI assays was followed, and after the strains were infected with λvir at an MOI of 0.1 and allowed to adsorb for 10 min at 37°C, the strains were centrifuged for 5 min at 13,000 rpm to pellet cells and adsorbed phages. One hundred microliters of the supernatant was removed, and dilutions were made in TM buffer; 10 μl of each dilution was added to 100 μl DJ480 (phage-sensitive) cells in TM buffer and incubated for 10 min at 37°C. This mixture was plated onto LB plates using top agar and incubated overnight at 37°C. The control titer was calculated using the same procedure described above with the same amount of phages required for an MOI of 0.1 added to 500 μl TM buffer (no bacteria). The percent adsorption was calculated as follows: (control titer − residual titer) × 100/control titer (77, 80).
Growth on minimal medium plates with different sugars.
For growth assays, M63 minimal medium plates with sugars (glucose, fructose, mannose, N-acetylglucosamine, and glucosamine) at a final concentration of 0.2% were prepared with or without 0.025 mM IPTG. The strains were streaked on the plates and incubated for 44 h at 37°C. By visual inspection, the strains were scored for growth, with +++ denoting growth of the control strain on each sugar, ++ and + denoting decreased growth compared to the control strain on the respective sugar, and − indicating no growth.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jeffrey Gardner, Andrei Kuzminov, Alan Davidson, and Sankar Adhya for their generous gifts of various phages that were critical for our experiments. We also thank Nadim Majdalani and John Cronan for providing strains. We are grateful to James Slauch for his help and advice with the design of phage experiments. We thank Sandy Pernitzsch from Scigraphix for graphic design of the model figure. Finally, we express our thanks to former and current Vanderpool and Slauch laboratory members for thought-provoking discussions.
This work was supported by the National Institutes of Health (R01 GM092830) and a University of Illinois Department of Microbiology Alice Helm Fellowship to P.T.R.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00475-19.
REFERENCES
- 1.Brüssow H, Hendrix RW. 2002. Phage genomics: small is beautiful. Cell 108:13–16. doi: 10.1016/s0092-8674(01)00637-7. [DOI] [PubMed] [Google Scholar]
- 2.Keen EC. 2015. A century of phage research: bacteriophages and the shaping of modern biology. Bioessays 37:6–9. doi: 10.1002/bies.201400152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayes S, Mahony J, Nauta A, van Sinderen D. 2017. Metagenomic approaches to assess bacteriophages in various environmental niches. Viruses 9:127. doi: 10.3390/v9060127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casjens S. 2003. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol 49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- 5.Harrison E, Brockhurst MA. 2017. Ecological and evolutionary benefits of temperate phage: what does or doesn’t kill you makes you stronger. BioEssays 39:1700112. doi: 10.1002/bies.201700112. [DOI] [PubMed] [Google Scholar]
- 6.Touchon M, Bernheim A, Rocha E. 2016. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J 10:2744–2754. doi: 10.1038/ismej.2016.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fortier L-C, Sekulovic O. 2013. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4:354–365. doi: 10.4161/viru.24498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brussow H, Canchaya C, Hardt W-D. 2004. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev 68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herold S, Karch H, Schmidt H. 2004. Shiga toxin-encoding bacteriophages—genomes in motion. Int J Med Microbiol 294:115–121. doi: 10.1016/j.ijmm.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 10.Freeman VJ. 1951. Studies on the virulence of bacteriophage-infected strains of Corynebacterium diphtheriae. J Bacteriol 61:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eklund MW, Poysky FT, Reed SM, Smith CA. 1971. Bacteriophage and the toxigenicity of Clostridium botulinum type C. Science 172:480–482. doi: 10.1126/science.172.3982.480. [DOI] [PubMed] [Google Scholar]
- 12.Edlin G, Lin L, Bitner R. 1977. Reproductive fitness of P1, P2, and Mu lysogens of Escherichia coli. J Virol 21:560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng Z, Liu X, Yao J, Guo Y, Li B, Li Y, Jiao N, Wang X. 2016. Cold adaptation regulated by cryptic prophage excision in Shewanella oneidensis. ISME J 10:2787–2800. doi: 10.1038/ismej.2016.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gödeke J, Paul K, Lassak J, Thormann KM. 2011. Phage-induced lysis enhances biofilm formation in Shewanella oneidensis MR-1. ISME J 5:613–626. doi: 10.1038/ismej.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asadulghani M, Ogura Y, Ooka T, Itoh T, Sawaguchi A, Iguchi A, Nakayama K, Hayashi T. 2009. The defective prophage pool of Escherichia coli O157: prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog 5:e1000408. doi: 10.1371/journal.ppat.1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bobay L-M, Touchon M, Rocha E. 2014. Pervasive domestication of defective prophages by bacteria. Proc Natl Acad Sci U S A 111:12127–12132. doi: 10.1073/pnas.1405336111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matos RC, Lapaque N, Rigottier-Gois L, Debarbieux L, Meylheuc T, Gonzalez-Zorn B, Repoila F, de Lopes MF, Serror P. 2013. Enterococcus faecalis prophage dynamics and contributions to pathogenic traits. PLoS Genet 9:e1003539. doi: 10.1371/journal.pgen.1003539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramisetty BCM, Sudhakari PA. 2019. Bacterial ‘grounded’ prophages: hotspots for genetic renovation and innovation. Front Genet 10:65. doi: 10.3389/fgene.2019.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Kim Y, Ma Q, Hong SH, Pokusaeva K, Sturino JM, Wood TK. 2010. Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1:147. doi: 10.1038/ncomms1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Béjar S, Bouché JP. 1985. A new dispensable genetic locus of the terminus region involved in control of cell division in Escherichia coli. Mol Gen Genet 201:146–150. doi: 10.1007/bf00425651. [DOI] [PubMed] [Google Scholar]
- 21.Bouché F, Bouché J-P. 1989. Genetic evidence that DicF, a second division inhibitor encoded by the Escherichia coli dicB operon, is probably RNA. Mol Microbiol 3:991–994. doi: 10.1111/j.1365-2958.1989.tb00249.x. [DOI] [PubMed] [Google Scholar]
- 22.Labie C, Bouché F, Bouché JP. 1989. Isolation and mapping of Escherichia coli mutations conferring resistance to division inhibition protein DicB. J Bacteriol 171:4315–4319. doi: 10.1128/jb.171.8.4315-4319.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Boer PA, Crossley RE, Rothfield LI. 1990. Central role for the Escherichia coli minC gene product in two different cell division-inhibition systems. Proc Natl Acad Sci U S A 87:1129–1133. doi: 10.1073/pnas.87.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tétart F, Bouché JP. 1992. Regulation of the expression of the cell-cycle gene ftsZ by DicF antisense RNA. Division does not require a fixed number of FtsZ molecules. Mol Microbiol 6:615–620. doi: 10.1111/j.1365-2958.1992.tb01508.x. [DOI] [PubMed] [Google Scholar]
- 25.Balasubramanian D, Ragunathan PT, Fei J, Vanderpool CK. 2016. A prophage-encoded small RNA controls metabolism and cell division in Escherichia coli. mSystems 1:e00021-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Labie C, Bouché F, Bouché JP. 1990. Minicell-forming mutants of Escherichia coli: suppression of both DicB- and MinD-dependent division inhibition by inactivation of the minC gene product. J Bacteriol 172:5852–5855. doi: 10.1128/jb.172.10.5852-5855.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson JE, Lackner LL, de Boer P. 2002. Targeting of (D)MinC/MinD and (D)MinC/DicB complexes to septal rings in Escherichia coli suggests a multistep mechanism for MinC-mediated destruction of nascent FtsZ rings. J Bacteriol 184:2951–2962. doi: 10.1128/JB.184.11.2951-2962.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson JE, Lackner LL, Hale CA, De Boer P. 2004. ZipA is required for targeting of DMinC/DicB, but not DMinC/MinD, complexes to septal ring assemblies in Escherichia coli. J Bacteriol 186:2418–2429. doi: 10.1128/jb.186.8.2418-2429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Béjar S, Bouché F, Bouché JP. 1988. Cell division inhibition gene dicB is regulated by a locus similar to lamboid bacteriophage immunity loci. Mol Gen Genet 212:11–19. doi: 10.1007/bf00322439. [DOI] [PubMed] [Google Scholar]
- 30.Faubladier M, Bouché JP. 1994. Division inhibition gene dicF of Escherichia coli reveals a widespread group of prophage sequences in bacterial genomes. J Bacteriol 176:1150–1156. doi: 10.1128/jb.176.4.1150-1156.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melson EM, Kendall MM. 2019. The sRNA DicF integrates oxygen sensing to enhance enterohemorrhagic Escherichia coli virulence via distinctive RNA control mechanisms. Proc Natl Acad Sci U S A 116:14210–14215. doi: 10.1073/pnas.1902725116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murashko ON, Lin-Chao S. 2017. Escherichia coli responds to environmental changes using enolasic degradosomes and stabilized DicF sRNA to alter cellular morphology. Proc Natl Acad Sci U S A 114:E8025–E8034. doi: 10.1073/pnas.1703731114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masuda H, Awano N, Inouye M. 2016. ydfD encodes a novel lytic protein in Escherichia coli. FEMS Microbiol Lett 363:fnw039. doi: 10.1093/femsle/fnw039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellis EL, Delbrück M. 1939. The growth of bacteriophage. J Gen Physiol 22:365–384. doi: 10.1085/jgp.22.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadas H, Einav M, Fishov I, Zaritsky A. 1997. Bacteriophage T4 development depends on the physiology of its host Escherichia coli. Microbiology 143:179–185. doi: 10.1099/00221287-143-1-179. [DOI] [PubMed] [Google Scholar]
- 36.Comeau AM, Tétart F, Trojet SN, Prère M-F, Krisch HM. 2007. Phage-antibiotic synergy (PAS): β-lactam and quinolone antibiotics stimulate virulent phage growth. PLoS One 2:e799. doi: 10.1371/journal.pone.0000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhillon EKS, Dhillon TS, Lai ANC, Linn S. 1980. Host range, immunity and antigenic properties of lambdoid coliphage HK97. J Gen Virol 50:217–220. doi: 10.1099/0022-1317-50-1-217. [DOI] [PubMed] [Google Scholar]
- 38.Cumby N, Reimer K, Mengin-Lecreulx D, Davidson AR, Maxwell KL. 2015. The phage tail tape measure protein, an inner membrane protein and a periplasmic chaperone play connected roles in the genome injection process of E. coli phage HK97. Mol Microbiol 96:437–447. doi: 10.1111/mmi.12918. [DOI] [PubMed] [Google Scholar]
- 39.Rybchin VN. 1984. Genetics of bacteriophage φ80—a review. Gene 27:3–11. doi: 10.1016/0378-1119(84)90233-6. [DOI] [PubMed] [Google Scholar]
- 40.Rotman E, Kouzminova E, Plunkett G, Kuzminov A. 2012. Genome of enterobacteriophage Lula/phi80 and insights into its ability to spread in the laboratory environment. J Bacteriol 194:6802–6817. doi: 10.1128/JB.01353-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Randall Hazelbauer L, Schwartz M. 1973. Isolation of the bacteriophage lambda receptor from Escherichia coli. J Bacteriol 116:1436–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boos W, Shuman H. 1998. Maltose/maltodextrin system of Escherichia coli: transport, metabolism, and regulation. Microbiol Mol Biol Rev 62:204–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Esquinas-Rychen M, Erni B. 2001. Facilitation of bacteriophage lambda DNA injection by inner membrane proteins of the bacterial phosphoenol-pyruvate: carbohydrate phosphotransferase system (PTS). J Mol Microbiol Biotechnol 3:361–370. [PubMed] [Google Scholar]
- 44.Scandella D, Arber W. 1974. An Escherichia coli mutant which inhibits the injection of phage λ DNA. Virology 58:504–513. doi: 10.1016/0042-6822(74)90084-1. [DOI] [PubMed] [Google Scholar]
- 45.Erni B, Zanolari B, Kocher HP. 1987. The mannose permease of Escherichia coli consists of three different proteins. Amino acid sequence and function in sugar transport, sugar phosphorylation, and penetration of phage lambda DNA. J Biol Chem 262:5238–5247. [PubMed] [Google Scholar]
- 46.Hancock REW, Braun V. 1976. Nature of the energy requirement for the irreversible adsorption of bacteriophages T1 and φ80 to Escherichia coli. J Bacteriol 125:409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braun V. 1985. The iron-transport systems of Escherichia coli, p 617–652. In Martonosi AN. (ed), The enzymes of biological membranes. Springer, Boston, MA. [Google Scholar]
- 48.Postma PWW, Lengeler JWW, Jacobson GRR. 1993. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev 57:543–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Plumbridge J. 2000. A mutation which affects both the specificity of PtsG sugar transport and the regulation of ptsG expression by Mlc in Escherichia coli. Microbiology 146:2655–2663. doi: 10.1099/00221287-146-10-2655. [DOI] [PubMed] [Google Scholar]
- 50.Curtis SJ, Epstein W. 1975. Phosphorylation of D-glucose in Escherichia coli mutants defective in glucosephosphotransferase, mannosephosphotransferase, and glucokinase. J Bacteriol 122:1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cam K, Béjar S, Gil D, Bouché JP. 1988. Identification and sequence of gene dicB: translation of the division inhibitor from an in-phase internal start. Nucleic Acids Res 16:6327–6338. doi: 10.1093/nar/16.14.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Margolin W. 2000. Themes and variations in prokaryotic cell division. FEMS Microbiol Rev 24:531–548. doi: 10.1111/j.1574-6976.2000.tb00554.x. [DOI] [PubMed] [Google Scholar]
- 53.Zhou H, Lutkenhaus J. 2005. MinC mutants deficient in MinD- and DicB-mediated cell division inhibition due to loss of interaction with MinD, DicB, or a septal component. J Bacteriol 187:2846–2857. doi: 10.1128/JB.187.8.2846-2857.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu Z, Mukherjee A, Pichoff S, Lutkenhaus J. 1999. The MinC component of the division site selection system in Escherichia coli interacts with FtsZ to prevent polymerization. Proc Natl Acad Sci U S A 96:14819–14824. doi: 10.1073/pnas.96.26.14819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pichoff S, Lutkenhaus J. 2001. Escherichia coli division inhibitor MinCD blocks septation by preventing Z-ring formation. J Bacteriol 183:6630–6635. doi: 10.1128/JB.183.22.6630-6635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lopian L, Elisha Y, Nussbaum-Shochat A, Amster-Choder O. 2010. Spatial and temporal organization of the E. coli PTS components. EMBO J 29:3630–3645. doi: 10.1038/emboj.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Govindarajan S, Elisha Y, Nevo-Dinur K, Amster-Choder O. 2013. The general phosphotransferase system proteins localize to sites of strong negative curvature in bacterial cells. mBio 4:e00443. doi: 10.1128/mBio.00443-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martins GB, Giacomelli G, Goldbeck O, Seibold GM, Bramkamp M. 2019. Substrate-dependent cluster density dynamics of Corynebacterium glutamicum phosphotransferase system permeases. Mol Microbiol 111:1335–1354. doi: 10.1111/mmi.14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Labrie SJ, Samson JE, Moineau S. 2010. Bacteriophage resistance mechanisms. Nat Rev Microbiol 8:317–327. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- 60.Bondy-Denomy J, Davidson AR. 2014. When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J Microbiol 52:235–242. doi: 10.1007/s12275-014-4083-3. [DOI] [PubMed] [Google Scholar]
- 61.Cumby N, Edwards AM, Davidson AR, Maxwell KL. 2012. The bacteriophage HK97 gp15 moron element encodes a novel superinfection exclusion protein. J Bacteriol 194:5012–5019. doi: 10.1128/JB.00843-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Susskind MM, Botstein D, Wright A. 1974. Superinfection exclusion by P22 prophage in lysogens of Salmonella typhimurium. III. Failure of superinfecting phage DNA to enter sieA+ lysogens. Virology 62:350–366. doi: 10.1016/0042-6822(74)90398-5. [DOI] [PubMed] [Google Scholar]
- 63.Hofer B, Ruge M, Dreiseikelmann B. 1995. The superinfection exclusion gene (sieA) of bacteriophage P22: identification and overexpression of the gene and localization of the gene product. J Bacteriol 177:3080–3086. doi: 10.1128/jb.177.11.3080-3086.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kliem M, Dreiseikelmann B. 1989. The superimmunity gene sim of bacteriophage P1 causes superinfection exclusion. Virology 171:350–355. doi: 10.1016/0042-6822(89)90602-8. [DOI] [PubMed] [Google Scholar]
- 65.Azam MS, Vanderpool CK. 2018. Translational regulation by bacterial small RNAs via an unusual Hfq-dependent mechanism. Nucleic Acids Res 46:2585–2599. doi: 10.1093/nar/gkx1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vanderpool CK, Gottesman S. 2004. Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol Microbiol 54:1076–1089. doi: 10.1111/j.1365-2958.2004.04348.x. [DOI] [PubMed] [Google Scholar]
- 67.Balasubramanian D, Vanderpool CK. 2013. Deciphering the interplay between two independent functions of the small RNA regulator SgrS in Salmonella. J Bacteriol 195:4620–4630. doi: 10.1128/JB.00586-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wadler CS, Vanderpool CK. 2007. A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc Natl Acad Sci U S A 104:20454–20459. doi: 10.1073/pnas.0708102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lloyd CR, Park S, Fei J, Vanderpool CK. 2017. The small protein SgrT controls transport activity of the glucose-specific phosphotransferase system. J Bacteriol 199:e00869-16. doi: 10.1128/JB.00869-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kosfeld A, Jahreis K. 2012. Characterization of the interaction between the small regulatory peptide SgrT and the EIICBGlc of the glucose-phosphotransferase system of E. coli K-12. Metabolites 2:756–774. doi: 10.3390/metabo2040756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu D, Ellis HM, Lee E-C, Jenkins NA, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chan W, Costantino N, Li R, Lee SC, Su Q, Melvin D, Court DL, Liu P. 2007. A recombineering based approach for high-throughput conditional knockout targeting vector construction. Nucleic Acids Res 35:e64. doi: 10.1093/nar/gkm163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miller J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 75.Sun Y, Vanderpool CK. 2013. Physiological consequences of multiple-target regulation by the small RNA SgrS in Escherichia coli. J Bacteriol 195:4804–4815. doi: 10.1128/JB.00722-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rotman E, Amado L, Kuzminov A. 2010. Unauthorized horizontal spread in the laboratory environment: the tactics of Lula, a temperate lambdoid bacteriophage of Escherichia coli. PLoS One 5:e11106. doi: 10.1371/journal.pone.0011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanders ME, Klaenhammer TR. 1980. Restriction and modification in group N streptococci: effect of heat on development of modified lytic bacteriophage. Appl Environ Microbiol 40:500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Domingues S, McGovern S, Plochocka D, Santos MA, Ehrlich SD, Polard P, Chopin M-C. 2008. The lactococcal abortive infection protein AbiP is membrane-anchored and binds nucleic acids. Virology 373:14–24. doi: 10.1016/j.virol.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 79.Moineau S, Durmaz E, Pandian S, Klaenhammer TR. 1993. Differentiation of two abortive mechanisms by using monoclonal antibodies directed toward lactococcal bacteriophage capsid proteins. Appl Environ Microbiol 59:208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stockdale SR, Mahony J, Courtin P, Chapot-Chartier M-P, van Pijkeren J-P, Britton RA, Neve H, Heller KJ, Aideh B, Vogensen FK, van Sinderen D. 2013. The lactococcal phages Tuc2009 and TP901-1 incorporate two alternate forms of their tail fiber into their virions for infection specialization. J Biol Chem 288:5581–5590. doi: 10.1074/jbc.M112.444901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.