

Abstract
Huntington’s disease (HD) is an inherited neurodegenerative disorder caused by a CAG repeat expansion within exon 1 of the huntingtin (HTT) gene. HTT mRNA contains 67 exons and does not always splice between exon 1 and exon 2 leading to the production of a small polyadenylated HTTexon1 transcript, and the full-length HTT mRNA has three 3′UTR isoforms. We have developed a QuantiGene multiplex panel for the simultaneous detection of all of these mouse Htt transcripts directly from tissue lysates and demonstrate that this can replace the more work-intensive Taqman qPCR assays. We have applied this to the analysis of brain regions from the zQ175 HD mouse model and wild type littermates at two months of age. We show that the incomplete splicing of Htt occurs throughout the brain and confirm that this originates from the mutant and not endogenous Htt allele. Given that HTTexon1 encodes the highly pathogenic exon 1 HTT protein, it is essential that the levels of all Htt transcripts can be monitored when evaluating HTT lowering approaches. Our QuantiGene panel will allow the rapid comparative assessment of all Htt transcripts in cell lysates and mouse tissues without the need to first extract RNA.
Subject terms: Neuroscience, Diseases of the nervous system, Huntington's disease
Introduction
Huntington’s disease (HD) is an autosomal dominant disorder that is characterised by movement disorders, as well as psychiatric and cognitive disturbances1. It is caused by the expansion of a CAG repeat in exon 1 of the huntingtin gene (HTT), which is translated to a huntingtin protein (HTT) with an abnormally long polyglutamine tract2. CAG repeats of 40 or more are fully penetrant, alleles of 36 to 39 result in an increased likelihood that HD will occur within a normal life span and individuals with 35 or less remain unaffected3. There is a negative correlation between the age of onset of motor symptoms and the length of the CAG repeat as measured in blood, with expansions of 70 and above resulting in onset before 20 years of age. Somatic expansion of the CAG repeat in neurons can lead to CAG repeat lengths in brain that are considerably longer than those present in blood4,5.
The mammalian HTT gene is approximately 180 kb in length and contains 67 exons. Two mature transcripts of approximately 10.3 and 13.7 kb were initially described that differed in the length of their 3′UTRs6. The recent identification of an additional polyadenylation (poly(A)) site in the 3’UTR results in a HTT transcript of approximately 12.5 kb, with the result that we now know that three full-length human HTT transcripts are produced7. In addition, we have previously shown that exon 1 of HTT does not always splice to exon 2, resulting in two small polyadenylated mRNAs (Httexon1)8. In mouse, these extend to cryptic poly(A) sites located at 680 and 1,145 bp into intron 18,9, and in human, to cryptic poly(A) sites extending to 2,710 and 7,327 bp into intron 18,10. The extent to which this incomplete splicing event occurs increases with increasing CAG repeat length8 and the Httexon1 transcripts are translated to generate the highly pathogenic exon 1 HTT protein8. These alternative mRNA processing events mean that at least five transcripts are produced from the human HTT and mouse Htt genes, which are translated to produce either full length HTT or the exon 1 HTT protein.
Strategies to lower the levels of HTT mRNA are a major focus for HD therapeutics and are at various stages of development. These were discussed in a recent review11 and include antisense oligonucleotides (ASOs), interfering RNAs (RNAi) and small molecule splicing modulators for targeting RNA, and zinc-finger transcription factors and CRISPR/Cas9 for targeting DNA. ASOs targeting HTT mRNA are the most advanced, and their intrathecal delivery has been shown to be tolerated in a phase II clinical trial and to result in the reduction of mutant HTT protein in cerebrospinal fluid (CSF)12. In some cases, these strategies target all HTT transcripts, but in others, they target the full length transcripts whilst leaving the Httexon1 transcripts intact. As the exon 1 HTT protein is highly pathogenic13,14, and the relative contribution of the mutant forms of the full length HTT and the exon 1 HTT proteins to disease pathogenesis is unknown, it is important that we can measure the extent to which the levels of all HTT transcripts are affected by these various strategies.
QuantiGene assays are based on branched DNA (bDNA) technology15–17, have a workflow that is easy and fast to perform in a multiplex 96-well plate format, are devoid of additional steps after homogenisation of the tissues of interest and are sensitive enough to detect low abundance transcripts. QuantiGene singleplex assays have been used in the past for quantifying huntingtin mRNA expression18–20. Therefore, we set out to establish a QuantiGene multiplex assay to provide a method for measuring all of the mouse Htt transcripts of interest concomitantly and validated this through the direct comparison with quantitative real-time PCR (qPCR). We applied this assay to measure Htt transcripts in the zQ175 knock-in model of HD21,22 that is used extensively for therapeutic target validation. We extended previous work8 to show that the Httexon1 transcript is present throughout the brain in this model and provide further evidence to confirm that it originates from the mutant Htt allele. In summary, the QuantiGene multiplex assay provides a rapid and reliable means of measuring the levels of mouse Htt transcripts directly in tissue homogenates.
Results
QuantiGene Htt multiplex assay custom design
In collaboration with Thermofisher, we set out to design a multiplex QuantiGene panel to detect the incompletely spliced (Httexon1) and full-length Htt (Htt-FL) transcripts (Fig. 1). This contained probes to intron 1 before the first cryptic poly(A) site at 680 bp (I1-pA1), to intron 1 sequences after this and before the second cryptic poly(A) site at 1,145 bp (I1–pA2), to sequences at the 3′ end of intron 1 (I1-3′), within intron 3 (I3) and to exons 50–53 for the fully-spliced coding sequence (E50-53). The two cryptic poly(A) sites are 465 bp apart, raising the possibility that the amplification trees corresponding to these two sequences might interfere due to their close proximity. To address this, we designed the 9-plex and 10-plex QuantiGene panels outlined in Table 1.
Figure 1.

Schematic representation of the location of the probe sets for the multiplex Htt QuantiGene assays. Custom probe sets were designed against Htt mRNA sequence to enable the measurement of all major Htt transcripts. The 9-plex assay contained probe sets to intron 1 sequences before the first cryptic polyA site at 680 bp to detect the Httexon1 transcripts, to the 3′ end of intron 1 and to intron 3 as controls for unprocessed mRNA, and to exons 50–53 for all Htt-FL transcripts. The 10-plex assay included a probe set to sequences between the first and second cryptic polyA sites, between 680 and 1145 bp. The 14-plex assay also included probe sets to each of the three 3′UTRs of the full-length transcript.
Table 1.
Design of the 9-plex and 10-plex QuantiGene assays for the detection of Htt transcripts.
| Transcript/gene symbol | Transcript/gene name | GOI/HK | 9-plex | 10-plex |
|---|---|---|---|---|
| (Htt)-I1-pA1 | Huntingtin – Intron 1- poly(A)1 | Htt | √ | √ |
| (Htt)-I1-pA2 | Huntingtin – Intron 1- poly(A)2 | Htt | X | √ |
| (Htt)- I1-3’ | Huntingtin – Intron 1–3’ | Htt | √ | √ |
| (Htt-)-I 3 | Huntingtin – Intron 3 | Htt | √ | √ |
| Htt-FL (E50-53) | Huntingtin – exons 50–53 | Htt | √ | √ |
| Eif4a2 | Eukaryotic Initiation factor 4a2 | HK | √ | √ |
| Rpl13a | Ribosomal protein L 13a | HK | √ | √ |
| Ubc | Ubiquitin C | HK | √ | √ |
| CanX | Calnexin | HK | √ | √ |
| Atp5b | ATP synthase subunit beta | HK | √ | √ |
GOI = gene of interest, HK = housekeeping reference gene.
The I1-pA1 probe set identifies the Httexon1 transcripts that terminate at both the first and the second cryptic poly(A) signals. The I1-pA2 probe set recognises only the Httexon1 transcript that terminates at the second poly(A) signal, while the I1-3′ probe set would identify incompletely spliced intron 1 sequences that have not terminated at cryptic poly(A) signals. The I3 probe set serves to control for any contaminating Htt pre-mRNA. The housekeeping reference genes chosen for the panels have been identified previously for their robustness and stability when used in brain tissues from HD mouse models23.
Establishment and optimisation of QuantiGene Htt multiplex assays
To examine the linearity of the probe set signals for each brain region, we used a pool of equimolar lysate from wild type (WT) and zQ175 heterozygous animals (n = 4/genotype). The two pools were then subjected to 2-fold serial dilutions and QuantiGene analysis using our 9-plex and 10-plex panels.
The lowest mean fluorescence intensity (MFI) signal for the striatum corresponded to the control I3 probe set, while the highest corresponded to that for the reference gene Ubc (Fig. 2c,h and Supplementary Fig. S1). The MFI signal for the I1–3′ probe set was comparable to the assay background (I3) even at the highest concentration used (Fig. 2b,g,c,h). The linear regression of the MFI signals for the reference genes Ubc and Eif4a2 in the striatum, for both the 9-plex and 10-plex QuantiGene panels, showed that the signal was saturated for the undiluted material (Supplementary Fig. S1). Thus, the undiluted material was excluded from the linear range for all probe sets in all other brain regions (Supplementary Figs S2–S5). Based on the linearity of the probe set signals and the coefficient of variance (% CV) we recommend a dilution range of the starting material of 1:3–1:4 and never more than 1:5 for all brain regions (Supplementary Table 1). Close evaluation of the Ubc signal suggested that saturation had been reached for all brain regions, even at the lowest dilution range, and therefore, Ubc was excluded from all further analyses.
Figure 2.
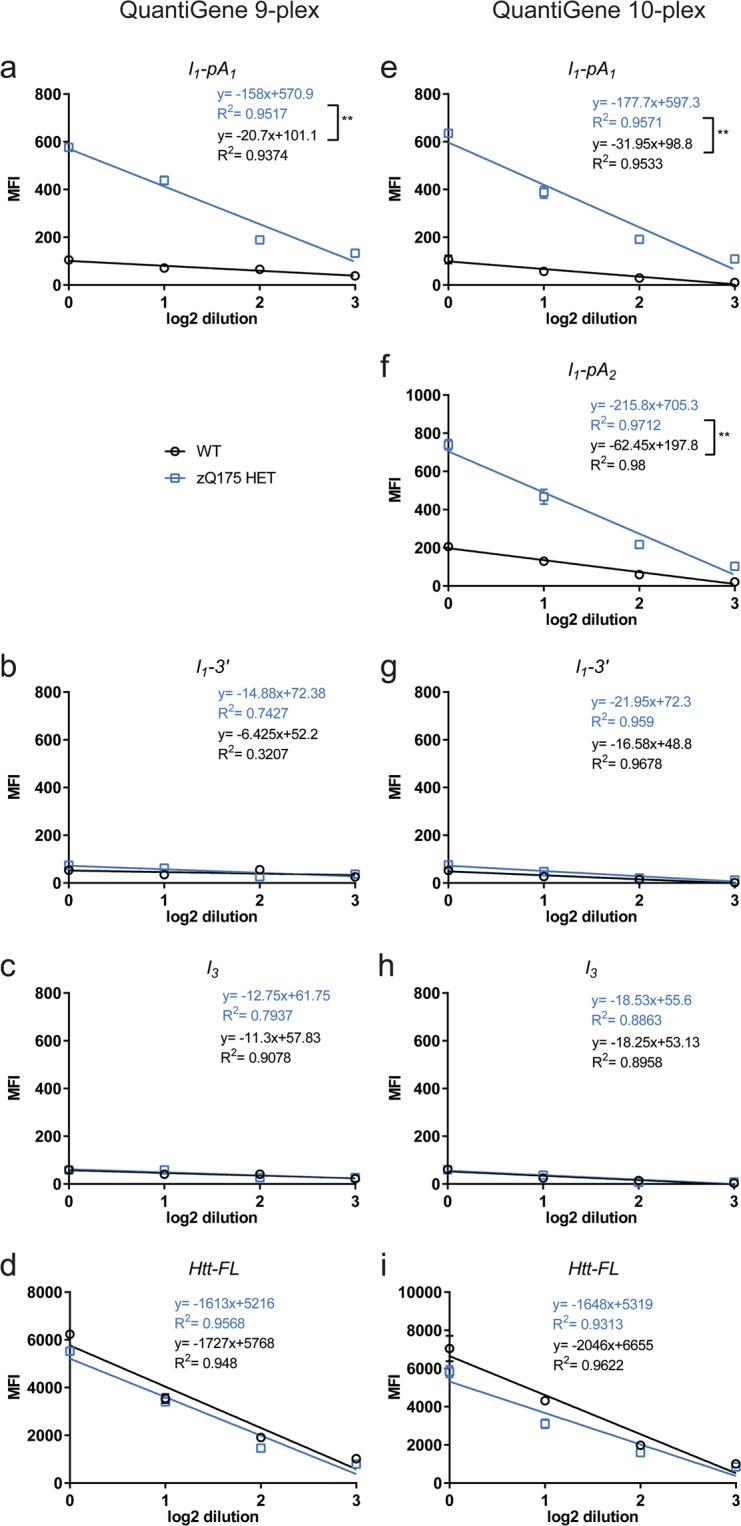
Establishment and optimisation of QuantiGene Htt multiplex assays in striatal tissue from WT and zQ175 heterozygous mice. The MFI signals corresponding to 2-fold serial dilutions of the QuantiGene striatal lysate from 2 month old animals are shown for each Htt transcript probe set. (a–d) 9-plex QuantiGene assay and (e–i) 10-plex QuantiGene assay. (a,e,f) The linear regression slopes of the Httexon1 transcript (I1-pA1 and I1–pA2) are significantly different between the two genotypes, with the WT MFI signal averaging background levels, as measured by the probe set for Htt I3. (b,g) The I1-3′ probe set showed that the un-spliced intron 1 sequences do not extend to the 3′ end of intron 1, with a signal similar to that observed for the assay background control, Htt I3. (c,h). The I3 probe set served as a control to indicate the level of unprocessed Htt mRNA. (d,i) Htt-FL probe set signals are shown. Data were from two technical replicates of equimolar pools of striatal samples (n = 4/genotype). Statistical analysis was by linear regression **p < 0.01. MFI = mean fluorescence intensity.
Importantly, the striatal MFI signal for the I1-pA1 probe set (for both plex panels) (Fig. 2a,e) and for the I1-pA2 probe set (for the 10-plex panel) (Fig. 2f) suggested that the genotypes behaved differently for these early intron 1 sequences. There was considerably more signal in the zQ175 pool than in the WT, as indicated by the difference in the linear regression slopes. The MFI signal for the I1-pA1 probe set showed a similar linear range of detection in both the 9- and 10-plex panels (Fig. 2a,e). The signal for the I1-pA2 probe set on the 10-plex panel (Fig. 2f) was significantly greater for the zQ175 heterozygotes than for WT. The MFI signal for the I1-3′ probe sets was comparable to I3 for both WT and zQ175 lysates (Fig. 2b,g,c,h for striatum and Supplementary Figs S2–S5 for other brain regions). The linear ranges for the probe sets for Htt-FL are shown in Fig. 2d,i, and Supplementary Figs S2–S5. Taken together, these data suggest that the 10-plex QuantiGene panel can be used to detect all Htt transcripts, without compromising the fluorescent signal due to the close proximity between the I1-pA1 and I1-pA2 probe sets. At the dilutions recommended, the housekeeping genes gave comparable signals for the two genotypes (Supplementary Figs S1–S5).
Validation of the QuantiGene Htt 10-plex assay
We set out to validate our 10-plex QuantiGene panel by comparing the expression levels of the Htt transcripts as measured by this approach with that determined by real time quantitative PCR (qPCR). The levels of Htt transcripts were measured in brain regions from zQ175 heterozygotes and WT littermates at 2 months of age (n = 4/genotype). The locations of the qPCR assays corresponded to the position of the QuantiGene probe sets, except that we did not include a qPCR assay for intron 3 (Supplementary Table S2).
The level of the Httexon1 transcript was shown to be abundant in the striatum, cortex, hippocampus, cerebellum and brain stem of zQ175 mice as compared to WT by both approaches (Fig. 3). Both assays confirm that the level of 3′ intron 1 sequences are at background levels (Fig. 3). This is in keeping with our previous RNAseq data indicating that transcription terminates at the second cryptic poly(A) site8. In the zQ175 samples, the signal from the QuantiGene I1-pA2 probe set is higher than that for I1-pA1 (Fig. 3a-d). As it is not possible for the level of the intron 1 sequences between poly(A)1 and poly(A)2 to be greater than that before poly(A)1, this may reflect differences in hybridisation efficiencies. This most likely accounts for the higher I1-pA2 signal in WT mice, which we consider to be background noise; the qPCR data suggest that any level of incomplete splicing that occurs in WT mice is below the level of detection (the readings for the I1-pA1 and I1-pA2 assays are not greater than for I1-3′).
Figure 3.

Comparison of the analysis of Htt transcripts in the striatum of zQ175 heterozygous animals using the 10-plex QuantiGene panel and qPCR assays. Expression of the Htt intronic sequences and Htt-FL transcripts was measured by our QuantiGene 10-plex assay (a–j) and qPCR (k-t) in striatum, cortex, hippocampus, cerebellum and brain stem of 2 month old mice, and presented relative to that for housekeeping genes. (a–e and k–o) Httexon1 transcripts can be detected in all brain regions by both approaches. (f–j and p–t) The levels of Htt-FL as measured by both approaches were comparable. Data were obtained from n = 4/genotype with two technical replicates for QuantiGene and three for qPCRs. Statistical analysis was by multiple t-test with Benjamini-Hochberg with an alpha of 0.05; *p < 0.05, **p < 0.01, ***p < 0.001. Tissue lysate dilutions were 1:3 for striatum and brain stem, 1:4 for cortex and cerebellum and 1:5 for hippocampus.
The Htt-FL transcript was also analysed in all five brain regions. In the striatum, the level of Htt-FL was reduced to approximately 70% – 80% of WT levels in the zQ175 mice as measured by both assays (Fig. 3f,p). For all of the other brain regions, the situation was less clear cut, although in all cases the mean level of Htt-FL expression was lower in the zQ175 than WT mice (Fig. 3). The qPCR assay gave tighter data for the cortex in which the reduction of Htt-FL could be detected (Fig. 3q). In comparison, the QuantiGene data was more variable, and so we would recommend that more than 4 samples per genotype should be analysed.
Design and application of a QuantiGene Htt 14-plex assay
We next extended our 10-plex assay to include probes against each of the three 3′UTR isoforms of Htt (short-, mid- and long- 3′UTR)7 and an additional housekeeping reference gene, Ppib18,20 (Fig. 1 and Supplementary Table S3). To ensure that the new 14-plex panel could detect all Htt transcripts and Ppib, we analysed the same zQ175 and WT lysates as had been used for the 10-plex panel (n = 4/genotype). The Httexon1 transcript levels in zQ175 mice were comparable to those detected with the 10-plex assay (Supplementary Fig. S6 and Fig. 3), except in brain stem, where they showed less variability, and consequently, the difference in the levels between zQ175 and WT samples was statistically significant. The E50-53 probe set, as well as each of the 3′UTR assays, showed a statistically significant reduction in Htt-FL levels in zQ175 as compared to WT samples for the striatum, cortex and hippocampus (Supplementary Fig. 6).
To further assess the application of the 14-plex QuantiGene panel, and to test the consistency of our data, we repeated our analysis of Htt transcripts in a different cohort of mice (n = 8/genotype) (Fig. 4). We could detect a statistically significant increase in the Httexon1 transcript in the zQ175 mice as compared to WT in all five brain regions tested (Fig. 4a-e). This correlated with a statistically significant reduction in Htt-FL levels as measured by all four of the Htt-FL assays for striatum (~23%), cortex (~22%) and hippocampus (~22%) (Fig. 4f-j).
Figure 4.
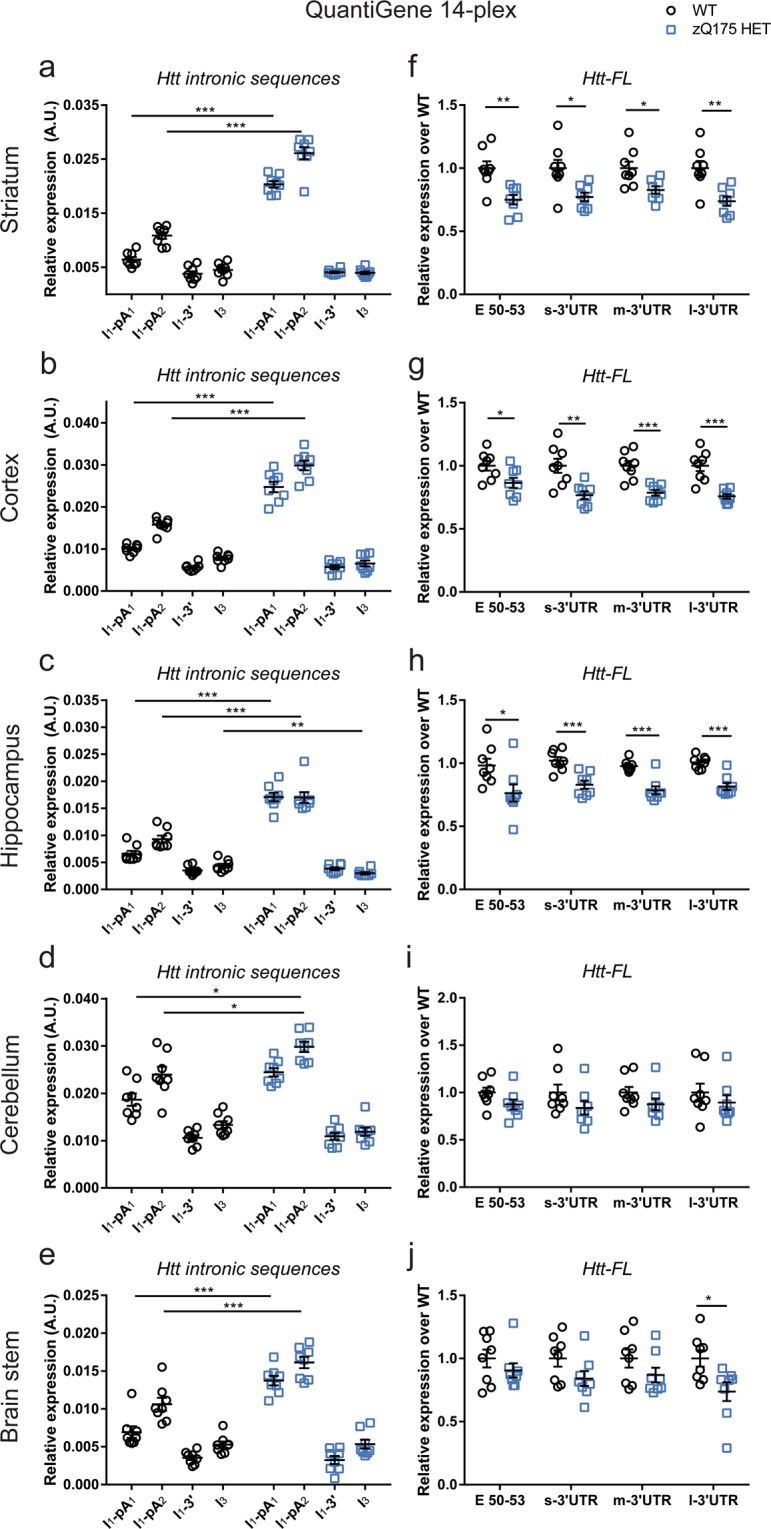
Expression analysis of Htt transcripts in five brain regions using the custom-made 14-plex QuantiGene assay. The expression of intronic Htt transcripts in 2 month old mice are presented relative to that for housekeeping genes. (a–e) The Httexon1 transcript is present in all zQ175 brain regions. (f–j) The level of the Htt-FL transcript is significantly decreased in the striatum, cortex and hippocampus as measured by the E50-53 and all 3′UTR probe sets. Data were from n = 4/genotype with two technical replicates. Statistical analysis was by multiple t-test with Benjamini-Hochberg with an alpha of 0.05, *p < 0.05, **p < 0.01, ***p < 0.001. Tissue lysate dilutions were 1:3 for striatum, hippocampus and brain stem, 1:4 for cortex and cerebellum.
Overall, we conclude that our 14-plex QuantiGene assay can reliably compare differences in the levels of the Httexon1 and Htt-FL transcripts between the zQ175 mouse model of HD and WT littermates.
The level of the Httexon1 transcript in zQ175 tissues corresponds to a reduction in mutant and not endogenous Htt-FL
We found that the level of the Htt-FL transcript was consistently decreased in zQ175 striatum, cortex and hippocampus (Fig. 4) and Supplementary Fig. S6). In order to determine whether this reflected a decrease in the endogenous transcript, the mutant transcript, or both, we employed an allele-specific qPCR assay (Fig. 5). We found that the level of endogenous Htt-FL was 50% of WT levels in all five brain regions for zQ175 mice. Therefore, any reductions in the level of Htt-FL must correspond to decreases in the mutant Htt-FL transcript. This is consistent with our failure to detect incomplete splicing of the endogenous Htt transcript in WT mice (Figs 2 and 4 and Sathasivam et al. 2013).
Figure 5.

The level of the endogenous Htt-FL transcript is not decreased in zQ175 tissues. Allele-specific qPCR analysis of the level of the endogenous Htt-FL transcript in five brain regions from 2 month old WT and zQ175 mice. The level of the endogenous Htt-FL transcript was present at 50% WT levels in the zQ175 samples for all five brain regions. Data were obtained from n = 4/genotype. Statistical analysis was by t-test, *p < 0.05, ***p < 0.001.
Discussion
In this study, we have assessed the expression of all known major Htt transcripts in the brains of the zQ175 mouse model of HD. We developed a multiplex QuantiGene assay based on the rapid, robust and high-throughput bDNA technology17 for this purpose and demonstrated that this can be used to replace qPCR for studies comparing Htt transcripts between samples. We applied the QuantiGene assay to the comparison of Htt transcripts in various brain regions between zQ175 and WT littermates at 2 months of age, and showed that incomplete slicing occurs throughout the brain for zQ175, but not WT, animals. Finally, we provide evidence to indicate that the Httexon1 transcript originates from the incomplete splicing of the mutant Htt allele.
We have developed and optimised a 14-plex QuantiGene assay to enable us to detect all major Htt transcripts directly from tissue lysates, obviating the need to prepare RNA and replacing the more labour-intensive qPCR. This assay includes probe sets targeting sequences extending to the first and second cryptic poly(A) sites in intron 1, to detect incomplete splicing; to the 3′ end of intron 1 and to intron 3, to give an indication of pre-mRNA background; to exons 50–53 and each of the 3′UTRs (short, mid and long) to detect Htt-FL and shifts in 3′UTR usage; as well as six housekeeping genes. We have applied this to the analysis of Htt transcripts in five brain regions from the zQ175 knock-in mouse model of HD and WT littermates. For each brain region, we calculated the lysate dilution required to ensure that mean fluorescence intensity (MFI) signals were in the linear range and recommend using dilutions at the lower end of the range detailed in Supplementary Table S1. We caution against using lysates that have been subjected to multiple freeze-thaw cycles, and a recent study suggested that the use of ‘RNAlater’, when preparing QuantiGene lysates, might better preserve long mRNAs, such as Htt-FL, which are more prone to degradation18.
QuantiGene analysis of Htt transcripts in brain regions of 2 month old zQ175 mice (Fig. 4) showed that intronic sequences 5′ to the cryptic poly(A) sites (I1-pA1 and I1-pA2) were present in zQ175 cortex, striatum, hippocampus, cerebellum and brain stem at higher levels than in WT, indicating that incomplete splicing of Htt occurs in all of these brain regions. The levels of intronic sequences at the 3′ end of intron 1 (I1-3′) were comparable to intron 3 (I3), indicative of pre-mRNA background. This is consistent with our previous RNAseq data showing that the incompletely spliced intron 1 sequence reads terminate at the second cryptic poly(A) site8. In all cases, the level of intron 1 sequences between the two poly(A) sites (I1-pA2) was higher than that before the first poly(A) site (I1-pA1). As it is not possible that the mRNA level between the poly(A) sites is higher than before the first poly(A), this must be a reflection of the differential efficiency of probe binding. The 14-plex analysis of WT tissues (I1-pA1 and I1-pA2) suggested that the incomplete splicing of Htt might be occurring in WT mice to a lesser extent. However, examination of the qPCR data (Fig. 3), for which the I1-pA1, I1-pA2 and I1-3′ assays give comparable data, indicates that this is unlikely, but arising instead from the differential efficiency of probe-set binding. Therefore, the 14-plex QuantiGene assay provides a valuable tool for comparative analyses between samples but, as used here, is not quantitative.
The level of Htt-FL transcript, as detected by all four probe sets (E50-53, s-3′UTR, m-3′UTR and l-3′UTR) is lower in zQ175 striatum, cortex and hippocampus than in WT mice. We employed an allele specific qPCR assay that selectively detects endogenous Htt in this knock-in model, and found that the level of the endogenous Htt-FL was not decreased in the heterozygous zQ175 mice. That the reduction in Htt-FL corresponds to a decrease in the mutant transcript, and that this is a consequence of incomplete splicing between exon 1 and exon 2 is consistent with our observation that incomplete splicing of endogenous Htt does not occur in WT mice and our previous RNAseq data8.
We have previously shown that the incomplete splicing of exon 1 to exon 2 of Htt results in the Httexon1 polyadenylated mRNA that is translated to produce the highly pathogenic exon 1 HTT protein8–10. The extent of incomplete splicing increases with increasing CAG repeat length, therefore, somatic CAG expansion would be expected to result in the increased production of the exon 1 HTT protein. Genome-wide association studies have identified a number of DNA repair genes as genetic modifiers of HD that have been shown to modulate somatic expansion levels24–26. Therefore, incomplete splicing produces a direct link between the genetic modifiers for HD and the production of a pathogenic fragment of HTT. As Htt lowering approaches are evaluated in mouse models, it is essential that their effect on the level of Httexon1 as well as Htt-FL transcripts can be assessed. Our QuantiGene assay provides a rapid method for screening Htt lowering agents targeting mouse Htt in cell culture and mouse models of HD.
Methods
Animal colony maintenance and breeding
All animal care and procedures were performed under a Home Office License in compliance with the Animals and Scientific Procedures Act, 1996, with approval by the University College London Ethical Review Process Committee. The zQ175 (delta neo)27 heterozygous colony21,22 was maintained by backcrossing to C57BL/6 J (Charles River). The animals had unconditional access to food and water, environmental enrichment in the form of two small aspen bricks and a play tunnel (Datesand Group) and were subjected to a 12-hour light/dark cycle. Animals were sacrificed at 2 months of age, brains dissected, frozen immediately in liquid nitrogen and stored at −80 °C.
DNA extractions, genotyping and repeat sizing
Genomic DNA isolation was performed after lysing an ear-punch in 50 mM Tris-HCl, 0.5% SDS, 100 mM EDTA pH = 8.0, with 0.1 mg/mL proteinase K at 50 °C overnight. Addition of 300 μL saturated 36% (w/v) NaCl solution was followed by thorough mixing and centrifugation at 21,000 g for 30 min. The DNA-containing supernatant was transferred to a new tube with 650 μL ice-cold 100% ethanol (EtOH) followed by mixing and centrifugation at 21,000 g for 20 min. The DNA-containing pellet was washed with 70% EtOH and centrifuged at 21,000 g for 15 min. The supernatant was discarded, the DNA pellet was left to air dry and lastly the DNA was resuspended in 60 μL 5 mM Tris-HCl, pH = 8.0. Genotyping of the zQ175 animals was performed as follows: in a total volume of 20 μL reaction, 20 ng of DNA was added to a mixture of 1x Go Taq Flexi buffer (Promega), 2.5 mM MgCl2, 0.2 mM dNTPs, 1 μM each primer (Forward: 5′-AGGAGCCGCTGCACCGA-3′ and Reverse: 5′-CTCTTCACAACAGTCATGTGCG-3′) and 0.05 U/ μl GoTaq2, and the following PCR programme was used: 98 °C for 30 s, followed by 34 cycles of 98 °C for 15 s, 64 °C for 15 s, 72 °C for 30 s, and a final elongation step at 72 °C for 5 min. The PCR products were loaded on a 2.5% agarose gel containing SYBR Safe DNA gel stain (Thermofisher), subjected to electrophoresis and visualised on a BioRad Gel Doc XR+ . The CAG repeat sizing was performed as previously described28 using the following primers: Forward: 5′-6FAM-ATGAAGGCCTTCGAGTCCCTCAAGTCCTTC-3′ and Reverse: 5′-GGCGGCTGAGGAAGCTGAGGA-3′, except that the DNA size standard was MapMarker 1000 ROX (BioVentures Inc.) and analysis of data was performed using the GeneMapper plate manager application. The CAG repeat size for the first cohort of the zQ175 mice was 197.75 ± 1.7 (S.D.) and for the second cohort was 207.5 ± 3.02.
Tissue lysis and QuantiGene assays
All Quantigene probe sets are available from ThermoFisher and the individual accession numbers and probe set regions are summarised in Supplementary Table S3; the sequences for the capture extenders, label extenders and blocking probes are available from ThermoFisher on request. The catalogue number for the 14-plex set validated in this manuscript is 21917. Investigators can select probe sets from this to customise their own multiplex assays, if they did not wish to include all of the Htt probe sets or all of the six housekeeping genes.
All QuantiGene reagents were from ThermoFisher. Homogenates of brain regions were prepared by adding 300 μL of working homogenisation solution (homogenisation solution (QG0517) with 1:100 (v/v) 50 mg/ mL Proteinase K (QS0512) for each 10 mg tissue) and using syringes with needles of 23 G and 18 G. Homogenates were then centrifuged at 15,000 g for 10 min and the supernatant was transferred to a fresh tube. For linearity probe set assessment, equal volumes of homogenates of the same genotype were pooled together and a 2-fold serial dilution was conducted for each pool. Otherwise, the homogenates were diluted so readings would fit in the linear range of the fluorescent signal for all probe sets in the QuantiGene panel (Supplementary Table S1). The QuantiGene assays were performed as indicated in the manufacturer’s protocol with the exception that the Streptavidin R-Phycoerythrin conjugate (SAPE) binding was done at 51 °C. Plates were read using a Magpix (Luminex). Data analysis was performed by normalising the signals obtained for the genes of interest to the geometric mean of the reference gene signals (Atp5b, Eif4a2, Rpl13a, Canx and Ppib as appropriate). All Htt-FL isoforms were expressed relative to the average expression of the WT animals. Technical duplicates of each sample were included in each experiment, with a minimum of two independent experiments for each cohort of animals.
RNA isolation and cDNA synthesis
Total RNA isolation was conducted using the RNeasy mini kit (Qiagen) following the manufacturer’s protocol, after homogenising the tissue in Qiazol lysis reagent (Qiagen) using syringes with needles of 23 G and 18 G. DNase I treatment was performed in-column using the RNA-free DNase kit (Qiagen) before the RNA was eluted in the appropriate amount of nuclease-free H2O (Sigma-Aldrich). The quality and quantity of RNA was measured using a NanoDrop 1000. Reverse transcription was performed on 0.5–1 μg of RNA using the M-MLV reverse transcriptase (RT) (Invitrogen) according to the company’s protocol and using oligo-dT(18) primers (Invitrogen). A negative control was included for each brain region in which no reverse transcriptase was added (-RT).
Real-time quantitative PCR assays
Real-time quantitative PCR (qPCR) was performed using hydrolysis probes with a single or double quencher (Supplementary Table S2). The reference gene assays are commercially available (Thermofisher Scientific) and comply with the MIQE guidelines29,30. Amplification efficiencies were evaluated by 4-fold serial dilutions of a pool of equal volumes of 4 cDNAs from different zQ175 heterozygous animals and are provided in Supplementary Table S2. A 1:10 dilution of the cDNA was used to avoid inhibition of the qPCR reaction. After determining the linear range of cDNA input and the amplification efficiency of the primer sets, a 1:50 dilution of the cDNA was made for all brain regions. Primers and probes for the custom Htt assays were from Eurofins and for the allele discrimination assays were from Integrated DNA Technologies. Samples were prepared in 1x TaqMan Fast Advanced Mix (Applied Biosystems), in a final volume of 15 μL, in 96-well thin wall Hard-Shell PCR plates (BioRad). The primer/probe mix for reference genes was used diluted to 1x and for our custom-designed assays, the primers and probes were used at concentrations of 0.3 mM and 0.2 mM respectively. Plates were sealed with Microseal ‘B’ seals (BioRad) and centrifuged at 800 g for 30 s. qPCRs were performed in a BioRad CFX96 qPCR machine with the following programme: 95 °C for 40 s, followed by 40 cycles of 95 °C for 7 s and 60 °C for 20 s. For the allele-discrimination assays, the following programme was used: 95 °C for 2 min, followed by 40 cycles of 95 °C for 10 s, 61.8 °C for 30 s, 72 °C for 30 s. Three technical replicates of each biological sample were used for all assays. Cq values of the triplicates were assessed and values deviating by 0.5 from the mean Cq value were excluded from further analysis. Normalisation of the data was done either by the 2−ΔΔCt method 31 or based on the amplification efficiency of the primer sets32 to the geometric mean of all the reference genes (Eif4a2, Atp5b, CanX, Ubc, Rpl13a). For the allele discrimination qPCR assays data were normalised to the geometric mean of two reference genes (Eif4a2, Atp5b).
Statistical analysis
Statistical analysis was performed using multiple two-tailed Student’s t-tests followed by Benjamini-Hochberg correction and a desired false discovery rate (q) adjusted to 0.05. All statistical analysis was performed using the GraphPad Prism software.
Supplementary information
Acknowledgements
We thank Rachel Flomen for providing technical information. This work was funded by the CHDI Foundation.
Author contributions
A.S.P., D.H. and G.P.B. designed the study; A.S.P., C.G.P., M.A.M. and B.A.T. performed the experiments; A.S.P. analysed the data and prepared the figures; A.S.P. and G.P.B. wrote the manuscript and all authors read and approved the manuscript.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-52411-2.
References
- 1.Bates GP, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- 2.The Huntignton’s Disease Collaborative Group A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntingtons-Disease Chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 3.Rubinsztein DC, et al. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am J Hum Genet. 1996;59:16–22. [PMC free article] [PubMed] [Google Scholar]
- 4.Kennedy L, et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. 2003;12:3359–3367. doi: 10.1093/hmg/ddg352. [DOI] [PubMed] [Google Scholar]
- 5.Gonitel R, et al. DNA instability in postmitotic neurons. Proc Natl Acad Sci USA. 2008;105:3467–3472. doi: 10.1073/pnas.0800048105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin B, et al. Differential 3′ poly(A)denylation of the Huntington disease gene results in two mRNA species with variable tissue expression. Hum Mol Genet. 1993;2:1541–1545. doi: 10.1093/hmg/2.10.1541. [DOI] [PubMed] [Google Scholar]
- 7.Romo L, Ashar-Patel A, Pfister E, Aronin N. Alterations in mRNA 3′ UTR Isoform Abundance Accompany Gene Expression Changes in Human Huntington’s Disease Brains. Cell Rep. 2017;20:3057–3070. doi: 10.1016/j.celrep.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sathasivam K, et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci USA. 2013;110:2366–2370. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neueder A, Dumas AA, Benjamin AC, Bates GP. Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat Commun. 2018;9:3955. doi: 10.1038/s41467-018-06281-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neueder A, et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci Rep. 2017;7:1307. doi: 10.1038/s41598-017-01510-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron. 2019;101:801–819. doi: 10.1016/j.neuron.2019.01.039. [DOI] [PubMed] [Google Scholar]
- 12.Tabrizi, S. J. et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med, 10.1056/NEJMoa1900907 (2019). [DOI] [PubMed]
- 13.Mangiarini L, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 14.Barbaro BA, et al. Comparative study of naturally occurring huntingtin fragments in Drosophila points to exon 1 as the most pathogenic species in Huntington’s disease. Hum Mol Genet. 2015;24:913–925. doi: 10.1093/hmg/ddu504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canales RD, et al. Evaluation of DNA microarray results with quantitative gene expression platforms. Nat Biotechnol. 2006;24:1115–1122. doi: 10.1038/nbt1236. [DOI] [PubMed] [Google Scholar]
- 16.Collins ML, et al. A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/ml. Nucleic Acids Res. 1997;25:2979–2984. doi: 10.1093/nar/25.15.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flagella M, et al. A multiplex branched DNA assay for parallel quantitative gene expression profiling. Anal Biochem. 2006;352:50–60. doi: 10.1016/j.ab.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Coles AH, et al. A High-Throughput Method for Direct Detection of Therapeutic Oligonucleotide-Induced Gene Silencing In Vivo. Nucleic Acid Ther. 2016;26:86–92. doi: 10.1089/nat.2015.0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alterman, J. F. et al. A High-throughput Assay for mRNA Silencing in Primary Cortical Neurons in vitro with Oligonucleotide Therapeutics. Bio Protoc, 7, 10.21769/BioProtoc.2501 (2017). [DOI] [PMC free article] [PubMed]
- 20.Alterman JF, et al. Hydrophobically Modified siRNAs Silence Huntingtin mRNA in Primary Neurons and Mouse Brain. Mol Ther Nucleic Acids. 2015;4:e266. doi: 10.1038/mtna.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heikkinen T, et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington’s disease. PLoS One. 2012;7:e50717. doi: 10.1371/journal.pone.0050717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menalled LB, et al. Comprehensive Behavioral and Molecular Characterization of a New Knock-In Mouse Model of Huntington’s Disease: zQ175. PLoS One. 2012;7:e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benn CL, Fox H, Bates GP. Optimisation of region-specific reference gene selection and relative gene expression analysis methods for pre-clinical trials of Huntington’s disease. Mol Neurodegener. 2008;3:17. doi: 10.1186/1750-1326-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Consortium G-H. Identification of Genetic Factors that modify clinical onset of Huntington’s disease. Cell. 2015;162:516–526. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goold R, et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. 2019;28:650–661. doi: 10.1093/hmg/ddy375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moss DJH, et al. Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol. 2017;16:701–711. doi: 10.1016/S1474-4422(17)30161-8. [DOI] [PubMed] [Google Scholar]
- 27.Franich, N. R. et al. Phenotype onset in Huntington’s disease knock-in mice is correlated with the incomplete splicing of the mutant huntingtin gene. J Neurosci Res Epub 10.1002/jnr.24493 (2019). [DOI] [PMC free article] [PubMed]
- 28.Sathasivam K, et al. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum Mol Genet. 2010;19:65–78. doi: 10.1093/hmg/ddp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bustin SA. Why the need for qPCR publication guidelines?–The case for MIQE. Methods. 2010;50:217–226. doi: 10.1016/j.ymeth.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Bustin SA, et al. MIQE precis: Practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. BMC Mol Biol. 2010;11:74. doi: 10.1186/1471-2199-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.