

Machine learning can be a powerful tool to prescreen new materials, thus accelerating the development of the OPV field.
Abstract
In the process of finding high-performance materials for organic photovoltaics (OPVs), it is meaningful if one can establish the relationship between chemical structures and photovoltaic properties even before synthesizing them. Here, we first establish a database containing over 1700 donor materials reported in the literature. Through supervised learning, our machine learning (ML) models can build up the structure-property relationship and, thus, implement fast screening of OPV materials. We explore several expressions for molecule structures, i.e., images, ASCII strings, descriptors, and fingerprints, as inputs for various ML algorithms. It is found that fingerprints with length over 1000 bits can obtain high prediction accuracy. The reliability of our approach is further verified by screening 10 newly designed donor materials. Good consistency between model predictions and experimental outcomes is obtained. The result indicates that ML is a powerful tool to prescreen new OPV materials, thus accelerating the development of the OPV field.
INTRODUCTION
Organic photovoltaic (OPV) cells provide a direct and economical way to transform solar energy into electricity. Recently, OPV research has undergone a rapid growth, and the power conversion efficiency (PCE) has exceeded 17% (1, 2). Until the present time, the mainstream of OPV research has focused on building up the relationship between a new OPV molecular structure and its photovoltaic properties. This process usually involves design and synthesis of photovoltaic materials, characterization of the optoelectronic properties of the material, as well as assembly and optimization of the photovoltaic cells. Such a traditional approach requires delicate control of chemical synthesis and device fabrication, laborious purification and experimental steps, substantial resource input, and a long research cycle. Thus, the OPV development is inefficient and slow, e.g., only less than 2000 OPV donor molecules have been synthesized and tested in a photovoltaic cell since the first report in 1973 (3). Nevertheless, these data, generated from decades of exploration, are priceless. Unfortunately, until now their potential value has not been fully exploited when searching for high-performance OPV materials.
To extract useful information from the data, a sophisticated program that can scan through a large dataset and extract relationships among the features is required. Machine learning (ML) (4) provides a set of computational tools that are capable of learning and recognizing patterns and relationships, predicting outcomes or making decisions, and reducing the size of a dataset, based on error (or loss function) minimization or probabilistic rules (e.g., maximizing a likelihood) using a training dataset (5). This data-driven approach enables ML to predict a wide range of material properties without the need for fundamental understanding of the chemistry or physics behind these properties (6). In recent years, ML-based methods have found great success in the prediction of the activity/properties of materials (7, 8), material discovery (9, 10), drug development (11), and material design (12). Appropriate expressions of chemical structures are another prerequisite for applying ML to materials discovery. In this regard, the development of cheminformatics (13) has established a useful toolbox that predates the application of ML. For example, molecular fingerprints emerged along with the development of similarity searching in medicinal chemistry in the 1980s (14).
Application of ML to the OPV field has also been explored in recent years (6, 15, 16). For example, Pyzer-Knapp et al. (17) trained an artificial neural network (ANN) to predict the frontier molecular orbitals and obtained a good accuracy. Their data were extracted from the Harvard Clean Energy Project (CEP) (18), in which the chemical structures of these molecules were generated from 26 basic building blocks by theoretical calculations. We used a convolutional neural network (CNN) and the data from the CEP to predict the PCE and achieved 91.02% prediction accuracy (19). We further proved that the CNN was capable of extracting features from pictures of chemical structures. Nagasawa et al. (20) established a database of polymer-fullerene–based OPV devices containing approximately 1000 experimentally tested materials. Using molecular access system (MACCS) fingerprints and the value of the highest occupied molecular orbital, the bandgap, and the molecular weight for the description of the molecules, their four-class classification model based on the random forest (RF) method for PCE prediction obtained an accuracy of 48%. Notably, the accuracy of the prediction could be substantially improved by using the xyz coordinates (21), combing the electronic and structural features (16), or using improved descriptors (15). For example, recently, Sahu et al. (15) adopted 13 microscopic properties of organic materials as descriptors for the prediction of PCE. Their model was able to build up a relationship between the molecule’s properties and PCE with a correlation of r = 0.79. However, there remain several drawbacks to the application of ML for screening OPV materials. For example, the chemical structures of realistic molecules reported in the literature are usually much more complicated when compared with those in the CEP (18). The differences in structure may lead to inaccurate ML predictions. Furthermore, the microscopic properties of molecules are primarily obtained from high-accuracy quantum calculations. The high computational costs of these calculations render them incompatible for large-scale fast virtual screening. Therefore, to achieve rapid screening and high prediction accuracy simultaneously, a sufficiently accurate and easily accessible programming language expression of molecules is urgently needed. In addition, a more general model is desirable to incorporate more realistic molecules, so that the results from ML can be used reliably for new material design. Moreover, the reliability of the ML methods should be verified by experiment using new materials, especially in the early stages of this new approach.
In this work, we established a database containing 1719 experimentally tested OPV donor materials collected from the literature. We first studied the importance of programming language expression of molecules for ML performance. To determine the most suitable one, we tested different types of expressions, including images, ASCII strings, two types of descriptors, and seven types of molecular fingerprints. The descriptors are used to classify materials into “low” and “high” performance based on the PCE value. Fingerprints led to the best performance (81.76% accuracy in predicting the PCE class), and their length had a notable influence on the accuracy of the predictions. Moreover, we used a variety of ML algorithms for the classification. RF models outperformed others when dealing with a small database in our scenario. Last, we independently verified the ML models by synthesizing 10 new OPV donor materials [9 of them have not been reported before, and the remaining one was reported very recently (22)]. The predictions of the model were in good agreement with the experimental results. Through this work, we set up a new methodology for OPV research, i.e., prescreening the designed OPV molecules by ML models and then only focusing on those that passed the ML virtual assessment in subsequent experiments. This approach will greatly accelerate the process of developing new, highly efficient organic semiconducting materials for OPV applications.
METHODS
ML algorithms
Five types of supervised ML algorithms were used in this study, including back propagation (BP) neural network (BPNN) (23), deep neural network (DNN) (24), deep learning (25), support vector machine (SVM) (26, 27), and RF (28, 29). These are advanced algorithms, and brief descriptions with additional details are provided in the Supplementary Materials. Among these methods, BPNN, DNN, and deep learning are based on the ANN (25, 30).
Database
The database contains 1719 realistic OPV donor materials collected from the literature. To obtain a more general model, polymers and small molecules were mixed together in the database. Whether the acceptor is a fullerene or a nonfullerene was also ignored. If a certain donor material has been reported several times, the highest PCE is chosen. All these criteria ensured the model can learn the maximum potential of a certain material.
In the established database, the median value of PCE is 2.82%, and the average value is 3.48%. As shown in Fig. 1A, the number of data points in the PCE range above 8% was small. To obtain an unbiased model, the number of data points in each of the two categories needs to be balanced, i.e., making the number of molecules in both categories roughly equal. Thus, we split the data into two categories (Fig. 1A) and selected 3% as a preliminary threshold. The molecules with a PCE in the range of 0 to 2.99% were regarded as “low performance” (represented in green in Fig. 1A), while those with a PCE higher than 3.00% (represented in purple in Fig. 1A) were regarded as “high-performance” OPV molecules.
Fig. 1. Information about our database of OPV donor materials.
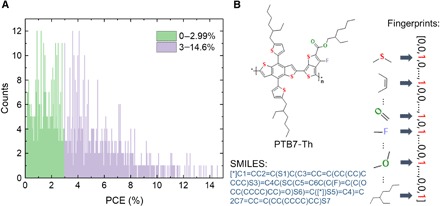
(A) Distribution of PCE values of the 1719 molecules in our database. (B) Schematics of expressions of a molecule, including image, simplified molecular-input line-entry system (SMILES), and fingerprints.
To discuss how the threshold of PCE influences the prediction accuracy, a higher threshold (10%) was selected to construct a new database. There are 48 molecules whose PCE values are more than 10%. To maintain a balance of the two categories, we selected 52 molecules randomly through stratified sampling from samples whose PCE values are below 10%. Consequently, the database with 10% as threshold contains 100 molecules.
Datasets for training and testing
When using 3% as threshold, around 90% (1549 molecules) and 10% (170 molecules) of the data were divided into independent training and testing subsets, respectively. The training subsets were used to train the models, i.e., establishing a relationship between the structure and the PCE. The testing subsets were used to test the models, i.e., to determine a prediction accuracy for the trained models. The two subsets are independent of each other. The same ratio (9, 1) was used to divide training and testing subsets when using 10% as threshold. To perform cross-validation while training our models, three databases containing different molecules in their respective training and testing subsets were used for each threshold.
Programming language expression of molecules
As shown in Fig. 1B and tables S1 and S2, various expressions of a molecule, including images, ASCII strings, two types of descriptors, and seven types of molecular fingerprints, were used as input for the ML models to predict the PCE. All the machine description language of molecules considered in this work is easily accessible, allowing for the rapid screening of a large number of donor materials.
Image is an intuitive expression of a material. The simplified molecular-input line-entry system (SMILES) (31) that describes the structure of chemical species using short ASCII strings was transformed from the monomer of a polymer or from the small molecule. Notably, this string is a sequence of characters composed of letters and symbols, which are not suitable for an ML algorithm. Thus, we converted each character to its corresponding ASCII value and then obtained a string of numbers. Technically, all strings should have the same length for the input of ML models. To this end, “0” was added at the end of the short strings. Descriptors (32) that contain molecular properties and fingerprints (33) that reflect the substructures as well as special patterns can be generated from SMILES. All the descriptors and fingerprints were obtained through ChemDes (an online transformer) (34).
RESULTS
Importance of programming language expressions for donor materials in modeling
The programming language expression is one of the important aspects in the ML approach as it transforms the raw data into a machine-readable representation (25). Various expressions for the same molecule comprise vastly different chemical information, or this information is presented in different abstract levels. A desirable form of expression should cover almost all the features of the molecule but contain no redundant information. Here, a set of ML models are used to explore the different expressions of a molecule by comparing their predicted accuracy for the PCE.
The image of a chemical structure is a direct and original expression of a molecule (Fig. 1B). However, features connected with PCE are not reflected in an image and are regarded as hidden features. To overcome this problem, we use deep learning, which can extract features from images. The confusion matrix shown in Fig. 2A indicates the performance of the deep learning model. The predicted accuracies of the best-performing deep learning model for the first (0 to 2.99%) and second (above 3.00%) categories are 70.79 and 67.90%, respectively. The overall accuracy is 69.41%. The unsatisfactory performance of the deep learning model with image as expression is attributed to the small size of our database (a typical feature of deep learning models is that they require large training sets). When the number of molecules in the database reaches 50,000, the accuracy of the deep learning model can exceed 90% (19). To fully train a deep learning model usually requires a large database containing millions of samples (35, 36). Here, each category only has hundreds of molecules, making it difficult for the model to extract enough information to achieve high accuracy. Fine-tuning a pretrained model (36) can considerably reduce the amount of data required, but thousands of samples are still needed to provide a sufficient number of features. Therefore, increasing the size of the database is one of the solutions when using images to express molecules.
Fig. 2. Testing results of ML models.
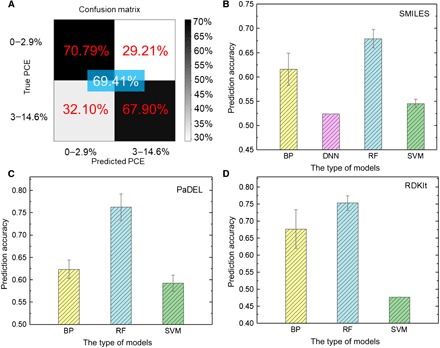
(A) Testing of the deep learning model using images as input. (B to D) Testing results of different ML models using (B) SMILES, (C) PaDEL, and (D) RDKIt descriptors as input.
The SMILES code provides another original expression for a molecule (Fig. 1B) (31). Through a traversal over the whole chemical structure, a string that contains the information on atoms, bonds, rings, aromaticity, and branches can be obtained based on established rules. The results of using SMILES as inputs for BP, DNN, RF, and SVM models are shown in Fig. 2B. The average accuracies through cross-validation of all the four methods are low; the highest one, achieved by the RF model, is only 67.84%. There are two possible reasons: (i) SMILES is still close to raw data, and unlike deep learning, the four classic ML methods do not have the ability to extract hidden features. As will be shown later, a further conversion, e.g., to fingerprints, is needed for these classic ML methods. (ii) As mentioned above, 0 is added to keep the length for SMILES for different molecules. These 0s may affect the process of building logical relationships in the models. Thus, SMILES performs worse than images as descriptors of the molecules for predicting the PCE class.
Molecular descriptors describe the properties of a molecule using an array of real numbers rather than expressing the chemical structure directly (32). Here, two kinds of descriptors (PaDEL and RDKIt) that have different sizes of data are used. The PaDEL descriptor (table S1) (37) consists of 1875 different types of descriptors, which can be defined as one-dimensional (1D) descriptors (i.e., the number of certain groups or atoms), 2D descriptors (i.e., graph invariants and molecular properties), and 3D descriptors (i.e., geometry). Figure 2C depicts the results using a PaDEL descriptor as input for the BP, RF, and SVM models. The RF model attained the best performance (average accuracy as high as 76.27%), far superior to the BP and SVM models. It needs to be noted that our DNN model cannot process a long array of real numbers in this experiment. The RDKIt descriptor (38) (196 bits; table S2) is much shorter than the PaDEL descriptor (1875 bits). The shorter length of the RDKIt descriptor implies it contains less information. The results of using RDKIt as the input are shown in Fig. 2D. The RF models again attain the best performance. However, the prediction accuracy of RDKIt (75.29%) is only 1% worse than that of PaDEL (76.27%). In contrast, the accuracy of RDKIt (67.65%) for the BPNN model is better than that of the PaDEL (62.35%). The accuracy of RDKIt for the SVM model is 47.65%, less than 50% (random classification), suggesting that SVM cannot establish a logical relationship between RDKIt descriptors and PCE. These results indicate that a large data size implies more descriptors that are not relevant to PCE, which will affect the ANN performance. In addition, a small data dimension means that the chemical information is insufficient to train SVM models effectively. Therefore, looking for appropriate descriptors directly related to the target object is the key when using molecular descriptors as inputs in ML approaches.
Molecular fingerprints are designed for large-scale database screening and take the form of an array of bits (39). They contain “1”s and “0”s to describe the presence or absence of particular substructures/patterns in the molecule. Here, seven types of fingerprints are used as inputs to train the BPNN, DNN, RF, and SVM models. The influence of the fingerprint length on the prediction performance of different models is also considered. The results of using different types of fingerprints as inputs are summarized in Fig. 3.
Fig. 3. Performance of ML models.
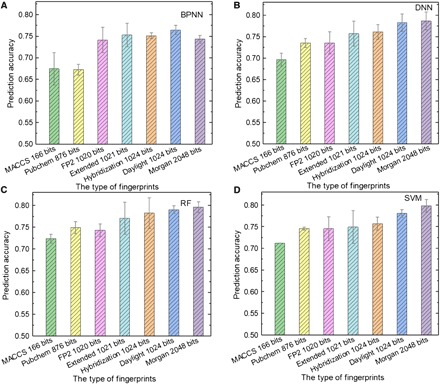
(A to D) The testing results of (A) BPNN, (B) DNN, (C) RF, and (D) SVM using different types of fingerprints as input.
MACCS fingerprints (40) have 166 bits, making them the shortest. Although it is short, the similarity of fingerprints among different molecules is relatively small. For example, both P3HT and PTB7 have 166 bits in total, and 26 bits of content in the fingerprints are different, leading to a “degree of difference” of 15.66% (the complete MACCS fingerprints are shown in table S3). However, the results of using MACCS fingerprints as the input are unsatisfactory (the highest average accuracy achieved by the RF model is only 72.35%) because of the limited information they contain. PubChem fingerprints (41) have 876 bits, longer than MACCS. However, the differences between molecules for PubChem are small. For instance, the degree of difference is 10.39% for P3HT and PTB7, implying most of the bits are the same for these two materials. The small difference among molecules suggests that the substructures described by PubChem exist in most of the molecules, and models will struggle to identify the difference among molecules. Although an RF model can obtain an average accuracy of 74.90%, we cannot conclude that the PubChem fingerprints are suitable as an expression of a molecule for screening OPV donor materials.
The FP2 fingerprint (42) has 1020 bits, and it is a path-based fingerprint that indexes small-molecule fragments based on linear segments up to seven atoms. The performances of the four ML methods are stable and satisfactory. The SVM model has the highest average accuracy of 74.51% (Fig. 3D). In addition, the Extended fingerprint (1021 bits) is an extension of the Chemistry Development Kit fingerprint (43), with additional bits describing ring features. The prediction results for the Extended fingerprints are similar to those for the FP2 fingerprints. The best-performing approach is obtained using the RF method (Fig. 3C), attaining an average accuracy of 77.06%.
Both Daylight (44) and Hybridization fingerprints (43) have 1024 bits, but the information expressed within these two fingerprints is quite different. Daylight fingerprints represents the pattern for each atom and its nearest neighbors. Hybridization fingerprints takes into account SP2 hybridization states rather than aromaticity. However, the verification results are similar for these two fingerprints used as inputs (Fig. 3). The highest average accuracies (obtained by the RF models) for the Daylight and Hybridization fingerprints are 79.02 and 78.24%, respectively. We point out that the best combination of programming language expression and ML algorithm over all models is obtained with the Hybridization fingerprint and RF, which achieves a prediction accuracy of 81.76%. Moreover, it is observed that the prediction performances of the FP2, Extended, Daylight, and Hybridization fingerprints are close to each other. These fingerprints are organized with different rules of representation but have similar lengths (around 1000 bits). The similar prediction performance of different fingerprints with almost the same length indicates that the fingerprint length, rather than the contents of the fingerprints, has a notable impact on the prediction of PCE.
The Morgan fingerprint (45) is the longest, having 2048 bits. For the BPNN model, the Morgan fingerprint performs poorer than most of the fingerprints with lengths around 1000 bits. Notably, the other ML models still have satisfactory results, and the highest average accuracy of 79.80% is obtained with the SVM model.
From the results described above, we can conclude that, generally, the performances of all ML models improve when the fingerprint length increases from 166 to 1024 bits. This is understandable since more chemical information is included in longer fingerprints. In particular, DNN, RF, and SVM models can establish an accurate relationship between the chemical structure and PCE when the length of the fingerprint exceeds 1000 bits, while BPNN performs the best with fingerprints whose length is around 1000 bits. This may be due to the relatively poor data processing capability of BPNN, as activation functions used in BPNN are imperfect (more details are described in the Supplementary Materials). A long fingerprint carries much more information than BPNN requires, which may “mislead” the model, causing too much pressure on computation (making the model difficult to converge). In addition, the overall results suggest that molecular fingerprints with lengths above 1000 bits are the most suitable and effective inputs for building ML models to predict the PCE, owing to their ease of accessibility and the abundance of chemical information they contain.
Considering that a higher threshold value of ML models is more meaningful when designing highly efficient materials, we increased the threshold from 3 to 10%. As mentioned earlier in Methods, an increase in the threshold will reduce the number of molecules in the database. We trained RF models with Daylight fingerprints as the input. When the threshold is set at 10%, the average prediction accuracy is 86.67%, but the SD is large (±11.58%), which may be due to the small database that contains only 100 molecules.
Screening for high PCE donor material via ML
To efficiently predict the PCE of donor materials, four ML methods are used, and their performance for different machine language expressions are summarized in Fig. 4A. The RF method performs the best, because its strategy is to choose multiple features rather than all features from the input for establishing the relationship (46), which is advantageous when dealing with complex and long inputs. For example, only the RF model performs well when using SMILES, PaDEL, and RDKIt descriptors to represent materials.
Fig. 4. Verification of ML models with experiment.
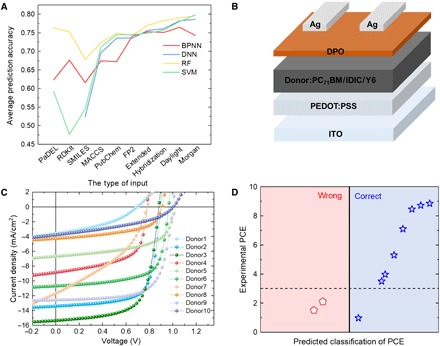
(A) Comparison of the results from four different models. (B) Schematic diagram of the cell architecture used in this study. (C) J-V curve of the solar cell with the active layer using the predicted donor material. (D) Prediction results versus experimental data for the predicted donor materials with the RF algorithm and Daylight fingerprints.
To further verify the reliability of our ML models, we designed 10 new small molecular donor materials (D1 to D10, whose chemical structures are available in fig. S2). The OPV fabrication process can be found in the Supplementary Materials. To the best of our knowledge, nine of them have not been reported yet, and one was published very recently (22). Originating from the well-studied A-π-D-π-A structure and the highly efficient BTR molecule developed by us (47), these 10 donor materials can be divided into three groups with variations in the A (end group), π (link), D (core), and side-chain groups. Donors D1, D2, D6, and D9 have the same π-D-π structure but different A moieties, while donors D3, D4, and D5 have chlorination or alkyl chain modification on the D part. In donors D7, D8, and D10, the π links were modified.
As shown in Fig. 4B, the OPV devices are based on a typical normal cell architecture. D3 and D7 used IDIC or Y3 as acceptors, respectively, while the other eight donors used PC71BM as acceptors. The donor:acceptor blend film is sandwiched in between a poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS)–coated indium tin oxide (ITO) transparent anode and a [2-(1,10-phenanthrolin-3-yl)naphth-6-yl]diphenylphosphine oxide (DPO) electron transport layer. Ag was used as a back cathode. After fabrication, these devices were tested under AM1.5G illumination in ambient to investigate their photovoltaic performance. The current density−voltage (J-V) curves of the OPV devices are displayed in Fig. 4C, and the photovoltaic performance parameters are summarized in table S4.
Before the experiment, we used our RF models with 3% as threshold to evaluate these 10 materials. Three representative fingerprints, i.e., FP2, Hybridization, and Daylight, were selected to express the chemical structure of the 10 new molecules. The results are displayed in table S5. The comparison between the prediction results by the RF model and the experimental PCE values is shown in Fig. 4D. Eight of 10 molecules are classified into the correct category, while two materials (D8 and D10) that exhibited low PCE (less than 3%) are classified into the category with the PCE range of above 3%. It is noted that the prediction result signifies the potential of a material for OPV application. So, these two materials may be further improved by optimizing the experimental conditions.
In addition, these 10 new materials have also been evaluated by the model using 10% as threshold. The prediction results are displayed in table S6 and fig. S3. The model with 10% as threshold can classify eight molecules into the correct category. In general, the predicted PCE classes are in good agreement with the experimental results. Experiment outcomes indicate that a minor change in structure can bring about a large difference in PCE values. Encouragingly, these minor modifications can be identified by an optimized ML model, thus leading to favorable prediction results. Although the ML model produces a prediction through comparing similarities, we believe the features of similarities learned by the models are complex. It is not merely the structural similarity, but perhaps it contains abstract features such as the location and connection of various substructures.
DISCUSSION
In summary, on the basis of a database containing realistic donor materials collected from the literature, various programming language expressions of donor molecules including images, ASCII strings, descriptors, and molecular fingerprints are used to build ML models to predict the corresponding OPV PCE class. The molecular fingerprints with lengths above 1000 bits provide the best programming language expressions of donor molecules due to their distinctness and ease of accessibility. The RF algorithm is found to be able to handle complex and long inputs, even in the presence of noise. This is because an RF chooses multiple features rather than the complete content of the input to establish the relationship. Last, an experiment was designed to prove the reliability of our ML approaches. We compared the prediction from the ML models and the results of the experiment for 10 new design small molecules. The ML predictions are consistent with experimental values with minor differences. We have developed a scheme to help OPV donor material design by combing ML approaches and experimental analysis. That is, a large number of donor materials could be screened through a preevaluation and classification by our ML model, and then the identified leading candidates will be synthesized and further tested by experiment. Our study on the relationship between the chemical structure of molecule and PCE of the molecule-based OPV could speed up new donor material design and hence accelerate the development of high PCE OPVs.
The greatest value of ML here and in other fields is the savings in time and resources. We envisage ML as an aid to guide experiments, e.g., rapidly evaluating very high numbers of new materials, which is not feasible with traditional experiments or ab initio models, to propose candidates for further laboratory analysis. ML is a tool that should be used in conjunction with the experiment, continually refined to incorporate new data. The use of the two together complimentarily is what can progress the material discovery.
Supplementary Material
Acknowledgments
We are grateful to the researchers who proposed and developed the theory of cheminformatics. We thank the programmers who developed the open-source cheminformatics software and ML tools. We thank K. Wang, W. Meng, C. Wang, W. Chen, F. Wu, J. Fu, R. Chen, K. Yang, L. Hu, Z. Xiong, J. Xi, and H. Han for assistance in constructing the database. Special thanks to academician Y. Li from the Institute of Chemistry, Chinese Academy of Sciences for the valuable suggestions. Funding: This research is supported by research grants from the National Youth Thousand Program Project (R52A199Z11), the National Special Funds for Repairing and Purchasing Scientific Institutions (Y72Z090Q10), the National Natural Science Foundation of China (21801238), CAS Pioneer Hundred Talents Program B (Y92A010Q10), and the Natural Science Foundation of Chongqing (cstc2018jcyjAX0556, cstc2017rgzn-zdyfX0030, cstc2017jcyjAX0451, cstc2017rgznzdyfX0023, and cstc2018jszx-cyzd0603). Author contributions: K.S. and W.S. conceived the idea and initiated this project. W.S., Q.Z., Y.S., Y.Z., and D.C. constructed and optimized the DNN, deep learning, SVM, RF, and BPNN models, respectively. S.L. and Z.X. designed and synthesized the OPV materials. K.Y. fabricated OPV devices. W.S., Y.Z., and K.S. wrote the manuscript. Y.L., Y.Z., L.F., Z.W., and A.A.S. contributed to the fruitful discussions and supervision of the project. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaay4275/DC1
Section S1. ML methods and machine language expressions of molecule
Section S2. Process of experiment and proof of the reliability of the ML model
Fig. S1. Introduction to different ML algorithms.
Fig. S2. Chemical structures of the 10 new donor materials.
Fig. S3. Prediction results versus experimental data for the 10 new donor materials.
Table S1. Details of PaDEL descriptors.
Table S2. Details of RDKIt descriptors.
Table S3. Complete MACCS fingerprint of P3HT and PTB7.
Table S4. Photovoltaic parameters of OPV devices fabricated with different donor materials.
Table S5. Prediction results from DNN, RF, and SVM using Hybridization and FP2 fingerprints as inputs, as well as DNN and RF using Daylight fingerprints.
Table S6. Prediction results from BPNN using Daylight fingerprints when classification threshold is 10%.
References and Notes
- 1.Meng L., Zhang Y., Wan X., Li C., Zhang X., Wang Y., Ke X., Xiao Z., Ding L., Xia R., Yip H.-L., Cao Y., Chen Y., Organic and solution-processed tandem solar cells with 17.3% efficiency. Science 361, 1094–1098 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Cui Y., Yao H., Zhang J., Zhang T., Wang Y., Hong L., Xian K., Xu B., Zhang S., Peng J., Wei Z., Gao F., Hou J., Over 16% efficiency organic photovoltaic cells enabled by a chlorinated acceptor with increased open-circuit voltages. Nat. Commun. 10, 2515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghosh A. K., Feng T., Rectification, space-charge-limited current, photovoltaic and photoconductive properties of Al/tetracene/Au sandwich cell. J. Appl. Phys. 44, 2781–2788 (1973). [Google Scholar]
- 4.Nasrabadi N. M., Pattern recognition and machine learning. J. Electron. Imaging 16, 049901 (2007). [Google Scholar]
- 5.E. Alpaydin, Introduction to machine learning (MIT press, 2009). [Google Scholar]
- 6.Cao B., Adutwum L. A., Oliynyk A. O., Luber E. J., Olsen B. C., Mar A., Buriak J. M., How to optimize materials and devices via design of experiments and machine learning: Demonstration using organic photovoltaics. ACS Nano 12, 7434–7444 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Isayev O., Oses C., Toher C., Gossett E., Curtarolo S., Tropsha A., Universal fragment descriptors for predicting properties of inorganic crystals. Nat. Commun. 8, 15679 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pilania G., Gubernatis J. E., Lookman T., Multi-fidelity machine learning models for accurate bandgap predictions of solids. Comp. Mater. Sci. 129, 156–163 (2017). [Google Scholar]
- 9.Raccuglia P., Elbert K. C., Adler P. D. F., Falk C., Wenny M. B., Mollo A., Zeller M., Friedler S. A., Schrier J., Norquist A. J., Machine-learning-assisted materials discovery using failed experiments. Nature 533, 73–76 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Gómez-Bombarelli R., Aguilera-Iparraguirre J., Hirzel T. D., Duvenaud D., Maclaurin D., Blood-Forsythe M. A., Chae H. S., Einzinger M., Ha D.-G., Wu T., Markopoulos G., Jeon S., Kang H., Miyazaki H., Numata M., Kim S., Huang W., Hong S. I. K., Baldo M., Adams R. P., Aspuru-Guzik A., Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater. 15, 1120–1127 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Liang L., Liu M., Martin C., Elefteriades J. A., Sun W., A machine learning approach to investigate the relationship between shape features and numerically predicted risk of ascending aortic aneurysm. Biomech. Model. Mechan. 16, 1519–1533 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu S., Zhou Q., Ouyang Y., Guo Y., Li Q., Wang J., Accelerated discovery of stable lead-free hybrid organic-inorganic perovskites via machine learning. Nat. Commun. 9, 3405 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.A. R. Leach, V. J. Gillet, An introduction to chemoinformatics (Springer Science & Business Media, 2007). [Google Scholar]
- 14.J. Willett, Similarity and clustering in chemical information systems (John Wiley & Sons, Inc., 1987). [Google Scholar]
- 15.Sahu H., Rao W., Troisi A., Ma H., Toward predicting efficiency of organic solar cells via machine learning and improved descriptors. Adv. Energy Mater. 8, 1801032 (2018). [Google Scholar]
- 16.Padula D., Simpson J. D., Troisi A., Combining electronic and structural features in machine learning models to predict organic solar cells properties. Mater. Horiz. 6, 343–349 (2019). [Google Scholar]
- 17.Pyzer-Knapp E. O., Li K., Aspuru-Guzik A., Learning from the harvard clean energy project: The use of neural networks to accelerate materials discovery. Adv. Funct. Mater. 25, 6495–6502 (2015). [Google Scholar]
- 18.Hachmann J., Olivares-Amaya R., Atahan-Evrenk S., Amador-Bedolla C., Sánchez-Carrera R. S., Gold-Parker A., Vogt L., Brockway A. M., Aspuru-Guzik A., The Harvard clean energy project: Large-scale computational screening and design of organic photovoltaics on the world community grid. J. Phys. Chem. Lett. 2, 2241–2251 (2011). [Google Scholar]
- 19.Sun W., Li M., Li Y., Wu Z., Sun Y., Lu S., Xiao Z., Zhao B., Sun K., The use of deep learning to fast evaluate organic photovoltaic materials. Adv. Theor. Simul. 2, 1800116 (2019). [Google Scholar]
- 20.Nagasawa S., Al-Naamani E., Saeki A., Computer-aided screening of conjugated polymers for organic solar cell: classification by random forest. J. Phys. Chem. Lett. 9, 2639–2646 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Jørgensen P. B., Mesta M., Shil S., García Lastra J. M., Jacobsen K. W., Thygesen K. S., Schmidt M. N., Machine learning-based screening of complex molecules for polymer solar cells. J. Chem. Phys. 148, 241735 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Duan T., Tang H., Liang R.-Z., Lv J., Kan Z., Singh R., Kumar M., Xiao Z., Lu S., Laquai F., Terminal group engineering for small-molecule donors boosts the performance of nonfullerene organic solar cells. J. Mater. Chem. A 7, 2541–2546 (2019). [Google Scholar]
- 23.R. Hecht-Nielsen, in Neural networks for perception. (Elsevier, 1992), pp. 65–93. [Google Scholar]
- 24.Montavon G., Samek W., Müller K.-R., Methods for interpreting and understanding deep neural networks. Digit. Signal Process. 73, 1–15 (2018). [Google Scholar]
- 25.LeCun Y., Bengio Y., Hinton G., Deep learning. Nature 521, 436–444 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Mammone A., Turchi M., Cristianini N., Support vector machines. WIREs Comput. Stat. 1, 283–289 (2009). [Google Scholar]
- 27.Chang C.-C., Lin C.-J., LIBSVM: A library for support vector machines. ACM transactions on intelligent systems and technology (TIST) 2, 27 (2011). [Google Scholar]
- 28.Liaw A., Wiener M., Classification and regression by random Forest. R news 2, 18–22 (2002). [Google Scholar]
- 29.A. Jaiantilal, Classification and regression by randomforest-matlab (2009) at URL http://code.google.com/p/randomforest-matlab.
- 30.Basheer I. A., Hajmeer M., Artificial neural networks: Fundamentals, computing, design, and application. J. Microbiol. Meth. 43, 3–31 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Weininger D., SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988). [Google Scholar]
- 32.R. Todeschini, V. Consonni, Handbook of molecular descriptors. (John Wiley & Sons, 2008), vol. 11. [Google Scholar]
- 33.Cereto-Massagué A., Ojeda M. J., Valls C., Mulero M., Garcia-Vallvé S., Pujadas G., Molecular fingerprint similarity search in virtual screening. Methods 71, 58–63 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Dong J., Cao D.-S., Miao H.-Y., Liu S., Deng B.-C., Yun Y.-H., Wang N.-N., Lu A.-P., Zeng W.-B., Chen A. F., ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Chem. 7, 60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erhan D., Manzagol P.-A., Bengio Y., Bengio S., Vincent P., The difficulty of training deep architectures and the effect of unsupervised pre-training. Artificial Intelligence and Statistics. (2009). [Google Scholar]
- 36.Tajbakhsh N., Shin J. Y., Gurudu S. R., Hurst R. T., Kendall C. B., Gotway M. B., Liang J., Convolutional neural networks for medical image analysis: Full training or fine tuning? IEEE Trans. Med. Imag. 35, 1299–1312 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Yap C. W., PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 32, 1466–1474 (2011). [DOI] [PubMed] [Google Scholar]
- 38.RDKit (http://sourceforge.net/projects/rdkit/).
- 39.Fingerprints - Screening and Similarity (www.daylight.com/dayhtml/doc/theory/theory.finger.html).
- 40.Durant J. L., Leland B. A., Henry D. R., Nourse J. G., Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 42, 1273–1280 (2002). [DOI] [PubMed] [Google Scholar]
- 41.E. E. Bolton, Y. Wang, P. A. Thiessen, S. H. Bryant, in Annual reports in computational chemistry. (Elsevier, 2008), vol. 4, pp. 217–241. [Google Scholar]
- 42.O'Boyle N. M., Banck M., James C. A., Morley C., Vandermeersch T., Hutchison G. R., Open Babel: An open chemical toolbox. J. Cheminform. 3, 33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinbeck C., Han Y., Kuhn S., Horlacher O., Luttmann E., Willighagen E., The Chemistry Development Kit (CDK): An open-source Java library for chemo-and bioinformatics. J. Chem. Inf. Comput. Sci. 43, 493–500 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daylight Fingerprints (www.daylight.com/meetings/summerschool01/course/basics/fp.html).
- 45.Rogers D., Hahn M., Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Breiman L., Bagging predictors. Machine learning 24, 123–140 (1996). [Google Scholar]
- 47.Sun K., Xiao Z., Lu S., Zajaczkowski W., Pisula W., Hanssen E., White J. M., Williamson R. M., Subbiah J., Ouyang J., Holmes A. B., Wong W. W. H., Jones D. J., A molecular nematic liquid crystalline material for high-performance organic photovoltaics. Nat. Commun. 6, 6013 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Y. Jia, E. Shelhamer, J. Donahue, S. Karayev, J. Long, R. Girshick, S. Guadarrama, T. Darrell, Caffe: Convolutional Architecture for Fast Feature Embedding. Proceedings of the 22nd ACM international conference on Multimedia. ACM (2014). [Google Scholar]
- 49.C. Szegedy, W. Liu, Y. Jia, P. Sermanet, S. Reed, D. Anguelov, D. Erhan, V. Vanhoucke, A. Rabinovich, Going deeper with convolutions. Proceedings of the IEEE conference on computer vision and pattern recognition. (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaay4275/DC1
Section S1. ML methods and machine language expressions of molecule
Section S2. Process of experiment and proof of the reliability of the ML model
Fig. S1. Introduction to different ML algorithms.
Fig. S2. Chemical structures of the 10 new donor materials.
Fig. S3. Prediction results versus experimental data for the 10 new donor materials.
Table S1. Details of PaDEL descriptors.
Table S2. Details of RDKIt descriptors.
Table S3. Complete MACCS fingerprint of P3HT and PTB7.
Table S4. Photovoltaic parameters of OPV devices fabricated with different donor materials.
Table S5. Prediction results from DNN, RF, and SVM using Hybridization and FP2 fingerprints as inputs, as well as DNN and RF using Daylight fingerprints.
Table S6. Prediction results from BPNN using Daylight fingerprints when classification threshold is 10%.