

Nanoparticles target inflammatory neutrophils to deliver doxorubicin for neutrophil apoptosis in resolution of inflammation.
Abstract
Human neutrophils are the most abundant circulating leukocytes and contribute to acute and chronic inflammatory disorders. Neutrophil apoptosis is programed cell death to maintain immune homeostasis, but inflammatory responses to infections or tissue injury disrupt neutrophil death program, leading to many diseases. Precise control of neutrophil apoptosis may resolve inflammation to return immune homeostasis. Here, we report a method in which doxorubicin (DOX)–conjugated protein nanoparticles (NPs) can in situ selectively target inflammatory neutrophils for intracellular delivery of DOX that induces neutrophil apoptosis. We showed that neutrophil uptake of NPs required their activation and was highly selective. DOX release was triggered by acidic environments in neutrophils, subsequently inhibiting neutrophil transmigration and inflammatory responses. In two disease models, DOX-conjugated NPs notably increased mouse survival in sepsis and prevented brain damage in cerebral ischemia/reperfusion, but the NPs did not suppress systemic immunity. Our studies offer a promising strategy to treat inflammatory diseases.
INTRODUCTION
Polymorphonuclear neutrophils (PMNs) are the most abundant white blood cells (50 to 70%) in humans (1), playing a central role in the innate immune response to infections or tissue injury (2, 3). It is shown that their defense mechanism involved in neutrophil infiltration and proinflammatory responses may be potentially detrimental to the host if neutrophils are dysregulated (4–6). Exaggerated activation and uncontrolled infiltration of neutrophils cause inflammatory and autoimmune diseases, such as acute lung inflammation/injury (7), ischemia/reperfusion (I/R) (8), rheumatoid arthritis (9), and sepsis (10). Anti-inflammatory agents are usually used to treat these diseases, for example, nonsteroidal anti-inflammatory drugs and anticytokine therapies (11). The off-targeting of these therapies may cause the systemic immune suppression leading to severe side effects and susceptibility to infections (5, 11). Depletion of neutrophils in blood and bone marrow by administration of antibodies shows reduced inflammatory responses (12), indicating that targeting neutrophils is an applicable strategy to alleviate inflammatory disorders. However, total loss of the immune sentinel neutrophils using antibodies renders the vulnerability to infections and impairs innate and adaptive immune systems (13). Therefore, developing new strategies to specifically target inflammatory neutrophils is needed.
Neutrophils have a short life span in circulation (8 to 20 hours), and their life span is precisely regulated by apoptosis (14). Apoptosis is a process of programmed cell death to maintain constant neutrophil numbers in circulation. Neutrophils undergo constitutive or spontaneous apoptosis that is a mechanism to preserve the immune homeostasis (15). Inflammation caused by harmful stimuli (microorganisms or damaged tissues) rapidly increases the numbers of neutrophils in blood, and their longevity extends. Subsequently, neutrophils are activated for transmigration and promote the cytokine release. Delayed/impaired apoptosis of neutrophils initiates acute and chronic inflammatory disorders, such as sepsis and ischemic stroke (5, 15). Therefore, specifically targeting inflammatory neutrophils to promote their apoptosis in time may be a strategy for improved therapies of inflammatory diseases (16, 17). Doxorubicin (DOX) is a widely used drug in cancer therapy. Intercalation of DOX into DNA double helices inhibits the progression of topoisomerase II, causing DNA damage to induce cell death (18).
Here, we report on the development of pH-responsive albumin nanoparticles (NPs) conjugated with DOX via a pH-labile bond, which can selectively target activated neutrophils in situ for intracellular delivery of DOX. We have found that DOX induces neutrophil apoptosis that inhibits neutrophil tissue transmigration and related inflammatory responses in bacterial toxin [lipopolysaccharide (LPS)]–induced mouse acute lung inflammation. To demonstrate translational potential of our drug delivery system, we performed LPS-induced sepsis and cerebral I/R in the mouse models. The results indicate that DOX-conjugated NPs alleviated inflammatory responses for improved mouse survival in sepsis and also diminished brain damage in ischemic stroke.
RESULTS
Figure 1A shows the concept of design of NPs that specifically target activated neutrophils in vivo to deliver DOX to induce neutrophil apoptosis for improved therapies of inflammatory disorders. To deliver DOX in activated neutrophils, we synthesized a pH-sensitive DOX prodrug by conjugating DOX to bovine serum albumin (BSA) via a hydrazone bond (called DOX-hyd-BSA) (Fig. 1B). The synthetic pathway and characterization of chemical structures of DOX-BSA conjugates are shown in figs. S1 and S2. DOX was first conjugated to polyethylene glycol (PEG) through hydrazine to produce DOX-hyd-PEG, followed by conjugating it to BSA. The BSA complexes were formed to NPs by desolvation, subsequently adding glutaraldehyde to cross-link BSA protein to make stable NPs. After intravenous administration of DOX-hyd-BSA NPs, the NPs can specifically target activated neutrophils in circulation and were internalized when neutrophils responded to infections or tissue injury. Hydrazone bonds were cleaved by acid in neutrophil environments to release DOX molecules, which were able to induce neutrophil apoptosis, thus mitigating neutrophil transmigration. In control, DOX was linked to BSA via a pH-insensitive amide bond (called DOX-ab-BSA); thus, DOX was not released from BSA.
Fig. 1. Scheme shows NP targeting of proinflammatory neutrophils to induce their apoptosis for treatment of inflammatory diseases.
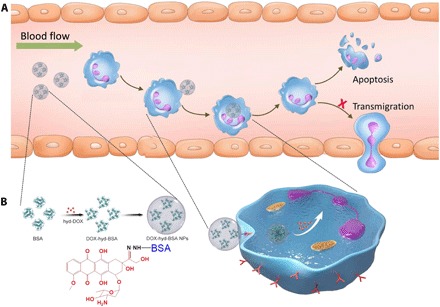
(A) The therapeutic process of DOX-conjugated BSA NPs includes neutrophil uptake of NPs in situ and cell apoptosis to prevent neutrophil transmigration and inflammatory responses. (B) DOX is conjugated to BSA via a hydrazone bond, followed by aggregating BSA conjugates to form DOX-hyd-BSA NPs, and DOX release from NPs triggered by low pH in neutrophils to promote neutrophil apoptosis.
BSA NPs were generated by adding ethanol to induce the self-assembling of BSA into an NP, and subsequently, BSA molecules were cross-linked using glutaraldehyde to form stable NPs (19). BSA NPs and two types of DOX-conjugated BSA NPs were characterized by dynamic light scattering (DLS) (Fig. 2A), showing that their sizes were about 130 nm in diameter. Transmission electron microscopy (TEM) showed a spherical shape of NPs (DOX-hyd-BSA shown in Fig. 2B, and BSA NPs and DOX-ab-BSA in fig. S3), and the size was consistent with the measurement by DLS. The surface charges of three types of BSA NPs were similar (Fig. 2C). Under alkaline conditions, the zeta potential of NPs was negative due to the deprotonation of amine residues in NPs. Under acidic conditions, surface charges of NPs were switched to positive because amine residues in NPs were protonated. The trend of zeta potentials of BSA NPs and DOX-hyd-BSA NPs was similar in acidic conditions, but DOX-ab-BSA NPs had the slightly higher surface charges because amine residues of DOX may contribute the additional protonation of amine residues. The results suggested that DOX may be released from DOX-hyd-BSA NPs at low pH. The serum stability of BSA-based NPs was evaluated in phosphate buffer solution (PBS) containing 20% fetal bovine serum (FBS) at pH 7.4. No aggregation was observed even after incubation for 4 days (fig. S4), displaying BSA NPs were very stable.
Fig. 2. Characterization of DOX-conjugated BSA NPs and DOX release from NPs at acidic condition to cause cell death.
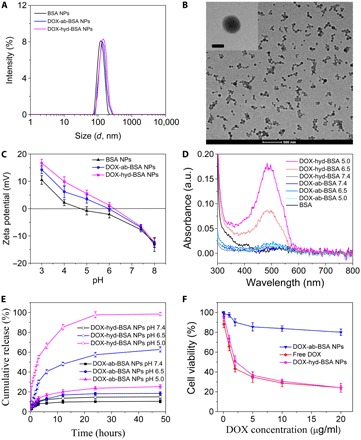
(A) Particle size of BSA NPs, DOX-ab-BSA NPs, and DOX-hyd-BSA NPs measured by DLS in PBS at pH 7.4. (B) TEM image of DOX-hyd-BSA NPs. Scale bars, 500 and 100 nm for inset. (C) Zeta potential of BSA NPs, DOX-ab-BSA NPs, and DOX-hyd-BSA NPs in PBS at different pH. (D) UV-vis spectra of their supernatants after NPs were incubated in PBS at different pH for 2 hours. a.u., arbitrary unit. (E) Cumulative DOX release from DOX-ab-BSA NPs or DOX-hyd-BSA NPs in PBS at pH 7.4, 6.5, or 5.0. (F) Cell death of differentiated HL-60 cells (neutrophil-like cells) induced by DOX. Cells were treated for 24 hours at different concentrations of DOX. Data are shown as means ± SD (n = 6 independent experiments).
Next, we addressed whether BSA NPs were responsive to acidic environments for DOX release. DOX-conjugated BSA NPs were incubated in PBS at pH 7.4 or at pH 5.0 to 6.5 (similar to neutrophil cytosol environments) (20). Two hours after incubation, each type of NP supernatants was collected, and their absorption spectra were measured using an ultraviolet-visible (UV-vis) spectrometer (Fig. 2D). Absorption of DOX molecules at 480 nm was observed when DOX-hyd-BSA NPs were incubated at pH 5.0 and 6.5, and the absorption was dependent on pH, indicating that DOX was able to be cleaved from BSA NPs. However, we did not observe DOX in the supernatant of DOX-hyd-BSA NPs at pH 7.4, suggesting that DOX could be tightly trapped in NPs in the physiological condition. DOX-ab-BSA NPs did not show the absorption of DOX in their supernatants from pH 5.0 to 7.4 because the bond between DOX and BSA was not pH responsive. We further measured a time course of drug release in DOX-BSA NPs and their pH dependence in vitro (Fig. 2E). DOX-ab-BSA NPs released 10 to 20% DOX at pH 5.0 to 7.4 in 48 hours. In contrast, DOX release from DOX-hyd-BSA NPs was rapid in the first 10 hours (e.g., 60% DOX release at pH 6.5 and 80% at pH 5.0), indicating that acidic environments in a cell may trigger DOX release after DOX-hyd-BSA NPs were internalized.
We next determined whether DOX can be released from NPs to promote cell death. We used HL-60 cells (a human promyelocytic leukemia cell line) because they are neutrophil like after their differentiation (21). The cytotoxicity was similar for both free DOX and DOX-hyd-BSA NPs, and their cytotoxicity was more efficient compared with that of DOX-ab-BSA NPs (Fig. 2F). The result showed that DOX can be released from BSA NPs via the cleavage of hydrazone bonds between DOX and BSA to cause cell death. Furthermore, BSA NPs alone did not have any toxicity to cells in a wide range of concentrations (0 to 400 μg/ml) (fig. S4), and BSA NPs were used at 100 μg/ml per mouse in our animal experiments.
Neutrophil activation is required for tissue infiltration that contributes inflammatory responses, so targeting activated neutrophils may increase drug delivery, avoiding systemic toxicity (2). We found that activated neutrophils can take up BSA NPs, but development of their responsive drug delivery systems to treat inflammatory disorders remains unknown (19). Furthermore, it is unclear whether inflammatory responses up-regulate Fcγ receptors to mediate the uptake of BAS NPs. We established intravital microscopy of mouse cremaster venules to study this correlation in a live mouse. First, we injected anti-CD16/32 (anti-Fcγ) to a mouse via the tail vein, and 3 hours later, we exposed the cremaster tissue under an intravital microscope and stained neutrophils by intravenous injection of anti-mouse LY-6G antibody (a mouse neutrophil marker). This experiment allowed us to study activities of resting (unstimulated) neutrophils in vivo. The intravital images (Fig. 3A, top, and movie S1) showed that there was no staining of anti-CD16/32 on neutrophils, suggesting that resting neutrophils did not highly express anti-CD16/32. To activate neutrophils in vivo, the mouse cremaster tissue was challenged with intrascrotal injection of tumor necrosis factor–α (TNF-α) 3 hours before imaging. The mouse was intravenously administered with anti-CD16/32 and anti–LY-6G antibodies (Fig. 3A, bottom, and movie S2). Colocalization of anti-CD16/32 and anti–LY-6G was observed, indicating that neutrophils were up-regulated to express Fcγ receptors after the mouse was challenged by TNF-α. We also analyzed the percentages of costaining of anti-CD16/32 and anti–LY-6G (Fig. 3B). The results showed that 95% neutrophils expressed anti-CD16/32 after the stimulation by TNF-α, but it was 10% when neutrophils were inactivated. However, the 10% neutrophils may be associated with their activation induced by cremaster surgery for intravital imaging (19). The results are consistent with the conclusion that Fcγ receptors play a central role in mediating neutrophil uptake of BSA NPs (19).
Fig. 3. Neutrophil activation up-regulates Fcγ receptors to mediate NP uptake.
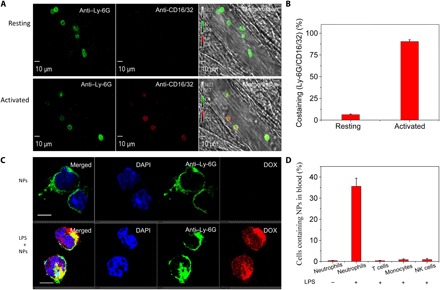
(A) Intravital microscopy of mouse cremaster muscle venules shows that neutrophil activation was associated with up-regulation of Fcγ receptors. The resting condition of neutrophils was established by no intrascrotal injection of TNF-α (0.5 μg per mouse) and the tail vein injection of Fcγ antibodies 3 hours before performing intravital microscopy (top). Intrascrotal injection of TNF-α (0.5 μg per mouse) activated neutrophils. Alexa Fluor 647–labeled anti-mouse CD16/32 (red) and Alexa Fluor 488–labeled anti-mouse LY-6G (green) antibodies were intravenously injected to stain Fcγ receptors and neutrophils, respectively (bottom). The images were taken using a Nikon A1R+ resonant-scanning confocal microscope at 488 and 640 nm. Scale bars, 10 μm. (B) Percentage of costaining between anti-mouse CD16/32 and anti-mouse LY-6G based on intravital images of (A). (C) Confocal laser scanning microscopy (CLSM) images of blood neutrophils from healthy mice or LPS-challenged mice. Four hours after intraperitoneal LPS injection, DOX-hyd-BSA NPs were intravenously administered to a mouse. Three hours later, the mouse blood was collected, and neutrophils were isolated using anti-mouse LY-6G beads. Alexa Fluor 488–labeled anti-mouse LY- 6G antibody was used to label neutrophils. Scale bars, 10 μm. (D) Uptake of BSA NPs by blood leukocytes analyzed by flow cytometry. Neutrophils, T cells, monocytes, and natural killer (NK) cells were isolated from blood and stained by Alexa Fluor 647–labeled anti-mouse LY-6G, CD3, CD115, and CD335 antibodies, respectively. All data are expressed as means ± SD (three mice per group).
The uptake of DOX-hyd-BSA NPs by differentiated HL-60 cells was evaluated in vitro. As shown in fig. S5, DOX-hyd-BSA NPs can be taken up efficiently by differentiated HL-60 cells. Next, we assessed the uptake of BSA-based NPs by neutrophils in vivo in LPS-induced inflammation mouse model. DOX-hyd-BSA NPs were intravenously administered to a mouse 4 hours after intraperitoneal injection of LPS (20 mg/kg) to induce systemic inflammation. Blood neutrophils were collected at 3 hours after injection of NPs, followed by staining anti-mouse LY-6G antibodies for analysis of NP uptake using confocal microscopy and flow cytometry. Without LPS challenge, we did not observe BSA NPs inside neutrophils, but neutrophil uptake was observed after LPS challenge (Fig. 3C). DOX fluorescence diffused in a whole cell, indicating that DOX may be released from NPs, because low pH (acidic environment) inside neutrophils induced the cleavage of a pH-labile linker between DOX and albumin. The flow cytometry result (Fig. 3D) showed that neutrophil activation (challenged with LPS) was required for NP uptake. We also analyzed the uptake of DOX-hyd-BSA NPs in other immune cells in blood (Fig. 3D), and the results showed that T cells (anti-mouse CD3 antibody to mark T cells), monocytes (anti-mouse CD115 antibody to mark monocytes), and natural killer (NK) cells (anti-mouse CD335 antibody to mark NK cells) (22) did not take up BSA NPs because of delayed activation of these adaptive immune cells. Together, BSA NPs can specifically target activated neutrophils for intracellular drug delivery during inflammation.
DOX is commonly used in cancer therapy. DOX induces cell death, but it is unknown whether the death of neutrophils is associated with apoptotic pathways. To address this question, we chose differentiated HL-60 cells because they are neutrophil-like cells. Phosphatidylserine on the outer leaflet of plasma membrane is a biomarker for cell apoptosis (23). Annexin V is commonly used to detect apoptotic cells by its binding to phosphatidylserine (24). 7-aminoactinomycin D (7AAD) is a fluorescent dye that is a membrane impermeant agent to identify dead cells (25). We stained differentiated HL-60 cells with annexin V–fluorescein isothiocyanate (FITC) and 7AAD after the cells were treated with free DOX or DOX-conjugated NPs. The confocal fluorescence microscopy showed that cells treated with free DOX or DOX-hyd-BSA NPs contained staining of annexin V–FITC, and less staining of 7AAD indicated that the cells were alive (Fig. 4A). In contrast, DOX-ab-BSA NPs did not induce cell apoptosis because DOX cannot be released from NPs. The flow cytometry analysis revealed that free DOX and DOX-hyd-BSA NPs caused cell apoptosis at 73 and 89%, respectively, whereas there were only 20% apoptotic cells after treatment with DOX-ab-BSA NPs (Fig. 4B and fig. S6). Furthermore, TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling), an assay to detect apoptotic cells, was used to confirm DOX-induced cell apoptosis. Figure 4C indicates that nearly 100% cells showed costaining of DOX and TUNEL after the cells were treated with free DOX, suggesting that DOX effectively promoted cell apoptosis. When DOX-hyd-BSA NPs were used, costaining of DOX and TUNEL was similar to that of free DOX, indicating that DOX can be released from NPs for intracellular delivery in neutrophils. In contrast, DOX-ab-BSA NP treatment showed DOX signal in cells, but a few cells had TUNEL signal, suggesting that cell uptake of DOX-ab-BSA NPs did not mediate cell apoptosis. Quantification of costaining of DOX and TUNEL in cells (Fig. 4D) suggests that DOX-hyd-BSA NPs can effectively induce neutrophil apoptosis.
Fig. 4. DOX-hyd-BSA NPs promote neutrophil apoptosis to inhibit inflammatory responses.
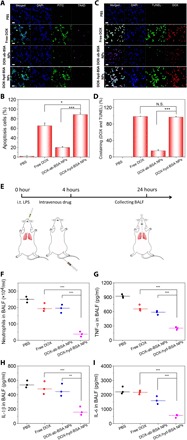
(A) CLSM images show the apoptosis of differentiated HL-60 cells. Annexin V–FITC is an apoptosis marker, and 7AAD (emission at 650 nm) is a fluorescent dye to mark dead cells. Cells were treated with PBS, free DOX, DOX-ab-BSA NPs, or DOX-hyd-BSA NPs for 24 hours, respectively. Scale bar, 20 μm. (B) Percentage of apoptotic cells analyzed by flow cytometry after HL-60 cells treated with various DOX formulations. (C) CLSM images of HL-60 cells stained with TUNEL after treatment with several DOX formulations for 24 hours. TUNEL and DOX were excited at 488 and 560 nm. Scale bar, 20 μm. (D) Percentage of costaining between DOX and TUNEL based on CLSM images of (C). N.S., not significant. (E) Diagram shows the experimental protocol in acute lung inflammation mouse model. Intratracheal (i.t.) administration of LPS causes local lung acute inflammation, and subsequently, neutrophils transmigrate from blood to airspace in the lung. Twenty-four hours after intravenous injection of several DOX formulations, BALF was collected to assess neutrophil number and cytokines. Number of neutrophils (F), inflammatory factors TNF-α (G), IL-1β (H), and IL-6 (I) in mouse BALF after i.t. LPS challenge (10 mg/kg). All data are expressed as means ± SD (three mice per group). *P < 0.05, **P < 0.01, and ***P < 0.001.
Acute lung inflammation is induced by LPS in bacterial infections and is associated with neutrophil recruitment to the lung (7, 26). Here, we asked whether targeted delivery of DOX to neutrophils in vivo could diminish neutrophil infiltration and related inflammatory responses in the mouse lung. Figure 4E shows the experimental design. Four hours after intratracheal (i.t.) LPS challenge to the mouse lung, the mouse was intravenously injected with various DOX formulations or PBS. Bronchoalveolar lavage fluid (BALF) was collected for the assessment of neutrophil infiltration from circulation to the lung airspace and cytokines. Neutrophils were measured by flow cytometry (Fig. 4F and fig. S7). After the treatment with DOX-hyd-BSA NPs at a very low dose of 0.2 mg/kg (DOX), neutrophils were markedly decreased compared with controls (free DOX– and DOX-ab-BSA NP–treated groups), suggesting that DOX-hyd-BSA NPs effectively inhibited the trafficking of activated neutrophils to the lungs in vivo because neutrophil death was induced by DOX. Lung neutrophil infiltration could enhance inflammatory responses to produce several inflammatory factors in BALF [such as TNF-α, interleukin-1β (IL-1β), and IL-6] (21). We subsequently measured the inflammatory factors and found that these cytokines (TNF-α, IL-1β, and IL-6) were decreased after treatment with DOX-hyd-BSA NPs compared with other treatments (such as free DOX and DOX-ab-BSA NPs) (Fig. 4, G to I). The results indicated that reduced cytokines may be associated with diminished neutrophil recruitment after neutrophil apoptosis.
Sepsis, characterized as a systemic inflammatory response syndrome, is a life-threatening organ dysfunction caused by a dysregulated host response to infections. Currently, the supportive care is a primary option, and no pharmacological therapies are available in clinic (27). The early stage of sepsis is strongly correlated to neutrophil tissue infiltration and related inflammatory responses (10). In an endotoxin-induced sepsis mouse model, intraperitoneal injection of LPS (50 mg/kg) caused acute and serious systemic inflammation, resulting in mouse death in a short period. We intravenously administered free DOX or DOX-hyd-BSA NPs 4 hours after LPS challenge. Free DOX showed a similar death rate in 72 hours to the PBS treatment, indicating that the treatment with free DOX did not protect mouse death from sepsis (Fig. 5A). By comparison, 70% mice survived in 72 hours when mice were treated with DOX-hyd-BSA NPs, showing the benefit of the NPs to sepsis. During the experiments, we monitored the mouse weight changes associated with metabolism and diet (Fig. 5, B to D). We found a feature in which dead mice markedly lost their weight. When mice survived, the initial weight loss was recovered and became stable. This phenomenon raised a question on how mice overcome sepsis. To address this question, we systemically investigated a time course of neutrophil numbers and cytokines in peripheral blood (Fig. 5, E to H) and lung BALF (Fig. 5, I to L). We observed that neutrophils were markedly decreased in blood and lung BALF in the first 16 hours after LPS challenge, and cytokines were also decreased accordingly when DOX-hyd-BSA NPs were administered compared with PBS treatment. The result suggests that DOX-hyd-BSA NPs may mediate apoptosis of inflammatory neutrophils in blood responsible for inhibition of neutrophil recruitment to the mouse lungs. Neutrophil death induced by DOX lastly inhibited the systemic inflammatory responses (Fig. 5, E to L). Neutrophil number and cytokine contents at 72 hours in mice treated with DOX-hyd-BSA NPs resumed to their levels as in healthy mice. The result indicates that DOX-hyd-BSA NPs do not inhibit the neutrophil production and their function in the bone marrow. We also checked the inflammatory responses in other organs in sepsis (fig. S8), finding that DOX-hyd-BSA NPs also significantly decreased inflammation in other organs. The result suggests that targeting proinflammatory neutrophils by DOX-hyd-BSA NPs can inhibit systemic inflammation in sepsis.
Fig. 5. DOX-hyd-BSA NPs protect mouse death from sepsis and does not impair the neutrophil generation in the bone marrow.
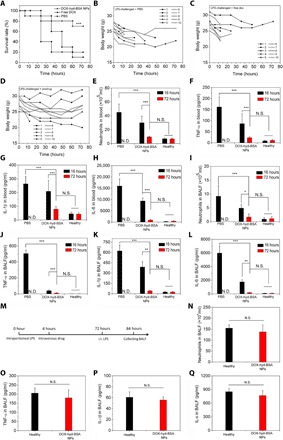
(A) Mouse survival rates in sepsis after treatments of NPs. Four hours after intraperitoneal LPS (50 mg/kg) challenge to mice, mice were treated with PBS, free DOX, and prodrug DOX-hyd-BSA NPs at 0.2 mg/kg of DOX, respectively. All data expressed are as means ± SD (10 mice per group). Statistical analysis was done using the Kaplan-Meier method. ***P < 0.001 compared to controls (PBS and free DOX). Mouse body weights were measured after treatments of PBS (B), free DOX (C), and DOX-hyd-BSA NPs (D) (equal to 0.2 mg/kg free DOX). Number of neutrophils (E), TNF-α (F), IL-1β (G), and IL-6 (H) in blood, and number of neutrophils (I), TNF-α (J), IL-1β (K), and IL-6 (L) in BALF at 16 and 72 hours after LPS challenge, respectively. N.D. (not detected) represents the mouse death. All data expressed as mean ± SD (five mice per group). (M) Diagram shows the experimental protocol to address whether DOX-conjugated BSA NPs impair neutrophil immune sentinel to the secondary infection. The mice were challenged with LPS (intraperitoneal, 50 mg/kg) or PBS (control). Four hours later, the LPS-challenged mice were intravenously treated with DOX-hyd-BSA NPs at 0.2 mg/kg of DOX. The control mice were not treated with LPS and NPs. Seventy-two hours later, all survival and control (healthy) mice were challenged with LPS [i.t. (intratracheal), 10 mg/kg)]. At 84 hours, BALF was collected to assess neutrophil number (N), TNF-α (O), IL-1β (P), and IL-6 (Q). All data are expressed as means ± SD [seven survival mice for the DOX-hyd-BSA NPs-treatment group (equal to 0.2 mg/kg free DOX), and three healthy mice for the control group]. Statistics were performed by a two-sample Student’s t test. Statistics were performed by a two-sample Student’s t test (*P < 0.05, **P < 0.01, and ***P < 0.001).
We further asked whether the treatment with DOX-conjugated BSA NPs impeded the innate immune responses of neutrophils when the second infection occurs. We designed the combinatory experiments in which a mouse was intraperitoneally challenged with LPS to cause sepsis, and then we treated the mouse with DOX-loaded NPs 4 hours after the LPS challenge. At 72 hours after the LPS challenge, the survival mouse was i.t. administered to examine whether neutrophils can transmigrate and respond to the second hit of LPS (experimental protocol shown in Fig. 5M). In the control, we i.t. challenged healthy mice with LPS. During the experiments, we monitored the mouse weight (fig. S9), finding that the mice that survived sepsis after the treatment with DOX-conjugated NPs returned their initial weight, indicating the therapeutic effect of NPs. We investigated the innate immune responses of neutrophils using an acute lung inflammation model, since this model allowed neutrophil mobility in vivo to be measured. The results (Fig. 5, N to Q) showed that the mice treated with pH-responsive DOX-conjugated NPs in sepsis can immediately respond to the challenge of LPS similar to what the healthy mice did. The data are evidence to support that DOX-conjugated NPs specifically regulate proinflammatory neutrophils but do not interfere with the production of new neutrophils in the bone marrow, thus avoiding the systemic immune suppression.
It is known that DOX shows severe cardiac toxicity in cancer therapy (28), so we addressed whether DOX-conjugated BSA NPs caused the side effect at a dose of 0.2 mg/kg used in our anti-inflammatory therapies. Following the protocol (Fig. 4E), we studied the histology of several organs (heart, liver, spleen, lung, and kidney) after administration of free DOX and several BSA-based NPs (fig. S10). We did not find any toxicity in heart, liver, spleen, lung, and kidney at 0.2 mg/kg DOX either in free DOX or in BSA NP formulations. However, at the high dose of free DOX treatment (4 mg/kg), the myocardial damage was apparent because of intensive vacuolization and myofibril loss (fig. S10) (28). The result indicated that the low-dose regime of DOX used in our anti-inflammatory therapy is tolerant, and there is no apparent organ toxicity in our current mouse experiments.
Stroke is a major cause of death and adult disability (8). Most strokes are related to ischemia, that is, blood vessel clogs in the brain. Currently, reperfusion is a surgery option to restore blood circulation, but reperfusion causes secondary tissue damage due to neuroinflammation. Neutrophils play a central role in this neuroinflammation, such as in cerebral I/R (8). We examined whether our DOX-hyd-BSA NPs can benefit the therapy for ischemic stroke. In the experiments, we established a middle cerebral artery occlusion (MCAO) mouse model to mimic cerebral I/R (Fig. 6A). One hour after inclusion with a filament in the internal carotid artery (ICA), reperfusion was performed by withdrawal of the filament. One hour after perfusion, NPs were intravenously administered, and brain tissues were collected for analysis of neutrophils and cytokine levels at 22 hours after injection of free DOX or NPs (Fig. 6, B to E). Compared with free DOX and PBS treatments, we observed that DOX-hyd-BSA NPs significantly reduced neutrophil numbers [MPO (myeloperoxidase), a marker of neutrophils] and cytokines in the brain, implying that inhibition of neutrophils by DOX-hyd-BSA NPs is useful to prevent the secondary brain damage from inflammatory responses of neutrophils induced by the surgery for ischemic stroke therapy. Neurological deficits related to cerebral I/R were evaluated after the treatment of DOX-hyd-BSA NPs (Fig. 6F and movies S3 to S6), indicating that DOX-hyd-BSA NPs enhanced mouse neurological recovery and improved mouse movements compared with several controls.
Fig. 6. DOX-hyd-BSA NPs mitigate neutrophil-induced neuroinflammation in cerebral I/R mouse model to restore neurological functions.
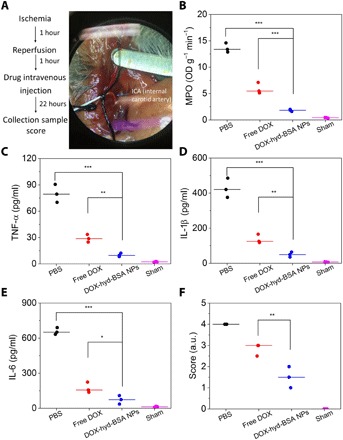
(A) Experimental design to examine the benefit of DOX-hyd-BSA NPs in cerebral I/R. OD, optical density. (B) MPO activity and (C) TNF-α, (D) IL-1β, and (E) IL-6 in brain damage tissues at 22 hours after administration of PBS, free DOX, and DOX-hyd-BSA NPs. (F) Mouse neurological behavior scores after treatments with PBS, free DOX, and DOX-hyd-BSA NPs at 0.2 mg/kg of DOX. All data are expressed as means ± SD (three mice per group). Statistics was performed by a two-sample Student’s t test (*P < 0.05, **P < 0.01, and ***P < 0.001). Photo Credit: Xinyue Dong, Washington State University.
DISCUSSION
Inflammation associated with the innate and adaptive immune systems is a defense to infections or tissue injury (4). However, when unchecked, inflammation may cause autoimmune or inflammatory disorders (5, 10), such as sepsis, stroke, aging, and even cancer. Anti-inflammatory agents have been developed to inhibit inflammation pathways (11), such as the nuclear factor κB pathway, but their off-targeting delivery causes systemic toxicity. In addition, anticytokine therapies have been used in clinics to neutralize cytokine storm during inflammatory responses (11). Cytokines, such as TNF-α and IL-1β, are mediators of diseases; thus, they are the targets for anticytokine therapy using anti–TNF-α and anti–IL-1β. Although blocking cytokines may reduce inflammation, it renders the host susceptible to infections (11). Selective targeting to immune cells for intracellular drug delivery may be a potential strategy to manage inflammatory responses to infections or tissue injury to maintain immune homeostasis.
Apoptosis is a natural process of cell death to maintain body homeostasis. For example, neutrophils have a short life span, and it is regulated by apoptosis to preserve constant numbers of neutrophils in circulation to warrant immune homeostasis, protecting the host damage from neutrophils. Inspired by this natural neutrophil apoptosis, we proposed a means to specifically target inflammatory neutrophils using NPs that deliver DOX to promote neutrophil apoptosis to treat inflammatory disorders (Fig. 1). This approach would enhance inflammation resolution without suppression of the host immune system. To prove this concept, we have developed DOX-conjugated BSA NPs that can specifically bind to activated neutrophils and control DOX release when NPs are taken up by neutrophils (Figs. 2 and 3). We have demonstrated that the uptake of NPs is required when neutrophils are activated during inflammation. Furthermore, the uptake of NPs is dependent on Fcγ receptors expressed on neutrophils as shown by intravital microscopy (Fig. 3), supported by our results that the uptake of BSA NPs is markedly decreased in Fcγ receptors–knockout mice. This is consistent with the function of Fcγ receptors to endocytose denatured BSA induced by the formation of NPs but not BSA molecules (19). However, other immune cells (such as monocytes, T cells, and NK cells) do not take up NPs in LPS-induced acute inflammation, and this may be associated with their adaptive characteristics. DOX has been studied to treat cancer because it can cause cell death. High doses of DOX administration in cancer therapy cause heart toxicity and lead to systemic inflammation. However, our study reveals that the low dose of DOX (0.2 mg/kg) administered to a mouse did not cause inflammation and heart toxicity (fig. S10). This is consistent with the tolerance of DOX used in cancer therapy. We show that DOX can promote neutrophil apoptosis (Fig. 4) and decrease inflammatory responses. In an acute lung inflammation mouse model, administration of DOX-hyd BSA NPs inhibited neutrophil transmigration into the mouse lung and also decreased cytokine levels compared with free DOX or PBS. The results suggest that neutrophil apoptosis prevents trafficking of neutrophils in response to LPS-induced lung inflammation.
Neutrophils are the most abundant circulating leukocytes in humans and a major component in response to infections or tissue damage. Neutrophil infiltration is the defense mechanism to eliminate pathogens and repair tissues (15), but dysregulation of neutrophil trafficking may cause inflammatory and autoimmune disorders (5). An emerging concept is to precisely control neutrophil apoptosis that is a balance between neutrophil defense activity and its clearance for inflammation resolution (16, 17). We have demonstrated that DOX-hyd-BSA NPs can mediate the apoptosis of proinflammatory neutrophils to increase inflammation resolution, thus preventing acute lung inflammation/injury (Fig. 4). Exaggerated neutrophil activation contributes to pathogenesis of sepsis and ischemic stroke, so we examined the usefulness of DOX-conjugated NPs to treat these two diseases. In the LPS-induced sepsis mouse model, administration of DOX-hyd-BSA NPs increased mouse survival to 70 versus 10 to 20% for controls (free DOX and PBS) (Fig. 5). Further studies show that neutrophil apoptosis decreased neutrophil numbers in circulation and in the lungs, thus inhibiting neutrophil trafficking to mitigate systemic inflammation. We discovered that administration of DOX-hyd-BSA NPs did not impair neutrophil production in the bone marrow when we compared neutrophil counts in healthy mice. In addition, the mice that survived sepsis after the treatment with DOX-conjugated NPs can normally respond to the second hit of LPS, similar to what healthy mice do. This result demonstrates a new concept to treat inflammatory diseases by specifically targeted delivery of therapeutics to proinflammatory neutrophils. This new approach avoids the systemic suppression caused by currently used anti-inflammatory agents. In the ischemic stroke mouse model, we have shown that inhibition of neutrophil trafficking by DOX-hyd-BSA NPs rescued mouse neurological damage during reperfusion therapy to ischemic stroke (Fig. 6). Collectively, our studies reveal a new concept to develop anti-inflammatory therapies by targeting immune cell apoptosis pathways using NPs. The advances in nanotechnology and biotechnology will enable the design of new NPs that can target inflammatory neutrophils by conjugating antibodies or targeting ligands to NPs (22, 29, 30) and deliver a wide range of therapeutics to regulate neutrophil functions (11).
We find that we can make human albumin NPs similar to BSA NPs; thus, our approaches for BSA NPs may be ready to test the translational possibilities of our technology. The coupling agent, glutaraldehyde, may cause immunogenicity in translation, but biocompatible coupling agents will be used in the future.
In summary, we have demonstrated a new approach to selectively target the apoptosis pathway in proinflammatory neutrophils using DOX-conjugated BSA NPs; thus, we can manage sepsis and decrease surgery-induced brain damage in ischemic stroke. Our NP design allows controlled release of DOX inside neutrophils, thus avoiding the systemic toxicity. Our study shows that DOX can increase neutrophil apoptosis for anti-inflammatory therapies, but there is no cardiac toxicity. This new concept of targeting neutrophil apoptosis pathways using NPs will be envisioned in a wide range of applications in inflammatory and autoimmune diseases.
MATERIALS AND METHODS
Materials
BSA, triethylamine (TEA; 99%), 1-ethyl-3-(3-(dimethylamino)propyl) carbodimide (EDC), N-hydroxysuccinimide (NHS), LPS (Escherichia coli 0111: B4), formaldehyde solution, and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). DOX hydrochloride was purchased from Wuhan Yuancheng Gongchuang Co. Dicarboxyl PEG (HOOC-PEG-COOH, Mw = 600) was purchased from Creative PEGWorks. Glutaraldehyde was obtained from Electron Microscopy Sciences (Hatfield, PA). RPMI 1640 and other media were purchased from Lonza (Walkersville, MD). Hanks’ balanced salt solution (HBSS) buffer (without Ca2+, Mg2+, and phenol red) was obtained from Corning (Corning, NY). Recombinant human and mouse TNF-α (carrier free, purity >98%); Alexa Fluor 647 anti-mouse CD16/32, CD3, CD115, and CD335 antibodies; Alexa Fluor 488/647 anti-mouse Ly-6G antibody; Cell Meter TUNEL apoptosis assay kit; and enzyme-linked immunosorbent assay (ELISA) kits for TNF-α, IL-1β, and IL-6 were purchased from BioLegend Inc. (San Diego, CA). Annexin V/Dead Cell Apoptosis kit and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Invitrogen (Carlsbad, USA). Human HL-60 cell lines were obtained from the American Type Culture Collection (Manassas, VA). Penicillin-streptomycin and glutamine (100×) were purchased from Life Technologies (Grand Island, NY). Pierce BCA protein assay kit was purchased from Thermo Fisher Scientific. All other chemical and biological reagents were used as they were received.
Synthesis of DOX-hyd-BSA
DOX-hyd-PEG-COOH was synthesized according to a previous study with some modifications, (31, 32). The synthetic pathway was shown in fig. S1. In brief, HOOC-PEG-COOH (2 mmol) was dissolved in DMSO at 1 mmol/ml and was activated by NHS/EDC (1:1 at the molar ratio) with the molar ratio of EDC to HOOC-PEG-COOH to be 3:1. Then, NH2NH2·H2O (1 eq relative to HOOC-PEG-COOH) and TEA were dropwise added into the solution. The reaction was carried out for 24 hours with gentle stirring at room temperature to obtain HOOC-PEG-CONHNH2, followed by adding 2.0 mmol DOX and 10 mmol TFA (trifluoroacetic acid) into the solution. The resulting mixture was stirred for 48 hours in the dark at room temperature. DOX-hyd-PEG-COOH conjugate was received after dialysis in deionized water and freeze dried. Last, DOX-hyd-PEG-COOH was conjugated to BSA through amino bonds. Briefly, 4.0 mmol of DOX-hyd-PEG-COOH was first dissolved in 20 ml of dioxane, and then NHS/EDC (4.0 mmol/4.0 mmol) and TEA (10 mmol) were added in the solution. The reaction was carried out for 12 hours in the dark at room temperature. Then, 1.0 mmol of BSA was dissolved in 20 ml of deionized water, and BSA solution was mixed with DOX-hyd-PEG-COOH. During the reaction process, the pH value of the reaction mixture was maintained at 7 by the dropwise addition of 0.1 mmol/ml NaOH solution. The solution was stirred for 4 hours, and then the pH was quickly adjusted to 9 by adding 0.1 mmol/ml NaOH solution. DOX-hyd-BSA powder was obtained after the solution was dialyzed in deionized water for 2 days, followed by 2 days of lyophilization. DOX-ab-PEG was synthesized as a negative control by using HOOC-PEG-COOH conjugated to DOX instead of NH2NH2·H2O.
Preparation of BSA NPs
BSA NPs were prepared by desolvation according to our previous studies (19, 26). DOX-hyd-BSA or DOX-ab-BSA was dissolved in deionized water at 2.5 mg/ml, but pure BSA NPs were formed at BSA solution (20 mg/ml). Thirty minutes later, 1.2 to 3.5 ml of ethanol was continuously added to the solution under stirring at 800 revolutions per minute (rpm) at room temperature. One hour later, stable BSA NPs were achieved by adding 20 to 80 μl of 2% glutaraldehyde to cross-link amine residues in BSA molecules. The resulting suspension was stirred overnight in the dark at room temperature. Last, the pellet of NPs was obtained after three times of centrifugation at 20,000g for 30 min at 4°C. After lyophilization, we found that the particle formation efficiency was 70 to 80% for DOX-hyd-BSA and 80 to 90% for BSA, respectively. The NP pellet was resuspended in PBS or 5% glucose for the study.
Characterization
1H nuclear magnetic resonance (NMR) spectra measurements were executed on a Bruker Avance III 500 (Switzerland) spectrometer operating at 500 MHz using deuterated chloroform (CDCl3-d) or D2O as solvent. To study the loading efficiency of DOX in BSA NPs, conjugated DOX was determined by UV-vis spectroscopy. One milligram of DOX-hyd-BSA NPs was dissolved in 1 ml of HCl (1 mol/liter) and stirred for 3 hours at 50°C. Afterward, the solution was centrifuged at 20,000g for 30 min at 4°C, and the supernatant was collected for DOX analysis at 480 nm. The drug conjugating content (DCC) was defined as the weight ratio of the drug conjugated to BSA. The DCC of DOX-hyd-BSA or DOX-ab-BSA was 2.4 or 2.5%, respectively.
Particle size and polydispersity indexes were measured using Malvern Zetasizer Nano ZS90 (Westborough, MA). The samples were incubated in PBS at pH 7.4 for 2 hours, and the measurements were conducted in a 1.0-ml quartz cuvette using a diode laser of 633 nm at 25°C, and scattering angle was fixed at 90°. To evaluate their serum stability, NPs were dispersed in 20% FBS PBS solution (pH 7.4) at a final concentration of 1 mg/ml. NP sizes were measured at the predefined time points. TEM of NPs was also performed using a FEI Tecnai G220 TWIN TEM (Hillsboro, OR).
To confirm the pH-triggered drug release property, BSA NPs, DOX-ab-BSA NPs, and DOX-hyd-BSA NPs were incubated in PBS at pH 7.4, 6.5, and 5.0 for 2 hours, respectively. After centrifugation, the supernatants were collected to determine DOX concentrations using a UV-vis spectrometer.
In vitro drug release profile
Release profiles of DOX from NPs were studied using the dialysis method at 37°C (33). Briefly, 3 mg of NPs was dispersed in 3 ml (Ve) of PBS at different pH and then was placed in a dialysis bag (molecular weight cutoff, 3500 Da). The dialysis bag was immersed in 47 ml of PBS (pH 7.4, 6.5, or 5.0) in a beaker. The beaker was then placed in a 37°C water bath and stirred at 110 rpm. The samples were drawn at desired time intervals, and the drug concentration was measured using UV-vis absorption. The accumulative drug release percent (Er) was calculated on the basis of Eq. 1. The experiments were carried out in triplicate at each pH value
| (1) |
where, mdrug represents the amount of DOX in NPs, V0 is the whole volume of release media (V0 = 50 ml), and Ci represents the concentration of DOX in the ith sample.
Cell culture condition
HL-60 cells were cultured in an RPMI 1640 medium containing 10% FBS, streptomycin (100 U/ml), and penicillin (100 U/ml) and differentiated into PMN-like cells by adding 1.3% (v/v) DMSO for 96 hours as previously reported (34, 35). Cells were maintained in an incubator in a humidified atmosphere containing 5% CO2 at 37°C.
In vitro cytotoxicity
Cytotoxic effects of BSA NPs on HL-60 cells were measured by Cell Counting Kit-8 assay. HL-60 cells were plated in 96-well plates (Costar, Corning, NY) at 5000 to 10,000 cells per well. After incubation for 24 hours, the culture medium was removed, and a complete medium with various concentrations of free DOX, BSA, and DOX-conjugated BSA NPs were used to incubate cells for 24 hours, respectively. Ten microliters of the solution cell proliferation reagent (Promega, Madison, WI) per well was added. Then, the cell viability was measured by a Synergy Neo fluorescence plate reader (BioTek, Winooski, VT) at 490 nm.
Apoptosis analysis
To investigate the apoptosis of neutrophils induced by DOX, annexin V–FITC and 7AAD were used to double stain differentiated HL-60 cells. The cells were seeded in a six-well plate at a density of 1 × 106 cells per well and treated with free DOX, DOX-hyd-BSA NPs, and DOX-ab-BSA NPs at a DOX concentration of 3 μg/ml for 24 hours at 37°C. The cells were resuspended in a binding buffer for staining of annexin V–FITC and 7AAD (emission at 650 nm) according to the manufacturer’s protocol (Invitrogen). Stained cells were analyzed using a flow cytometer and imaged by a Nikon A1R+ confocal laser scanning microscope.
To further confirm the apoptosis of differentiated HL-60 cells induced by DOX, TUNEL assay was also performed using Cell Meter TUNEL (green fluorescence) Apoptosis Assay Kit (AAT Bioquest Inc.) according to the manufacturer’s protocol, and the images were taken by a Nikon A1R+ confocal laser scanning microscope.
Mice
Adult CD1 (male, 22 to 30 g, 4 to 6 weeks) were purchased from Harlan Laboratories (Madison, WI). The mice were maintained in polyethylene cages with stainless steel lids at 20°C with a 12-hour light/dark cycle and covered with a filter cap. Animals were fed with food and water ad libitum. All animal care and experimental protocols used in this study were approved by the Washington State University Institutional Animal Care and Use Committee. All experiments were made under anesthesia using intraperitoneal injection of the mixture of ketamine (100 mg kg−1) and xylazine (5 mg kg−1) in saline.
NP targeting to neutrophils
To investigate whether DOX-hyd-BSA NPs can bind to activated neutrophils in vivo, we first studied the expression of Fcγ receptors on neutrophils using intravital microscopy. TNF-α (500 ng, 250 μl of saline) was intrascrotally injected into a mouse (C57BL/6). At 3 hours after injection of TNF-α, the mouse was anesthetized with a mixture of ketamine and xylazine as described above and maintained at 37°C on a thermo-controlled rodent blanket. A tracheal tube was inserted, and a right jugular vein was cannulated for injection of antibodies Alexa Fluor 488–labeled anti-mouse LY-6G and Alexa Fluor 647–labeled anti-mouse CD16/32. A scrotum was incised, and the testicle and surrounding cremaster muscles were exteriorized onto an intravital microscopy tray. The cremaster preparation was perfused with thermo-controlled (37°C) and aerated (95% N2, 5% CO2) bicarbonate-buffered saline throughout the experiment. Images were recorded using a Nikon A1R+ laser scanning confocal microscope with a resonant scanner. In studies on resting neutrophils in vivo, a mouse was not treated with TNF-α. Three hours after injection of Alexa Fluor 647–labeled anti-mouse CD16/32 antibody via the tail vein, the mouse cremaster tissue was exposed for intravital imaging after intravenous administration of Alexa Fluor 488–labeled anti-mouse LY-6G antibody to stain neutrophils. The images were recorded using a Nikon A1R+ laser scanning confocal microscope with a resonant scanner.
To investigate cell uptake specificity of BSA NPs in circulation, we isolated blood cells and studied them using confocal laser scanning microscopy (CLSM) and flow cytometry. Briefly, 4 hours after the LPS challenge (intraperitoneally, 20 mg kg−1), DOX-hyd-BSA NPs were intravenously injected into mice. Healthy mice were used as control. Three hours later, whole blood was harvested in a heparinized tube from the heart. Neutrophils in the blood were isolated by pluriSelect anti-mouse LY-6G S-pluriBeads according to the manufacturer’s protocol (pluriSelect, Spring Valley, CA). The cells were fixed with 2 ml of 4% paraformaldehyde for 30 min and were stained with Alexa Fluor 488–labeled anti-mouse LY-6G antibody. A slide smear of cell solution was prepared by 7620 Cytopro Cytocentrifuge (ELITech, Princeton, NJ). A drop of ProLong Gold Antifade reagent with DAPI (Invitrogen, Eugene, OR) was added on the cells, and a coverslip was applied on the slide. Four hours later, the cells were observed using a Nikon A1R+ confocal laser scanning microscope.
We also performed flow cytometry to quantify neutrophil uptake of NPs. Blood leukocytes were isolated to determine the specificity of uptake of NPs in the blood. In brief, 3 ml of Histopaque 10771 was carefully layered on top of 3 ml of Histopaque 11191 in a 15-ml centrifuge tube. Mouse whole blood was decanted on the top, followed by centrifugation at 890g for 30 min at 22°C with a gentle acceleration. Leukocytes were located between plasma and the Histopaque 10771 layer. The leukocyte suspension was collected and dissolved in PBS without Ca2+ and Mg2+. After the resulting cell suspension was centrifuged at 870g for 5 min at 4°C, T cells, monocytes, and NK cells were labeled by Alexa Fluor 647–labeled anti-mouse CD 3, CD 115, and CD 335 antibodies, respectively, for flow cytometric analysis.
Acute lung inflammation mouse model
Mice were anesthetized, and placed in a supine position head up on a board tilted at 15°. Afterward, LPS (10 mg/kg) in HBSS was intratracheally administrated to mice with a FMJ-250 High-Pressure Syringe (Penn-Century, Wyndmoor, PA). Mice were held upright for 2 min after administration.
BALF collection and cell counts
Mice were challenged with i.t. injection of LPS (10 mg/kg). Four hours later, PBS, free DOX (0.2 or 4 mg/kg), DOX-ab-BSA NPs, or DOX-hyd-BSA NPs (equal to 0.2 mg/kg of DOX) were intravenously injected. At 20 hours after drug administration, the mice were anesthetized with intravenous injection of a mixture of ketamine and xylazine. The BALF was collected by inserting a needle into the upper trachea. HBSS (0.9 ml) was infused into the lungs and carefully withdrawn to obtain BALF. This process was repeated three times. BALF was collected and centrifuged at 350g for 10 min at 4°C. The supernatant was collected for ELISA analysis. Afterward, the cells were resuspended in 0.5 ml of HBSS. The total cell number was determined with a hemocytometer.
Flow cytometry was used to quantify neutrophils in BALF. In detail, neutrophils from BALF were washed with 1 ml of 5% BSA in HBSS and centrifuged at 350g for 10 min at 4°C for three times, which was lastly resuspended in 400 μl of 5% BSA in HBSS. Three microliters of Alexa Fluor 647–labeled anti-mouse LY-6G antibody was added and incubated for 20 min in the dark, followed by washing with 1 ml of 0.1% BSA in HBSS under centrifugation for three times. Samples were then resuspended in 400 μl of 0.1% BSA in HBSS and filtered by a 100-μm filter, and then analyzed by a flow cytometer (Accuri C6 Flow Cytometer, BD Biosciences, San Jose, CA).
Cytokines
BALF was collected and centrifuged at 350g for 10 min as described above. Supernatants from BALF were harvested for ELISA analysis. Concentrations of TNF-α, IL-6, and IL-1β in supernatants were determined with commercial ELISA kits according to the manufacturer’s instructions (BioLegend, San Diego, CA). The triplicate experiment was conducted.
Hematoxylin and eosin staining
After different treatments (healthy, PBS, 4 mg/kg of free DOX, 0.2 mg/kg of free DOX, DOX-hyd-BSA NPs, and DOX-ab-BSA NPs, equal to 0.2 mg/kg of DOX for NPs), mice were euthanized by carbon dioxide asphyxiation. Organs were removed and fixed with 10% formalin, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E) for pathology (RTPH 360 Rapid Tissue Processor Operator Manual and SS-2030 Linear Slide Stainer, General Data, and Leica RM 2145, Leica Microsystems). The samples were imaged by a microscope (ZEISS, Observer Z1, USA).
Survival study in the sepsis mouse model
Adult CD1 mice were intraperitoneally injected with LPS (50 mg/kg) in the mouse sepsis model. Four hours later, the LPS-challenged mice were grouped randomly (10 mice per group) and treated intravenously with PBS, free DOX (0.2 mg/kg), and prodrug DOX-hyd-BSA NPs (equal to 0.2 mg/kg of free DOX). The animals were monitored every 6 hours in the first 12 hours, followed by monitoring mice every 12 hours in 72 hours.
Therapeutic efficacy in the sepsis mouse model
Adult CD1 mice were intraperitoneally administrated with LPS (50 mg/kg). Four hours later, the mice were grouped randomly (10 mice per group) and intravenously injected with PBS, free DOX, and DOX-hyd-BSA NPs (equal to 0.2 mg kg−1 of free DOX), respectively. The healthy mice were used as positive control. At predetermined time points (16 and 72 hours after LPS challenge), mouse BALF, blood, and major organs were collected. Cell number and inflammatory factors (TNF-α, IL-6, and IL-1β) in BALF were determined by a hemocytometer and ELISA, respectively, as aforementioned. Blood was collected as described above. The plasma was harvested for ELISA assay after the blood was centrifuged at 1500g for 20 min. Cell number was counted as above. The organs were homogenized with PBS (100 mg/ml) to obtain the pipettable homogenate for MPO activity and ELISA assay.
MCAO mouse model
CD1 mice were used. Mice were anesthetized using ketamine (100 mg/kg) and xylazine (5 mg/kg). They were positioned in the supine position on a heating pad. A carotid artery (CA) was exposed via the midline neck incision. An external CA (ECA) was separated and oculated with two knots. Next, the ICA was isolated, and the CA and ICA were clipped. A small hole was cut in the ECA above the second knot. A 6-0 medium MCAO suture was then introduced into ICA. Mice were kept for 60 min after occlusion in a heated cage, and the suture was withdrawn for reperfusion. Last, the skin was closed, and the mice were returned to their individual cages.
MPO activity
The damaged brain tissues were collected 24 hours after MCAO and homogenized in PBS with 5% hexadecyltrimethylammonium bromide. The homogenate was sonicated and centrifuged at 13,000 rpm for 10 min. Next, 10 μl of supernatant was loaded into each well of a 96-well plate. A solution of o-dianisidine dihydrochloride with 0.0005% hydrogen peroxide in potassium phosphate buffer was added to the samples. Absorbance was measured at 450 nm. MPO activity was expressed as change in absorbance per minute per gram of tissue.
Cytokine quantification
The damaged brain tissues of mice were collected 24 hours after MCAO surgery and homogenized in PBS buffer. The level of cytokines (TNF-α, IL-1β, and IL-6) was quantified using commercial ELISA assay as aforementioned.
Assessment of I/R injury by neurological deficit score
Videos were taken 24 hours after MCAO surgery, and neurological deficit scores were given by two people. The scores were divided into five grades for neurological scores of mice after cerebral ischemia (36–38): grade 0, normal and no neurological defect; grade 1, mild circling when a mouse is picked up via the tail and attempts to rotate to the contralateral side; grade 2, consistent strong and immediate circling or a mouse only turns to the surgery contralateral side while the animal is suspended by the tail; grade 3, severe rotation or lacking of walking abilities; and grade 4, animals do not walk spontaneously and lose the response.
Statistical analysis
The experimental data were presented with average values, expressed as means ± SD. Statistical analysis was conducted using one-way analysis of variance (ANOVA) or Student’s t test of origin 8.5. A value of P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
Funding: This research was supported by the National Institute of Health grant RO1GM116823 to Z.W. Author contributions: C.Y.Z. and Z.W. designed the research, analyzed the data, and wrote the paper. C.Y.Z., X.D., J.G., W.L., and Z.L. performed the experiments and analyzed the data. All authors offered the input in the manuscript. Z.W. conceived and supervised this project. Competing interests: Z.W. and C.Y.Z. are inventors on a patent related to this work filed by the Washington State University (no. 62/884,839, 8/9/2019). The authors declare no other competing interest. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax7964/DC1
Fig. S1. Synthetic pathways of DOX-hyd-BSA.
Fig. S2. 1H NMR analysis.
Fig. S3. TEM images of BSA NPs and DOX-ab-BSA NPs.
Fig. S4. The stability of BSA NPs in serum.
Fig. S5. Quantitative analysis of cellular uptake of DOX by differentiated HL-60 cells.
Fig. S6. Flow cytometric analysis on apoptosis of differentiated HL-60 cells 24 hours after treatments with free DOX (0.2 mg/kg), DOX-ab-BSA, or DOX-hyd-BSA NPs (equal to DOX of 0.2 mg/kg), respectively.
Fig. S7. Flow cytometric analysis of neutrophils in BALF.
Fig. S8. DOX-hyd-BSA NPs decrease systemic inflammation in the mouse sepsis model.
Fig. S9. Mouse body weights were monitored following the experiment as shown in Fig. 5M.
Fig. S10. Toxicity of DOX-conjugated BSA NPs evaluated by histological analysis.
Movie S1. Intravital microscopy of a cremaster venule shows resting neutrophils in circulation.
Movie S2. Intravital microscopy of a cremaster venule shows activated neutrophils in circulation.
Movie S3. Movement of a sham mouse in the cerebral I/R model.
Movie S4. Movement of a mouse with cerebral I/R after PBS treatment.
Movie S5. Movement of a mouse with cerebral I/R after DOX treatment.
Movie S6. Movement of a mouse with cerebral I/R after treatment of DOX-hyd-BSA NPs.
REFERENCES AND NOTES
- 1.Mayadas T. N., Cullere X., Lowell C. A., The multifaceted functions of neutrophils. Annu. Rev. Pathol. 9, 181–218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kolaczkowska E., Kubes P., Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Nauseef W. M., Borregaard N., Neutrophils at work. Nat. Immunol. 15, 602–611 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Nathan C., Ding A., Nonresolving inflammation. Cell 140, 871–882 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Soehnlein O., Steffens S., Hidalgo A., Weber C., Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol. 17, 248–261 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Hidalgo A., Chang J., Jang J.-E., Peired A. J., Chiang E. Y., Frenette P. S., Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat. Med. 15, 384–391 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthay M. A., Zemans R. L., The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. 6, 147–163 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becher B., Spath S., Goverman J., Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 17, 49–59 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Wright H. L., Moots R. J., Edwards S. W., The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 10, 593–601 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Brown K. A., Brain S. D., Pearson J. D., Edgeworth J. D., Lewis S. M., Treacher D. F., Neutrophils in development of multiple organ failure in sepsis. Lancet 368, 157–169 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Dinarello C. A., Anti-inflammatory agents: Present and future. Cell 140, 935–950 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato T., Shinzawa H., Abe Y., Takahashi T., Arai S., Sendo F., Inhibition of Corynebacterium parvum-primedand lipopolysaccharide-induced hepatic necrosis in rats by selective depletion of neutrophils using a monoclonal antibody. J. Leukoc. Biol. 53, 144–150 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Jaeger B. N., Donadieu J., Cognet C., Bernat C., Ordoñez-Rueda D., Barlogis V., Mahlaoui N., Fenis A., Narni-Mancinelli E., Beaupain B., Bellanné-Chantelot C., Bajénoff M., Malissen B., Malissen M., Vivier E., Ugolini S., Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J. Exp. Med. 209, 565–580 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotzin J. J., Spencer S. P., McCright S. J., Kumar D. B. U., Collet M. A., Mowel W. K., Elliott E. N., Uyar A., Makiya M. A., Dunagin M. C., Harman C. C. D., Virtue A. T., Zhu S., Bailis W., Stein J., Hughes C., Raj A., Wherry E. J., Goff L. A., Klion A. D., Rinn J. L., Williams A., Flavell R. A., Henao-Mejia J., The long non-coding RNA Morrbid regulates Bim and short-lived myeloid cell lifespan. Nature 537, 239–243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nathan C., Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 6, 173–182 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Rossi A. G., Sawatzky D. A., Walker A., Ward C., Sheldrake T. A., Riley N. A., Caldicott A., Martinez-Losa M., Walker T. R., Duffin R., Gray M., Crescenzi E., Martin M. C., Brady H. J., Savill J. S., Dransfield I., Haslett C., Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 12, 1056–1064 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Kennedy A. D., DeLeo F. R., Neutrophil apoptosis and the resolution of infection. Immunol. Res. 43, 25–61 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Tewey K., Rowe T., Yang L., Halligan B., Liu L., Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 226, 466–468 (1984). [DOI] [PubMed] [Google Scholar]
- 19.Wang Z., Li J., Cho J., Malik A. B., Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat. Nanotechnol. 9, 204–210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jankowski A., Scott C. C., Grinstein S., Determinants of the phagosomal pH in neutrophils. J. Biol. Chem. 277, 6059–6066 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Gao J., Chu D., Wang Z., Cell membrane-formed nanovesicles for disease-targeted delivery. J. Control. Release 224, 208–216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu D., Dong X., Zhao Q., Gu J., Wang Z., Photosensitization priming of tumor microenvironments improves delivery of nanotherapeutics via neutrophil infiltration. Adv. Mater. 29, 1701021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mariño G., Kroemer G., Mechanisms of apoptotic phosphatidylserine exposure. Cell Res. 23, 1247–1248 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vermes I., Haanen C., Steffens-Nakken H., Reutellingsperger C., A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184, 39–51 (1995). [DOI] [PubMed] [Google Scholar]
- 25.Zembruski N. C. L., Stache V., Haefeli W. E., Weiss J., 7-Aminoactinomycin D for apoptosis staining in flow cytometry. Anal. Biochem. 429, 79–81 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Chu D., Gao J., Wang Z., Neutrophil-mediated delivery of therapeutic nanoparticles across blood vessel barrier for treatment of inflammation and infection. ACS Nano 9, 11800–11811 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fink M. P., Warren H. S., Strategies to improve drug development for sepsis. Nat. Rev. Drug Discov. 13, 741–758 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Aries A., Paradis P., Lefebvre C., Schwartz R. J., Nemer M., Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc. Natl. Acad. Sci. U.S.A. 101, 6975–6980 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang C. Y., Gao J., Wang Z., Bioresponsive nanoparticles targeted to infectious microenvironments for sepsis management. Adv. Mater. 30, e1803618 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitragotri S., Anderson D. G., Chen X., Chow E. K., Ho D., Kabanov A. V., Karp J. M., Kataoka K., Mirkin C. A., Petrosko S. H., Shi J., Stevens M. M., Sun S., Teoh S., Venkatraman S. S., Xia Y., Wang S., Gu Z., Xu C., Accelerating the translation of nanomaterials in biomedicine. ACS Nano 9, 6644–6654 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye W.-L., Zhao Y.-p., Li H.-q., Na R., Li F., Mei Q.-b., Zhao M.-g., Zhou S.-y., Doxorubicin-poly (ethylene glycol)-alendronate self-assembled micelles for targeted therapy of bone metastatic cancer. Sci. Rep. 5, 14614 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arote R. B., Hwang S.-K., Lim H.-T., Kim T.-H., Jere D., Jiang H.-L., Kim Y.-K., Cho M.-H., Cho C.-S., The therapeutic efficiency of FP-PEA/TAM67 gene complexes via folate receptor-mediated endocytosis in a xenograft mice model. Biomaterials 31, 2435–2445 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Zhang C. Y., Yang Y. Q., Huang T. X., Zhao B., Guo X. D., Wang J. F., Zhang L. J., Self-assembled pH-responsive MPEG-b-(PLA-co-PAE) block copolymer micelles for anticancer drug delivery. Biomaterials 33, 6273–6283 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Gao J., Wang S., Wang Z., High yield, scalable and remotely drug-loaded neutrophil-derived extracellular vesicles (EVs) for anti-inflammation therapy. Biomaterials 135, 62–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C., Ling C.-l., Pang L., Wang Q., Liu J.-x., Wang B.-s., Liang J.-m., Guo Y.-z., Qin J., Wang J.-x., Direct macromolecular drug delivery to cerebral ischemia area using neutrophil-mediated nanoparticles. Theranostics 7, 3260–3275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maas A. I., Stocchetti N., Bullock R., Moderate and severe traumatic brain injury in adults. Lancet Neurol. 7, 728–741 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Zhang B., Song C., Feng B., Fan W., Neuroprotection by triptolide against cerebral ischemia/reperfusion injury through the inhibition of NF-κB/PUMA signal in rats. Ther. Clin. Risk Manag. 12, 817–824 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haapaniemi E., Tatlisumak T., Hamel K., Soinne L., Lanni C., Opgenorth T. J., Kaste M., Plasma endothelin-1 levels neither increase nor correlate with neurological scores, stroke risk factors, or outcome in patients with ischemic stroke. Stroke 31, 720–725 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax7964/DC1
Fig. S1. Synthetic pathways of DOX-hyd-BSA.
Fig. S2. 1H NMR analysis.
Fig. S3. TEM images of BSA NPs and DOX-ab-BSA NPs.
Fig. S4. The stability of BSA NPs in serum.
Fig. S5. Quantitative analysis of cellular uptake of DOX by differentiated HL-60 cells.
Fig. S6. Flow cytometric analysis on apoptosis of differentiated HL-60 cells 24 hours after treatments with free DOX (0.2 mg/kg), DOX-ab-BSA, or DOX-hyd-BSA NPs (equal to DOX of 0.2 mg/kg), respectively.
Fig. S7. Flow cytometric analysis of neutrophils in BALF.
Fig. S8. DOX-hyd-BSA NPs decrease systemic inflammation in the mouse sepsis model.
Fig. S9. Mouse body weights were monitored following the experiment as shown in Fig. 5M.
Fig. S10. Toxicity of DOX-conjugated BSA NPs evaluated by histological analysis.
Movie S1. Intravital microscopy of a cremaster venule shows resting neutrophils in circulation.
Movie S2. Intravital microscopy of a cremaster venule shows activated neutrophils in circulation.
Movie S3. Movement of a sham mouse in the cerebral I/R model.
Movie S4. Movement of a mouse with cerebral I/R after PBS treatment.
Movie S5. Movement of a mouse with cerebral I/R after DOX treatment.
Movie S6. Movement of a mouse with cerebral I/R after treatment of DOX-hyd-BSA NPs.