

A deep learning–based model predicts activity of a widely used genome editing tool in a highly accurate manner.
Abstract
We evaluated SpCas9 activities at 12,832 target sequences using a high-throughput approach based on a human cell library containing single-guide RNA–encoding and target sequence pairs. Deep learning–based training on this large dataset of SpCas9-induced indel frequencies led to the development of a SpCas9 activity–predicting model named DeepSpCas9. When tested against independently generated datasets (our own and those published by other groups), DeepSpCas9 showed high generalization performance. DeepSpCas9 is available at http://deepcrispr.info/DeepSpCas9.
INTRODUCTION
CRISPR-Cas, a prokaryotic adaptive immune system, has been harnessed for genome editing in various species and cell types, including human cells (1–6). The ability to accurately predict SpCas9 activity is important for applications of genome editing (7–12). So far, several computational models that predict SpCas9 activity have been developed on the basis of datasets of phenotypic changes of gene-edited cells (7, 9, 11–17) or medium-sized datasets of SpCas9-induced indel frequencies obtained by episomal plasmid–based library-on-library approaches (10, 18, 19). However, the generalization performances of these models have been limited (20), possibly because the quality and size of the training datasets were not ideal. Most of the models were developed using Cas9 activity datasets generated by phenotypic analysis of gene knockouts (7, 9, 11–17), which can be biased by the function of the corresponding genes and can include false negatives in which indels are introduced at the target sequences but do not induce functional knockouts (11); furthermore, for three models, the sizes of the SpCas9-induced indel frequency datasets were just medium-sized (10, 18, 19).
We recently reported a deep learning–based computational model called DeepCpf1, which predicts AsCpf1 (Cpf1 from Acidaminococcus sp. BV3L6) activity with a high generalization performance (21). Our high-throughput evaluation of Cpf1 activity using lentiviral libraries of guide RNA–encoding and target sequence pairs (22) enabled the generation of a large dataset of Cpf1-induced indel frequencies, which was used as the training data for DeepCpf1. Although similar paired library-based methods have recently been used to develop computational models that predict the indel sequence patterns generated by Cas9-induced double-strand breaks (23–25), a large dataset of Cas9-induced indel frequencies has not been generated, preventing the development of a Cas9 activity–predicting computational model with high generalization performance. Here, we developed a high-throughput method for evaluating SpCas9-induced indel frequencies at tens of thousands of target sequences by modifying our previously developed methods for Cpf1 (22), which enabled the development of DeepSpCas9, a deep learning–based model that accurately predicts SpCas9 activities with a high generalization performance.
RESULTS AND DISCUSSION
Generation of large datasets of SpCas9 activities through a high-throughput evaluation
For a high-throughput evaluation of SpCas9 activities, we first prepared a lentiviral library of 15,656 guide RNA–encoding and target sequence pairs using a modification of the approach that we previously used for Cpf1 evaluation (21, 22). The target sequences were selected from the human genome and synthetic sequences without any information about the activity of the corresponding single-guide RNAs (sgRNAs) (detailed information is available in Materials and Methods). Oligonucleotides containing these 15,656 target sequences were array-synthesized in a way such that each oligonucleotide contained a target sequence and a corresponding guide sequence for the sgRNA (fig. S1A). This pool of oligonucleotides containing pairs of guide and target sequences was polymerase chain reaction (PCR)–amplified and cloned into a lentiviral plasmid using Gibson assembly (fig. S1B). Then, we cut the resulting library plasmids at the 3′ end of the guide sequence using Bsm BI and inserted the sgRNA scaffold sequence at the cut site (fig. S1B). This two-step approach for generating plasmid libraries was similarly used for generating double-guide RNA libraries (26–31) and libraries of guide RNA–encoding and target sequence pairs for the analysis of SpCas9-induced mutation patterns (23–25). Lentivirus was first generated from this plasmid library and then used to treat human embryonic kidney (HEK) 293T cells to make a cell library, in which each cell contains a synthetic target sequence in its genome and expresses the corresponding sgRNA (fig. S1C). Next, the cell library was treated with SpCas9-encoding lentivirus, which led to sgRNA-directed cleavage and indel formation at the integrated target sequences with frequencies that depended on the sgRNA activity. The target sequences were PCR-amplified and subjected to deep sequencing to measure indel frequencies (21, 22). This high-throughput experiment generated two datasets named HT_Cas9_Train and HT_Cas9_Test (tables S1 and S2).
Indel frequencies at the integrated target sequences are highly correlated with those at the endogenous target sites
We next evaluated whether the indel frequencies at the integrated synthetic target sequences correlated with those at the corresponding endogenous target sites. For this determination, we generated a dataset, named Endo_Cas9, of SpCas9 activities at 124 endogenous target sites with different chromatin accessibility properties [50 targets at deoxyribonuclease I (DNase I) hypersensitive (DHS) regions and 74 targets at non-DHS regions; see table S3] because we previously found that Cpf1 activity is significantly affected by chromatin accessibility (22). We observed a strong correlation between indel frequencies at integrated target sequences and those at endogenous sites (Spearman R = 0.70, Pearson R2 = 0.53; Fig. 1A), which is higher than the correlation previously reported using a medium-scale library-on-library approach (18). Furthermore, we observed a weak tendency for SpCas9-induced indel frequencies at DHS sites to be marginally or merely higher than those at non-DHS sites (P = 0.018; Fig. 1B). In this respect, SpCas9 differs from Cpf1, which elicited markedly higher levels of indels at DHS versus non-DHS sites (Fig. 1B) (22). When we calculated the correlations between indel frequencies at integrated sites and a subset of endogenous sites with similar chromatin accessibility, the correlations were comparable regardless of chromatin accessibility information (Fig. 1, C and D). This observation also contrasts with previous observations of Cpf1, for which there was a much higher correlation between target site subsets with similar chromatin accessibility (22).
Fig. 1. Correlations between indel frequencies at endogenous and integrated sites and effect of chromatin accessibility on indel frequencies.
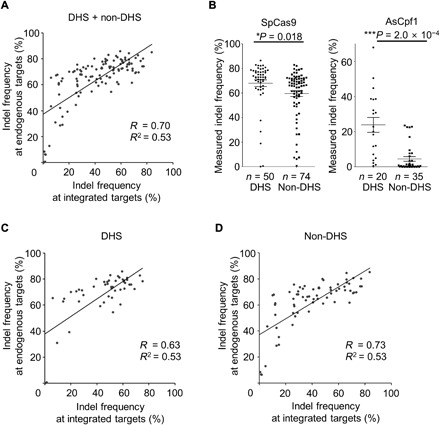
(A) Correlation between indel frequencies at 120 endogenous and corresponding integrated target sequences. The Spearman correlation coefficients (R) and squared Pearson correlation coefficients (R2) are shown. (B) Effect of chromatin accessibility on the activities of SpCas9 (left) and AsCpf1 (right) at endogenous sites. Indel frequencies at endogenous sites were evaluated after transfection of plasmids encoding SpCas9 or AsCpf1 and guide RNAs. Indel frequencies at the target sites were compared after being divided into two groups, DHS sites and other sites (non-DHS). The numbers of analyzed target sites are as follows: SpCas9, n = 50 for DHS target sites and n = 74 for non-DHS target sites; AsCpf1, n = 20 for DHS target sites and n = 35 for non-DHS target sites. The HEK-plasmid dataset from (20) was used for drawing this graph. Error bars represent SEM. Statistical significances determined by Student’s t test are shown. (C and D) Correlation between indel frequencies at endogenous and corresponding integrated target sequences at 50 DHS sites (C) and 70 non-DHS sites (D). The Spearman correlation coefficients (R) and squared Pearson correlation coefficients (R2) are shown.
Development of DeepSpCas9, a deep learning–based computational model predicting sgRNA efficacy
We next attempted to develop an accurate computational model for predicting SpCas9 activity. Using deep learning–based training on a large dataset, we previously developed a computational model named DeepCpf1 that predicts AsCpf1 activity in a highly accurate manner (21). In this study, we used HT_Cas9_Train (tables S1 and S2), a dataset of SpCas9 activities at 12,832 integrated target sequences, which do not include target sequences used to generate Endo_Cas9 (tables S2 and S3). By training on HT_Cas9_Train using an end-to-end deep learning framework (fig. S2) (32–34), which is a modification of what we previously used to generate DeepCpf1, we developed DeepSpCas9, a deep learning–based regression model that predicts SpCas9 activity based on target sequence composition. As a base model architecture, we used a convolutional neural network (CNN) comprising one convolutional layer and three fully connected layers. As the input sequence, we used 30–nucleotide (nt) sequences, which include 4–base pair (bp) left neighbor, 20-bp protospacer, 3-bp protospacer adjacent motif (PAM), and 3-bp right neighbor sequences. The input sequences were converted into a four-dimensional binary matrix by one-hot encoding. Given that multiple filter sizes often improve the performance of CNNs (33), we used a total of 210 multiple sized filters (100 3-nt filters, 70 5-nt filters, and 40 7-nt filters) instead of the equal-sized filters that we previously used (fig. S2) (21). We conducted 10-fold cross-validation with HT_Cas9_Train to evaluate the generalization performance of model selection and training. We tested a total of 324 different combinations of model hyperparameters (table S5) and selected as the final combination that led to the highest validation score calculated using Spearman correlation coefficients between the experimentally measured and predicted activity levels. As the size of the training dataset for the cross-validation increased, the average Spearman correlation coefficients between experimentally obtained indel frequencies and predicted scores from DeepSpCas9 steadily increased up to 0.77 (Fig. 2A). When compared to conventional machine learning algorithms such as support vector machine (SVM), L1-regularized linear regression, L2-regularized linear regression, L1L2-regularized linear regression, AdaBoost, random forest, and gradient-boosted regression trees, which include those that previously showed competent performance for SpCas9 activity prediction (7, 18), the Spearman correlations of DeepSpCas9 in the cross-validation were significantly higher than those of these conventional machine learning algorithms (versus the second best model, SVM: P = 7.2 × 10−3, 7.5 × 10−4, 1.9 × 10−3, 1.1 × 10−4, 7.0 × 10−2, 5.9 × 10−6, 1.5 × 10−2, 2.0 × 10−4, 1.6 × 10−3, and 3.9 × 10−2 for each fold; versus the third best model, L2 regression: P = 2.5 × 10−7, 8.3 × 10−10, 1.9 × 10−8, 2.5 × 10−9, 1.9 × 10−3, 2.1 × 10−11, 2.0 × 10−7, 1.4 × 10−13, 8.8 × 10−10, and 2.0 × 10−4 for each fold) and were similar to that of the equal-sized filter-based deep learning model (P = 4.2 × 10−2, 2.9 × 10−1, 9.6 × 10−1, 9.7 × 10−1, 6.1 × 10−1, 9.0 × 10−1, 5.7 × 10−1, 2.9 × 10−1, 4.1 × 10−1, and 5.1 × 10−1 for each fold) (Fig. 2B). Furthermore, when these algorithms were examined using the test dataset Endo_Cas9 (derived using target sequences that were never included in the training dataset HT_Cas9_Train) and two previously published datasets of Cas9 activities at endogenous sites [Hart 2015 (35) and Xu 2015 (16)], the Spearman correlation of DeepSpCas9 was also higher than those of the conventional machine learning algorithms and that of the equal-sized filter-based deep learning model (Fig. 2C), indicating that DeepSpCas9 exhibited the best performance among all of the models.
Fig. 2. Evaluation of machine learning–based computational models predicting Cas9 activities.
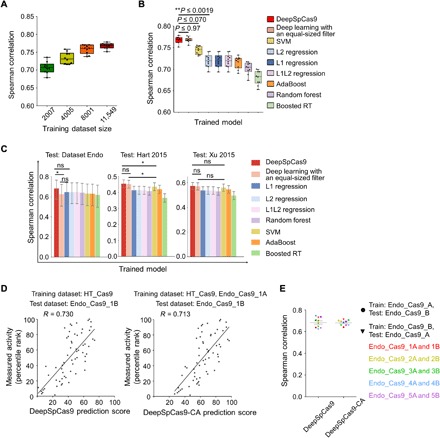
(A) Cross-validation of DeepSpCas9 models trained on datasets of varying sizes. Each dot represents the Spearman correlation coefficient between the measured indel frequency and the predicted activity from 10-fold cross-validation (total n = 10 correlation coefficients). (B) Cross-validation of SpCas9 activity prediction models based on previously reported machine learning–based approaches. Each dot represents the Spearman correlation coefficient between the measured indel frequency and the predicted activity from 10-fold cross-validation (total n = 10 correlation coefficients). Statistical significances between the best, next-best, and third-best models are shown (Steiger’s test). In (A) and (B), the top, middle, and bottom lines in the boxes represent the 25th, 50th, and 75th percentiles, respectively. Whiskers indicate the minimum and maximum values. The confidence intervals are described in table S6. RT, regression trees. (C) Performance comparison of DeepSpCas9 with other prediction models using dataset Endo_Cas9 (n = 124 independent target sites) and two published datasets (n = 4207 and 2060 independent target sites for datasets Hart 2015 and Xu 2015, respectively) as the test datasets. Error bars represent 95% confidence intervals, which are described in detail in table S6. For clarity, results from statistical testing are shown only for DeepSpCas9 versus deep learning with an equal-sized filter, DeepSpCas9 versus the best conventional machine learning–based model, and deep learning with an equal-sized filter versus the best conventional machine learning–based model (left to right: *P = 1.4 × 10−2, DeepSpCas9 versus deep learning with an equal-sized filter; *P = 1.1 × 10−2, DeepSpCas9 versus SVM; *P = 4.6 × 10−2, deep learning with an equal-sized filter versus SVM; Steiger’s test). ns, not significant. (D) Performance comparison of DeepSpCas9 and DeepSpCas9-CA (chromatin accessibility). The DeepSpCas9-CA model was developed by fine-tuning the DeepSpCas9 model using the Endo-1A dataset. DeepSpCas9 (left) and DeepSpCas9-CA (right) models were evaluated with the Endo-1B dataset. The Spearman correlation coefficients (R) are shown. (E) Results from 10 iterations of fine-tuning and evaluation. Each dot represents the Spearman correlation coefficient between the measured indel frequency and the predicted activity. A total of 10 (= 2 × 5) rounds of fine-tuning and subsequent testing results are shown.
Considering chromatin accessibility information barely improves SpCas9 activity prediction
We previously improved the prediction of Cpf1 activities at endogenous target sites by considering chromatin accessibility (21). To determine whether such a consideration would also improve SpCas9 activity prediction, we first divided the Endo_Cas9 dataset into paired subsets (table S3; detailed information is available in Materials and Methods). Then, we fine-tuned DeepSpCas9 using a data subset such as Endo_Cas9_1A and binary chromatin accessibility information from the Encyclopedia of DNA Elements (ENCODE) (36), leading to the development of a fine-tuned model predicting SpCas9 activity based on both target sequence information and chromatin accessibility. When evaluated with the other data subset, Endo_Cas9_1B, as the test dataset, the fine-tuned model showed performance comparable to that of DeepSpCas9 (Fig. 2D). We next repeated this fine-tuning and subsequent testing after exchanging the training and test datasets: We used Endo_Cas9_1B as the training dataset for fine-tuning and Endo_Cas9_1A as the test dataset. We also conducted these analyses using the other four pairs of datasets. This total of 10 (= 2 × 5) rounds of fine-tuning and subsequent testing revealed that the Spearman correlations of these fine-tuned models are comparable to those of DeepSpCas9 (Fig. 2E), suggesting that fine-tuning with chromatin accessibility information barely improves the accuracy of DeepSpCas9 in predicting indel frequencies at endogenous sites. This result is in line with the finding that SpCas9 activity is only slightly affected by chromatin accessibility (Fig. 1B) and in strong contrast to DeepCpf1, which showed markedly improved performance when chromatin accessibility information was considered (21).
DeepSpCas9 shows high generalization performance
To assess its generalization performance, we next evaluated DeepSpCas9 using other sufficiently large published datasets (number of target sequences, >100), derived from different studies from independent laboratories (seven datasets generated using U6 promoter–driven sgRNAs and three datasets generated using in vitro transcribed sgRNAs) (7, 10, 14, 16, 18, 35, 37–39), as test data and compared the results with those of other SpCas9 activity–predicting programs (7, 10, 13–16, 18, 39). For a fair comparison of generalization performances, we excluded correlations of models tested against their own training datasets (20). We found that the Spearman correlations of DeepSpCas9 were the highest among those of nine previously published models in all seven tests against datasets generated using U6 promoter–driven sgRNAs and that statistical significance was observed for five of the seven tests when compared with the second best models (Fig. 3), suggesting that DeepSpCas9 has the highest generalization performance compared to any of the other computational models predicting SpCas9 activity. DeepCRISPR (13), a recently reported deep learning computational model trained using datasets of phenotypic changes of cells containing Cas9-induced gene edits, showed a lower generalization performance as compared to Doench 2016 (rule set 2 or sgRNA designer), which was developed before DeepCRISPR. When tested against the three datasets generated using in vitro transcribed sgRNA, the Spearman correlations of DeepSpCas9 were the highest together with those of CRISPRscan, which was generated for the prediction of in vitro transcribed sgRNA activities. Neither Doench 2016 (7) nor CRISPRscan (10) showed the highest Spearman correlations for datasets of both U6 promoter–driven and in vitro transcribed sgRNAs. Together, these data suggest that the generalization performance of DeepSpCas9 is high.
Fig. 3. Comparison of generalization performances of computational models predicting Cas9 activities.
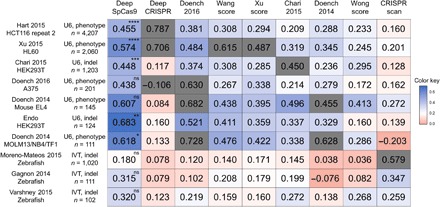
The heat map shows Spearman correlation coefficients determined from DeepSpCas9 and previously reported models, which are arranged horizontally. The names of the vertically placed test datasets include information about the cell line or species used. Other related parameters, such as the guide RNA expression method [U6 promoter–driven (U6) versus in vitro transcribed (IVT)], the Cas9 activity analysis method [phenotypic change (phenotype) versus indel], and the number of analyzed sites, are also shown. Each gray box indicates the correlation of a model tested against a test dataset that includes its own training dataset. In the evaluation against each test dataset, the statistical significance between the two best models is indicated for the best model (from the top: ****P = 5.3 × 10−9, ****P = 1.8 × 10−10, ****P = 3.4 × 10−8, ****P = 1.1 × 10−13, ****P = 2.9 × 10−11, ****P = 3.9 × 10−8, ***P = 2.5 × 10−4, *P = 3.7 × 10−2, and *P = 3.9 × 10−2; Steiger’s test).
We provide a web tool that enables accurate prediction of SpCas9 activity by DeepSpCas9 at http://deepcrispr.info/DeepSpCas9 and provide code for incorporation of DeepSpCas9 into existing tools (Supplementary Code). Given that DeepSpCas9 has high generalization performance, we expect that it will greatly facilitate genome editing using SpCas9.
MATERIALS AND METHODS
Oligonucleotide library design
A pool of 17,840 oligonucleotides was array-synthesized by and purchased from Twist Bioscience (San Francisco, CA). We designed each oligonucleotide to contain a 20-nt guide sequence for the sgRNA, a BsmBI restriction site, a 20-nt barcode sequence (barcode 1), a second BsmBI site, a 15-nt barcode sequence (barcode 2), and the corresponding 30-nt target sequence including a PAM (fig. S1A). Barcode 1 was inserted between the two BsmBI sites to reduce template switching during PCR amplification of the oligonucleotide library (29). Barcode 2, placed upstream of the target sequence, was used to identify each guide RNA and target sequence pair after deep sequencing.
For the target sequences for the oligonucleotide pool, we extracted sequences from the human genome and generated random synthetic sequences without any information about the activity of the corresponding sgRNAs. We first randomly extracted 9824 target sequences from the GeCKOv1 library (40), excluding those with BsmBI sites in their sequences. From 1841 target sequences from the coding sequences of three human and six mouse cell surface marker–encoding genes (14) and 2549 sequences from genes related to resistance against vemurafenib, selumetinib, and 6-thioguanine (7), we obtained 1804 and 2484 target sequences, respectively, after excluding sequences containing BsmBI sites. For training the algorithm with guide sequences with extreme GC content, we randomly generated synthetic input sequences containing an NGG PAM with a total length of 30 nt using in-house Python scripts (Supplementary Code) and subsequently selected 998 input sequences containing guide sequences with extremely low or high GC content (≤20% or ≥80%). In addition, 546 endogenous target sequences from human coding and noncoding genes of interest designed for other studies in our laboratory were included; for this group, five unique barcodes per target sequence were used to yield fivefold coverage for each target site. Together, the set of 17,840 oligonucleotides is composed of 9824 + (1804 + 2484) + 998 + (546 × 5) oligonucleotides.
Plasmid library preparation
Preparation of the plasmid library containing guide RNA and corresponding target sequence pairs involved a two-step cloning process: Gibson assembly followed by restriction enzyme–induced cutting and ligation (fig. S1). This multistep procedure effectively prevented uncoupling between guide RNA and target sequence pairs during PCR amplification of the oligonucleotide pool (29). The multistep cloning protocol was adapted and modified from a previously described process (31).
Step I: Generation of the initial plasmid library containing guide and target sequence pairs
The Lenti-gRNA-Puro plasmid (#84752, Addgene) (22) was linearized with Bsm BI enzyme [New England Biolabs (NEB), Ipswich, MA] at 55°C for 6 hours. After digestion, the vector was treated with 2 μl of calf intestinal alkaline phosphatase (NEB) at 37°C for 30 min and then gel-purified using a MEGAquick-spin Total Fragment DNA Purification kit (iNtRON Biotechnology, Seongnam, South Korea).
The oligonucleotide pool was PCR-amplified using Phusion Polymerase (NEB); the primers are described in table S4. The amplicons were gel-purified on a 4% agarose gel and assembled with the cut Lenti-gRNA-Puro plasmid described above using a NEBuilder HiFi DNA Assembly kit (NEB). After incubation at 50°C for 1 hour, the assembled products were purified using a MEGAquick-spin Total Fragment DNA Purification kit (iNtRON Biotechnology) and transformed into electrocompetent cells (Lucigen, Middleton, WI) with a MicroPulser electroporator (Bio-Rad, Hercules, CA). Transformed cells were seeded onto Luria-Bertani (LB) agar plates supplemented with carbenicillin (50 μg/ml) and incubated at 37°C for 16 hours. A small fraction (20 μl) of the culture was separately spread to calculate the library coverage; the resulting library coverage ranged from 200× to 220× the initial number of oligonucleotides (i.e., 17,840). Total colonies were harvested, and plasmids were extracted using a Plasmid Maxiprep kit (Qiagen, Hilden, Germany).
Step II: Insertion of the sgRNA scaffold
The initial plasmid library generated in Step I was digested with Bsm BI (NEB) for 9 hours and treated with 2 μl of calf intestinal alkaline phosphatase (NEB) at 37°C for 30 min. The digested product was size-selected via 0.8% agarose gel electrophoresis and purified using a MEGAquick-spin Total Fragment DNA Purification kit (iNtRON Biotechnology).
Separately, a synthesized insert fragment containing the sgRNA scaffold was cloned into a TOPO vector (T-blunt vector, SolGent, Daejeon, South Korea). The insert fragment sequence is as follows (the sgRNA scaffold with a poly-T sequence is in bold, and the BsmBI cut sites are underlined): CGTCTCTGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTGGGAGACG.
Subsequently, the TOPO vector containing the insert fragment was digested with BsmBI (NEB), and the 83-nt insert fragment was gel-purified on a 4% agarose gel. A ligation reaction was performed using 40 ng of this purified insert and 100 ng of the cut initial plasmid library vector described above (fig. S1). Following an overnight incubation at 16°C, the reaction product was heat-inactivated at 65°C for 10 min and column-purified. The purified product was transformed into electrocompetent cells (Lucigen) with a MicroPulser electroporator (Bio-Rad). Transformed cells were seeded onto LB agar plates supplemented with carbenicillin (50 μg/ml) and incubated for 16 hours at 37°C. A small fraction of the culture was separately spread on an LB plate with carbenicillin (50 μg/ml) to calculate the library coverage. Accordingly, we obtained a final plasmid library coverage of 25× to 30× the initial number of oligonucleotides (i.e., 17,840). Colonies were harvested and plasmids were extracted using a Plasmid Maxiprep kit (Qiagen).
Lentivirus production
HEK293T cells (American Type Culture Collection) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco, Waltham MA). For lentivirus production, transfer plasmids containing the gene of interest, psPAX2, and pMD2.G were mixed at a weight ratio of 4:3:1 to yield a total of 20 μg of the plasmid mixture, which was then delivered to 80 to 90% confluent HEK293T cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). At 12 hours after transfection, cells were refreshed with 10 ml of growth medium. The supernatant containing the virus was collected at 36 hours after the initial transfection, filtered through a Millex-HV 0.45-μm low protein–binding membrane (Millipore, Darmstadt, Germany), divided into aliquots, and frozen at −80°C until use. To determine the virus titer, viral aliquots were serially diluted and transduced into HEK293T cells in the presence of polybrene (8 μg/ml). The untransduced cells and serially diluted virus-treated cells were cultured in the presence of puromycin (2 μg/ml) or blasticidin S (20 μg/ml) (InvivoGen). When almost all of the untransduced cells had died, the number of surviving cells in the virus-treated population was counted to estimate the viral titer as previously described (40).
Cell library generation
HEK293T cells (9.0 × 106) were seeded into a 150-mm tissue culture dish and incubated overnight. The cells were transduced with the lentiviral library at a multiplicity of infection (MOI) of 0.3 in the presence of polybrene (8 μg/ml) and incubated overnight (15 to 18 hours). To remove untransduced cells, the cells were cultured in the presence of puromycin (2 μg/ml). To preserve its diversity, the cell library was maintained at a quantity of at least 9.0 × 106 cells throughout the study.
Cas9 delivery into the cell library
A total of 1.8 × 107 cells (two 150-mm culture dishes with 9.0 × 106 cells per dish) from the cell library were transduced with SpCas9-encoding lentiviral vectors at an MOI of 5 in DMEM supplemented with 10% FBS and polybrene (8 μg/ml). After overnight incubation (15 to 18 hours), the culture medium was replaced with DMEM supplemented with 10% FBS and blasticidin S (20 μg/ml) (InvivoGen). The cultures were harvested at 2.9 days after transduction.
Measurement of indel frequencies at endogenous sites
A total of 124 target sites were selected from the 546 endogenous targets, which are described above in the oligonucleotide library design section, by stratified random sampling (50 targets for DHS regions and 74 targets for non-DHS regions). HEK293T cells were transfected with a mixture of 100 ng of plasmid encoding sgRNA (pRG2; #104174, Addgene) and 100 ng of plasmid encoding SpCas9 (pRGEN-Cas9-CMV/T7-Puro-RFP; purchased from ToolGen, Seoul, Korea) at a density of 1.0 × 105 cells per well in a 96-well plate using TransIT-X2 (Mirus Bio, Madison, WI) according to the manufacturer’s instructions. Following an overnight incubation, the culture medium was replaced with DMEM containing puromycin (2 μg/ml). Cells were harvested and subjected to deep sequencing 3.7 days after the transfection. The average value of indel frequencies from the triplicate studies was used as the representative indel frequency for each target site.
Deep sequencing
Genomic DNA was extracted from cell pellets using a Wizard Genomic DNA Purification kit (Promega, Fitchburg, WI). For the high-throughput experiment, integrated target sequences were PCR-amplified using 2X Taq PCR Smart mix (SolGent). A total of 576 μg of genomic DNA was used for the first PCR to achieve over 3000× coverage over the library (assuming 10 μg of genomic DNA for 106 cells) (22). We performed 288 independent 50-μl PCRs with an initial genomic DNA concentration of 2 μg per reaction. The PCR products were then combined into a single pool and purified with a MEGAquick-spin Total Fragment DNA Purification kit (iNtRON Biotechnology); 20 ng of purified product was subsequently PCR-amplified using primers containing both Illumina adaptor and barcode sequences (table S4). For the cells transfected with SpCas9- and sgRNA-encoding plasmids, we carried out the independent first PCRs in a 20-μl reaction volume containing 40 ng of initial genomic DNA template per sample. Then, a second PCR to attach the Illumina adaptor and barcode sequences was conducted in a 20-μl reaction volume using 0.2 μl of the unpurified product from the first PCR. The resulting amplicons were gel-purified and analyzed using HiSeq or MiniSeq (Illumina, San Diego, CA). The primers used for PCRs are shown in table S4.
Analysis of indel frequencies
Deep sequencing data were analyzed using in-house Python scripts (Supplementary Code), which were modified from previously used code (22). Each guide RNA and target sequence pair were identified using the unique 15-nt barcode sequence located upstream of the target sequence. Insertions or deletions located around the expected cleavage site (i.e., the 8-nt region centered on the middle of the cleavage site) were considered to be Cas9-induced mutations. To exclude the background indel frequencies originating from array synthesis and PCR amplification procedures, we normalized the observed indel frequency by subtracting the background indel frequency determined in the absence of Cas9 delivery according to the function
To increase the accuracy of the analysis, deep sequencing data were filtered; target sequences with deep sequencing read counts below 200 and background indel frequencies above 8% were excluded as similarly performed previously (21).
Calculation of chromatin accessibility
DNase-sequencing (DNase-seq) narrow peak data from ENCODE (36) were used to calculate chromatin accessibility as previously described (21). For each target site, 23 bases of the PAM plus protospacer sequence were aligned to the hg19 human reference genome using bowtie (41). Only the target sites that overlapped with DNase-seq narrow peaks were considered as DNase I hypersensitive target sites.
Generation of paired subsets of the Endo_Cas9 dataset
We divided the Endo_Cas9 dataset into paired subsets by stratified random sampling from strata of DHS and non-DHS sites so that a similar ratio of DHS/non-DHS sites was assigned to each subset. We named the resulting data subset pairs Endo_Cas9_1A and Endo_Cas9_1B. We then repeated this stratified random sampling to generate four more pairs of data subsets, designated Endo_Cas9_2A and Endo_Cas9_2B, etc. (table S3).
Convolutional neural network
CNNs (32) are one of the most robust deep learning architectures applicable to locally correlated data and have been successfully implemented in DNA sequence–related research such as the prediction of CRISPR-Cpf1 guide RNA activity (21), transcription factor binding affinity (42), and DNA sequence accessibility (43). CNNs consist of three types of layers: a convolutional layer, a pooling layer, and a fully connected layer. In the convolutional layer, various filters are applied to the data, which allows the network to obtain features from local regions rather than the whole. In the pooling layer, several operations (max, average, etc.) are used to effectively decrease the dimensions and “pool out” useful features from local features extracted from the previous convolutional layer. Convolutional layers and pooling layers are usually interchanged at the initial steps of the CNN, and fully connected layers are constructed in the latter phases. The fully connected layer combines the pooled features by weighted sum and a nonlinear function to get the final function as the solution. Compared to simple multilayer perceptron models, CNNs exploit strong heuristics for locally related data. This characteristic has resulted in CNN-based models outperforming the majority of the previously used models in various fields of research.
Multiple filter sizes
In CNNs, for each layer, the filter size should be experimentally determined during the model selection phase, as the optimal filter size for the best performance is unknown. In GoogLeNet (33), an inception module was used to overcome this manual process. In an inception module, various sizes of filters are used in one layer as shown in fig. S2. Along with several other techniques, GoogLeNet demonstrated a significant gain in performance compared to the original CNN. Accordingly, we adopted the multiple filter size technique from the inception module as our basic module for DeepSpCas9.
Development of DeepSpCas9
DeepSpCas9 is a deep learning–based computational model for SpCas9 activity prediction. The training data consisted of the high-throughput dataset (HT_Cas9_Train; table S1) and is used for 10-fold cross-validation during the model selection phase. Thirty-nucleotide-long input sequences, which include 4-bp left neighbor, 20-bp protospacer, 3-bp PAM, and 3-bp right neighbor sequences, were converted into a four-dimensional binary matrix by one-hot encoding (fig. S2). DeepSpCas9 has one convolutional layer and one pooling layer at the front, as well as three fully connected layers with a dropout rate of 0.3 in each layer. The adopted convolutional layer includes an inception module with a total of 210 filters (100, 70, and 40 filters at 3, 5, and 7 nt in length, respectively). The pooling layer and three fully connected layers use ReLU activation functions. We tested a total of 324 different models (details in table S5) and selected the model and training epoch that produced the highest validation score calculated using Spearman correlation coefficients between the experimentally measured and predicted activity levels. After selecting the optimal hyperparameters, we used the full training dataset with selected hyperparameters to train the final model.
For the development of DeepSpCas9-CA (chromatin accessibility), we fine-tuned DeepSpCas9 using a data subset generated by stratified random sampling of the Endo dataset (e.g., Endo-1A) and binary chromatin accessibility information. We added a fully connected layer with 60 U that transformed the binary chromatin accessibility information into a 60-dimensional vector, which enabled the integration of the sequence feature vector and chromatin accessibility information through element-wise multiplication. The regression output layer performs a linear transformation of the outputs and calculates the prediction scores for SpCas9 activity. We applied a dropout rate of 0.3, a mean squared error, as the objective function, and an Adam optimizer with a learning rate of 10−3 in both layers. DeepSpCas9 and DeepSpCas9-CA were implemented using TensorFlow (44).
Training of conventional machine learning–based models
We trained seven models based on conventional machine learning algorithms, i.e., SVM, L1-regularized linear regression, L2-regularized linear regression, L1L2-regularized linear regression, AdaBoost, random forest, and gradient-boosted regression trees. All of the models were implemented using scikit-learn (version 0.19.1) (45). A total of 627 features, which included position-independent and position-dependent nucleotides and dinucleotides, melting temperature, GC counts, and the minimum self-folding free energy, were extracted as previously described (7, 21). We performed 10-fold cross-validation for model selection among the regularization parameters and hyperparameter configurations, the number of which is comparable to the number of hyperparameter configurations used for the development of DeepSpCas9 (324). For L1-, L2-, and L1L2-regularized linear regression, over 250 points that were evenly spaced between 10−6 and 106 in log space were searched to optimize the regularization parameter. For SVM, we searched over 225 models from the following hyperparameters: penalty parameter C and kernel parameter γ, 15 points that were evenly spaced between 10−3 and 103. For random forest, AdaBoost, and gradient-boosted regression tree, we searched over 192 models selected from the following hyperparameter configurations: the number of base estimators (chosen from [50, 100, 200, 400]), the maximum depth of the individual regression estimators (chosen from [50, 100, 200, expanded until all leaves are pure]), the minimum number of samples to split an internal node (chosen from [2, 4]), the minimum number of samples to be at a leaf node (chosen from [1, 2]), and the maximum number of features to consider when looking for the best split (chosen from [all features, the square root of all features, the binary logarithm of all features]).
Performance comparison of DeepSpCas9 with other models
We compared the prediction performance of DeepSpCas9 with those of the conventional machine learning–based models trained on the high-throughput dataset and other previously reported prediction models (7, 10, 13–16, 18, 39). The performance of each prediction model was evaluated by the Spearman correlation coefficients between experimentally measured sgRNA activities and prediction scores from each model. We used the Endo dataset generated in this study and the other 14 published datasets from other groups that were large enough (number of target sequences, >100) (7, 10, 14, 16, 18, 35, 37–39) collected by Haeussler et al. (20). In these test datasets, the target sequences included in the HT_Cas9_Train dataset were excluded. Furthermore, for a fair comparison of generalization performances, we excluded correlations of models tested against their own training datasets (20).
Statistical significance
To compare the indel frequencies between DHS and non-DHS sites, we used the two-tailed Student’s t test under the null hypothesis that the indel frequencies of the two groups are the same (Fig. 1B). To compare the Spearman correlation between prediction scores from two models (Fig. 2, B to E), we used Steiger’s test, which is used for testing two dependent correlation coefficients from exactly the same dataset. Statistical significance was determined using PASW Statistics (version 18.0, IBM) and Microsoft Excel (version 16.0, Microsoft Corporation).
Supplementary Material
Acknowledgments
Funding: This work was supported by the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (HI17C0676 to H.H.K.). Author contributions: H.K.K. and Y.K. performed experiments to build high-throughput datasets of SpCas9 indel frequencies. H.K.K., Y.K., and J.Y.B. performed experiments to build datasets of SpCas9 indel frequencies at endogenous sites. S.L., S.M., and S.Y. developed the framework and carried out the model training and computational validation. J.W.C., J.P., and D.J. significantly contributed to bioinformatics analyses. H.H.K. conceived and designed the study. H.K.K., Y.K., S.L., S.M., S.Y., and H.H.K. analyzed the data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The deep sequencing data from this study have been submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA; www.ncbi.nlm.nih.gov/sra/) under accession number SRP150719. The datasets used in this study are provided in tables S1 and S2. Source code for DeepSpCas9 and custom Python scripts used for the indel frequency calculations are available in Supplementary Code and on github (at https://github.com/MyungjaeSong/Paired-Library and https://github.com/CRISPRJWCHOI/IndelSearcher). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax9249/DC1
Fig. S1. Development of a high-throughput evaluation system for Cas9-induced indel frequencies.
Fig. S2. Overview of DeepSpCas9 development.
Table S1. Datasets generated from the results of the high-throughput experiments.
Table S2. Datasets used for this study.
Table S3. Datasets generated from the results of the experiments at endogenous target sites.
Table S4. Oligonucleotides used in this study.
Table S5. Model selection results.
Table S6. Confidence intervals for the values shown in the graphs.
Supplementary Code
REFERENCES AND NOTES
- 1.Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M., RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho S. W., Kim S., Kim J. M., Kim J.-S., Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J., RNA-programmed genome editing in human cells. eLife 2, e00471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang W. Y., Fu Y., Reyon D., Maeder M. L., Tsai S. Q., Sander J. D., Peterson R. T., Yeh J.-R., Joung J. K., Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang W., Bikard D., Cox D., Zhang F., Marraffini L. A., RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doench J. G., Fusi N., Sullender M., Hegde M., Vaimberg E. W., Donovan K. F., Smith I., Tothova Z., Wilen C., Orchard R., Virgin H. W., Listgarten J., Root D. E., Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meier J. A., Zhang F., Sanjana N. E., GUIDES: sgRNA design for loss-of-function screens. Nat. Methods 14, 831–832 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuan P. F., Powers S., He S., Li K., Zhao X., Huang B., A systematic evaluation of nucleotide properties for CRISPR sgRNA design. BMC Bioinformatics 18, 297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreno-Mateos M. A., Vejnar C. E., Beaudoin J. D., Fernandez J. P., Mis E. K., Khokha M. K., Giraldez A. J., CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12, 982–988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson L. O. W., Reti D., O'Brien A. R., Dunne R. A., Bauer D. C., High activity target-site identification using phenotypic independent CRISPR-Cas9 core functionality. CRISPR J. 1, 182–190 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Labuhn M., Adams F. F., Ng M., Knoess S., Schambach A., Charpentier E. M., Schwarzer A., Mateo J. L., Klusmann J. H., Heckl D., Refined sgRNA efficacy prediction improves large- and small-scale CRISPR-Cas9 applications. Nucleic Acids Res. 46, 1375–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chuai G., Ma H., Yan J., Chen M., Hong N., Xue D., Zhou C., Zhu C., Chen K., Duan B., Gu F., Qu S., Huang D., Wei J., Liu Q., DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 19, 80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doench J. G., Hartenian E., Graham D. B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B. L., Xavier R. J., Root D. E., Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong N., Liu W., Wang X., WU-CRISPR: Characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 16, 218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu H., Xiao T., Chen C. H., Li W., Meyer C. A., Wu Q., Wu D., Cong L., Zhang F., Liu J. S., Brown M., Liu X. S., Sequence determinants of improved CRISPR sgRNA design. Genome Res. 25, 1147–1157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng H., Zheng Y., Blumenstein M., Tao D., Li J., CRISPR/Cas9 cleavage efficiency regression through boosting algorithms and Markov sequence profiling. Bioinformatics 34, 3069–3077 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Chari R., Mali P., Moosburner M., Church G. M., Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods 12, 823–826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chari R., Yeo N. C., Chavez A., Church G. M., sgRNA scorer 2.0: A species-independent model to predict CRISPR/Cas9 activity. ACS Synth. Biol. 6, 902–904 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haeussler M., Schonig K., Eckert H., Eschstruth A., Mianne J., Renaud J. B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., Joly J. S., Concordet J.-P., Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim H. K., Min S., Song M., Jung S., Choi J. W., Kim Y., Lee S., Yoon S., Kim H. H., Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity. Nat. Biotechnol. 36, 239–241 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Kim H. K., Song M., Lee J., Menon A. V., Jung S., Kang Y. M., Choi J. W., Woo E., Koh H. C., Nam J. W., Kim H., In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat. Methods 14, 153–159 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Allen F., Crepaldi L., Alsinet C., Strong A. J., Kleshchevnikov V., De Angeli P., Palenikova P., Khodak A., Kiselev V., Kosicki M., Bassett A. R., Harding H., Galanty Y., Munoz-Martinez F., Metzakopian E., Jackson S. P., Parts L., Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 37, 64–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen M. W., Arbab M., Hsu J. Y., Worstell D., Culbertson S. J., Krabbe O., Cassa C. A., Liu D. R., Gifford D. K., Sherwood R. I., Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakrabarti A. M., Henser-Brownhill T., Monserrat J., Poetsch A. R., Luscombe N. M., Scaffidi P., Target-specific precision of CRISPR-mediated genome editing. Mol. Cell 73, 699–713.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vidigal J. A., Ventura A., Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nat. Commun. 6, 8083 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong A. S. L., Choi G. C., Cui C. H., Pregernig G., Milani P., Adam M., Perli S. D., Kazer S. W., Gaillard A., Hermann M., Shalek A. K., Fraenkel E., Lu T. K., Multiplexed barcoded CRISPR-Cas9 screening enabled by CombiGEM. Proc. Natl. Acad. Sci. U.S.A. 113, 2544–2549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu S., Li W., Liu J., Chen C. H., Liao Q., Xu P., Xu H., Xiao T., Cao Z., Peng J., Yuan P., Brown M., Liu X. S., Wei W., Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol. 34, 1279–1286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du D., Roguev A., Gordon D. E., Chen M., Chen S. H., Shales M., Shen J. P., Ideker T., Mali P., Qi L. S., Krogan N. J., Genetic interaction mapping in mammalian cells using CRISPR interference. Nat. Methods 14, 577–580 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Najm F. J., Strand C., Donovan K. F., Hegde M., Sanson K. R., Vaimberg E. W., Sullender M. E., Hartenian E., Kalani Z., Fusi N., Listgarten J., Younger S. T., Bernstein B. E., Root D. E., Doench J. G., Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 36, 179–189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen J. P., Zhao D., Sasik R., Luebeck J., Birmingham A., Bojorquez-Gomez A., Licon K., Klepper K., Pekin D., Beckett A. N., Sanchez K. S., Thomas A., Kuo C. C., Du D., Roguev A., Lewis N. E., Chang A. N., Kreisberg J. F., Krogan N., Qi L., Ideker T., Mali P., Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 14, 573–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lecun Y., Bottou L., Bengio Y., Haffner P., Gradient-based learning applied to document recognition. Proc. IEEE 86, 2278–2324 (1998). [Google Scholar]
- 33.C. Szegedy, L. Wei, J. Yangqing, P. Sermanet, S. Reed, D. Anguelov, D. Erhan, V. Vanhoucke, A. Rabinovich, Going deeper with convolutions, in 2015 IEEE Conference on Computer Vision and Pattern Recognition (CVPR) (IEEE, 2015), pp. 1–9. [Google Scholar]
- 34.LeCun Y., Bengio Y., Hinton G., Deep learning. Nature 521, 436–444 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K. R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S., Mero P., Dirks P., Sidhu S., Roth F. P., Rissland O. S., Durocher D., Angers S., Moffat J., High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526 (2015). [DOI] [PubMed] [Google Scholar]
- 36.The ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gagnon J. A., Valen E., Thyme S. B., Huang P., Akhmetova L., Pauli A., Montague T. G., Zimmerman S., Richter C., Schier A. F., Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLOS ONE 9, e98186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varshney G. K., Pei W., LaFave M. C., Idol J., Xu L., Gallardo V., Carrington B., Bishop K., Jones M., Li M., Harper U., Huang S. C., Prakash A., Chen W., Sood R., Ledin J., Burgess S. M., High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 25, 1030–1042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang T., Wei J. J., Sabatini D. M., Lander E. S., Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelsen T. S., Heckl D., Ebert B. L., Root D. E., Doench J. G., Zhang F., Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alipanahi B., Delong A., Weirauch M. T., Frey B. J., Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Kelley D. R., Snoek J., Rinn J. L., Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 26, 990–999 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.M. Abadi, P. Barham, J. Chen, Z. Chen, A. Davis, J. Dean, M. Devin, S. Ghemawat, G. Irving, M. Isard, M. Kudlur, J. Levenberg, R. Monga, S. Moore, D. G. Murray, B. Steiner, P. Tucker, V. Vasudevan, P. Warden, M. Wicke, Y. Yu, X. Zheng, paper presented at the Proceedings of the 12th USENIX Conference on Operating Systems Design and Implementation, Savannah, GA, USA, 2016. [Google Scholar]
- 45.D. Kingma, J. Ba, Adam: A method for stochastic optimization. arXiv:1412.6980 [cs.LG] (22 December 2014).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax9249/DC1
Fig. S1. Development of a high-throughput evaluation system for Cas9-induced indel frequencies.
Fig. S2. Overview of DeepSpCas9 development.
Table S1. Datasets generated from the results of the high-throughput experiments.
Table S2. Datasets used for this study.
Table S3. Datasets generated from the results of the experiments at endogenous target sites.
Table S4. Oligonucleotides used in this study.
Table S5. Model selection results.
Table S6. Confidence intervals for the values shown in the graphs.
Supplementary Code