

Summary
Compared with SNV&indel-based neoantigens, fusion-based neoantigens are not well characterized. In the present study, we performed a comprehensive analysis of the landscape of tumor fusion neoantigens in cancer and proposed a score scheme to quantitatively assess their immunogenic potentials. By analyzing three large-scale tumor datasets, we demonstrated that (1) the tumor fusion candidate neoantigen burden is not related to the immunotherapy outcome; (2) fusion neoantigens tend to have notably higher immunogenic potentials than SNV&indel-based candidate neoantigens, making them better candidates for cancer vaccines; (3) fusion candidate neoantigens distribute sparsely between individual patients. Although several recurrent candidate neoantigens exist, they usually have extremely low immunogenic potentials, suggesting that vaccination-based cancer immunotherapy must be personalized; (4) compared with fusion mutations involving tumor passenger genes, fusion mutations involving oncogenic genes have remarkably low immunogenic potentials, indicating that they undergo selection pressure during tumorigenesis.
Subject Areas: Genomics, Bioinformatics, Cancer
Graphical Abstract
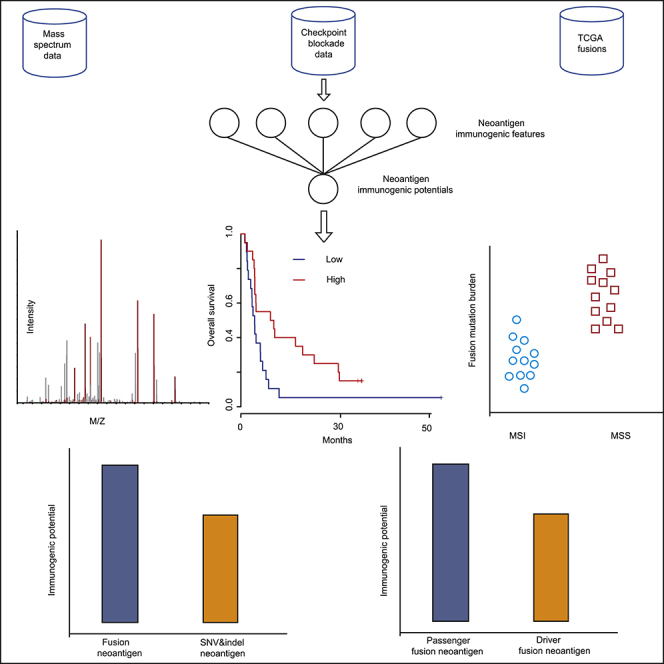
Highlights
-
•
A score scheme is presented to evaluate the immunogenicity of fusion neoantigen
-
•
Tumor fusion neoantigen burden is not related to the immunotherapy outcome
-
•
Compared with SNV&indel neoantigen, fusion neoantigen has higher immunogenicity
-
•
Oncogene fusion has lower immunogenicity than passenger gene fusion
Genomics; Bioinformatics; Cancer
Introduction
Vaccination therapy to fight cancer by boosting the response of the human immune system to cancer cells is a highly promising treatment strategy. Neoantigens are peptides generated from somatically mutated genes that play an important role in vaccination-based cancer immunotherapy (Liu and Mardis, 2017, Vitiello and Zanetti, 2017). It has been reported that predicted neoantigen load is strongly correlated with the clinical response to immunotherapy. Fusion is a hybrid gene formed from two previously separate genes, and it has long been known to play an important role in tumorigenesis (Mertens et al., 2015). Because of the ability to create new open reading frame (ORF) and produce plentiful neo-peptides, neoantigens can be generated from fusions (Yang et al., 2019). Current studies of neoantigen sources mainly focus on single nucleotide variants (SNV) and small insertions and deletions (indel), whereas fusion-based neoantigens across different cancers are not yet well characterized (Zhou et al., 2019). Therefore, in the present study, we comprehensively characterized the landscape of fusion candidate neoantigens presented by major histocompatibility complex class I (MHC I) in three cohort datasets, i.e., the dataset of cancer cell lines with MHC I mass spectrum (MS), the dataset of sequencing data from immune checkpoint blockade (ICB) trials, and The Cancer Genome Atlas dataset (TCGA). Specifically, we characterized the tumor fusion neoantigens by taking the T-cell receptor (TCR) recognition mechanisms into consideration. TCR is a molecule found on the surface of T cells, or lymphocytes, that is responsible for recognizing fragments of antigen as peptides bound to MHC molecules. To activate a T-cell response, peptide-MHC complex (pMHC) must be recognized by T-cell receptors. However, in the process of mature T-cell generation, negative selection mechanism removes T cells that are capable of strongly binding with self-peptides. As a result, the TCR repertoire has intrinsic biases in their generation probabilities (Murugan et al., 2012). Furthermore, due to the T-cell cross-reactivity (the ability of the T cell to recognize more than one pMHC) and negative selection, predicted neoantigens may differ greatly in their immunogenic potentials. Therefore, how to quantitatively and unbiasedly evaluate the neoantigen immunogenic potentials remains a challenge issue. We hereby proposed a score scheme to evaluate fusion neoantigen immunogenic potentials, taking two factors, i.e., the likelihood of peptide presentation by MHC (Bjerregaard et al., 2017) and pMHC subsequent recognition by T cells (Łuksza et al., 2017), into consideration. Through applying our score scheme to the MS, ICB, and TCGA cohort datasets, several findings are presented, providing useful clues for personalized cancer vaccination-based immunotherapy.
Results
Analysis of the MS Cohort Dataset
Fusion Peptides Can Be Processed and Presented on the Tumor Cell Surface
High resolution mass-spectrometry enables the identification and quantification of MHC ligands that are naturally processed and presented. To demonstrate that gene fusion is able to generate neo-peptides that can be presented by the MHC I, fusion candidate neoantigens in 10 human breast cancer cell lines were predicted following our computational workflow (Methods). In our study, mutation burden is defined as the total number of somatic mutations detected in a tumor sample and neoantigen burden is defined as the total number of neoantigens produced by those mutations. The tumor fusion candidate neoantigen burden varies from 24 to 147 in cancer cell lines, with a median value of 63 (Figure 1A). Two fusion candidate neoantigens, i.e., TAISPIAVLPR produced by OTUB1-CDC20 (by CDC20 frameshift transcript) in HCC1806 cell line and APKSSSGFSL produced by MTSS1L-RPS15A (by RPS15A frameshift transcript) in HCC1428 cell line, were discovered in complex with MHC I via mass spectrometry with high confidences (Figure 1B, Percolator q-value<0.01, Table S1). These results provide direct evidences of the processing and presentation of fusion peptides through the MHC I, which were also recently experimentally demonstrated by Yang (Yang et al., 2019).
Figure 1.

Analysis of the MS Cohort Dataset
(A) Ten breast cancer cell line RNA sequencing data were analyzed according to our computational pipeline neoFusion. Two fusion neo-peptides, i.e., TAISPIAVLPR produced by OTUB1-CDC20 in HCC1806 cell line and APKSSSGFSL produced by MTSS1L-RPS15A in HCC1428 cell line, were experimentally validated to be presented by MHC I using the MS data.
(B) Peptide-spectrum matches with q-value < 0.01 were extracted by mzR R package and then visualized by xiSPEC software.
See also Table S1.
Fusion candidate neoantigens were further prioritized according to their score as defined in the Methods. It should be noted that when scoring those predicted fusion neoantigens, only the likelihood of peptide presented by MHC I (Methods) was calculated, as those peptides are eluted from pMHC complexes. The fusion candidate neoantigens TAISPIAVLPR in HCC1806 cell line (92 fusion candidate neoantigens in total) and APKSSSGFSL in HCC1428 cell line (29 fusion candidate neoantigens in total) rank 6/92 and 2/29, respectively, suggesting that candidate neoantigens with higher ranks are presented by the MHC I with high priorities (p value<0.05, Methods).
Analysis of the ICB Cohort Dataset
Overview of the Landscape of Fusion Candidate Neoantigens in the ICB Dataset
Two melanoma ICB cohorts with whole-exome and RNA sequencing data were analyzed according to our computational workflow (Methods). In the Van Allen cohort, the total number of fusions per sample varied from 0 to 25, with a median value of 5. In the Hugo cohort, the total number of fusions per sample varied from 0 to 9, with a median value of 1. In these two cohorts, tumor fusion candidate neoantigens were notably lower than SNV&indel candidate neoantigens, in terms of both burden and score (Figure 2A, Table S2).
Figure 2.

Analysis of the ICB Cohort Dataset
(A) The overview of the tumor candidate neoantigen burden and score in the two ICB melanoma cohorts. Y axis values were log2 transformed.
(B) The tumor fusion candidate neoantigen score*CTL could not separate patients in both cohorts.
(C) The overall tumor candidate neoantigen score*CTL significantly separated patients in both cohorts. Samples were split by the median value cutoff in C and D. See also Figures S1 and S2 and Table S2.
(D) In the Van Allen cohort, the overall tumor candidate neoantigen score of the response group was significantly higher than that of the no response group. In the Hugo cohort, there was no difference between the response group and no response group. Boxplots show the first, median, and third quartiles, and whiskers extend to 1.5X the interquartile range.
The Tumor Fusion Candidate Neoantigen Burden Is Not Associated with the Immunotherapy Outcome
Given that the tumor SNV&indel neoantigen burden closely correlates with the response to checkpoint inhibitors (Van Allen et al., 2015, Hugo et al., 2017, Lauss et al., 2017), we next examined whether the tumor fusion candidate neoantigen burden is similarly associated with the immunotherapy response. The tumor microenvironment, including the surrounding immune cells and fibroblasts, should be taken into consideration in predicting the immunotherapy outcome (Church and Galon, 2015). According to Rooney et al., cytolytic activity is a biomarker of the immune response (Rooney et al., 2015). Similarly, Balachandran et al. reported that tumors having both the highest neoantigen burden and the most abundant CD8+ T-cell infiltrates, but not either alone, stratified patients with the longest survival (Balachandran et al., 2017). In other words, high-quality neoantigens and sufficient T cells are needed simultaneously to elicit a T-cell response. To this end, the cytotoxic lymphocyte score (CTL, Methods) was incorporated to amplify the tumor candidate neoantigen score (score∗CTL, Methods) for survival analysis. In both cohorts, the tumor fusion candidate neoantigen burden, neoantigen score, and the tumor fusion candidate neoantigen score∗CTL were not associated with the checkpoint inhibitor response (Figures S1A, S1B, and 2B), suggesting that the tumor fusion candidate neoantigen burden is not a biomarker for the immunotherapy outcome.
The Overall Tumor Candidate Neoantigen Score∗CTL Is Associated with the Immunotherapy Outcome
Since the response to checkpoint inhibitors is associated with the neoantigen burden, we reasoned that neoantigens generated by all mutations should be taken into account. Therefore, we summed the SNV&indel candidate neoantigen burden and fusion candidate neoantigen burden to obtain the overall tumor candidate neoantigen burden. Similarly, we summed the SNV&indel candidate neoantigen score and fusion candidate neoantigen score to obtain the overall tumor candidate neoantigen score. In the Van Allen cohort, the overall tumor neoantigen burden, score, and CTL, respectively were not related to the immunotherapy outcome (Figures S1C, S2A, and S2B). Survival, however, was significantly improved in patients with a higher overall tumor candidate neoantigen score∗CTL (Figure 2C, log rank p value = 0.021) and overall tumor candidate neoantigen burden∗CTL (Figure S2C, log rank p value = 0.032) in the Van Allen cohort. In the Hugo cohort, the overall tumor candidate neoantigen burden∗CTL and CTL respectively were not related to the immunotherapy outcome (Figures S2C and S1C). Survival was significantly improved in patients with a higher overall tumor candidate neoantigen burden, score, and higher overall tumor candidate neoantigen score∗CTL (Figures S2A, S2B, and 2C, log rank p value<0.05) in the Hugo cohort. Taking together, all the metrics except the overall tumor candidate neoantigen score∗CTL have their limitations in immunotherapy response prediction in these two cohorts, indicating the rationality and effectiveness of our proposed score scheme.
It can be seen that in these two ICB cohorts, tumor fusion candidate neoantigen burden and score were notably lower than tumor SNV&indel candidate neoantigen burden and score respectively; as a result, adding the fusion candidate neoantigen burden and score to the overall tumor candidate neoantigen burden and score does not affect the p value in predicting the immunotherapy outcome.
Moreover, we examined the overall tumor candidate neoantigen score∗CTL, together with age and sex, in a multivariable model. Our results indicated that the overall tumor candidate neoantigen score∗CTL was associated with the response to checkpoint inhibitors, independent of age and sex (Figure S1D, Van Allen cohort: hazard ratio [HR] 0.44, 95% confidence interval [CI] 0.22–0.89, log rank p value = 0.022; Hugo cohort: HR 0.24, 95% CI 0.064–0.92, log rank p value = 0.038).
It should be noted that patients in these two cohorts were previously stratified into two groups according to the RECIST criteria (Methods). In the Van Allen cohort, the overall tumor candidate neoantigen score of the response group was significantly higher than the no response group (Figure 2D, p value = 0.013, Mann-Whitney U test). In the Hugo cohort, however, there was no significant difference between the response and no response group (Figure 2D, p value = 0.061, Mann-Whitney U test), possibly due to the small sample size.
Incorporating Fitness Score into Our Score Scheme Boosts the Predictive Power in the Survival Analysis
Previously, Luksza et al. (Łuksza et al., 2017) presented a fitness model to calculate the likelihood of pMHC recognized by T-cell receptors (fitness score, Methods) by alignment neo-peptides with a set of epitopes retrieved from IEDB. To activate a T-cell response, pMHC must be recognized by T-cell receptors. Therefore, we evaluated whether incorporating the fitness score into our score scheme could boost the predictive power in the survival analysis. Our results showed that taking the fitness score into consideration significantly separated patients in both cohorts in predicting the immunotherapy outcome (Figure 2C). Although the patients could not be separated in the Hugo cohort without the incorporation of the fitness score (Figure S2D), indicating that fitness model does boost predictive power in the survival analysis.
Analysis of the TCGA Cohort Dataset
Overview of the Landscape of Fusion Candidate Neoantigens in the TCGA Dataset
In total, there are 67,502 predicted fusion candidate neoantigens among the 6552 TCGA samples. The most common number of mismatches between the candidate neoantigen and the corresponding wild type peptide is 3, and frameshift fusion produces up to 145 candidate neoantigens. In our study, the tumor SNV&indel candidate neoantigen burden strongly correlated with the tumor SNV&indel mutation burden (pearson R = 0.89, p value<2.2 × 10−16). As for fusion, the correlation was slightly weaker (pearson R = 0.74, p value<2.2 × 10−16). Similar to the tumor candidate SNV&indel neoantigen burden (Thorsson et al., 2018), the overall tumor candidate neoantigen score∗CTL is not a prognostic factor for overall survival (Figure S3A) except for TCGA BLCA. Previously, based on the mechanism of T-cell central tolerance, Turajlic defined an SNV&indel candidate neoantigen with a half maximal inhibitory concentration (IC50) of less than 50 nM and the corresponding wild-type peptide with an IC50 greater than 50 nM as the specific candidate neoantigen (Turajlic et al., 2017). Because of self-immune tolerance, compared with nonspecific candidate neoantigens, specific candidate neoantigens tend to have higher immunogenic potentials. Following this definition, we applied this idea to fusion candidate neoantigens. Because we used binding affinity percent rank metric to filter peptides in the present study, a neoantigen with binding affinity percent rank ≤2 and a corresponding wild-type peptide with a binding affinity percent rank >2 was defined as a specific candidate neoantigen (Jurtz et al., 2017). For different cancer types, the fusion-specific candidate neoantigen burden per sample varied from 0 to 205, with a median value of 0. In our study, fusion mutation burden is the total number of fusions detected in a sample. The fusion mutation burden per sample varied from 0 to 55, with a median value of 1. The fusion candidate neoantigen burden per sample varied from 0 to 360, with a median value of 0. Breast invasive carcinoma (BRCA) was the cancer type with the highest fusion mutation burden per sample. Kidney chromophobe (KICH), kidney renal cell carcinoma (KIRC), and kidney renal papillary cell carcinoma (KIRP) were the three cancer types with the lowest fusion mutation burden, fusion candidate neoantigen burden, and fusion-specific candidate neoantigen burden, each with a median of 0 (Figure 3A).
Figure 3.

Analysis of the TCGA Cohort Dataset
(A) The landscape of the fusion mutation burden, fusion candidate neoantigen burden, and fusion-specific candidate neoantigen burden in the 20 cancer types.
(B) The landscape of the fusion mutation burden, fusion candidate neoantigen burden, and fusion-specific candidate neoantigen burden ratio in the 20 cancer types.
(C) The overview of the fusion and SNV&indel candidate neoantigen scores in the 20 cancer types. Except for PAAD, READ, and THCA, the fusion candidate neoantigen scores were significantly higher than that of the SNV&indel candidate neoantigen scores. Black star indicates p value>0.01.
(D) A fusion mutation was able to generate much more and high-quality candidate neoantigens than an SNV&indel mutation.
(E) In contrast to the SNV&indel mutation burden, the fusion mutation burden was significantly higher in microsatellite stable tumors.
Boxplots show the first, median, and third quartiles, and whiskers extend to 1.5X the interquartile range. Outlier points are not shown. See also Tables S3 and S4.
Fusion Candidate Neoantigens Tend to Have Higher Immunogenic Potentials Than SNV&Indel Candidate Neoantigens
For TCGA cohort data, the fusion mutation burden, candidate neoantigen burden, and specific candidate neoantigen burden were notably lower than the SNV&indel mutation, candidate neoantigen, and specific candidate neoantigen burden in all cancer types (Figure 3B). Except for pancreatic adenocarcinoma (PAAD), rectum adenocarcinoma (READ), and thyroid carcinoma (THCA), the score of fusion putative neoantigen was significantly higher than the score of SNV&indel candidate neoantigen in all cancer types (Figure 3C, Mann-Whitney U test, p value<0.01). Compared with SNV&indel, a fusion is able to produce more putative neoantigens (Figure 3D, p value<0.01, Mann-Whitney U test). Intriguingly, candidate neoantigens generated by fusion are much more likely to be specific candidate neoantigens (Figure 3D). Of 6,552 TCGA tumor samples, 3,161 simultaneously harbored tumor fusion and SNV&indel putative neoantigens. In 1,018 of 3,161 tumor samples, the putative neoantigen with highest immunogenic potentials were generated by fusion. Given that the mean fusion candidate neoantigen burden per sample was notably lower than the SNV&indel candidate neoantigen burden, this result further supports the fact that the fusion candidate neoantigens have significantly higher immunogenic potentials (p value<0.001, binomial test).
The Fusion Mutation Burden Is Significantly Higher in Microsatellite Stable Tumors
Microsatellite instability (MSI), a pattern of hypermutation that occurs at genomic microsatellites, is caused by defects in the mismatch repair system. The US Food and Drug Administration approved the high MSI phenotype as a biomarker for immunotherapy (Le et al., 2015). MSI is highly positively correlated with the tumor SNV&indel mutation burden (Bonneville et al., 2017). In this study, we further investigated the relationship between MSI and the tumor fusion mutation burden in the TCGA cohort data. The proportion of the MSI sample, as measured by the MANTIS score (Reeser et al., 2016), varied substantially across 20 cancer types. Because only colon adenocarcinoma (COAD), stomach adenocarcinoma (STAD), and uterine corpus endometrial carcinoma (UCEC) had sufficient MSI samples for analysis (samples ≥15), we focused on these three cancer types. Consistent with previous studies, MSI tumors had a significantly higher SNV&indel mutation burden (Figure 3E). In contrast to the SNV&indel mutation burden, however, the fusion mutation burden was notably lower in MSI tumors (STAD, UCEC p value<0.01, COAD p value = 0.012, Mann-Whitney U test). Furthermore, we investigated the relationship between the category of fusion and the status of microsatellite. Fusion mutations were separated into two categories with respect to the gene involved, i.e., driver gene fusion and passenger gene fusion (Gao et al., 2018; Table S3). The proportion of MSS tumor harboring driver fusion is higher than that of MSI tumor (STAD, COAD 3.2% vs 1.2%, UCEC 5% vs 0.6%). The driver fusion mutation burden was higher in MSS tumor (STAD, UCEC p value<0.01, Mann-Whitney U test). These may be explained by the fact that as a result of mismatch repair system deficiency, MSI tumors harbored notably more SNV&indel mutations, and these tumors are primarily driven by SNV&indel mutations (Vaish and Mittal, 2002). By contrast, because fewer SNV&indel mutations exist, microsatellite stable (MSS) tumor cells are likely to rely on other mechanism to gain a growth advantage such as producing driver fusion mutations.
Frameshift Fusion Candidate Neoantigens Tend to Have Higher Immunogenic Potentials Than Inframe Fusion Candidate Neoantigens
In our study, fusions were also separated into three categories with respect to the frame of the 3′ gene, i.e., noframe fusions, inframe fusions, and frameshift fusions. Among the 25,664 TCGA fusions, there were 9,284 noframe fusions, 7,738 frameshift fusions, and 8,642 inframe fusions, respectively. Not surprisingly, frameshift fusions can produce more candidate neoantigens due to the ability to create new ORF (Figure 4A, p value<0.01, Mann-Whitney U test). On average, a frameshift fusion generates 6 candidate neoantigens, and an inframe fusion generates 2.46 candidate neoantigens. Moreover, frameshift fusion candidate neoantigens tend to have higher immunogenic potentials than inframe fusion candidate neoantigens (Figure 4B, p value<0.01, Mann-Whitney U test). It should be noted that compared with inframe fusion neoantigens, frameshift neoantigens could increase the nonsense-mediated decay (NMD) mechanism, thereby decreasing its own immunogenic potential. The main function of NMD is to reduce errors in gene expression by eliminating mRNA transcripts that contain premature stop codons. NMD will potentially reduce the expression level of frameshift fusion transcripts and thus their immunogenic potential. Our results indicated that compared with samples without frameshift fusion mutation, NMD activity in samples harboring frameshift fusion is slightly higher (Methods). Estimating NMD efficiency and taking expression level into consideration when evaluating fusion peptides immunogenic potentials should make our score scheme and conclusion more reliable. However, this factor was not incorporated in our score scheme in our present study, because the expression information of fusion genes is unavailable.
Figure 4.

Analysis of the TCGA Cohort Dataset
(A) A frameshift fusion was able to generate much more candidate neoantigens than that of an inframe fusion.
(B) Frameshift fusion candidate neoantigens have notably higher immunogenic potentials. ** indicates p value<=0.01 and * indicates p value<=0.05.
(C) The inframe ratio of Onco and kinase fusion were significantly higher than that of passenger fusion. Onco, oncogenic; TSG, tumor suppressor gene.
(D) Candidate fusion neoantigens were extremely sparse.
(E) Several fusions occurred in most cancer types while several fusions only occurred in specific cancer types. Of 498 PRAD tumor samples, 190 harbored the TMPRSS2-ERG fusion. Only 15 most recurrent fusions were displayed. Size in the plot corresponds to the number of samples harbor that fusion.
Boxplots show the first, median and third quartiles and whiskers extend to 1.5X the interquartile range. Outlier points are not shown. See also Tables S3 and S4.
Furthermore, we examined the relationship between the fusion frame's status and the fusion's category. The TCGA fusions were separated into four categories with respect to the gene involved, i.e., oncogene (Onco) fusion, tumor suppressor gene (TSG) fusion, kinase fusion, and passenger gene fusion (Gao et al., 2018; Table S3). In total, 2,104 kinase fusions, 522 Onco fusions, 436 TSG fusions, and 23,115 passenger fusions were observed. Onco fusions and kinase fusions are more likely to be inframe than those of passenger fusions, as preserving the ORF is required to keep their oncogenic function (Figure 4C, p value<0.01, chi-squared test). TSG fusion during creating a new ORF will reduce or lose its function, which leads to tumorigenesis. In other words, TSG fusion is not required to maintain their function during tumorigenesis. Therefore, the inframe ratio does not differ between TSG fusion and passenger fusion.
Onco Fusion Mutations Tend to Have Lower Immunogenic Potentials Than Passenger Fusion Mutations
Immunoediting, a dynamic process comprising immunosurveillance and tumor progression, describes the relation between tumor cells and the immune system (Schreiber et al., 2011). Although the immune system exerts negative selective pressure on tumors, it also helps to sculpt the tumor genotype. Mutations in the driver gene, while conferring cells a selective growth advantage, render cells vulnerable to the immune system as a result of generating neoantigens. In addition, driver mutations are necessary in the development of cancers. As a consequence, driver mutations detected in tumors should be biased toward with lower immunogenic potentials (Marty et al., 2018, Sun et al., 2017). The fusion score defined as the sum of candidate neoantigen scores generated by that fusion (Methods) showed that the Onco fusion score was significantly lower than the passenger fusion score but not others (Methods).
Highly Recurrent Candidate Neoantigens Tend to Have Extremely Low Immunogenic Potentials
Only 5.8% fusion candidate neoantigens in the TCGA cohort data were shared between patients (Figure 4D). We found that highly recurrent candidate neoantigens have extremely low immunogenic potentials. For example, the neoantigen score of KMALNSEAL, a candidate neoantigen generated by TMPRSS2-ERG, which presents in 38% PRAD, only ranks at the 92nd percentile (Figure 4E). The low immunogenic potential of highly recurrent fusion candidate neoantigens clearly suggests that neoantigen-based cancer vaccination immunotherapy must be personalized.
Discussion
Fusion, which is an important class of somatic mutations, is an ideal source of tumor-derived neoantigens for creating an ORF. Compared with SNV&indel-based neoantigens, however, fusion-based neoantigens are not well characterized. A comprehensive literature review indicated that INTEGRATE-neo is the only existing in silico tool for fusion neoantigen prediction; however, it cannot assess their immunogenic potentials, which can be substantially different due to a single nucleotide mismatch (Bjerregaard et al., 2017). In this study, we propose an effective tool neoFusion for fusion neoantigen identification (Figure 5). Furthermore, a rational score scheme to quantitatively assess the identified fusion neoantigen immunogenic potentials is presented (Figure 5).
Figure 5.

neoFusion Workflow Overview
Gene fusions were detected by STAR-Fusion with RNA sequencing data. Translated fusion proteins output by STAR-Fusion were chopped up into 9–11 kmers peptides until a stop codon. The pMHC binding affinity and binding affinity percent rank were determined by NetMHCpan in binding affinity mode. Peptides with binding affinity percent rank ≤2 were reported as candidate neoantigens. Fusion candidate neoantigens were scored according to our score scheme and ranked according to their scores. See also Figure S4.
By analyzing the ICB cohort dataset, we found that (1) neither the tumor fusion candidate neoantigen burden nor the tumor fusion candidate neoantigen score∗CTL was associated with the immunotherapy outcome in the two melanoma ICB cohorts, indicating that the tumor fusion candidate neoantigen burden may not be a predictive biomarker for the immunotherapy response; (2) in the Van Allen cohort, only the overall tumor candidate neoantigen score∗CTL and burden∗CTL significantly separated patients. In the Hugo cohort, only the overall tumor candidate neoantigen score∗CTL, score, and burden separated the patients. Taking together, all the metrics except the overall tumor candidate neoantigen score∗CTL have their limitations in immunotherapy response prediction in these two cohorts, indicating the rationality and effectiveness of our score scheme; (3) so far, a higher PD-1 or PD-L1 expression (Garon et al., 2015), a higher neoantigen load, the microsatellite instability, and a higher peripheral baseline TCR diversity (Postow et al., 2015) are all reported to be associated with a better immunotherapy outcome; therefore, prediction of the response to immunotherapy is still an open question and a comprehensive model to accurately predict patient response is still lacking, likely requiring much more data to train and refine. We believe that the neoantigen score, tumor microenvironment such as CTL score, and other types of neoantigens besides SNV&indel based should be taken into consideration in predicting the immunotherapy outcome; (4) in these two ICB cohorts, tumor fusion candidate neoantigen burden and score were notably lower than tumor SNV&indel candidate neoantigen burden and score, respectively; therefore, adding them to the overall tumor candidate neoantigen burden and score does not improve the prediction accuracy of immunotherapy response. However, recently Yang et al. identified a patient exhibited complete response to anti-PD1 immunotherapy despite a low SNV&indel mutation burden and demonstrated that the patient elicited a T-cell response to neoantigen generated by fusion (Yang et al., 2019). Therefore, in certain cancer types such as BLCA, since the tumor fusion candidate neoantigens contribute to a relatively high proportion of the overall tumor candidate neoantigens, taking tumor fusion candidate neoantigens into consideration may improve the prediction of immunotherapy outcome.
Through comparing the TCGA fusion candidate neoantigens with the TCGA SNV&indel candidate neoantigens, we presented the following findings: (1) fusion, which is able to create novel ORF, generate 6-fold more candidate neoantigens and 11-fold more specific candidate neoantigens as SNV&indel. Compared with the SNV&indel candidate neoantigen burden, the fusion candidate neoantigen burden per sample was notably lower. Nevertheless, fusion candidate neoantigens tend to have notably higher immunogenic potentials. In 32.2% TCGA patients, candidate neoantigens with the highest immunogenic potentials were produced by fusion, making fusion neoantigens a better source for cancer vaccines; (2) similar to the SNV&indel candidate neoantigen burden, the fusion candidate neoantigen burden strongly correlated with the fusion mutation load. Furthermore, both types of candidate neoantigens were extremely sparse. Although several recurrent fusion candidate neoantigens exist, they usually have extremely low immunogenic potentials, further indicating that cancer vaccination strategies based on neoantigens must be personalized (Schreiber et al., 2011). To be recurrent, mutations must confer tumor cells a selective advantage. Producing neo-peptides that do not attract the attention of the human immune system confer such an advantage. Therefore, those highly recurrent fusion peptides such as KMALNSEAL in PRAD usually have low immunogenic potentials.
The comparison between passenger fusion mutations and other types of fusion mutations indicated that (1) Onco fusion mutations tend to have lower immunogenic potentials than passenger fusion mutations. Onco fusion mutations, while conferring cells a selective growth advantage, render cells vulnerable to the immune system as a result of generating neoantigens. Cancer cells that harbor Onco fusion mutations poorly bound to MHC are thus positively selected during tumorigenesis. As tumor cells grow and activate mechanisms to evade the immune system, passenger mutations are acquired regardless of their affinities to the MHC complex (Marty et al., 2018). Therefore, Onco fusion mutations tend to have lower immunogenic potentials than passenger fusion mutations; (2) similar to Onco fusion mutations, TSG fusion mutations should have lower immunogenic potentials than passenger fusion mutations. However, the immunogenicity score did not differ between passenger fusion mutations and TSG fusion mutations. These may be explained by the fact that, in contrast to Onco fusions, TSG fusions tend to be under-expressed and thus insufficient to generate a T-cell response (Gao et al., 2018). In conclusion, neoantigens produced by Onco and TSG fusion mutations are less likely to induce a T-cell response, and passenger fusion neoantigens may have particular relevance for vaccine.
Limitations of the Study
Our study presents the first comprehensive profile of fusion neoantigens from a pan-cancer perspective, which provides useful clues for personalized cancer vaccination-based immunotherapy. Several limitations should be noted: (1) 8 kmer and 12 kmer and above peptides can also be displayed by MHC I; however, in the present study, only the most common 9–11 kmer peptides were considered; (2) the fusion expression level factor was not incorporated in our score scheme in the present study, because such information was absent; however, knowledge accumulated in immunotherapy community will make the accurate and objective evaluation of the peptides immunogenic potential feasible in the future.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Quantification and Statistical Analysis
Survival analysis was performed using the Kaplan-Meier method provided by survival R (version 3.4.4) packages. The log rank test and Cox proportional hazard model were used to assess the correlation between metrics and overall survival. We used a one-sided nonparametric Mann-Whitney U test for non-normally-distributed variables to assess the difference in mean or median for a continuous variable between two groups. All statistical analyses were performed with the Python 3 SciPy, and NumPy libraries. The parameters of software used in our study were set as default without explicitly stated.
Data and Code Availability
neoFusion is available at https://github.com/bm2-lab/neoFusion, with a Docker version at https://hub.docker.com/r/bm2lab/neoFusion/.
The mass spectrum data: ten breast cancer cell line RNA-sequencing data were downloaded fromSequence Read Archive (NCBI: SRP026537). The corresponding MS proteomics data were downloaded from ProteomeXchange Consortium (proteomecentral.proteomexchange.org, PXD006406).
The immune checkpoint blockade cohort data: two cohorts of melanoma datasets were downloaded from the database of Genotypes and Phenotypes (dbGaP: phs000452.v2.p1) and SRA (NCBI: SRP070710), respectively. The overall survival and progression-free survival data and other data needed were retrieved from the original article supplementary.
The TCGA cohort data: TCGA fusion, oncogene, kinase gene, and tumor suppressor gene lists were retrieved from Gao et al. (Gao et al., 2018). The TCGA whole-exome sequencing (WES) VCFs and corresponding expression profile files were downloaded from TCGA website. HLA allele information was requested from The Cancer Immunome Atlas (https://tcia.at/home; Thorsson et al., 2018). The landscape of microsatellite instability of TCGA tumor samples were obtained from Bonneville et al.
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant No. 2017YFC0908500, 2016YFC1303205), National Natural Science Foundation of China (Grant No. 61572361), Shanghai Rising-Star Program (Grant No. 16QA1403900), Shanghai Municipal Health Commission Innovative integration for molecular oncology (Grant No. 2019CXJQ03) and Shanghai Natural Science Foundation Program (Grant No. 17ZR1449400).
Author Contributions
Q.L., Z.M.L., and C.Z. conceived the study. Z.T.W., C.Z., Z.B.Z., and M.G. analyzed the tumor sample data. Z.T.W., Q.L., Z.M.L., and C.Z. wrote the manuscript with assistance from other authors.
Declaration of Interests
The authors declare no competing interests.
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.028.
Contributor Information
Chao Zhang, Email: zhangchao@tongji.edu.cn.
Zhongmin Liu, Email: liu.zhongmin@tongji.edu.cn.
Qi Liu, Email: qiliu@tongji.edu.cn.
Supplemental Information
References
- Balachandran V.P., Łuksza M., Zhao J.N., Makarov V., Moral J.A., Remark R., Herbst B., Askan G., Bhanot U., Senbabaoglu Y. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:S12–S16. doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerregaard A.M., Nielsen M., Hadrup S.R., Szallasi Z., Eklund A.C. MuPeXI: prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother. 2017;66:1123–1130. doi: 10.1007/s00262-017-2001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneville R., Krook M.A., Kautto E.A., Miya J., Wing M.R., Chen H.Z., Reeser J.W., Yu L., Roychowdhury S. Landscape of microsatellite instability across 39 cancer types. JCO Precision Oncol. 2017;2017:1–15. doi: 10.1200/PO.17.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church S.E., Galon J. Tumor Microenvironment and Immunotherapy: the whole picture is better than a glimpse. Immunity. 2015;43:631–633. doi: 10.1016/j.immuni.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Gao Q., Liang W.W., Foltz S.M., Mutharasu G., Jayasinghe R.G., Cao S., Liao W.W., Reynolds S.M., Wyczalkowski M.A., Yao L. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018;23:227–238.e3. doi: 10.1016/j.celrep.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garon E.B., Rizvi N.A., Hui R., Leighl N., Balmanoukian A.S., Eder J.P., Patnaik A., Aggarwal C., Gubens M., Horn L. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G. Genomic and transcripomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2017;168:542. doi: 10.1016/j.cell.2017.01.010. [DOI] [PubMed] [Google Scholar]
- Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauss M., Donia M., Harbst K., Andersen R., Mitra S., Rosengren F., Salim M., Vallon-Christersson J., Törngren T., Kvist A. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017;8:1–10. doi: 10.1038/s41467-017-01460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le D.T. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.S., Mardis E.R. Applications of immunogenomics to cancer. Cell. 2017;168:600–612. doi: 10.1016/j.cell.2017.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty R., Thompson W.K., Salem R.M., Font-Burgada J., Zanetti M., Carter H. Evolutionary pressure against MHC class II binding cancer mutations. Cell. 2018;175:416–428.e13. doi: 10.1016/j.cell.2018.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens F., Johansson B., Fioretos T., Mitelman F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 2015;15:371–381. doi: 10.1038/nrc3947. [DOI] [PubMed] [Google Scholar]
- Murugan A. Statistical inference of the generation probability of T-cell receptors from sequence repertoires. Proc. Natl. Acad. Sci. U S A. 2012;109:16161–16166. doi: 10.1073/pnas.1212755109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow M.A., Manuel M., Wong P., Yuan J., Dong Z., Liu C., Perez S., Tanneau I., Noel M., Courtier A. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J. ImmunoTherapy Cancer. 2015;3:23. doi: 10.1186/s40425-015-0070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeser J.W. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget. 2016;8:7452–7463. doi: 10.18632/oncotarget.13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney M.S., Shukla S.A., Wu C.J., Getz G., Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: integrating immunitys roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Sun Z., Chen F., Meng F., Wei J., Liu B. MHC class II restricted neoantigen: a promising target in tumor immunotherapy. Cancer Lett. 2017;392:17–25. doi: 10.1016/j.canlet.2016.12.039. [DOI] [PubMed] [Google Scholar]
- Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A. The immune landscape of cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S., Litchfield K., Xu H., Rosenthal R., McGranahan N., Reading J.L., Wong Y.N.S., Rowan A., Kanu N., Al Bakir M. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18:1009–1021. doi: 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- Vaish M., Mittal B. DNA mismatch repair, microsatellite instability and cancer. Indian J. Exp. Biol. 2002;40:989–994. [PubMed] [Google Scholar]
- Van Allen E.M., Miao D., Schilling B., Shukla S.A., Blank C., Zimmer L., Sucker A., Hillen U., Foppen M.H.G., Goldinger S.M. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350 doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello A., Zanetti M. Neoantigen prediction and the need for validation. Nat. Biotechnol. 2017;35:815. doi: 10.1038/nbt.3932. [DOI] [PubMed] [Google Scholar]
- Yang W., Lee K.W., Srivastava R.M., Kuo F., Krishna C., Chowell D., Makarov V., Hoen D., Dalin M.G., Wexler L. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019;25:767–775. doi: 10.1038/s41591-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Zhu C., Liu Q. Toward in silico identification of tumor neoantigens in immunotherapy. Trends Mol. Med. 2019 doi: 10.1016/j.molmed.2019.08.001. [DOI] [PubMed] [Google Scholar]
- Łuksza M., Riaz N., Makarov V., Balachandran V.P., Hellmann M.D., Solovyov A., Rizvi N.A., Merghoub T., Levine A.J., Chan T.A. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551:517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
neoFusion is available at https://github.com/bm2-lab/neoFusion, with a Docker version at https://hub.docker.com/r/bm2lab/neoFusion/.
The mass spectrum data: ten breast cancer cell line RNA-sequencing data were downloaded fromSequence Read Archive (NCBI: SRP026537). The corresponding MS proteomics data were downloaded from ProteomeXchange Consortium (proteomecentral.proteomexchange.org, PXD006406).
The immune checkpoint blockade cohort data: two cohorts of melanoma datasets were downloaded from the database of Genotypes and Phenotypes (dbGaP: phs000452.v2.p1) and SRA (NCBI: SRP070710), respectively. The overall survival and progression-free survival data and other data needed were retrieved from the original article supplementary.
The TCGA cohort data: TCGA fusion, oncogene, kinase gene, and tumor suppressor gene lists were retrieved from Gao et al. (Gao et al., 2018). The TCGA whole-exome sequencing (WES) VCFs and corresponding expression profile files were downloaded from TCGA website. HLA allele information was requested from The Cancer Immunome Atlas (https://tcia.at/home; Thorsson et al., 2018). The landscape of microsatellite instability of TCGA tumor samples were obtained from Bonneville et al.