

Summary
Precise control of developmental processes is encoded in the genome in the form of gene regulatory networks (GRNs). Such multi-factorial systems are difficult to decode in vertebrates owing to their complex gene hierarchies and dynamic molecular interactions. Here we present a genome-wide in vivo reconstruction of the GRN underlying development of the multipotent neural crest (NC) embryonic cell population. By coupling NC-specific epigenomic and transcriptional profiling at population and single-cell levels with genome/epigenome engineering in vivo, we identify multiple regulatory layers governing NC ontogeny, including NC-specific enhancers and super-enhancers, novel trans-factors, and cis-signatures allowing reverse engineering of the NC-GRN at unprecedented resolution. Furthermore, identification and dissection of divergent upstream combinatorial regulatory codes has afforded new insights into opposing gene circuits that define canonical and neural NC fates early during NC ontogeny. Our integrated approach, allowing dissection of cell-type-specific regulatory circuits in vivo, has broad implications for GRN discovery and investigation.
Keywords: neural crest; gene regulatory network; chick; enhancers; ATAC; super-enhancers; Capture-C; transcription factors, scRNA-seq; scATAC-seq
Graphical Abstract
Highlights
-
•
Profiling chromatin dynamics uncovers complete repertoire of neural crest (NC) enhancers
-
•
NC super-enhancers act additively to sustain network robustness
-
•
Enhancer clustering reveals early split between neural-NC and canonical NC programs
-
•
Global construction of cranial NC gene regulatory network
Using chromatin and transcriptional profiling of cranial neural crest (NC) at population and single-cell levels, Williams et al. reverse engineer and functionally validate global cranial NC gene regulatory network. Interrogation of a full cohort of NC enhancers and upstream inputs reveals distinct regulatory codes governing neural and canonical NC programs.
Introduction
Gene regulatory networks (GRNs) are information-processing systems embedded in genomes, responsible for orchestrating cell cycle, homeostasis, physiological processes, and development (Levine and Davidson, 2005, Wyrick and Young, 2002). Cis-regulatory elements (CREs) form the core of GRNs by integrating extrinsic signaling cues and endogenous transcription factors (TFs), which together control the spatiotemporal dynamics of complex developmental programs such as self-renewal and cell-fate decisions (Gouti et al., 2017, Levine and Davidson, 2005, Wyrick and Young, 2002). Pioneering work on GRNs was conducted in several systems, including yeast (Lee et al., 2002), Drosophila (Sandmann et al., 2007), sea urchin embryos (Smith et al., 2007), vertebrate T-lymphocytes (Georgescu et al., 2008), and developing nervous system (Meulemans and Bronner-Fraser, 2004, Sauka-Spengler and Bronner-Fraser, 2008, Gouti et al., 2017), yielding valuable insights into the architecture, logic, modularity, and connectivity of developmental circuitries and their pivotal role in evolutionary dynamics (Davidson and Erwin, 2006). Our current understanding of GRNs was implied from either studies of transcriptional regulation in unicellular organisms (Lee et al., 2002, Milo et al., 2002) or candidate gene approaches to infer cis-regulatory and epistatic relationships in the sea urchin, Drosophila, and vertebrate embryos (Levine and Davidson, 2005, Sauka-Spengler and Bronner-Fraser, 2008). However, an unbiased genome-wide representation of vertebrate GRNs has remained elusive.
Vertebrate GRNs use both proximal and distal acting CREs known as transcriptional enhancers (Levine et al., 2014, Levine and Davidson, 2005) that can be located hundreds of kilobases away from their target promoters (Gonen et al., 2018) or embedded within large and often repeat-rich genomes (Prescott et al., 2015), thus posing a challenge for GRN reverse engineering. Nonetheless, single-cell RNA sequencing recently enabled the interrogation of mouse trunk neural and mesodermal networks (Gouti et al., 2017) as well as those involved in neuronal and cancer circuitries (Aibar et al., 2017), yielding new regulatory hierarchies within GRNs despite their lack of epigenomic detail. Furthermore, high-resolution genome-wide mapping of TF binding, chromatin accessibility, and histone modifications adapted to low cell numbers allow enhancer characterization in vivo (Georgescu et al., 2008, Smith et al., 2007). Thus, a systems-level approach combining epigenomic and transcriptional characterization of specific cell types at population and single-cell levels is poised to resolve complex embryonic GRNs.
The neural crest (NC) is an emblematic embryonic population of multipotent cells that gives rise to a wide range of neural and mesenchymal derivatives including the peripheral nervous system and craniofacial skeleton (Le Douarin and Kalcheim, 1999, Sauka-Spengler and Bronner-Fraser, 2008), thus representing an attractive system to study cell-fate decisions and regeneration. Following induction at the neural plate border (NPB), premigratory NC cells transiently reside within the dorsal neural tube, where they undergo an epithelial-to-mesenchymal transition (EMT), migrate to distant sites in the developing embryo, and ultimately differentiate into a plethora of derivatives (Bronner and Simões-Costa, 2016, Sauka-Spengler and Bronner-Fraser, 2008). Mistakes and mutations in these processes lead to debilitating birth defects, such as Hirschsprung disease (Carter et al., 2012), craniofacial abnormalities (Trainor, 2010), and CHARGE syndrome (Schulz et al., 2014) as well as a number of malignancies including melanoma, neuroblastoma, and gliomas (Karunasena et al., 2015), highlighting the importance of understanding the global GRN controlling NC ontogeny.
Previous studies have described a number of important NC genes (Simões-Costa et al., 2014), including evolutionarily conserved TFs and downstream effectors, leading to a description of a putative NC-GRN underlying key processes during NC ontogeny (Betancur et al., 2010a, Meulemans and Bronner-Fraser, 2004, Sauka-Spengler and Bronner-Fraser, 2008, Simões-Costa and Bronner, 2015). However, synergistic relationships between components of such GRN were implied from gene-centric knockdown approaches, where directionality remained largely unvalidated and enhancers of only a handful of factors have been identified (Barembaum and Bronner, 2013, Betancur et al., 2010b, Simões-Costa et al., 2012, Vadasz et al., 2013). Furthermore, other regulatory layers and context-dependent dynamics were not integrated into the GRN architecture, such as recently described bimodal activity of key NC regulators (Lukoseviciute et al., 2018).
It has been a matter of intense debate whether the NC is a homogeneous population of multipotent cells or a population of fate-restricted, transcriptionally heterogeneous neural-NC and mesenchymal progenitors (Dupin et al., 2018, Le Lievre et al., 1980, Nitzan et al., 2013). Single-cell lineage tracing in chick and mice have clearly established multipotency of some NC cells (Baggiolini et al., 2015, Bronner-Fraser and Fraser, 1988), and a stem-cell niche has been proposed (Basch et al., 2006). However, NC transiency and dynamics in vivo have imposed a considerable challenge to the unbiased interrogation of these conflicting results and, more broadly, of the complex emergent properties of the developing NC.
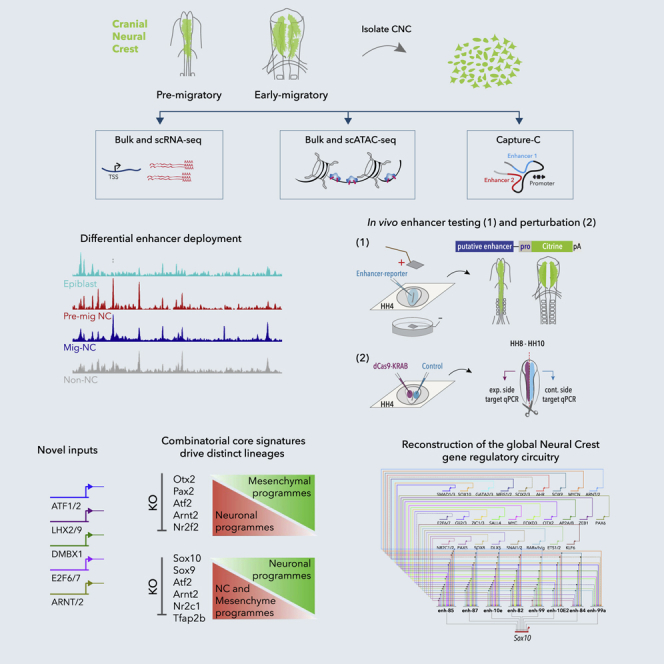
Here, we tackle the challenge of reconstructing global NC-GRN by adapting genome-wide epigenome and transcriptome profiling to small numbers of chick NC cells and conducting an unbiased, systems-level functional study of the NC regulome in vivo. By exploring the spatial and temporal chromatin accessibility dynamics, we identified rewiring of cis-regulatory landscapes and deployment of specific enhancers during NC development. Assigning NC enhancers to cognate promoters using high-resolution targeted chromatin conformation capture (Capture-C) (Davies et al., 2016) and positional association to expressed genes, together with the definition of upstream TF inputs using scRNA-seq and motif analysis, has enabled comprehensive reverse engineering of the GRN underlying early NC ontogeny.
By characterizing transcriptional signatures and cis-regulatory landscapes at the global and single-cell level, we demonstrate NC subpopulations that (1) express Msx2, Alx1, and other stem cell-like factors; (2) express neural markers NR2F2, Pax2, Otx2, and Gli2; and (3) express canonical NC factors Sox10, TFAP2s, Cxcr4, and Pax3 are already segregated at the premigratory stage. De novo identification of core transcriptional networks revealed a combinatorial cis-regulatory code comprising Sox9, Otx2, NR2F2, and Pax2 that orchestrated NC-derived neural programs and the canonical NC regulators Sox10 and TFAP2, together with novel regulators Arnt2 and ATF2 as emergent canonical/mesenchymal NC signature. Using CRISRP/Cas9 genome engineering techniques recently developed in the chick (Williams et al., 2018), we knocked out these core factors and examined their causal links to downstream CREs. We reveal direct feedforward loops controlled by the heterotypic binding of these TFs, as well as the negative cross-regulation of these two identities, which function as core logical features of the NC-GRN. Furthermore, by functionally dissecting the Sox10 super-enhancer, we highlight enhancer redundancy and cis-regulatory hierarchies as determinants of transcriptional robustness in the NC. Our integrative approach, allowing definition and interrogation of the GRN underlying a system as complex as the NC in vivo and dissection of its gene regulatory circuits, has broad implications for vertebrate GRN discovery and study. The assembled comprehensive NC-GRN provides an interactive resource for exploring new regulatory hierarchies.
Results
Transcriptional Profiling of Early Cranial NC
Following electroporation of the cranial NC-specific enhancer NC1 driving Citrine (Figures 1A and 1B), fluorescence-activated cell sorting (FACS) was used to collect Citrine+ NC and environing (Citrine−) non-NC cells from chicken embryos at two stages: premigratory, 5-6ss (ss, somite-stage, HH9; Hamburger and Hamilton, 1951), when epithelial NC cells reside within the dorsal neural tube, and early-migratory, 8-10ss (HH10), when NC cells have undergone EMT and commenced migration.
Figure 1.

Transcriptional Profiling of Early NC
(A) Schematic representation of Citrine+ NC cells (green) and Citrine− non-NC cells (gray) in early chicken embryos. Red dashed line indicates dissected cranial regions.
(B) In vivo NC-specific activity of FoxD3 enhancer, NC1, at 5-6ss and 8-10ss.
(C) Number of differentially expressed (enriched and depleted) genes in 5-6ss and 8-10ss NC compared to their respective non-NC controls and between stages.
(D and E) Volcano plots showing enriched genes (LogFoldChange >1, base mean >50) in 5-6ss (D) and 8-10ss (E) compared to corresponding non-NC cells; magenta, transcription factors; green, cell-surface molecules; yellow, signaling molecules; blue, differentiation genes. Differential expression was determined using DESeq2 with a negative binomial model, p-values calculated using Wald test, corrected for multiple testing using the Benjamin-Hochberg method (padj).
(F) GO terms associated with NC-enriched genes. Fold enrichments were obtained using statistical overrepresentation test, p-values calculated with binomial distributions and Bonferroni correction for multiple hypothesis testing.
(G) Co-expression clusters of highly correlated genes identified by WGCNA; representative genes are shown. Two replicates per stage for NC and non-NC cells are shown on the x axis.
(H) Cellular co-localization of cluster-iii genes Zfhx4, TFAP2B, Pax7, and Sox10 at 6ss (top panel) and 8ss (bottom panel), detected by HCR.
(I) Co-localization of cluster-iv genes Otogl, Arnt2, and Col9a3 at 8ss.
(J) Co-localization of cluster-vii genes Lmo4, Mef2C, and Col2a1 at 10ss.
Differential gene expression analysis comparing Citrine+ NC cells and the corresponding Citrine− controls revealed approximately 400–500 significantly enriched genes per stage (Figures 1C and S1A). Bona fide NC TFs (Msx1, Pax7, TFAP2A/B, and Sox9/10), cell-adhesion molecules (Cdh6, Cdh13, Tmem132c/d), and signaling components (Wnt3a/8b, Bmp5, Bmper) were enriched at 5-6ss (Figure 1D). At 8-10ss, in addition to bona fide NC TFs and other TFs (such as Tle1, Sox5/8, TFAP2C, Pax3), genes involved in cell migration (EdnrB, Cxcr4), extracellular matrix (ECM) remodeling (Adamts1/20/l1), and differentiation into various NC derivatives (Col9a3, Enc1, Ltk, RXRG) were added to the program (Figures 1E and S1B). Consistent with gene ontology (GO) analysis, these results suggest that at the premigratory stage (5-6ss) NC is engaged in stem cell development and specification processes, whereas differentiation programs (cartilage and connective tissue, myelination, and skeletal muscle development) commence by 8-10ss stage (Figure 1F). Thus, we observed a clear shift from homophilic cell-to-cell communication based on cadherins and protocadherins (Cdh6/13, Pcdh8) to heterophilic cell-ECM communication (EdnrB, Cxcr4, PlxnA4, Adamts1/20), revealing active rewiring of the GRN as NC cells undergo EMT and commence migration. Genes enriched in regulatory control of non-NC reflect predominantly extrinsic signaling cues such as Bmp pathway components or development of other embryonic structures (heart, ear, etc.), consistent with their presence in other cranial tissues surrounding the NC (Figure S1C).
We next used weighted gene co-expression network analysis (WGCNA) (Langfelder and Horvath, 2008, Langfelder et al., 2008) to characterize gene co-expression clusters and expand the previously proposed NC-GRN (Figures 1G and S1D). We discovered further subdivision into early NC specification modules featuring Snai2, Sox8, Msx1, and Noelin1 (cluster i) and Sox5, Chd7, and type II cadherins, Cdh6 and Cdh10 (cluster ii), as well as bona fide specification module (cluster iii), featuring canonical NC TFs Sox10, Pax7, and TFAP2A, maintained from 5ss onward. NC migration module (cluster iv) included ECM structural and remodeling molecules Adamts20, AdamtsL1, and Col9a1/a3 while cluster v featured genes predominantly enriched in non-NC cells including signaling factors, in agreement with their role as cues emanating from the environing tissues (Figures 1G and S1C). Such cluster assignment reflecting co-expression with known NC genes provided an excellent framework for inferring the role of novel regulators and classifying them within the NC-GRN. For instance, the zinc-finger homeobox 4 protein Zfhx4, recently shown to regulate multipotency, self-renewal, and migration of therapy-resistant glioblastomas (NC-derived brain cancer) (Chudnovsky et al., 2014), was recovered in the bona fide NC specification module (cluster iii). Zfhx4 expression overlapped with bona fide NC specifiers Pax7, Sox10, and TFAP2A/B at pre-migratory stage (6ss) but was confined to the dorsal neural tube at 8ss, suggesting a role in NC specification, but not migration (Figure 1H). Cluster iv showed novel factors Otogl, Col9a3, and Arnt2 first expressed in a broad pattern encompassing the neural tube and surrounding ectoderm at 6ss (Figure S1E), enriched in migrating NC cells by 8ss (Figure 1I), and maintained through to 11ss (Figure S1E). The LIM adaptor protein Lmo4, previously described as an essential regulator of NC induction (Ochoa et al., 2012) and EMT (Ferronha et al., 2013), was recovered in the late GRN module (cluster vii) harboring downstream effectors driving migration or differentiation into distinct NC lineages, such as myocyte-specific enhancer factors MEF2A/C, and late non-canonical Wnt factors (Wnt7A/B), required in NC for proper craniofacial development and during migration (De Calisto et al., 2005, Verzi et al., 2007). Lmo4 transcripts colocalized with its cluster counterparts (Mef2C, Col2a1) in premigratory (7ss) and migrating NC (10ss) (Figures S1E and S1J), suggesting a role even after EMT has occurred.
Chromatin Dynamics Reflects Spatiotemporal Enhancer Activities In Vivo
We next sought to identify genomic regulatory features controlling NC programs. To this end, we generated high-resolution maps of chromatin accessibility using ATAC-seq of NC and non-NC cells at 5-6ss and 8-10ss. ATAC-seq was also performed on dissociated cells from dissected epiblast regions at HH4 and somites from 10ss embryos, such that NC could be compared to either more naive or developmentally diverse population, respectively. Linear regression analysis showed a high reproducibility between biological replicates (Figures S2A–S2C), but we observed dynamic changes in chromatin accessibility between different cellular populations. For example, in the vicinity of the NC specifier Snai2 (Figure 2A), we identified a number of differentially accessible elements, open specifically in NC cells and not in the naive epiblast, neighboring tissues, or somites (Figure 2B), and hypothesized these could represent putative NC enhancers. To determine physical interactions between such distal regulatory elements and their cognate promoters, we performed Capture-C (Davies et al., 2016) using dorsal neural tubes dissected from 6ss embryos and peripheral blood cells from HH36 embryos as controls. We defined a broad NC-specific topologically associating domain (TAD) spanning ∼700 kb, located predominantly downstream of the Snai2 locus and encompassing a number of open chromatin elements that interact with the promoter with high statistical significance (Figure 2C). Using in vivo reporter assays, we identified five active enhancers within the Snai2 TAD, two distal (enh-332 and enh-334), and three proximal (enh-13, enh-239, enh-242), (Figures 2D–2H and S3D). Enh-13 (Figures 2G–2H′) was active in the cranial neural tube including NC at 5ss, and was confined to NC cells by 9ss, thus representing the first NC-specific Snai2 enhancer described to date. Enhancers 332, 334, 242, and 239 were active in both NC and non-NC at 6-10ss (Figures 2D–2F′ and S3D), consistent with these regions being accessible in both NC and non-NC cells. A region specifically open in the somites displayed somite-specific activity (enh-241, Figure S3D). Overall, these data suggest that chromatin accessibility dynamics across multiple cell types and developmental stages reflects tissue-specific enhancer activity.
Figure 2.

Profiling Chromatin Accessibility Dynamics during Early NC Development
(A and B) Genome browser views of (A) 5-6ss NC RNA-seq data and (B) ATAC-seq profiles in NC and non-NC cells at the Snai2 locus. 5-6ss NC ATAC is shown in green and 8-10ss in blue; corresponding non-NC samples are shown in gray. ATAC data from HH4 and somite tissue are shown in light blue and pink, respectively (data shown is normalized to read count and shown relative to promoter peaks across all samples). Boxes indicate putative enhancers, tested in vivo.
(C) Capture-C tracks showing the TAD at the Snai2 locus in NC (blue track) and control red blood cells (RBC, gray track). Differential interactions were determined using DESeq2, hypothesis tested with Wald test and corrected for multiple testing using the Benjamin-Hochberg method. Wald statistics track (stat, in red) represents ratio of LogFoldChange values and their standard errors.
(D–H) Enhancer driven in vivo reporter (Citrine) expression of tested enhancers. (D′–H′) Transverse sections of (D)–(H), immunostained for Citrine; red line indicates approximate location of section; blue, DAPI.
Dynamics of cis-Regulatory Landscapes
To expand the analysis of chromatin dynamics globally and attain the regulatory information encoded in enhancers, we performed k-means clustering of our ATAC-seq datasets using the seqMINER platform (Zhan and Liu, 2015), which reiteratively assigned all open chromatin peaks genome-wide into patterns of chromatin accessibility dynamics. We focused on the top seven clusters based on their density and patterns of accessibility in NC cells (Figures 3A and S2E). Elements recovered in k-Clusters were predominantly located in intergenic or intronic regions (Figure S2D), consistent with their cis-regulatory activity and correlated with unimodal transcription (Figure S2F). Broad signal elements constitutively accessible across all samples were recovered in k-Cl4 (Spearman correlation coefficient rs = 0.94) (Figures 3A and 3B), and ∼20% of those represented promoter regions (Figure S2D). Elements specifically accessible in NC cells were found in k-Cl3 (rs = 0.74, p < 2.2 × 10−16) (Figures 3A and 3B), thus assigned as the bona fide NC enhancer group. Accessibility in NC cells also characterized the k-Cl1 elements (rs = 0.71, p < 2.2 × 10−16) (Figures 3A and 3B); however, these were also open in non-NC cells, indicating activity in the environing neuroepithelial ectoderm. K-Cl6 elements accessible from the early (HH4) epiblast stage and open in premigratory NC define the portion of the NC regulatory landscape possibly established during gastrulation (Basch et al., 2006, Buitrago-Delgado et al., 2015). As k-Cl6 accessibility was reduced in NC cells at 8-10ss, these elements were likely involved in the early establishment of NC programs but are later decommissioned (Figure 3A). Elements more prominently open in non-NC with some accessibility in NC were recovered in k-Cl2, while early (HH4) acting elements were found in k-Cl5 and k-Cl7 (Figure 3A). These results thus establish the dynamic cis-regulatory landscapes across different stages and cell types, likely underlying NC ontogeny.
Figure 3.

K-Means Clustering and Differential Analysis of Chromatin Accessibility during Early NC Development
(A) Read-count-normalized ATAC-seq samples obtained from NC and non-NC control cells at HH4, 5-6ss, and 8-10ss were reiteratively assigned into cohesive groups of elements showing similar patterns of chromatin accessibility dynamics genome-wide using k-means algorithm.
(B) Log-scaled scatter plots of normalised ATAC-seq counts of selected k-means clusters, quantifying differential accessibility by calculating slopes and Spearman correlation coefficients in NC and control (non-NC) cells.
(C) MA plot depicting differentially accessible elements as determined by DiffBind using a negative binomial distribution model (FDR < 0.1, Fold enrichment > 1). A selection of putative enhancers to key NC genes are color coded; numbered enhancers have been tested in vivo (Figures 2D–2H, 3F, 3F′, 4E–4J, and S3).
(D and D′) Venn diagrams showing overlap of k-Cluster and DiffBind elements.
(E and E′) Plots representing genes assigned to k-Cl3 (E) and k-Cl1 (E′) elements, ranked by the number of associated elements. Inserts show mean merged accessibility profiles per cluster.
(F and F′) Genome browser snapshots of Lmo4 (F) and Zfhx4 (F′) loci showing Capture-C (blue, NC cells; gray, RBC; purple, differential mean between experimental and control samples; red, Wald statistics track (stat) indicating ratio of LogFoldChange values and their standard errors, determined using DESeq2, with p-values calculated with Wald test and Benjamin-Hochberg correction) and ATAC (green, 5-6ss; blue, 8-10ss; gray, negative non-NC) tracks; k-means element assignment is shown. In vivo activity of selected enhancers, boxed in gray, is shown at the bottom. Zfhx4 and Lmo4 featured NC-specific upstream regulatory apparatus. Zfhx4 expression is solely controlled by k-Cl1 enhancers, with NC-specific activity (enh-5 and enh-316) as well as non-NC regulation (enh-11 and enh-12). Lmo4 NC-specific expression is governed by k-Cl3 enh-17 and non-NC activity k-Cl1 enh-15.
In parallel, we employed a stringent statistical analysis of differential accessibility in purified NC cells compared to neighboring non-NC cells using the DEseq2 algorithm, part of the DiffBind package (Stark, 2011). We identified 1,605 statistically significant NC-specific elements (from here on referred to as DiffBind elements) at 5-6ss and 5,734 at 8-10ss (FDR < 0.1, Log2FoldChange >1) (Figures 3C and 3C′), suggesting the progressive establishment of the bona fide NC cis-regulatory landscape. DiffBind elements were enriched at key NC loci (Figures 3C and 3C′) and differentially overlapped individual k-Clusters. Consistent with their early opening, DiffBind elements at 5-6ss were mainly shared with k-Cl1 (∼27%) and k-Cl6 (∼14%), and to a lesser extent with k-Cl3 (∼7%). DiffBind 8-10ss elements predominantly overlapped with bona fide NC k-Cl3 elements (∼47%), and to a lesser degree with k-Cl1 (∼12%) and k-Cl6 (∼6%) (Figures 3D and 3D′).
Assignment of CREs from k-Cl3 and k-Cl1 to the nearest expressed gene indicated that key NC genes were controlled by multiple regulatory elements. We further distinguished genes predominantly regulated by elements from specific k-Clusters from those with regulation involving two or more k-Clusters. For example, canonical NC factors Sox10, EdnrB, and Myc, along with less-well-characterized NC genes Lrrc4c and Epha5 (Murko et al., 2018, Robinson et al., 1997), were predominantly controlled by k-Cl3 elements (Figure 3E), while genes involved in neural development, such as Sox11, Grid2, and MycN, and cell migration and guidance (Fgfrl1, Wnt5a, Sema3a) showed enrichment in k-Cl1 elements (Figure 3E′). However, other bona fide NC and EMT factors, such as TFAP2B, Snai2, Zeb2, and Lmo4, were driven by a combination of k-Cl3 and k-Cl1 elements (Figures 3E and E′). Thus, deciphering distinct contributions of different enhancer clusters is critical for understanding how NC lineages are defined.
We next validated genome-wide enhancer predictions by assessing the spatial and temporal activity of an extensive cohort of putative enhancers in vivo. We selected a number of canonical bona fide NC loci (Sox10, Pax7, Sox9, Snai2, Ets1, TFAP2A, and TFAP2B), as well as Lmo4 and Zfhx4, and determined their TADs by Capture-C. TADs varied in size and distribution, for example, the Lmo4 TAD encompassed ∼500 kb downstream and ∼200 kb upstream of the promoter (Figure 3F), whereas the Zfhx4 TAD spanned ∼700 kb predominantly upstream of the coding region (Figure 3F′). Pax7 TAD (∼500 kb), located in a gene-rich region, predominantly encompassed the body of the Pax7 gene and included mostly intronic and downstream CREs (Figure S4A), whereas the Sox9 TAD, located within a gene desert, spanned a large (∼1.5 Mbp) region upstream and downstream of the promoter (Figure S4C). The ∼750 kb TFAP2B TAD was spread predominantly downstream (Figure S4B), and the large (2.3 Mb) Ets1 TAD spanned evenly upstream and downstream regions around the promoter (Figure S4D). We developed a novel high-throughput screening assay that employs a modified highly efficient GoldenGate cloning strategy and combined fluorescent and “Nanotag” reporters (Nam and Davidson, 2012) that can be analyzed by Nanostring (see STAR Methods and Figures S3A–S3C). We tested the activity of a number of novel enhancers regulating Lmo4 (enh-17) (Figure 3F), Zfhx4 (enh-5, enh-11, enh-12, enh-316) (Figure 3F′), Sox9 (enh-283, enh-345, enh-249), Pax7 (enh-194, enh-195, enh-199, enh-216, enh-218), FoxD3, TFAP2A, and many others (Figure S3D). These results validated our integrative framework for identification of cell-type- and stage-specific NC enhancers and provided an excellent resource for the study of NC gene regulation in vivo.
Taken together, our results show that differential chromatin accessibility analysis and k-means clustering capture complementary aspects of the NC cis-regulatory landscape, providing novel insights into enhancer deployment during NC ontogeny and shared regulation with other cell types.
Enhancer Hierarchies within a Super-enhancer Dynamically Controlling Sox10
Spatial organization of dynamically accessible regions, as well as the increased ratio of enhancers per gene at developmental loci (Figures 3, S2, and S4) suggested that enhancers form clusters of elements within TADs, constituting locus control regions (Spitz et al., 2003, Talbot et al., 1989) also known as super-enhancers (SEs) (Hnisz et al., 2013). Importantly, SE-like regions function as the core regulatory circuits within GRNs, specifying the cell-type identity, and their dysregulation can impair cellular programming and be involved in the emergence of neoplasias (Boeva et al., 2017, Hay et al., 2016, Hnisz et al., 2013). Given their functional importance, we performed ChIP-seq in NC cells for H3K27Ac, as SE-like regions are differentially enriched for this histone modification (Lovén et al., 2013). Using the ROSE (rank ordering of super enhancers) algorithm (Lovén et al., 2013, Whyte et al., 2013) on H3K27Ac ChIP-seq profiles, we identified 1,379 SEs (Figure 4A) at the delaminating NC stage (8-10ss) and 1,288 SEs at the premigratory NC stage (5-6ss) (Figure S4E). Highly ranked SE-like clusters were associated to the loci of canonical NC regulators including Sox10, Fzd3, TFAP2A, TFAP2B, Sox5, and Snai2 (Figure 4A) at 8-10ss, but not at the earlier premigratory stage, suggesting bona fide NC cis-regulatory landscapes get rewired at the delaminating or actively migrating stage, when the majority of enhancers are deployed. Moreover, ∼30% of bona fide NC elements belonging to DiffBind 8-10ss cluster, which features statistically significant differential accessibility, are occupied by the Mediator complex (hallmark of SE-like identity) as inferred by ChIP-seq for the transcriptional regulator Brd4 (Figure 4B).
Figure 4.

Super-enhancer-like Clusters Regulate Key NC Genes
(A) Super-enhancers ranked by H3K27ac signal in 8-10ss NC, using the ROSE algorithm; top-ranked NC genes are annotated.
(B) Mean merged profile of DiffBind elements at 8-10ss and control non-NC elements occupied by Brd4.
(C) Capture-C genome browser tracks at Sox10 locus (blue, NC cells; gray, RBC; red, stat, representing ratio of LogFoldChange values and their standard errors, determined using DESeq2, p-values calculated with Wald test and Benjamin-Hochberg correction) and ATAC data (green, 5-6ss; blue, 8-10ss; gray, non-NC). DiffBind elements at 5-6ss (green) and 8-10ss (blue), Brd4-bound peaks (orange), and k-means elements are indicated. Selected putative enhancers are boxed.
(D) Endogenous Sox10 expression pattern detected by HCR.
(E–J) In vivo activity of novel Sox10 enhancers, (E) distal k-Cl1 enh-99 at 7ss, (F) enh-87 at 7ss, (G) enh-99 at 8ss, (H) enh-85 at 7ss, (I) k-Cl6 enh-89 at 8ss, (J) enh-84 at 9ss.
(K) Schematic of bilateral electroporation assay for epigenome engineering experiments.
(L) Schematic representation of decommissioned enhancers.
(M) HCR for Sox10, TFAP2B, and FoxD3 following decommissioning of targeted enhancers 84, 85, and 99.
(N) Table describing effect on endogenous Sox10 expression following epigenome modulation of Sox10 enhancers (single arrow, weak [< 20% decrease in Sox10 expression]; double arrow, moderate [20%–40% decrease]; triple arrow, strong [> 40% decrease]; n/a, not analyzed; −, no effect).
Given the pivotal role of Sox10 in NC ontogeny (Kelsh, 2006, Sauka-Spengler and Bronner-Fraser, 2008) and its high SE ranking (Figure 4A), we sought to thoroughly test the Sox10 SE locus. We identified a cluster of open elements spanning an ∼20 kb region, 2 kb upstream of the promoter, within the defined Sox10 TAD (Figure 4C). Each of these elements were present in our DiffBind sets, and the majority were recovered in k-Cl3 cluster, with the exception of the two most distal peaks (enh-99 and enh-89), belonging to k-Cl1 and k-Cl6, respectively (Figure 4C). In vivo enhancer reporter assays showed these elements have differing spatiotemporal patterns of activity in NC cells (Figures 4E–4J), collectively reconstituting the endogenous Sox10 expression pattern (Figure 4D). Enh-99 identified in k-Cl1 and co-occupied by Brd4 is the earliest acting Sox10 enhancer (detected from 5ss) that likely controls the onset of the Sox10 transcription (Figures 4E and 4G). Interestingly, we found that a region within the enh-99 (Enh-99a) predominantly drove specific reporter activity in the trunk NC (Figure S3D). Enh-99 activity was followed by enhancers enh-87, enh-85, and enh-84, which onset at 7ss (Figures 4F and 4H) and continued throughout early NC migration, with enh-84 activity being particularly prominent in migrating NC (8-10ss) (Figure 4J), consistent with the occupancy of these elements by Brd4 and their assignment to the bona fide k-Cl3. Surprisingly, enh-89, a k-Cl6 element accessible from the epiblast stage, was active at 8ss, predominantly in the hindbrain (Figure 4I), providing further evidence of the role of enhancer pleiotropy (Preger-Ben Noon et al., 2018) in NC gene regulation. Conversely, enh-87, a k-Cl3 element, was active in a complimentary domain (anterior cranial region, Figure 4F), suggesting that enh-89 and enh-87 collectively controlled cranial antero-posterior Sox10 expression in a manner redundant to enh-99, possibly acting as shadow enhancers (Cannavò et al., 2016). All five new enhancers displayed statistically significant interactions with the Sox10 promoter (Figure 4C). The most proximal open element, enh-82, which did not display any in vivo enhancer activity at the stages tested, possibly represents a cis-regulatory repressor or a late-acting enhancer. Interestingly, although both previously described Sox10 enhancers (Betancur et al., 2010b) were recovered by our DiffBind analysis, only cranial enhancer 10E2, and not the trunk enhancer 10E1, showed significant interaction with the Sox10 promoter, suggesting an instructive model (de Laat and Duboule, 2013) in which de novo formed loops may control Sox10 transcription at different axial levels.
To determine the functional contribution of each element within the Sox10 SE region, we modulated endogenous enhancer activity using CRISPR-mediated epigenome engineering (Williams et al., 2018). Five guide RNAs (gRNAs) were employed to target the transcriptional repressor dCas9-Krab to each individual enhancer. We used an in vivo bilateral electroporation assay, in which the left side of the embryo received target gRNAs with dCas9-Krab and the right side received a scrambled control gRNA with dCas9-Krab (Figure 4K). Left and right dorsal neural tubes from individual embryos were dissected at different stages (5-8ss), and qPCR was performed to determine the effect of enhancer perturbation on endogenous Sox10 expression (Figure S4F). We found that loss of enh-99 alone affected Sox10 expression early (5ss), whereas targeting other enhancers only disrupted Sox10 expression from 6-7ss (Figures 4L and 4N). However, decommissioning any individual enhancer on its own was insufficient to cause a sustained effect on Sox10 transcription (past 7ss), probably due to compensation by other concomitantly active elements. Sox10 expression indeed increases as NC development progresses, and such enhancement is most likely due to multi-enhancer usage. We therefore targeted other Sox10 enhancers in conjunction with enh-99 and found that simultaneous repression of enh-84 and enh-99 was sufficient to disrupt Sox10 expression from 5ss through to 8ss (Figures 4L, 4N, and S4F). Furthermore, when enh-84, enh-85, and enh-99 were decommissioned concurrently, Sox10 transcription was continually repressed through to the 8ss (Figures 4L–4N and S4F). Furthermore, we examined expression of several NC factors following the decommissioning of enhancers enh-84, enh-85, and enh-99. In addition to strong reduction in Sox10 expression, consistent with our qPCR analysis, we also detected reduction in expression of FoxD3 and TFAP2B on the experimental (left) side of the embryo as compared to control (right), suggesting that collective Sox10 enhancer inactivation results in perturbation of NC development, either by reducing the amount of Sox10 protein acting upstream of FoxD3 and TFAP2B or by affecting overall numbers of delaminating NC cells (Figure 4M).
Collectively, our results suggested that additive or sub-additive combinatorial action of enhancers (Bothma et al., 2015, Hay et al., 2016) was critical for sustained Sox10 expression during NC EMT. Furthermore, these data demonstrated the role of functional redundancy between individual enhancers in establishing transcriptional robustness, as previously described in other systems (Osterwalder et al., 2018). Moreover, our results offer an example of the global dynamics of NC transcriptional regulation at a key SE, by providing evidence that (1) k-Cl1 elements may be required and sufficient for initiation, but not for maintenance of NC-specific program, as in the case of enh-99 partially shared with neural program (enh-99a); (2) early regulatory elements from stem cells are (re)used in NC cells (Buitrago-Delgado et al., 2015), as revealed by k-Cl6 member enh-89; and (3) multiple k-Cl3 elements (enh-84, enh-85, enh-87, and 10E2) represent bona fide NC enhancers essential for transcriptional robustness within the NC-GRN.
Early Heterogeneity and NC Lineage Decisions
Heterogeneity at the Regulatory Level
The observed cis-regulatory dynamics provided evidence of early heterogeneity at the epigenomic level, consistent with previous reports suggesting that regulatory mechanisms limit NC potential and thus guide NC lineage decisions (Dupin et al., 2018, Nitzan et al., 2013). Consistently, GO term enrichment analysis of genes associated to each group of clustered elements revealed that pathways associated with canonical NC development and differentiation programs (p < 0.01, Binomial test with Bonferroni correction) were exclusively associated with k-Cl3 and DiffBind elements (Figure 5A, red box). Given that k-Cl3 and DiffBind enhancers only became accessible from premigratory NC stages (Figure 5B), this suggested that they represented linchpin elements mediating bona fide NC programs. Conversely, programs associated with neural lineages are mediated by the successive use of k-Cl6, k-Cl1 and k-Cl3 elements (Figure 5A, green box), revealing an early regulatory split between mesenchymal and neural progenitors at the cis-regulatory level. In addition, DiffBind elements at 5-6ss suggested the existence of a stem cell-like niche within bona fide premigratory NC, likely maintained at least until 8-10ss (Figure 5A, red box).
Figure 5.

Functional Dissection of k-Cluster Elements, Assignment to Single-Cell Transcriptomes, and Correlation with scATAC Clusters
(A) GO terms associated to k-means and DiffBind clusters. Fold enrichments were obtained using statistical overrepresentation test, p-values calculated with binomial distributions and Bonferroni correction for multiple hypothesis testing. K-Cl3 and DiffBind elements exclusively reflect canonical NC and mesenchymal cell development, and specific NC differentiation programs (sympathetic nervous system), while sharing roles in homophilic cell adhesion, gliogenesis, and axonogenesis with k-Cl6 and k-Cl1 (∗∗p < 0.01, Binomial test with Bonferroni correction, fold change >4). Only late-acting elements (k-Cl3, k-Cl1, and DiffBind 8-10ss) correlate to heterophilic cell-cell adhesion, while k-Cl6 elements play a role in early regulation of neuronal NC lineages, ectodermal placode formation, and cell-cell adhesion.
(B) Mean merged density profiles of k-Clusters.
(C) De novo TF binding motifs enriched in k-means and DiffBind clusters were identified using Homer. Binominal p-testing was used to determine motifs with p < 1 × 10−11.
(D) scRNA-seq heatmap visualizing hierarchical clustering of single NC cells at 6-7ss (total 124 cells, 74 6ss, and 63 7ss), sc-Cluster-1 (early NC, in red), sc-Cluster-3 (late NC, in blue), sc-Cluster-2 (neuronal-NC, in green). Top 50 differentially expressed genes are shown.
(E) PCA of top 100 genes from scRNA-seq. p values reflecting statistical significance of the single cell RNA-seq and k-means cluster associations were calculated using two-tailed hypergeometric test.
(F) tSNE plot depicting clustering of1509 NC cells at 7ss obtained by 10X Chromium scRNA-seq.
(G) Genes enriched in distinct clusters are labeled.
(H) Clusters of cells with differential chromatin accessibility as determined by scATAC.
(I) Chromatin accessibility patterns at differentially regulated loci. Colors correspond to (H).
(J) Enriched TF motifs across scATAC clusters.
In addition to sharing NC terms with k-Cl3 and k-Cl1, DiffBind elements yielded terms indicative of current identity as well as highlighted the future state of NC (early multipotent, premigratory NC at 5-6ss, and early differentiating, migratory NC at 8-10ss). For instance, terms including cartilage development, regulation of ossification, and CNS neuron development, more reflective of later NC behavior, were over-represented at 5-6ss, whereas terms such as gliogenesis and neuron migration were enriched at 8-10ss, indicating that the cis-regulatory landscapes are primed before the onset of corresponding gene expression and observable phenotypes. Interestingly, early-opening k-Cl6 elements and k-Cl1 elements that are fully accessible by 5-6ss (Figure 5B) function in the upstream regulation of neuronal differentiation processes (pre-synapse assembly, synaptic membrane adhesion, and gliogenesis), suggesting that at the regulatory level, neural-NC fates begin to be primed from as early as gastrulation.
We next investigated the upstream TF codes driving the observed functional heterogeneity between enhancer groups. Using the Homer algorithm (Heinz et al., 2010), we identified enriched de novo TF binding motifs within our clustered elements with high statistical significance (p < 10−12, Binomial test) (Figure 5C). We detected trans-regulatory inputs encrypted in cis elements, including binding signatures of early acting factors, canonical NC factors, and inputs specifically correlated to either neural or mesenchymal lineages (Figure 5C). A canonical NC signature highly enriched in k-Cl3 and late DiffBind elements (Figure 5C, red box) provided genome-wide evidence of the pivotal role of TFAP2s, Sox10, and Fox factors in the NC cis-regulatory landscapes, suggesting a role for feedforward loops in the global regulation of bona fide NC. Moreover, we also found factors suggestive of mesenchymal fates, such as cardiac specifier Pax3, pigment cell driver MITF, myoblast regulator MyoD, and mesenchymal stem regulator NFAT, some of which are known to act downstream of Sox10, TFAP2, and FoxD3 (Choi et al., 1990, Conway et al., 1997, Sauka-Spengler and Bronner-Fraser, 2008, Zhu et al., 2014). Among other k-Cl3 motifs, we also found sub-groups of exclusive inputs (e.g., Sox3) and those shared with k-Cl6 (Tead1/2 and Meis1/2) and k-Cl1 (i.e., ATF, Maf, retinoid acid receptors, Zic, Zfp, and GATA), possibly reflecting the role for some of these factors in stem-cell-like and neural lineages, respectively.
The k-Cl1 motif signature (Figure 5C, green box) was consistent with the putative role of these elements in neural lineages. For instance, high enrichment in Sox9 and FoxD3 motifs is in line with the postulated role for these factors in driving the switch from neural to mesenchymal lineages in early trunk NC (Nitzan et al., 2013). Intriguingly, we also found significant inputs from Otx2 and Pax2, factors known to play canonical roles in the early neural specification and neural differentiation (Finkelstein and Perrimon, 1991), whose placement within the NC-GRN has been long hypothesized (Le Douarin and Kalcheim, 1999). Moreover, crucial NC nuclear receptors NR2C2 and NR2F2 (Rada-Iglesias et al., 2012) were also found to drive k-Cl1 elements, as well as SoxC/SoxD factors (Sox4/5), consistent with the role of k-Cl1 in the early onset of NC programs. We also uncovered a k-Cl6-specific niche of upstream inputs including self-renewal genes Oct2/4 and Klf, EMT regulator Zeb1, and the insulator protein CTCF, possibly suggesting that the establishment of NC TADs commences from gastrulation or insulator elements reused through development. Finally, a signature of premigratory NC-associated TFs (e.g., Sox2/9, Msx2, Lef1/Tcf, and Pax7) was associated with DiffBind elements at 5-6ss, whereas TF enrichment within DiffBind elements at 8-10ss indicated the role of a Sox8/Sox10/TFAP2-dependent feedforward loop in the global regulation of migratory NC (Figure 5C). Remarkably, these results allow us to conclude that our DiffBind element analysis captures the temporal dynamics of pre- and migratory bona fide NC, whereas k-Cl3, k-Cl6, and k-Cl1 elements reveal a regulatory interconnection of canonical and non-canonical NC programs to different cell types and lineages, including naive stem cells, neural and mesenchymal progenitors, and their lineages.
Heterogeneity at the Transcriptional Level
To inquire whether the uncovered heterogeneous regulatory logic was reflected at the transcriptional level, we performed single-cell RNA-seq on FAC-sorted NC cells within a narrow dynamic window capturing the intrinsic transcriptional dynamics of NC before and during delamination (6-7ss). SmartSeq2 strategy yielded 137 deep NC single cell transcriptomes (Figure 5D), and principle component analysis (PCA) revealed three distinct populations: sc-cluster-1, sc-cluster-2, and sc-cluster-3 (Figures 5D and 5E). Sc-cluster-1 and sc-cluster-3 presented clear NC identity, expressing hallmark NC genes (Pax7, Sox9, Snai2, Sox10, TFAP2A/B, Ets1), but segregating according to developmental stage. Sc-cluster-1 was uniquely characterized by a number of early stage-specific factors (Brn3, Alx1, Cldn1, and Sfrp2) and sc-cluster-3 by later stage-specific, mesenchymal-related NC genes (Col9A3, Elk3, Ltk, ItgA9, and Cxcr4). In contrast, sc-cluster-2 contained cells collected at both 6ss and 7ss that, in addition to sharing some of the NC factors with sc-cluster-1 and sc-cluster-3, also uniquely expressed genes indicative of a neural phenotype, such as NR2F2, Sox3, CXCL12, Cdh2, etc., but also a number of key neural genes exclusively driven only by k-Cl1 elements (Vimentin, Nbeal1, Pax6, etc.) or in concert with k-Cl6 (Otx2, Gli2, MylK, Nlgn1, and retinoic acid signaling players Nav2 and Cyp1b). Interestingly, while initiating neural differentiation programs, sc-cluster-2 cells were also characterized by expression of NPB specification factors (Sall1, Zic1, Msx2, Meis1/2, Frzb, etc.) involved in the maintenance of NC programs that were mainly driven by k-Cl6 elements but never expressed majority of canonical, bona fide NC specifiers (Figure 5D).
To expand the dynamic range of our single-cell characterization, we used the 10X Chromium platform to resolve transcriptomes of 3,091 NC cells at 7ss. Following quality-control processing, we identified eight distinct clusters (2,359 cells) (Figures S5A and S5B), from which we removed two clusters that expressed mostly heat shock or stress response genes, resulting in five sub-clusters (1,509 cells) (Figure 5F). 10X-cluster-1 was characterized by expression of genes previously identified as specific of sc-cluster-1 and sc-cluster-3 (Sox10, FoxD3, TFAP2A/B, Ets1), thus representing bona fide canonical NC phenotype. Neural-NC sc-cluster-2 markers (Sox3, Pax6, Cdh2, Mylk) were enriched in 10X-cluster-0. Moreover, we identified additional clusters displaying neuro-epithelial-like genes (Cldn1/3, Dlx5, Cxcl12) in 10X-cluster-4 and a more mesenchymal signature (Olfm1, Zeb1, Elk3) in 10X-cluster-3. 10X-cluster-2 expressed higher levels of cell-cycling genes likely representing a transitionary population (Figure 5F). In summary, SmartSeq2 and10X single-cell transcriptional analyses fully confirmed the dichotomy within the early cranial NC population and early split into neural-NC and canonical NC identities.
Identified single-cell clusters significantly linked to the heterogeneity uncovered at the regulatory level (ATAC k-Clusters) (Figure 5E). Premigratory NC sc-cluster-1 links only to the k-Cl3 elements (∗p = 0.02), whereas bona fide NC sc-cluster-3 is significantly associated to both canonical k-Cl3 and the early neural k-Cl1 elements (∗∗∗∗p = 2.23 × 10−6 and ∗∗p = 0.003, respectively; two-tailed hypergeometric p test) (Figures 5B and 5E). Strikingly, the singular neural-NC cluster sc-cluster-2 was clearly controlled only by early k-Cl6 and neural-NC k-Cl1 enhancers (∗p = 0.018 and ∗∗∗p = 4.0 × 10−4, respectively) (Figures 5B and 5E). When similar correlations were calculated for differentially expressed gene sets obtained in bulk RNA-seq experiments, we found that NC-specific transcriptional signature at 8-10ss, and to a lesser degree at 5-6ss, was strongly associated with k-Cl3 and k-Cl1-controlled regulation (Figure S2H). Such a strong correlation between k-Cluster enhancers and single-cell gene expression clusters indicates the role of early cis-regulatory heterogeneity in establishing diverse NC progenitor lineages.
In order to examine the chromatin accessibility of cranial NC cells at a single-cell level, we performed single-cell ATAC (scATAC) of FAC-sorted NC cells at 7ss, using the 10X Chromium platform. Using k-means algorithm, we identified four discreet clusters of cells depicting differential chromatin accessibility (Figure 5H). To characterize these clusters, we first assigned and analyzed genes proximal to the open chromatin elements. We found that bona fide premigratory NC genes (FoxD3, Pax7, Sox8, Msx1) were evenly associated with the four clusters, particularly scATAC-1 and scATAC-2 (Figure S5D), whereas later bona fide NC factors were differentially enriched in scATAC-1 (Sox10, p = 5.34 × 10−60; Col9a3, p = 2.18 × 10−44; Mef2C, p = 5.57 × 10−33) (Figure S5D). When scATAC clusters were analyzed in the context of our bulk ATAC k-Clusters, k-Cl3 elements were overrepresented in scATAC-1 (∗∗∗∗p = 0.000 two-tailed hypergeometric p test), whereas k-Cl1 elements were more dispersed, but with a higher tendency to be accessible in scATAC-2 cells (p = 0.164) (Figure S5E). The majority of k-Cl3 elements (∼60%) were recovered as accessible at 7ss in the scATAC clusters -1 and -2. To explore the dynamics of scATAC elements, we surveyed the differential regulation of notable NC genes (Figure 5I). Sox10 enhancers (enh-99, enh-89, enh-87, enh-85, enh-84; Figure 4) were recovered in scATAC-1 and scATAC-3, with some of the elements accessible in smaller proportion of the cells in the scATAC-2 cluster, while Col9a3 was predominantly controlled by scATAC-1 elements (Figure S5F). Other examples of differential regulation were the novel NC gene, Map4k4, which was singularly regulated by scATAC-1 elements; Lhx2, which was governed by scATAC-2 (Figures 5I and S5G); Cxcr4 by both scATAC-1 and scATAC-2 elements; and BMP2, that was predominantly regulated by scATAC-4 elements (Figure 5I). Many NC genes (e.g., Sox8, Ets2) were regulated by elements open across all scATAC clusters, consistent with their uniform expression in 7ss premigratory NC cells (Figure S5F). Interestingly, TF motif enrichment within scATAC clusters showed a predominant restriction of significant TF motifs to scATAC-2 and scATAC-3 (Figure 5J), including several (Pax2, Otx2, Ets1) previously found as enriched and characterizing the k-Cl1 cluster (Figure 5C), whereas TF motifs identified as controlling k-Cl3 elements (Sox9, Pax3, Sox8) were more dispersed (Figure 5J).
A Combinatorial cis-Regulatory Code Programs NC Fate Restriction
The establishment of developmental regulatory programs requires combinatorial TF activity (Cunha et al., 2010, Junion et al., 2012, Zinzen et al., 2009). We therefore examined putative TF co-binding patterns at NC enhancers as a means of identifying the core transcriptional networks underlying these distinctive identities. Screening all two-way combinations of enriched de novo TF motifs in our enhancer clusters allowed us to detect a cohort of significantly enriched co-occurring TFs (∗p < 0.05, two-sided chi-squared test with Bonferroni correction) (Figures 6A, 6B, and S6).
Figure 6.

Combinatorial Motif Analysis and Perturbation of the NC-GRN
(A and B) TF co-binding relationships predicted for k-Cl3 (A) and k-Cl1 (B) elements. p values were calculated using two-tailed Chi-squared test, with Bonferroni correction for multiple hypothesis.
(C) Schematic of bilateral electroporation assay for CRISPR-mediated GRN perturbation experiment.
(D and E) GO terms associated with mis-regulated genes following knockout of indicated TFs associated with k-Cl3 (D) and k-Cl1 (E). Enriched GO terms were obtained using statistical overrepresentation test, p-values calculated with binomial distributions and Bonferroni correction for multiple hypothesis testing.
(F and I) Heatmap comparing gene expression in left (experimental) and right (control) dissected dorsal neural tubes from three representative embryos where k-Cl3 (F) or k-Cl1 (I) core TFs were targeted. Differential expression was determined using DESeq2 with a negative binomial model, p-values calculated using Wald test, corrected for multiple testing using the Benjamin-Hochberg method (padj<0.1, Log2Foldchange >1).
(G and J) Expression patterns of perturbed genes following knockout of k-Cl3 (G) or k-Cl1(J) core TFs obtained by HCR.
(H) Sub-circuitry of gene regulatory interactions governing Cxcr7, PlxnA4, and PdgfrA, all downregulated in perturbation of k-Cl3 core.
(K) Sub-circuitry of gene regulatory interactions governing Bche, Bmpr1, Ptprm, RarB, and TFAP2C, all downregulated in perturbation of k-Cl1 core.
In k-Cl3, we found a striking enrichment of canonical co-occupancies specific to bona fide NC (Figures 6A and S6C), such as Sox10-TFAP2 (p = 1.6 × 10−94), Sox10-Sox9 (p = 5.8 × 10−13), Sox10-Sox8 (p = 4.8 × 10−148), and Sox10-Sox/Fox (p = 7.7 × 10−85) (Sauka-Spengler and Bronner-Fraser, 2008), as well as novel ones, such as ATF-Sox10 (p = 1.07 × 10−150) and Arnt/Arnt2-Sox10 (p = 1.4 × 10−80), suggesting a dynamic interplay of TF combinations in the regulation of NC-specific identity. Moreover, analysis of DiffBind at 5-6ss and 8-10ss enabled the further identification of temporally restricted, NC-specific combinations controlling premigratory (e.g., Lhx-Sox2, Fox-Sox2, Dmbx1-Lhx, p < 2.7 × 10−28) (Figures S6A and S6C) and early migrating (i.e., Sox10-TFAP2s, Smad3-Sox/Fox, Sox10-Smad3, TFAP2-Sox/Fox, p < 2.3 × 10−32) (Figures S6B and S6C) identities, respectively (Sauka-Spengler and Bronner-Fraser, 2008). K-Cl1, however, showed an enrichment in non-canonical NC TF interactions (Figures 6B and S6C), such as co-binding of Otx2-Pax2 (p = 6.7 × 10−50), Otx2-Sox9 (p = 4.2 × 10−30), Pax2-Sox9 (p = 8.5 × 10−24), NR2F2-Otx2 (p = 1.4 × 10−22), Otx2-Zic1/2/3 (p = 2.9 × 10−11), and NR2F2-Sox9 (p = 1.6 × 10−10) among others, highlighting known and integrating new core transcriptional circuitry underlying NC-derived neural progenitors into our current view of the NC-GRN (Betancur et al., 2010a, Sauka-Spengler and Bronner-Fraser, 2008, Simões-Costa and Bronner, 2015). Importantly, these results place Otx2 and Pax2 alongside the canonical NC regulators Sox9, Zic1-3, Smad2/3, and NR2F2, as putative core regulators of the NC-GRN, displaying previously unappreciated connectivity to downstream, neural effector genes.
To functionally probe the core transcriptional circuits governing divergent NC identities (canonical/mesenchymal versus neural NC), we used CRISPR-Cas9 (Williams et al., 2018) to endogenously knock out combinations of upstream core co-acting factors. Using in vivo bilateral co-electroporation assay, we delivered the target gRNAs with Cas9 to the left and the control gRNA with Cas9 to the right side of the embryo and performed RNA-seq on left and right dissected dorsal neural tubes at 7ss, to assess the global effect of the perturbation on NC-GRN (Figures 6C).
Perturbation of the factors constituting the k-Cl3 minimal upstream core proposed to underlie NC canonical/mesenchymal identities (Sox10, Sox9, TFAP2B, ATF2, Arnt2, and NR2C1; Figure 6A) caused specific downregulation of 241 and upregulation of 62 genes (FDR < 0.1, Log2Foldchange >1). Downregulated genes included key premigratory NC regulators such as Msx1 and Draxin, further reinforcing the role of feedback loops in this transcriptional network, as well as downstream effector genes (PlxnA4, PdgfrA, Cxcr7, Col9A1/A3, Pcdh18, and Cdh13) (Figures 6F, 6G, and S7A), some of which were previously implicated in NC migration (Olesnicky Killian et al., 2009, Smith and Tallquist, 2010, Waimey et al., 2008). Conversely, the upregulated genes were involved in neural programs (FoxG1, Hesx1, Lhx2/9, Six3/6, ALDH1A1) (Aguiar et al., 2014, Andoniadou et al., 2007, Peukert et al., 2011), consistent with the GO terms associated to them (∗p < 0.05, Binomial test with Bonferroni correction) (Figure 6D).
Conversely, perturbation of the factors constituting the k-Cl1 minimal upstream core proposed to govern neural-NC program (Pax2, Otx2, NR2F2, ATF2, and Arnt2; Figures 3, 5, and 6B) resulted in downregulation of neural genes (BCHE, BMPR1a, Lrrn1, PCDH7, Sema6, Cxcr4, PTRM, RarB, TFAP2C, Pax3) involved in cell migration, neurogenesis, cell adhesion, and neural differentiation (∗p < 0.05, Binomial test with Bonferroni correction) (Figures 6E, 6I, 6J, and S7B). Interestingly, key TFs and nuclear receptors governing mesenchymal programs (TFAP2A/B, Twist1/2, FoxD1/3, Wnt4, FGFR1/3, Bmp2) were modestly but significantly upregulated, alongside other neural-related pathways dependent on NKX6.2, Sox2/11/21, PTEN, and Sema3b, known to govern neural differentiation (Figures 6I, S7C, and S7D).
In conclusion, perturbing the upstream transcriptional core of k-Cl3 caused the direct suppression of NC-specific mesenchymal progenitor programs with concurrent upregulation of neural signature genes, whereas disruption of k-Cl1 regulatory core appeared to have an opposite effect on the global transcriptional landscape. Thus, our knockout strategy disrupted the balance between neural and mesenchymal progenitor states, suggesting that the uncovered dichotomised NC transcriptional cores, mediated by k-Cl3 and k-Cl1 regulatory elements, controlled the shift between NC-derived mesenchymal and neural lineages, likely through mutual negative cross-regulation.
Integrating Regulatory Information into GRN Circuitry
Current state-of-the-art approaches to reverse engineer global vertebrate GRNs have either used the empirical reconstruction of GRN hierarchies by screening motif binding sites around promoters, together with high-resolution single cell profiles (Aibar et al., 2017), or have employed mathematical modeling to identify best-fit topologies of epistatic interactions and developmental trajectories determined at the single-cell transcriptomic level (Gouti et al., 2017). Here, we build on these strategies by benefiting from unbiased, high signal-to-noise, low-input epigenomes, and high-resolution single-cell transcriptomes of NC cells. We unraveled the co-binding dynamics of TFs directly regulating the NC programs by performing a comprehensive, database-driven screen to identify all known vertebrate motifs (p < 0.0001, Binomial test) present in our enhancers (k-Cl3, k-Cl1, and DiffBind sets) as a means of identifying the full ensemble of TF inputs. We further filtered motif occurrences by integrating TF co-expression data from the single-cell profiles (Figure 5D, ShinyApp). We next produced comprehensive, genome-wide reconstructions of the k-Cl3 and k-Cl1-dependent GRNs (Files S1 and S2), allowing us to identify the hierarchical position of k-Cl3 (Sox10, Sox9, TFAP2B, ATF2, Arnt2, and NR2C1) and k-Cl1 (Otx2, Pax2, NR2F2, Atf2, and Arn2) minimal core factors within their respective NC-GRNs, as well to predict their downstream targets.
The extracted GRNs that control representative genes downregulated upon k-Cl3 TF core knockout (Figure 6D) feature contributions from multiple enhancers within these loci varying between six (PlxnA4) and two (PdgfrA) per gene (Figure 6H), further highlighting the importance of enhancer redundancy for the robustness of transcriptional networks. These enhancers primarily mediate heterotypic binding of the core TFs, TFAP2B, and Sox9/10 as well as additional inputs from other co-binding partners, including Arnt2, NR2C2, MafF, retinoid acid receptors, Zeb1, and Zic3 (Figure 6H). Some of these inputs form the k-Cl3 core circuits (Figure 6A) and are themselves regulated by other targeted factors (NR2C1 and Arnt2, for instance), again highlighting feedforward loops as core features of the NC-GRN (Files S1, S2, S3, and S4).
The extracted GRNs controlling representative genes downregulated upon k-Cl1 TF core knockout (Figure 6E) identify the direct inputs of disrupted TFs, such as Otx2-Lhx2 and Smad3-Pax2, into BCHE, BMPER1, and TFAP2C, and Otx2-Zic1/3 into RXRb, with additional inputs (NR2F2, Arnt2, Meis1/2, Sox8-10) contributing to individual enhancers (Figure 6K). These results suggested that the core transcriptional network mediated by k-Cl1 elements acts upstream of RA signaling, through Otx2-dependent activation of RXRb, which may dynamically control the response of these cells to gradients of RA, a critical process in the patterning of neuronal lineages (Gouti et al., 2017). Taken together, our results corroborate the idea that k-Cl1 elements orchestrate the early establishment of the neural progenitor state shared between NC and non-NC cells.
Generating sub-circuits governing a small number of genes allows representation of regulatory dynamics and demonstrates that patterns of co-regulation can be resolved for functionally related genes. Moreover, combinatorial patterns of TF inputs suggest the existence of regulatory modules within the broader NC-GRN, possibly related to the previously identified co-expression gene networks (Figure 1G). As such circuitry requires cellular co-expression of all molecules involved, we used our single-cell profiles to assess co-expression of upstream TF inputs and downstream genes in order to further validate our networks (Figure S7D). This approach validated novel regulatory relationships and elucidated the core components of the global NC-GRN, highlighting the importance of enhancer regulation by combinatorial TF binding for normal NC development.
Database Resource
Finally, we have assembled our data into a user-friendly, interactive Shiny App (https://livedataoxford.shinyapps.io/Chick_NC_GRN-TSS-Lab/) (Figure 7), thus providing a searchable interface whereby individual or multiple genes can be queried for (1) gene expression, (2) WGCNA cluster association, (3) regulatory elements and retrieval of upstream regulatory inputs, (4) k-Cluster dependent GRN visualization, and (5) visualization of single-cell co-expression profiles. This unique tool offers a valuable resource for the NC and wider GRN communities, as it allows exploration of the wealth of novel regulatory sequences and interactions reported here. In-depth analysis of the NC-GRNs of interest that are easily retrievable from our databases is poised to provide critical insight into precise gene regulatory events underlying NC ontogeny.
Figure 7.

Shiny App Tool for Exploration of NC Gene Expression and Regulation
(A) Searchable interface for gene of interest (GOI).
(B) Gene expression tab allows to explore levels (shown in FPKMs) as well as dynamics and differential gene expression stats across samples analyzed.
(C) WGCNA cluster tab shows assignment to co-expression clusters.
(D) Gene co-expression at the single-cell level is visualized in a searchable heatmap; features adjustable cell percentage overlap and multiple gene-name searches.
(E) Regulatory tab enables exploration of assigned CREs (genome co-ordinates in galgal4) and inherent TF motifs (exact location within element provided).
(F) Network tab enables visualization of individual regulatory circuits assembled based on either k-Cl3 and k-Cl1 enhancers and their respective TF inputs. TF occurrence frequency and activating/repressing interactions are shown.
Discussion
GRNs control key regulatory events that ensure accurate development. Over many years of investigation, a wealth of knowledge about NC formation has been accumulated through bottom-up, candidate-gene approach studies, providing essential insights into developmental processes governing NC ontogeny (Bronner and Simões-Costa, 2016, Le Douarin and Kalcheim, 1999, Sauka-Spengler and Bronner-Fraser, 2008). Here we presented a top-down, unbiased approach to interrogate NC gene regulatory mechanisms genome-wide that has allowed us to reverse engineer a comprehensive representation of the NC-GRN, encompassing the full complement of NC enhancers and their upstream TF inputs. Specifically, we uncovered cis-regulatory interactions driving different NC lineages, including a number of novel factors, super-enhancer regions regulating NC master regulators, and single-cell signatures depicting epigenomic and transcriptomic heterogeneity within the NC. This represents the first high-resolution, genome-wide reconstruction of a vertebrate GRN in vivo, highlighting the potential of our integrated method to decode complex biological networks underlying ontogeny.
Chromatin Dynamics Infers NC Regulatory Architecture
Investigation of NC regulation at the epigenomic level has uncovered dynamic changes in chromatin accessibility of distal regulatory elements employed in the NC and other embryonic cell types at different stages of NC ontogeny. Our global analysis of chromatin dynamics has revealed (1) an NC-independent class of ubiquitously accessible elements, enriched at promoters (k-Cl4); (2) a class of elements specifically open only in the NC, at either premigratory and/or migrating stages (k-Cl3); (3) a class of elements dynamically open in both NC and neuroepithelial cells (k-Cl1); and (4) a class of elements accessible in naive epiblast and premigratory NC cells that become inaccessible by migrating stages (k-Cl6). Dissection of the specific enhancer clusters by TF binding and co-binding motif enrichment analysis, target gene assignment by position and high-resolution Capture-C analysis of enhancer-promoter contacts, and functional classification of assigned target genes has indicated that three key clusters (k-Cl3, k-Cl6, and k-Cl1) embed the regulatory logic of NC development.
The k-Cl3 elements were mainly associated with the bona fide NC loci, such as TFAP2A/B and Sox10, whereas k-Cl6 and k-Cl1 elements were enriched at loci related to stem cells and neural progenitors, respectively, such as Oct4 and Otx2. We propose that k-Cl6 elements initiate NC specification, likely through cis-regulatory pleiotropy, for example, reusing enhancer elements from epiblast cells in the premigratory NC. Establishment of an early premigratory neural-NC program is captured by k-Cl1 elements, shared with non-NC neural progenitors, whereas k-Cl3 elements may mediate canonical NC circuits restricted to bona fide NC progenitors. DiffBind clusters at 5-6ss and 8-10ss capture the temporal dynamics of canonical premigratory and early delaminating bona fide NC, revealing a global picture of NC spatiotemporal heterogeneity at the epigenomic level.
Functional interrogation of multiple CREs from the Sox10 super-enhancer region using CRISPR/dCas9-mediated epigenome editing has revealed the hierarchical contribution of different classes of enhancers to the NC-GRN. As expected, decommissioning individual enhancers caused little effect. Only disruption of the earliest acting element, enh-99, that belonged to the neural-NC k-Cl1 cluster, caused moderate, albeit not sustained, disruption of endogenous Sox10 transcription. This suggested that the establishment of the early neural regulatory landscape may be essential for the onset of bona fide NC programs. However, when multiple enhancers were decommissioned, a robust effect was achieved, highlighting the role of enhancer redundancy in the robustness of transcriptional networks.
Taken together, our data suggest that the regulatory logic of NC development is hierarchically embedded in the dynamic cis-regulatory landscapes. This approach has thus allowed us to not only infer the full complement of active NC enhancers but also to garner the information about the upstream inputs and downstream regulatory outputs, from which the NC-GRN could be reconstructed.
NC Heterogeneity and the Regulatory Basis for NC Lineages
By analyzing circuitries mediated by distinct clusters of NC regulatory elements (k-Cl3 versus k-Cl1), we have revealed the heterogeneity of cis-regulatory landscapes within cranial NC cells. Furthermore, we clearly linked this regulatory dichotomy to the heterogeneity uncovered at the transcriptional level using single-cell profiling. Our results show that identified single-cell NC clusters featured either canonical NC or neural-NC identity and associated to the corresponding cis-regulatory clusters with high-statistical support, thus linking their cis-regulatory and intrinsic transcriptional dynamics. Furthermore, our scATAC data revealed cluster-specific enrichment of k-Cl3 elements, whereas k-Cl1 elements were omnipresent, which is consistent with our previous observations that k-Cl3 elements are open in only a portion of NC cells at 5-7ss. However, genes associated to scATAC elements were less dynamic owing to the lack of temporal resolution in this particular assay. This highlights the necessity to use multiple stages and control cells in order to reconstruct diverse and dynamic developmental GRNs. Thus, our epigenomic analysis, considering temporal and spatial dynamics of enhancer activation, is currently a preferable choice to attain the TF/enhancer/gene expression information and reverse engineer NC gene regulatory circuits.
Combinatorial Regulatory Codes for Specification of NC Fates
We have identified unique combinatorial cis-regulatory codes underlying either canonical NC identity featuring bona fide NC interactions such as Sox10-Sox8/9 and Sox10-TFAP2, as well as co-activities with critical new players ATF and Arnt (Bogeas et al., 2018) or neural-NC identity enriched for non-canonical interactions including Otx2-Pax2, Otx2-NR2F2, Pax2-Sox9, and Otx2-Sox9. Our results are in line with previously proposed minimal network underlying the switch from neural to mesenchymal fates, involving Sox9 and FoxD3 (Nitzan et al., 2013). Furthermore, our approach has enabled us to build genome-wide circuits and resolve novel hierarchies such as previously unappreciated implication of Otx2, Pax2, and NR2F2 in the NC-GRN. While the involvement of Otx2 gene within the NC-GRN has long been hypothesized (Le Douarin and Kalcheim, 1999), given the craniofacial abnormalities that accompanied heterozygous Otx2+/− mice (Acampora et al., 1995), we now position Otx2 within the network, by identifying its upstream regulators (Sox9/10, Sox2/3, Pax2, Msx1/2, Arnt/Arnt2, Lhx, RXRg, Pax2, etc.) and numerous downstream targets (including Adamts19, Cdh6/9/10/13, Col4a2/6, EdnrB, Pcdh8/9/15/19, PlxnA4, Robo1, Sema3D/5b/6A/6D, Smad3, Sox11, Zfhx4, and Zic1, among many others).
By performing CRISPR/Cas9 knockout of top regulators of both programs in vivo, we showed that the activation of cis-regulatory landscape specific to canonical NC and mesenchymal progenitors, mediated by k-Cl3 elements, appears vulnerable to perturbation, as the disruption of the corresponding upstream core factors leads to a robust change toward a neural progenitor state. However, the rewiring of the early neural cis-regulatory landscape, represented by strongly interconnected k-Cl1 elements robustly coupled by auto-regulatory and feedforward loops and active in both NC and neuroepithelial progenitors, appears more difficult to achieve. Such a strong, early relationship between NC and non-NC neuroepithelial programs may, to some extent, explain the NC-derived neural progenitors’ resilience to reprograming (Simoes-Costa and Bronner, 2016). Nonetheless, the modest yet significant effect in shifting from neural to mesenchymal fates (as revealed by upregulation of Twist1/2 and TFAP2A/B, among others), primarily dictated by Otx2 and Pax2 disruption, indeed suggests that these two core networks mutually negatively regulate one another and thus dynamically balance neural and canonical/mesenchymal NC identities.
A Genome-wide View of the NC-GRN
Our integrated approach, using the intermediary of active NC-specific enhancers, has enabled the assembly of comprehensive NC-GRNs, linking predicted upstream regulatory factors to their downstream target genes (Files S1, S2, S3, and S4). Our approach has revealed new hierarchies and modular relationships and has recapitulated previously known interactions, highlighting its utility to interrogate the complex regulatory logic controlling early NC ontogeny. Moreover, the fact that at least 57% of the genes downregulated upon specific network core perturbations were directly regulated by at least one k-Cl3 or k-Cl1 element, as per our analysis, confirms the direct causal links within the proposed NC-GRNs. With the recent and ongoing development of novel, low-input methods to profile TF binding in vivo (Skene and Henikoff, 2017), we expect to soon be able to circumvent the key limitation of our approach, which is relying only on cis-regulatory dissection through the use of known, high-resolution motifs.
In summary, our data provide a whole-genome framework for understanding the early regulatory events underlying essential NC cell functions. We expect that our resource will be useful for the interrogation of the circuitries underlying essential processes during NC ontogeny, such as maintenance of stemness and multipotency, as well as broader cellular processes and behaviors such as EMT, migration, differentiation, and cell fate restriction.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K27ac antibody | Abcam | Cat#Ab4729; RRID: AB_2118291 |
| IgG antibody | Millipore | Cat#12-370; RRID: AB_145841 |
| Brd4 antibody | Bethyl Laboratories | Cat#A301-985A; RRID: AB_1576498 |
| GFP antibody (rabbit) | Torrey Pines Biolabs | Cat#TP401; RRID: AB_10013661 |
| Donkey anti-rabbit IgG (AlexaFluor-488-conjugated) | ThermoFisher Scientific | Cat#A21206; RRID: AB_141708 |
| Critical Commercial Assays | ||
| RNAqueous Micro Total RNA isolation kit | Ambion | Cat#AM1931 |
| SmartSeq2 V4 kit | Takara Clontech | Cat#634889 |
| Nextera XT DNA library preparation kit | Illumina | Cat#FC-131-1024 |
| Chromium Single Cell 3’ Library and Gel Bead Kit v2 | 10X Genomics | Cat#120267 |
| Chromium Chip E single cell ATAC kit | 10X Genomics | Cat#1000086 |
| Chromium Single Cell ATAC Library and Gel Bead Kit | 10X Genomics | Cat#PN-1000111 |
| Qubit High sensitivity DNA kit | ThermoFisher Scientific | Cat#Q32854 |
| Tapestation High sensitivity D1000 | Agilent | Cat#5067-5584 |
| KAPA Library Quantification Kit | Kapa Biosystems | Cat#KK4835 |
| Nextera DNA kit | Illumina | Cat#FC-121-1030 |
| NEB Next High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat#M0543S |
| NextSeq 500/550 High Output Kit v2 (75 cycles) | Illumina | Cat#FC-404-2005 |
| NextSeq 500/550 High Output Kit v2.5 (150 cycles) | Illumina | Cat#20024907 |
| Dynabeads Protein A | Life Technologies | Cat#10006D |
| NEBNext Ultra DNA Library Prep kit | New England Biolabs | Cat#E7370S |
| Agencourt AMPure XP beads | Beckman Coulter | Cat#A63880 |
| Hybloc Competitor DNA, a Cot-1 DNA equivalent | Applied Genetics Laboratories | Cat#CHB |
| Nimblegen SeqCap EZ hybridization and wash kit | Roche | Cat#05634261001 |
| V2 Nimblegen SeqCap EZ accessory kit | Roche | Cat#07145594001 |
| Dynabeads Streptavidin M270 | Life Technologies | Cat#65305 |
| 300-cycle MiSeq Reagent Kit v2 | Illumina | Cat#MS-102-2002 |
| Long Range HotStart PCR kit | KAPA Biosystems | Cat#KK3501 |
| E.Z.N.A. Endo Free Plasmid Mini Kit II | Omega BioTek | Cat#D6950-02 |
| Endofree maxi prep kit | Qiagen | Cat#12362 |
| Vectashield with DAPI | Vector Laboratories | Cat#H-1200 |
| Deposited Data | ||
| RNA-seq data (bulk) | This paper | ShinyApp GEO: GSE121526 |
| ChIP-seq data | This paper | GEO: GSE121329 |
| Capture-C data | This paper | GEO: GSE121324 |
| ATAC-seq data (bulk) | This paper | ShinyApp; GEO: GSE121318 |
| sc-RNA (SmartSeq2) | This paper | GEO: GSE130500 |
| sc-RNA and sc-ATAC (10X Chromium) | This paper | GEO: GSE131688 |
| RNA-seq following TF knockouts | This paper | GEO: GSE121331 |
Lead Contact and Materials Availability
All plasmids generated and used in this study are readily available from Addgene (https://www.addgene.org/Tatjana_Sauka-Spengler/). Further information and requests for reagents and protocols may be directed to the lead contact, Tatjana Sauka-Spengler (tatjana.sauka-spengler@imm.ox.ac.uk).
Experimental Model and Subject Details
Chick Embryos
Fertilized Bovans Brown chicken eggs (Henry Stewart & Co) were incubated at 37°C with approximately 40% humidity. Embryos were staged according to (Hamburger and Hamilton, 1951). All experiments were performed on chicken embryos younger than 12 days of development, and as such were not regulated by the Animals (Scientific Procedures) Act 1986.
Method Details
Embryo Preparation and Ex Ovo Electroporation
Hamburger and Hamilton (HH4) embryos were captured using the filter paper based ‘easy-culture’, method. Eggs were opened after desired incubation period, albumin was removed and embryos were removed from yolk using punctured filter paper and kept in Ringers solution until electroporation, as previously described (Betancur et al., 2010b, Sauka-Spengler and Barembaum, 2008, Simões-Costa et al., 2012, Williams et al., 2018). Plasmid(s) were injected across the whole the epiblast and electroporated: 5 pulses of 5V, 50ms on, 100ms off. We used the bilateral electroporation technique for perturbation experiments, whereby control and experimental regents are delivered to opposite sides of the primitive streak, providing ideal internal, stage matched controls for each experiment. Embryos were cultured on albumin at 37°C/5% CO2 overnight to the desired stage.
Cell Dissociation and FAC-Sorting
Following electroporation with the NC specific Foxd3 enhancer NC1 (Simões-Costa et al., 2012), driving a Citrine reporter (2μg/μl), we used fluorescence activated cell sorting (FACS) to collect Citrine-positive NC and Citrine-negative control cells at two distinct stages of cranial NC development (5-6ss and 8-10ss). Dissected cranial regions from electroporated embryos were dissociated with dispase (1.5mg/mL in DMEM/10mM HEPES pH 7.5) at 37°C for 15 min with intermittent pipetting to achieve a single cell suspension and with 0.05% Trypsin at 37°C for a final 3 min dissociation step. The reaction was stopped and cells were re-suspended in an excess of Hanks (1X HBSS, 0.25% BSA, 10mM HEPES pH8) buffer. Cells were centrifuged at 500 g for 10min, re-suspended in Hanks buffer, passed through a 40-μm cell strainers, and centrifuged at 750 g for 10min. Pelleted cells were re-suspended in 500μl Hanks buffer. Citrine positive cells were sorted and collected using BD FACS-Aria Fusion. We collected ∼300 and ∼600 NC cells per embryo at 5-6ss and 8-10ss, respectively. FACS isolated cells were used for RNA-seq and epigenome profiling experiments (bulk and single-cell).
Bulk RNA Extraction, Library Preparation and Sequencing
FAC-sorted cells were centrifuged at 2000 g for 5mins at 4°C, washed with PBS, and resuspended in lysis buffer and stored at 80°C. RNA was extracted using Ambion RNAqueous Micro Total RNA isolation kit (Cat. #AM1931, ThermoFisher Scientific), Turbo-DNase treatment was performed for 1 h to ensure removal of plasmid. RNA integrity was checked using Bioanalyser, and only samples with RIN >7 were used. Sequencing libraries were prepared using Takara SMARTer low input RNA kit and sequenced using 100bp paired-end reads on the Illumina Hiseq2000 platform. Minimum of three biological replicas for each stage were used for analysis.
Single Cell RNA Preparation, Library Preparation and Sequencing
Individual NC cells were collected by FACS. For SmartSeq2 cDNA was generated and sequencing libraries were prepared as previously described (Picelli et al., 2014). Briefly, mRNAs were primed with oligo-dT and reverse transcribed using an LNA-containing template switching oligo. Libraries were generated from amplified cDNA by Tagmentation with Tn5. Libraries were sequenced using 50bp single end reads for 96 cells. A 4 x107 dilution of ERCC spike-in control was used.
For 10X Genomics droplet-based single-cell RNA-seq the Chromium Single Cell 3′ Library and Gel Bead Kit v2 (Cat. #120267) was used following manufacturers protocol. Briefly, NC cells were collected by FACS into 2ul Hanks buffer, approximately 8000 cells were loaded onto the Chromium. Libraries were quantified using Qubit and Kappa qPCR and 4nM 10X scRNA-seq library was sequenced on a NextSeq500 platform (Illumina) using high output v2.5 150-cycle kit in 26 × 8 x 0 × 98 mode.
ATAC, Library Preparation and Sequencing
ATAC was performed using a protocol adapted from (Buenrostro et al., 2013). FAC-sorted cells were lysed (10mM Tris-HCl, pH7.4, 10mM NaCl, 3mM MgCl2, 0.1% Igepal) and tagmented using Illumina Nextera DNA kit (FC-121-1030) for 30mins at 37°C. Tagmented DNA was amplified using NEB Next High-Fidelity 2X PCR Master Mix (Cat. #M0543S) for 11 cycles. Tagmentation efficiency was assessed using Agilent Tapestation.
Single-cell ATAC was performed using the 10X Genomics Chromium platform. Nuclei were prepared using demonstrated protocol (CG000169 | Rev A) from 10X Genomics. Briefly, pelleted cells (500 g, 5mins, 4°C) were washed with PBS supplemented with BSA (0.04%) and resuspended in nuclei lysis buffer (10mM TrisHCL-pH7.4, 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 0.1% Noidet P40 substitute, 0.01% Digitonin, and 1% BSA), lysis efficiency was determined using Countess II FL automated cell counter. Tagmentation was conducted on bulk nuclei and libraries were generated using the Chromium Chip E Single Cell ATAC Kit and Chromium Single Cell ATAC Library & Gel Bead Kit (PN-1000111) before loading (approximately 5000 nuclei) into Chromium and following supplier’s protocol (CG000168). ATAC-seq libraries were sequenced using paired-end 40bp reads on the Illumina NextSeq500 platform. 4nM 10X scATAC-seq library was sequenced on a NextSeq500 platform (illumina) using high output v2.5 75-cycle kit in 36 × 8 x 16 × 36 mode. 5% of PhiX control was spiked in and 1.8pM loaded onto the flow cell according to manufacturer’s instructions.
H3K27Ac ChIP, Library Preparation and Sequencing
FAC-sorted cells were cross-linked with 1% formaldehyde for 9mins. Fixation was quenched with 125mM of glycine for 5min. Cross-linker was washed out by 3x pellet washes with 1xPBS ((supplemented with 1X PI (protease inhibitors)), 1mM DTT and 0.2mM PMSF) centrifuging at 2000 g for 4mins at 4°C. Pellets were re-suspended in isotonic nuclei extraction buffer (NEB: 0.5% NP40, 0.25% Triton-X, 10mM Tris-HCl-pH 7.5, 3mM CaCl2, 0.25M sucrose, 1mM DTT, 0.2mM PMSF, 1X PI). Cell nuclei were expulsed with 20 strokes using pestle B in a glass homogenizer, pelleted and washed with 1xPBS (with 1X PI, 1mM DTT and 0.2mM PMSF). Nuclei were lysed in SDS lysis buffer (0.7% SDS, 10mM EDTA, 50mM Tris-HCl (pH 7.5), 1x PI). Cross-linked chromatin was sonicated at 11A 10x (10 s on 30 s off) followed by 6A 8x (30 s on 30 s off) using a probe sonicator, to achieve 300-800bp fragments. Pre-blocked Protein A Dynabeads were incubated with antibody (H3K27ac, Abcam Ab4729, Brd4, Bethyl Laboratories A301-985A). Sonicated DNA-protein complexes were applied to beads overnight at 4°C. IgG antibody (Millipore 12-370) was used as control and an input sample was taken. Samples were washed 8x with RIPA buffer (50mM HEPES-KOH (pH 8.0), 500mM LiCl, 1mM EDTA, 1% NP40, 0.7% Na-Deoxycholate, 1x PI) and 1x NaCl TE wash (1x TE, 50mM NaCl) at 4°C. Chromatin was eluted from the beads with SDS ChIP elution buffer (50mM Tris-HCl (pH 7.5), 10mM EDTA, 1% SDS). Cross-linking was reversed overnight at 65°C at 1300 rpm. Cellular RNA was digested with RNaseA (0.2mg/mL) at 37°C for 1 h, and cellular proteins were removed with Proteinase-K (0.4mg/mL) at 55°C for 1 h. Samples were purified by standard phenol-chloroform extraction and ethanol precipitation. ChIP-ed DNA was amplified using published protocol (Adli and Bernstein, 2011) for small cell number ChIP, whereby Seqeunase was used to prime ChIP DNA with custom oligos containing PCR primers and restriction enzyme (BciVI) sites allowing amplification and subsequent digestion yielding library prep ready fragmented DNA. Libraries were prepared using NEBNext Ultra DNA library prep kit following manufacturers protocol. ChIP libraries were sequenced using 50bp paired-end reads using Illumina Hiseq2500 platform. Three biological replicates for each stage and antibody were obtained.
Capture-C
Capture-C was performed according to the published protocol (Davies et al., 2016), with a few minor adaptions. To collect the material, dorsal neural tubes were dissected from 6-7ss embryos and dissociated in nuclei extraction buffer (NEB, as above), 20 strokes with pestle A in a glass homogenizer. Cells were cross-linked with 2% formaldehyde for 8min. Fixation was quenched with cold 1M glycine. Pelleted cells (1500rpm, 4°C, 5min) were washed with 1xPBS, re-pelleted and re-suspended in lysis buffer (10mM Tris-HCl (pH8), 10mM NaCl, 0.2% Igepal-NP40, 1x cOmplete protease inhibitor (PI)), and left on ice for 20min before pelleting. Supernatant was removed and pellets were stored at −80°C. On the day of the Capture, cross-linked cells were re-suspended in water on ice and homogenized with pestle B, 45 strokes, and centrifuged at 14,000 rpm for 5min. Pelleted cells were re-suspended in water, restriction enzyme buffer and SDS (0.25% final) and incubated at 37°C with 1400 rpm shaking for 1 h. Triton X-100 (8.25% final) was added and samples were incubated again with shaking for 1 h. 3x 125U aliquots of restriction enzyme (DpnII, NEB High-concentration 50,000 U/mL), were added several hours apart, such that samples were digested for 16-24 h. Restriction enzyme was inactivated by 20min incubation qt 65°C. To ligate fragmented DNA, water, ligation buffer and 240 U T4 ligase (high-concentration 30U/μl; Cat. #EL0013, ThermoFisher Scientific) was added and samples were incubated overnight at 16°C with 1400rpm mixing. Samples were uncrosslinked overnight at 65°C in presence of 50ug of Proteinase-K and then treated with 300U RNaseA (Cat. #1119915, Roche) for 30min at 37°C. DNA was extracted using standard Phenol:Chloroform method and ethanol precipitation. Thus, obtained 3C libraries were then sonicated to 200bp, using Covaris S220 with the following settings: duty cycle 10%, intensity 5, cycles per burst 200, 6 cycles each of 60 s and set mode. Sonicated samples were cleaned using Ampure XP beads, quantified by Qubit (ThermoFisher Scientific) and checked using 2200 Agilent Tapestation System. Libraries were prepared using NEBNext Ultra library prep kit and NEBNext Multiplex oligos for Illumina according to the manufacturer’s protocol with the exception of using Herculase enhanced DNA polymerase (Agilent). Libraries were cleaned up with Ampure XP beads and again assessed using Tapestation and Qubit.
Libraries were prepared for hybridization by adding 5μg Chicken Hybloc competitor DNA (Applied Genetics Laboratories) to 1.5-2μg 3C library, 1nmol Nimblegen-HE universal oligo, and 1nmol Nimblegen-HE index oligo (specific to the indeces used in library preparation) (Nimblegen SeqCap EZ hybridization and wash kit, Cat. #05634261001, Roche). The assembled reaction was dried by vacuum centrifuge at 50°C. Hybridization buffer and component were added and the DNA was carefully reconstituted. Target specific biotinylated oligos were preheated to 47°C in 0.2ml PCR tubes. 3C libraries and blocking oligos were denatured at 95°C for 10min before adding to the preheated biotinylated oligos. The mixture was then incubated at 47°C for 64-72 h. Hybridized samples were washed and M-270 Streptavidin Dynabeads (Cat. #65305, ThermoFisher Scientific) were prepared according to Nimblegen SeqCap EZ hybridization and wash kit protocol. Hybridized samples we applied to streptavidin beads and incubated at 47°C for 45min with 600rpm mixing. Washing steps were performed as per kit’s instructions. Amplification of captured multiplex DNA sample was done using the V2 Nimblegen SeqCap EZ accessory kit (Cat. #07145594001, Roche), followed by an Ampure XP bead clean up. As recommended (Davies et al., 2016), to maximize the capture, we repeated the hybridization reaction (24 h), as well as subsequent washes and amplification steps. Finally, captured libraries were sequenced using 150bp paired-end reads on the Illumina Miseq platform (300-cycle MiSeq Reagent Kit v2, Cat. #MS-102-2002, Illumina). A full version protocol is available at http://www.tsslab.co.uk/resources.
Generating Nanotag Reporter Vectors
pTK Citrine/Cerulean/mCherry reporter vectors (Simões-Costa et al., 2012) were modified to generate an ‘easy-insert’ integration site enabling highly efficient cloning of putative enhancers using rapid simultaneous ligation/digestion reaction in a single tube. To this end, a lacZ expression cassette flanked with restriction sites for the type IIs restriction endonuclease BsmBI (similar to (Cermak et al., 2011)) was cloned upstream of theof the minimal Thymidine Kinase (TK) promoter. To enable multiplexing and simultaneous detection of reporter activity from different enhancers, we further modified pTK reporter vectors to include unique ‘‘barcoded’’ DNA-tags (“nanotags”) (Nam and Davidson, 2012) downstream of fluorescent reporters, within their 3′UTR regions and upstream of their polyadenylation signal. We thus generated 48 new Nanotag reporter vectors (16 each for pTK-Citrine, pTK-Cerulean and pTK-mCherry). pTK Nanotag vector collection is available from Addgene (https://www.addgene.org/Tatjana_Sauka-Spengler/).
Enhancer Cloning and Testing
Multiplexed, high-throughput enhancer cloning pipeline is abridged below, the full technical protocol is available at http://www.tsslab.co.uk/resources.
Putative enhancers were amplified from chick gDNA using primers containing specific sequence tails (see supplementary excel file) to facilitate subsequent cloning into nanotagged reporter vector using a modified GoldenGate (Engler et al., 2009) protocol. Briefly, gel-purified amplicons were combined with modified pTK nanotag reporter vector with T4 DNA ligase and BsmBI restriction enzyme and subjected to a cycling reaction that allows simultaneous BsmBI digestion and T4-mediated ligation of amplicon into the reporter vector. Endotoxin-free plasmid preparations (E.Z.N.A. Endo Free Plasmid Mini Kit II, Cat. #D6950-02, Omega Bio-Tek or QIAGEN endo-free maxi prep kit, Cat. #12362, QIAGEN) were prepared for electroporation. Putative enhancers in reporter plasmids were pooled at 0.2μg/μl each, 10-12 plasmids including positive, NC1, and negative (short-oligos, Table S2) controls, and electroporated into the entire epiblast of the early chick gastrula embryos (HH4) (Sauka-Spengler and Barembaum, 2008, Simões-Costa et al., 2012, Williams et al., 2018). Embryos were allowed to develop to desired stages using ex ovo culture in thin albumin. Cranial regions were dissected and extracted RNA was hybridized with Nanostring reporter code-set oligos designed to detect nanotag transcripts. Nanostring assay was conducted as manufacturers protocol and absolute transcript counts were recorded. Enhancers with a count >50 were electroporated individually at 2μg/μl and fluorescent reporter activity was imaged throughout early NC developmental stages. An extended set of ‘electroporation-ready’ novel positive NC enhancers cloned into the fluorescent reporters is available from Addgene (https://www.addgene.org/Tatjana_Sauka-Spengler/).
Imaging Analysis
Embryos were imaged on an Olympus MVX10 stereomicroscope with 2.5X objective using Axio Vision 4.8 software. Zeiss 780 Upright confocal microscope was used for imaging at high cellular resolution
Cryosectioning and Immunostaining
Embryos selected for cryosectioning were fixed in 4% PFA (paraformaldehyde) for 1 h at room temperature (RT) or at 4°C overnight, then washed 3x 10min in 1x PBS. Embryos were cryoprotected in 15% sucrose/ PBS overnight at 4°C, followed by overnight incubation in 7.5% gelatine/15% sucrose /PBS at 37°C. Following ∼4-hour incubation in 20% gelatine/PBS embryos were embedded in 20% gelatine/PBS, snap frozen in LN2 and stored at −80°C. Cryosectioning was performed at 10 μm thickness.
Prior to immunocytochemistry, excess gelatine was removed from the slides by a brief rinse in pre-warmed PBS (37°C). Sections were rinsed 3x 5min in PBT (2% DMSO, 0.5% Triton X-100 in PBS), blocked in 10% donkey serum in PBT (block solution) for 1 h at RT, and incubated overnight at 4°C with primary antibody (1:200 dilution of rabbit anti-GFP in block solution, Cat. #TP401, Torrey Pines Biolabs). Sections were then washed in PBT at RT 5x 10min, followed by incubation with secondary antibody (AlexaFluor-488-conjugated donkey anti-rabbit IgG diluted at 1:1000 in PBT, Cat. #A21206, ThermoFisher Scientific) for 2 h at RT. Sections were washed 6-8x 10min in PBT at RT then overnight at 4°C and mounted using Vectashield with DAPI (Cat. #H-1200, Vector Laboratories).
Hybridization Chain Reaction Fluorescent In Situ
In situ v3 HCR for Sox10 was performed as previously described (Choi et al., 2018, Williams et al., 2018). Embryos were fixed in 4% PFA for 1 h at room temperature, dehydrated in methanol and stored at −20°C. Following rehydration embryos were treated with Proteinase-K (20μg/mL) for 2.5 min at room temperature and post-fixed with 4% PFA for 20 min at room temperature. Embryos were washed in PBST for 2x 5 min on ice, then 50% PBST / 50% 5X SSCT (5X sodium chloride sodium citrate, 0.1% Tween-20) for 5 min on ice and 5X SSCT alone on ice for 5 min. Following washes embryos were pre-hybridized for 5 min on ice, then for 30 min at 37°C in fresh hybridization buffer. Probes were prepared at 4pmol/mL (in hybridization buffer) and hybridization buffer was replaced with probe mixture overnight at 37°C. Excess probes were removed with probe wash buffer for 4x 15 min at 37°C. Embryos were pre-amplified in amplification buffer for 5 min at room temperature. Hairpins were prepared by snap-cooling 30pmol (10μl of 3μM stock hairpin) individually at 95°C for 90 s and cooled to room temperature for minimum 30 min, protected from light. Cooled hairpins were added to 500ml amplification buffer. Pre-amplification buffer was removed from embryos and hairpin solution was added overnight at room temperature, protected from light. Excess hairpins were removed by washing in 5X SSCT 2x 5 min, 2x 30 min and 1x 5 min at room temperature. Embryos were mounted on slides and imaged using Zeiss 780 Upright confocal microscope.
Imaging of Enhancer-Reporter Assays and HCR
Imaging of wholemount embryos in enhancer-reporter activity assays and HCR was performed on a Zeiss 780 Upright confocal microscope, using a 10x objective. Z-stacks scans were collected across approximately 50-70um and a 2x2 tile scan was used (2x3 for embryos 10ss and above). Images were processed using Zeiss Zen microscope software, tile scans were stitched using bidirectional stitching mode, with overlap of 10%. The maximum intensity projections of embryo z-stacks are presented in the manuscript. For sectioned samples, images were obtained on a Zeiss 780 Upright confocal with a 25x oil immersion objective and single z-slices are shown.
In Vivo CRISPR-Mediated Perturbation Assays
sgRNAs were cloned into a chicken pU6-3 vector using a BsmBI mediated approach as previously described (Williams et al., 2018) and used in conjunction with dCas9-Krab (Addgene #92361) (Williams et al., 2018) to decommission endogenous enhancers or Cas9-2A-Citrine (Addgene #92358) (Williams et al., 2018) to knockout transcription factors. Following bilateral electroporation, left and right dorsal neural tubes were dissected separately; RNA was extracted using Ambion RNAqueous Micro Total RNA isolation kit (Cat. #AM1931, ThermoFisher Scientific). For enhancer decommissioning experiments, oligo-dT-primed cDNA was synthesized using Superscript III (Cat. #18080093, ThermoFisher Scientific) and qPCR performed using Fast SYBR Green reagent (Cat. #4385612, ThermoFisher Scientific) on the Applied Biosystems 7500 Fast Real-Time PCR System. The standard curve method was used to quantify gene expression, ∼10 embryos per targeted enhancer were analyzed. For TF knockout experiments RNA-seq libraries were prepared using SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Cat. #634889, Takara Bio Clontech) and sequenced using 40bp paired-end reads on Illumina NextSeq500. 6 individual embryos and associated RNA-sew libraries were analyzed.
Quantification and Statistical Analysis
NGS Data Processing
Sequencing files from each sequencing lane were de-multiplexed and the resulting files were merged. Reads were trimmed for quality using sickle (Joshi, 2011) (v.1.33) quality control was performed using fastQC (v.0.10.1) and deeptools (Ramírez et al., 2016) (v.2.2.2). Reads were mapped to the chicken genome galGal4 assembly using bowtie (Langmead and Salzberg, 2012) (v.1.0.0), except for RNA-seq files, which were mapped using RNA-STAR (2.4.2a) (Dobin et al., 2013). In the CRISPR knockout experiments, reads were mapped to k-Cl1. Sequence alignment/map files were compressed to the binary version (BAM) for downstream analysis. Differential expression analysis was carried out using in DESeq2 (Love et al., 2014) (v.1.14.1). Peaks were called for each ATAC-seq sample using MACS2 (v.2.0.10) (Feng et al., 2012) and ATAC-seq consensus peaks were generated using bedtools (Langmead et al., 2009) (v. 2.25.0) intersect command. For RNA- and ATAC-seq reproducibility analyses, BAM files were first normalized for sequencing depth using samtools (Li et al., 2009) (v.1.3) and count tables were retrieved using Subread (Liao et al., 2013) (v.1.4.5) featureCounts (Liao et al., 2014) tool from an Ensembl gene list in GTF format (for RNA-seq) and from a consensus set of 99,583 accessible regions (for ATAC-seq), respectively. Count tables were analyzed in R (v.3.2.3) and principal component analysis (PCA) was performed using the DEseq2 (v1.4.5) package in the R (v.3.2.1) environment. A custom Perl script was used to generate smoothened genome browser tracks in BigWig format for ATAC-seq data visualization.
WGCNA
Weighted correlation network analysis (WGCNA) (Langfelder and Horvath, 2008) was performed on normalized gene count tables generated by DESeq2, according to the pipeline detailed in the online tutorial (https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/#WGCNAIntro).
Single-Cell Analysis
SmartSeq2 scRNA-seq Reads were mapped using RNA-STAR (2.4.2a) (Dobin et al., 2013) to the chicken genome Galgal4 assembly and the ERCC spike-in controls using the default parameters. Subread package with default parameters. Reads that aligned to more than one locus as well as ambiguous fragments were excluded from all further analysis. To remove cells with low quality sequencing, cells with a) less than 100,000 sequenced reads or b) less than 50% uniquely mapped reads were excluded from any further analysis. Further filtering was done based on the distributions of a) the number of expressed genes per cell, b) the proportion of reads on mitochondrial genes or c) the proportion of reads on ERCC spike-ins, requiring that the cells are within 3 MADs (Median Absolute Deviation) for each distribution (McCarthy et al., 2017). In the remaining 124 cells, 10,291 genes were detected using the following criteria: a) they had any reads in more than 5 cells and b) had an average number of counts above 1. The cell-based factors from the deconvolved pool-based size factors were used for the normalization of the gene counts, as described in (Lun et al., 2016), using pool sizes of 10, 20, 40 and 60 cells. The clustering was done with the pagoda2 package (https://github.com/hms-dbmi/pagoda2) using the infomap and the walktrap community methods. There was a 96% concordance between the two methods and the infomap method was used to obtain the final clusters. An analysis for differentially expressed genes was performed using the SCDE package (Kharchenko et al., 2014) to find cluster specific gene markers. For each cluster a one-versus-one approach was applied between all pairs of clusters using the default parameters.
10X Chromium scRNA-seq Single-cell RNA-seq raw base call files were demultiplexed using Cellranger (v3.0.0) (Zheng et al., 2017) and mapped to the galGal5 transcriptome that contained the Citrine transcript and compiled using the Cellranger “mkref” function. 3,108 estimated number of cells were obtained with mean reads per cell of 152,318, median genes per cell of 1,501, median UMI counts per cell of 4,968 and total genes detected was 15,511. 97.7% of reads had valid barcodes with a Q30 of 97.5% and 83.6% of the reads mapped confidently to the chicken genome. Downstream analysis was carried out using Seurat package (v3.0.0)(Butler et al., 2018) in R. Matrices were filtered to remove barcodes with fewer than 200 genes, more than 3000 genes expressed and carried a high percentage of UMIs from mitochondrial features (greater than 10%) (Figure S5A). Expression values for the total UMI counts per cell were normalized and permutation test carried to determine the significant principal components and dimensions in the data before performing linear dimensional reduction (resolution = 0.45, dims = 1:15). Seven cell clusters were obtained and visualized on a t-SNE plot. After identifying cell markers, we removed two clusters that contained mostly housekeeping or cycling genes and perform further subclustering of the remaining 5 clusters with a total of 1509 cells (resolution = 0.45, dims = 1:20).
10X Chromium scATAC-seq Single cell ATAC-seq raw base call files were demultiplexed using Cellranger-ATAC (v1.1.0). A custom galGal5 genome was built using the mkref function with the configuration file containing: 1) galGal5 fasta file, 2) v94 GTF file filtered to only include annotated genes, 3) Jaspar2018 core vertebrate motif file, 4) primary contigs of chromosomes containing only annotated genes and 4) mitochondrion contigs. Following successful reference creation, demultiplexed files were analyzed with the “count” function using the custom generated gg5 genome. 1,259 estimated number of cells were recovered with 5,923 median fragments per cell, 75.1% fraction of transposition events in peaks in cell barcodes an 17.0% fraction of fragments overlapping any targeted region (this value is much lower than expected due to the custom gg5 genome reference that does not include well annotated targeting files related to other functional domains). 59.4% of fragments were in nucleosome-free regions with 30.9% fragments flanking a single nucleosome and 17.0% fraction of fragments overlapping TSS (Figure S5C). Downstream analysis was carried out from the generated ‘cloupe’ file and imported into the Loupe Cell Browser (v3.1.0). Three clusters were obtained and enriched peaks per cluster with a log2FC of >0 and p-adjusted value of < 0.05 were used.
K-Means Clustering of ATAC Data
Clustering of downsampled ATAC-seq datasets was performed with the seqMINER platform (v.1.3.4) (Ye et al., 2011) on a consensus set of 99,583 accessible regions using the k-means enrichment linear clustering normalization algorithm set to k = 10 and a window of ± 1.0 kb from the peak center. Heatmaps and merged profiles were plotted using the Deeptools (v.2.2.2) package (Ramírez et al., 2016). K-Cl specific count tables were then used for correlation and linear regression analyses and visualization in R (v.3.2.3).
Differential Accessibility
Differential accessibility analysis was carried out in R (v.3.2.1) using the DiffBind package (v1.10.2). Differential accessibility across samples was calculated using a negative binomial distribution model implemented in DEseq2 (v1.4.5). A stringent threshold (FDR < 0.1, Fold enrichment >1) was used to define high-confidence differentially accessible ATAC-seq peaks by comparing the enrichment of accessible regions in NC cells at two stages (5-6ss and 8-10ss) over non-NC cells. Normalized read counts were clustered and visualized using pheatmap (v. 1.0.10) (Kolde, 2018) with default settings.
Next-Generation Capture-C Analysis
NG Capture-C was performed as described previously (Davies et al., 2016) on dissected dorsal neural tube tissue and chicken erythrocytes. Samples were indexed for multiplexing and co-capture of promoters using biotinylated 120-mers (Integrated DNA technologies, IDT), designed with the CapSequm webtool (http://apps.molbiol.ox.ac.uk/CaptureC/cgi-bin/CapSequm.cgi) (Hughes et al., 2014) and pooled to a final concentration of 2.9nM (Table S1). Captured material was pooled and sequenced using 150bp paired-end reads on the Illumina Miseq platform (300-cycle MiSeq Reagent Kit v2, Cat. #MS-102-2002, Illumina). Reads were mapped using Capture-C scripts (https://github.com/Hughes-Genome-Group/CCseqBasicF/releases), and analyzed as previously described (Hay et al., 2016) using intra-house R-scripts and DESeq2.
Functional Annotation of CREs
A custom Python (v.2.7.5) script was used to assign peaks to nearest expressed genes. Ensembl identifiers were converted to gene names using the Chicken Ensembl Gene ID converter (https://www.biotools.fr/). Genes associated to two or more enhancers were used for functional analysis and GRN assembly. High-resolution TADs were determined as described in (Davies et al., 2016). The Panther package (Mi et al., 2017) (v.11) was used to calculate statistical overrepresentation tests using default settings. P-values were calculated with binomial distributions with Bonferroni correction for multiple hypothesis testing.
Motif Analysis
De novo motif discovery was performed using Homer (Heinz et al., 2010) (v.4.7) findMotifsGenome.pl script, using clusters of ATAC-seq peaks identified by differential accessibility analysis and k-means clustering. Motifs of 8–22bp in length were searched in ± 250bp windows from the peak center and a constant background containing a set of 99,583 peaks was used. Non-redundant matrices with p < 1 × 10−11 (Binomial P-test) were retained. De novo motifs were annotated using Homer (v.4.7), STAMP (Mahony and Benos, 2007), TOMTOM (Gupta et al., 2007) and manual comparisons. Log10 transformed P-values were clustered and visualized using pheatmap (v. 1.0.10) in R (v. 3.2.1) with default settings.
Combinatorial Binding
All possible pairs of cluster-specific de novo motif combinations were computed in R (v. 3.2.1). Positive controls were defined by using a set of 99,583 consensus peaks subtracted to each cluster of elements using bedtools (v. 2.25.0) intersect –v command. Homer (v.4.7) annotatePeaks.pl script was used to screen all de novo motifs occurring in windows of ± 250bp from the peak center. Motif occurrences were converted to a matrix of motif presence (1) or absence (0) in R (v.3.2.3), and a custom Python3 script using the Pandas package was used to calculate motif co-occurrences. Combinations enriched at α = 5% (two-tailed Chi-square test) with Bonferroni correction for multiple hypothesis (m) testing were retained for P-values < α/m (Noble, 2009). Motif combination frequencies in differentially accessible and in k-Clusters groups of elements versus that of positive controls were used as a metric of combinatorial enrichment. Co-binding networks were plotted using the Circlize package in R (v.3.2.3).
Identification and Ranking of Super-Enhancer-like Clusters
Super-enhancers were identified using the ROSE (Ranked Order of Super Enhancers) pipeline (http://younglab.wi.mit.edu/super_enhancer_code.html). This algorithm groups (stitches) identified individual enhancers positioned within a defined distance into putative super-enhancers (SEs) and ranks them by their input-subtracted H3K27Acetylation signal. We used the default options with 12.5 kb as the maximum grouping (stitching) distance, the reads per million normalization for H3K27Ac ChIP signal from 5-6ss and 8-10ss FAC-sorted NC and a promoter exclusion zone of 1,000 or 2,500 basepairs. The enhancer clusters were ranked by cumulative normalized H3K27Ac signal (H3K27ac signal versus enhancer rank) and super-enhancers were identified as clusters lying beyond the identified inflection point on the ranking curve (Whyte et al., 2013). Super-enhancers cluster were associated to the NC enhancers identified in this study and assigned to the closest expressed gene(s). The identification of groups of NC enhancers enriched in Brd4 occupancy on was achieved by k-means clustering (Seqminer platform) (Ye et al., 2011) of the Brd4 ChIP datasets using 8-10ss DiffBind enhancers as a reference.
GRN Assembly
A comprehensive database collection of high-resolution TF binding models from vertebrates (HOCOMOCO v.11) (Kulakovskiy et al., 2018) was downloaded in Homer (v.4.7) format. Position weight matrixes (PWMs) with a stringent Binomial 3-value < 0.0001 were selected, resulting in a list of 769 PWMs. These TF binding models were first filtered based on PWM redundancy and were further eliminated by applying a minimum expression level threshold (1 > FPKM) to their cognate TFs, resulting in a list of 169 motifs. scRNA-seq was used to define TFs co-expressed in clusters of single cells. This list was further refined by eliminating ubiquitous TFs and by combining TF paralogs (e.g., TFAP2A and C; TFAP2B and D etc. based on high similarity of their binding motifs). This resulted in a final list of 49 PWMs being retained. Homer (v.4.7) annotatePeaks.pl script was used to screen these PWMs in all differentially accessible and in k-Clusters groups of elements, allowing the inference of the upstream (TF) inputs putatively binding to such elements. Matrices containing the list of CREs and upstream TF inputs were merged with another matrix containing the CREs assignments to their target promoters, which were determined either by Capture-C or by positional information from expressed genes. These elements were grouped based in the differential accessibility analysis and k-means clustering, which facilitated the retaining of the GRN spatiotemporal dynamics. The combination of such matrixes linked the upstream (TF) inputs and the downstream outputs (target genes), resolving GRN hierarchies and modules in a genome-wide fashion, at single-cell resolution and with epigenomic details depicted. GRN hierarchical relationships were assembled and used for network analysis and visualization in BioTapestry (Longabaugh et al., 2005) (v. 7.1.1). Sub-networks for specific genes or groups of genes were retrieved from the matrixes containing all hierarchies and can be interactively surveyed in the ShinyApp.
Data and Code Availability
The ShinyApp allowing access to the data and network construction can be accessed at https://livedataoxford.shinyapps.io/Chick_NC_GRN-TSS-Lab/. The beta version of the ShinyApp and the associated code can be downloaded from Github: https://github.com/tsslab/chick_NC-GRN. The data and images associated with the ShinyApp can be downloaded from Figshare: https://figshare.com/articles/chick_NC-GRN_RData/6953294 and https://Figureshare.com/articles/chick_NC-GRN_images/6953306. The raw and processed data generated in this study have also been submitted to GEO: GSE121527).
Acknowledgments
We thank all members of the T.S.-S. lab for helpful discussions and Kevin Clark from the WIMM Flow Cytometry Facility for cell sorting assistance. Imaging was performed at the Wolfson Imaging Centre Oxford, Next-Generation sequencing at the MRC WIMM Sequencing Facility and the Oxford Genomics Centre, and single-cell RNA-seq at MRC WIMM Single-Cell Core Facility with help of Dr Neil Ashley. We are grateful to Toros Tasgin, Francesco Camera, and Joyce Lee for technical assistance. This work was supported by MRC (G0902418), The Lister Institute for Preventative Medicine Research Prize, John Fell Fund (131/038), and Leverhulme Trust (grant RPG-2015-026) to T.S.-S.. I.C.-F. received funds from the British Genetics Society “Genes and Development” studentship and a Brazilian Science Without Borders Fellowship (CNPq 205008/2013-5) and is currently funded by the Oxford-Angus McLeod-St. John’s College graduate fellowship and the WIMM prize studentship. I.T.C.L. was funded by NIHR Academic Clinical Fellowship in partnership with Oxford University Clinical Academic Graduate School (OUCAGS).
Author Contributions
Conceptualization, R.M.W., T.S.-S.; Methodology, R.M.W. and U.S.; Software, I.C.-F., E.R., D.G., J.T., and I.T.C.L; Validation, R.M.W.; Formal Analysis, R.M.W., I.C.-F., and T.S.-S.; Investigation, R.M.W.; Writing, R.M.W., I.C.-F., and T.S.-S.; Writing – Review & Editing, all authors; Visualization, R.M.W., I.C.-F., D.G., E.R., and T.S.-S.; Supervision, T.S.-S.; Funding Acquisition, T.S.-S.
Declaration of Interests
The authors declare no competing interests.
Published: October 21, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2019.10.003.
Supplemental Information
References
- Acampora D., Mazan S., Lallemand Y., Avantaggiato V., Maury M., Simeone A., Brûlet P. Forebrain and midbrain regions are deleted in Otx2-/- mutants due to a defective anterior neuroectoderm specification during gastrulation. Development. 1995;121:3279–3290. doi: 10.1242/dev.121.10.3279. [DOI] [PubMed] [Google Scholar]
- Adli M., Bernstein B.E. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat. Protoc. 2011;6:1656–1668. doi: 10.1038/nprot.2011.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguiar D.P., Sghari S., Creuzet S. The facial neural crest controls fore- and midbrain patterning by regulating Foxg1 expression through Smad1 activity. Development. 2014;141:2494–2505. doi: 10.1242/dev.101790. [DOI] [PubMed] [Google Scholar]
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoniadou C.L., Signore M., Sajedi E., Gaston-Massuet C., Kelberman D., Burns A.J., Itasaki N., Dattani M., Martinez-Barbera J.P. Lack of the murine homeobox gene Hesx1 leads to a posterior transformation of the anterior forebrain. Development. 2007;134:1499–1508. doi: 10.1242/dev.02829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggiolini A., Varum S., Mateos J.M., Bettosini D., John N., Bonalli M., Ziegler U., Dimou L., Clevers H., Furrer R., Sommer L. Premigratory and migratory neural crest cells are multipotent in vivo. Cell Stem Cell. 2015;16:314–322. doi: 10.1016/j.stem.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Barembaum M., Bronner M.E. Identification and dissection of a key enhancer mediating cranial neural crest specific expression of transcription factor, Ets-1. Dev. Biol. 2013;382:567–575. doi: 10.1016/j.ydbio.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basch M.L., Bronner-Fraser M., García-Castro M.I. Specification of the neural crest occurs during gastrulation and requires Pax7. Nature. 2006;441:218–222. doi: 10.1038/nature04684. [DOI] [PubMed] [Google Scholar]
- Betancur P., Bronner-Fraser M., Sauka-Spengler T. Assembling neural crest regulatory circuits into a gene regulatory network. Annu. Rev. Cell Dev. Biol. 2010;26:581–603. doi: 10.1146/annurev.cellbio.042308.113245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur P., Bronner-Fraser M., Sauka-Spengler T. Genomic code for Sox10 activation reveals a key regulatory enhancer for cranial neural crest. Proc. Natl. Acad. Sci. USA. 2010;107:3570–3575. doi: 10.1073/pnas.0906596107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V., Louis-Brennetot C., Peltier A., Durand S., Pierre-Eugène C., Raynal V., Etchevers H.C., Thomas S., Lermine A., Daudigeos-Dubus E. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017;49:1408–1413. doi: 10.1038/ng.3921. [DOI] [PubMed] [Google Scholar]
- Bogeas A., Morvan-Dubois G., El-Habr E.A., Lejeune F.X., Defrance M., Narayanan A., Kuranda K., Burel-Vandenbos F., Sayd S., Delaunay V. Changes in chromatin state reveal ARNT2 at a node of a tumorigenic transcription factor signature driving glioblastoma cell aggressiveness. Acta Neuropathol. 2018;135:267–283. doi: 10.1007/s00401-017-1783-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothma J.P., Garcia H.G., Ng S., Perry M.W., Gregor T., Levine M. Enhancer additivity and non-additivity are determined by enhancer strength in the Drosophila embryo. eLife. 2015;4 doi: 10.7554/eLife.07956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner M.E., Simões-Costa M. The neural crest migrating into the twenty-first century. Curr. Top. Dev. Biol. 2016;116:115–134. doi: 10.1016/bs.ctdb.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner-Fraser M., Fraser S.E. Cell lineage analysis reveals multipotency of some avian neural crest cells. Nature. 1988;335:161–164. doi: 10.1038/335161a0. [DOI] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitrago-Delgado E., Nordin K., Rao A., Geary L., LaBonne C. NEURODEVELOPMENT. Shared regulatory programs suggest retention of blastula-stage potential in neural crest cells. Science. 2015;348:1332–1335. doi: 10.1126/science.aaa3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavò E., Khoueiry P., Garfield D.A., Geeleher P., Zichner T., Gustafson E.H., Ciglar L., Korbel J.O., Furlong E.E. Shadow enhancers are pervasive features of developmental regulatory networks. Curr. Biol. 2016;26:38–51. doi: 10.1016/j.cub.2015.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter T.C., Kay D.M., Browne M.L., Liu A., Romitti P.A., Kuehn D., Conley M.R., Caggana M., Druschel C.M., Brody L.C., Mills J.L. Hirschsprung’s disease and variants in genes that regulate enteric neural crest cell proliferation, migration and differentiation. J. Hum. Genet. 2012;57:485–493. doi: 10.1038/jhg.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Costa M.L., Mermelstein C.S., Chagas C., Holtzer S., Holtzer H. MyoD converts primary dermal fibroblasts, chondroblasts, smooth muscle, and retinal pigmented epithelial cells into striated mononucleated myoblasts and multinucleated myotubes. Proc. Natl. Acad. Sci. USA. 1990;87:7988–7992. doi: 10.1073/pnas.87.20.7988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H.M.T., Schwarzkopf M., Fornace M.E., Acharya A., Artavanis G., Stegmaier J., Cunha A., Pierce N.A. Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development. 2018;145:dev165753. doi: 10.1242/dev.165753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudnovsky Y., Kim D., Zheng S., Whyte W.A., Bansal M., Bray M.A., Gopal S., Theisen M.A., Bilodeau S., Thiru P. ZFHX4 interacts with the NuRD core member CHD4 and regulates the glioblastoma tumor-initiating cell state. Cell Rep. 2014;6:313–324. doi: 10.1016/j.celrep.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway S.J., Henderson D.J., Copp A.J. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development. 1997;124:505–514. doi: 10.1242/dev.124.2.505. [DOI] [PubMed] [Google Scholar]
- Cunha P.M., Sandmann T., Gustafson E.H., Ciglar L., Eichenlaub M.P., Furlong E.E. Combinatorial binding leads to diverse regulatory responses: Lmd is a tissue-specific modulator of Mef2 activity. PLoS Genet. 2010;6:e1001014. doi: 10.1371/journal.pgen.1001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson E.H., Erwin D.H. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- Davies J.O., Telenius J.M., McGowan S.J., Roberts N.A., Taylor S., Higgs D.R., Hughes J.R. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods. 2016;13:74–80. doi: 10.1038/nmeth.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Calisto J., Araya C., Marchant L., Riaz C.F., Mayor R. Essential role of non-canonical Wnt signalling in neural crest migration. Development. 2005;132:2587–2597. doi: 10.1242/dev.01857. [DOI] [PubMed] [Google Scholar]
- de Laat W., Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502:499–506. doi: 10.1038/nature12753. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupin E., Calloni G.W., Coelho-Aguiar J.M., Le Douarin N.M. The issue of the multipotency of the neural crest cells. Dev. Biol. 2018;444(Suppl 1):S47–S59. doi: 10.1016/j.ydbio.2018.03.024. [DOI] [PubMed] [Google Scholar]
- Engler C., Gruetzner R., Kandzia R., Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J., Liu T., Qin B., Zhang Y., Liu X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferronha T., Rabadán M.A., Gil-Guiñon E., Le Dréau G., de Torres C., Martí E. LMO4 is an essential cofactor in the Snail2-mediated epithelial-to-mesenchymal transition of neuroblastoma and neural crest cells. J. Neurosci. 2013;33:2773–2783. doi: 10.1523/JNEUROSCI.4511-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein R., Perrimon N. The molecular genetics of head development in Drosophila melanogaster. Development. 1991;112:899–912. doi: 10.1242/dev.112.4.899. [DOI] [PubMed] [Google Scholar]
- Georgescu C., Longabaugh W.J., Scripture-Adams D.D., David-Fung E.S., Yui M.A., Zarnegar M.A., Bolouri H., Rothenberg E.V. A gene regulatory network armature for T lymphocyte specification. Proc. Natl. Acad. Sci. USA. 2008;105:20100–20105. doi: 10.1073/pnas.0806501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen N., Futtner C.R., Wood S., Garcia-Moreno S.A., Salamone I.M., Samson S.C., Sekido R., Poulat F., Maatouk D.M., Lovell-Badge R. Sex reversal following deletion of a single distal enhancer of Sox9. Science. 2018;360:1469–1473. doi: 10.1126/science.aas9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouti M., Delile J., Stamataki D., Wymeersch F.J., Huang Y., Kleinjung J., Wilson V., Briscoe J. A gene regulatory network balances neural and mesoderm specification during vertebrate trunk development. Dev. Cell. 2017;41:243–261. doi: 10.1016/j.devcel.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V., Hamilton H.L. A series of normal stages in the development of the chick embryo. J. Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Hay D., Hughes J.R., Babbs C., Davies J.O.J., Graham B.J., Hanssen L., Kassouf M.T., Marieke Oudelaar A.M., Sharpe J.A., Suciu M.C. Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet. 2016;48:895–903. doi: 10.1038/ng.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J.R., Roberts N., McGowan S., Hay D., Giannoulatou E., Lynch M., De Gobbi M., Taylor S., Gibbons R., Higgs D.R. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet. 2014;46:205–212. doi: 10.1038/ng.2871. [DOI] [PubMed] [Google Scholar]
- Joshi N.F.J. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. 2011. https://github.com/najoshi/sickle
- Junion G., Spivakov M., Girardot C., Braun M., Gustafson E.H., Birney E., Furlong E.E. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell. 2012;148:473–486. doi: 10.1016/j.cell.2012.01.030. [DOI] [PubMed] [Google Scholar]
- Karunasena E., Mciver L.J., Bavarva J.H., Wu X., Zhu H., Garner H.R. ‘Cut from the same cloth’: Shared microsatellite variants among cancers link to ectodermal tissues-neural tube and crest cells. Oncotarget. 2015;6:22038–22047. doi: 10.18632/oncotarget.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsh R.N. Sorting out Sox10 functions in neural crest development. BioEssays. 2006;28:788–798. doi: 10.1002/bies.20445. [DOI] [PubMed] [Google Scholar]
- Kharchenko P.V., Silberstein L., Scadden D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods. 2014;11:740–742. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde, R. (2018). pheatmap: Pretty Heatmaps. R package version 1.0.10.
- Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018;46(D1):D252–D259. doi: 10.1093/nar/gkx1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Zhang B., Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008;24:719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin N.M., Kalcheim C. Second Edition. Cambridge University Press; 1999. The Neural Crest. [Google Scholar]
- Le Lievre C.S., Schweizer G.G., Ziller C.M., Le Douarin N.M. Restrictions of developmental capabilities in neural crest cell derivatives as tested by in vivo transplantation experiments. Dev. Biol. 1980;77:362–378. doi: 10.1016/0012-1606(80)90481-9. [DOI] [PubMed] [Google Scholar]
- Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K., Hannett N.M., Harbison C.T., Thompson C.M., Simon I. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Levine M., Davidson E.H. Gene regulatory networks for development. Proc. Natl. Acad. Sci. USA. 2005;102:4936–4942. doi: 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M., Cattoglio C., Tjian R. Looping back to leap forward: transcription enters a new era. Cell. 2014;157:13–25. doi: 10.1016/j.cell.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Longabaugh W.J., Davidson E.H., Bolouri H. Computational representation of developmental genetic regulatory networks. Dev. Biol. 2005;283:1–16. doi: 10.1016/j.ydbio.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R., Bradner J.E., Lee T.I., Young R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoseviciute M., Gavriouchkina D., Williams R.M., Hochgreb-Hagele T., Senanayake U., Chong-Morrison V., Thongjuea S., Repapi E., Mead A., Sauka-Spengler T. From pioneer to repressor: bimodal foxd3 activity dynamically remodels neural crest regulatory landscape in vivo. Dev. Cell. 2018;47:608–628. doi: 10.1016/j.devcel.2018.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T., Bach K., Marioni J.C. Pooling across cells to normalize single-cell rNA sequencing data with many zero counts. Genome Biol. 2016;17:75. doi: 10.1186/s13059-016-0947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony S., Benos P.V. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007;35:W253–W258. doi: 10.1093/nar/gkm272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D.J., Campbell K.R., Lun A.T., Wills Q.F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017;33:1179–1186. doi: 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulemans D., Bronner-Fraser M. Gene-regulatory interactions in neural crest evolution and development. Dev. Cell. 2004;7:291–299. doi: 10.1016/j.devcel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., Thomas P.D. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45(D1):D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- Murko C., Vieceli F.M., Bronner M. Transcriptome dataset of trunk neural crest cells migrating along the ventral pathway of chick embryos. Data Brief. 2018;21:2547–2553. doi: 10.1016/j.dib.2018.11.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J., Davidson E.H. Barcoded DNA-tag reporters for multiplex cis-regulatory analysis. PLoS One. 2012;7:e35934. doi: 10.1371/journal.pone.0035934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitzan E., Krispin S., Pfaltzgraff E.R., Klar A., Labosky P.A., Kalcheim C. A dynamic code of dorsal neural tube genes regulates the segregation between neurogenic and melanogenic neural crest cells. Development. 2013;140:2269–2279. doi: 10.1242/dev.093294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble W.S. How does multiple testing correction work? Nat. Biotechnol. 2009;27:1135–1137. doi: 10.1038/nbt1209-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa S.D., Salvador S., LaBonne C. The LIM adaptor protein LMO4 is an essential regulator of neural crest development. Dev. Biol. 2012;361:313–325. doi: 10.1016/j.ydbio.2011.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesnicky Killian E.C., Birkholz D.A., Artinger K.B. A role for chemokine signaling in neural crest cell migration and craniofacial development. Dev. Biol. 2009;333:161–172. doi: 10.1016/j.ydbio.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder M., Barozzi I., Tissières V., Fukuda-Yuzawa Y., Mannion B.J., Afzal S.Y., Lee E.A., Zhu Y., Plajzer-Frick I., Pickle C.S. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature. 2018;554:239–243. doi: 10.1038/nature25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peukert D., Weber S., Lumsden A., Scholpp S. Lhx2 and Lhx9 determine neuronal differentiation and compartition in the caudal forebrain by regulating Wnt signaling. PLoS Biol. 2011;9:e1001218. doi: 10.1371/journal.pbio.1001218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Preger-Ben Noon E., Sabarís G., Ortiz D.M., Sager J., Liebowitz A., Stern D.L., Frankel N. Comprehensive analysis of a cis-regulatory region reveals pleiotropy in enhancer function. Cell Rep. 2018;22:3021–3031. doi: 10.1016/j.celrep.2018.02.073. [DOI] [PubMed] [Google Scholar]
- Prescott S.L., Srinivasan R., Marchetto M.C., Grishina I., Narvaiza I., Selleri L., Gage F.H., Swigut T., Wysocka J. Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell. 2015;163:68–83. doi: 10.1016/j.cell.2015.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A., Bajpai R., Prescott S., Brugmann S.A., Swigut T., Wysocka J. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell. 2012;11:633–648. doi: 10.1016/j.stem.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44(W1):W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson V., Smith A., Flenniken A.M., Wilkinson D.G. Roles of Eph receptors and ephrins in neural crest pathfinding. Cell Tissue Res. 1997;290:265–274. doi: 10.1007/s004410050931. [DOI] [PubMed] [Google Scholar]
- Sandmann T., Girardot C., Brehme M., Tongprasit W., Stolc V., Furlong E.E. A core transcriptional network for early mesoderm development in Drosophila melanogaster. Genes Dev. 2007;21:436–449. doi: 10.1101/gad.1509007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauka-Spengler T., Barembaum M. Gain- and loss-of-function approaches in the chick embryo. Methods Cell Biol. 2008;87:237–256. doi: 10.1016/S0091-679X(08)00212-4. [DOI] [PubMed] [Google Scholar]
- Sauka-Spengler T., Bronner-Fraser M. A gene regulatory network orchestrates neural crest formation. Nat. Rev. Mol. Cell Biol. 2008;9:557–568. doi: 10.1038/nrm2428. [DOI] [PubMed] [Google Scholar]
- Schulz Y., Wehner P., Opitz L., Salinas-Riester G., Bongers E.M., van Ravenswaaij-Arts C.M., Wincent J., Schoumans J., Kohlhase J., Borchers A., Pauli S. CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum. Genet. 2014;133:997–1009. doi: 10.1007/s00439-014-1444-2. [DOI] [PubMed] [Google Scholar]
- Simões-Costa M., Bronner M.E. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142:242–257. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes-Costa M., Bronner M.E. Reprogramming of avian neural crest axial identity and cell fate. Science. 2016;352:1570–1573. doi: 10.1126/science.aaf2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Costa M.S., McKeown S.J., Tan-Cabugao J., Sauka-Spengler T., Bronner M.E. Dynamic and differential regulation of stem cell factor FoxD3 in the neural crest is Encrypted in the genome. PLoS Genet. 2012;8:e1003142. doi: 10.1371/journal.pgen.1003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Costa M., Tan-Cabugao J., Antoshechkin I., Sauka-Spengler T., Bronner M.E. Transcriptome analysis reveals novel players in the cranial neural crest gene regulatory network. Genome Res. 2014;24:281–290. doi: 10.1101/gr.161182.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene P.J., Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife. 2017;6:e21856. doi: 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.L., Tallquist M.D. PDGF function in diverse neural crest cell populations. Cell Adhes. Migr. 2010;4:561–566. doi: 10.4161/cam.4.4.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J., Theodoris C., Davidson E.H. A gene regulatory network subcircuit drives a dynamic pattern of gene expression. Science. 2007;318:794–797. doi: 10.1126/science.1146524. [DOI] [PubMed] [Google Scholar]
- Spitz F., Gonzalez F., Duboule D. A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell. 2003;113:405–417. doi: 10.1016/s0092-8674(03)00310-6. [DOI] [PubMed] [Google Scholar]
- Stark R.B., G. D. DiffBind: differential binding analysis of ChIP-seq peak data. 2011. http://bioconductor.org/packages/release/bioc/html/DiffBind.html Bioconductor.
- Talbot D., Collis P., Antoniou M., Vidal M., Grosveld F., Greaves D.R. A dominant control region from the human beta-globin locus conferring integration site-independent gene expression. Nature. 1989;338:352–355. doi: 10.1038/338352a0. [DOI] [PubMed] [Google Scholar]
- Trainor P.A. Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am. J. Med. Genet. A. 2010;152A:2984–2994. doi: 10.1002/ajmg.a.33454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadasz S., Marquez J., Tulloch M., Shylo N.A., García-Castro M.I. Pax7 is regulated by cMyb during early neural crest development through a novel enhancer. Development. 2013;140:3691–3702. doi: 10.1242/dev.088328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi M.P., Agarwal P., Brown C., McCulley D.J., Schwarz J.J., Black B.L. The transcription factor MEF2C is required for craniofacial development. Dev. Cell. 2007;12:645–652. doi: 10.1016/j.devcel.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waimey K.E., Huang P.H., Chen M., Cheng H.J. Plexin-A3 and plexin-A4 restrict the migration of sympathetic neurons but not their neural crest precursors. Dev. Biol. 2008;315:448–458. doi: 10.1016/j.ydbio.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.M., Senanayake U., Artibani M., Taylor G., Wells D., Ahmed A.A., Sauka-Spengler T. Genome and epigenome engineering CRISPR toolkit for in vivo modulation of cis-regulatory interactions and gene expression in the chicken embryo. Development. 2018;145:dev160333. doi: 10.1242/dev.160333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrick J.J., Young R.A. Deciphering gene expression regulatory networks. Curr. Opin. Genet. Dev. 2002;12:130–136. doi: 10.1016/s0959-437x(02)00277-0. [DOI] [PubMed] [Google Scholar]
- Ye T., Krebs A.R., Choukrallah M.A., Keime C., Plewniak F., Davidson I., Tora L. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011;39:e35. doi: 10.1093/nar/gkq1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzen R.P., Girardot C., Gagneur J., Braun M., Furlong E.E. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- Zhan X., Liu D.J. SEQMINER: an R-Package to Facilitate the Functional Interpretation of Sequence-Based Associations. Genet. Epidemiol. 2015;39:619–623. doi: 10.1002/gepi.21918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G.X., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Zhang S., Jin Y. Foxd3 suppresses NFAT-mediated differentiation to maintain self-renewal of embryonic stem cells. EMBO Rep. 2014;15:1286–1296. doi: 10.15252/embr.201438643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ShinyApp allowing access to the data and network construction can be accessed at https://livedataoxford.shinyapps.io/Chick_NC_GRN-TSS-Lab/. The beta version of the ShinyApp and the associated code can be downloaded from Github: https://github.com/tsslab/chick_NC-GRN. The data and images associated with the ShinyApp can be downloaded from Figshare: https://figshare.com/articles/chick_NC-GRN_RData/6953294 and https://Figureshare.com/articles/chick_NC-GRN_images/6953306. The raw and processed data generated in this study have also been submitted to GEO: GSE121527).