

Abstract
Breast cancer is the most common cancer in women, and some patients develop recurrence after standard therapy. Effective predictors are urgently needed to detect recurrence earlier. The activation of Hedgehog signaling in breast cancer is correlated with poor prognosis. PTCH1 is an essential membrane receptor of Hedgehog. However, there are few reports about mutations in Hedgehog genes in breast cancer. We conducted a comprehensive study via an experimental and bioinformatics approach to detect mutated genes in breast cancer. Twenty-two breast cancer patients who developed recurrence within 24 months postoperatively were enrolled with 22 control cancer patients. Targeted deep sequencing was performed to assess the mutations among individuals with breast cancer using a panel of 143 cancer-associated genes. Bioinformatics and public databases were used to predict the protein functions of the mutated genes. Mutations were identified in 44 breast cancer specimens, and the most frequently mutated genes were BRCA2, APC, ATM, BRCA1, NF1, TET2, TSC1, TSC2, NOTCH1, MSH2, PTCH1, TP53, PIK3CA, FBXW7, and RB1. Mutation of these genes was correlated with protein phosphorylation and autophosphorylation, such as peptidyl-tyrosine and protein kinase C phosphorylation. Among these highly mutated genes, mutations of PTCH1 were associated with poor prognosis and increased recurrence of breast cancer, especially mutations in exons 22 and 23. The public sequencing data from the COSMIC database were exploited to predict the functions of the mutations. Our findings suggest that mutation of PTCH1 is correlated with early recurrence of breast cancer patients and will become a powerful predictor for recurrence of breast cancer.
Subject terms: Breast cancer, Molecular medicine, Breast cancer
Introduction
According to the Cancer Statistics Report in 2018, breast cancer constitutes 30% of new cancer cases diagnosed in American women1. The Centers for Disease Control and Prevention estimated that cancer will surpass heart disease to become the primary source of U.S. mortality by 2020. In 2014, the Taiwan Cancer Registry reported a breast cancer incidence of 11,769 cases per 100,000 females in Taiwanese women2. Although many therapeutics have been applied to treat breast cancer patients, some patients developed recurrence rapidly after standard treatment. Besides traditional classification, no effective predictors can be used to detect recurrence in chemoresistant breast cancer patients. There is also a lack of suitable treatment for these patients.
Cancer is a complex disease arising from the sequential accumulation of somatic mutations leading to the transformation of normal cells to cancerous cells. Detection of novel somatic mutations is important for examining the mechanism of carcinogenesis and to seek proper treatments3,4. Because of cancer heterogeneity, identifying somatic mutations from tumor samples is more difficult than detecting germline mutations using purified peripheral blood. Intercellular and intratumor genetic heterogeneity are the main sources of complications for identifying low-frequency mutations. However, somatic mutations of cancer samples are also important in clinical applications, including initial diagnosis, long-term monitoring, and relapse identification. All of these are essential components of cancer treatment. Detection of low-frequency mutations is also desirable in newly developed applications5–7.
Cancer is a genetic disease caused by both acquired and inherited mutations8. Incorporation of genome sequencing of cancer samples and online information from large data sets may accelerate the process of detecting new predictors. Solid evidence has been provided from cancer samples and sufficient references from online information9,10. It is important to integrate genetic alterations, cancer phenotypes and clinical outcomes of patients11,12. The present study involved two main steps. First, we analyzed the variations of 143 genes from 44 samples of breast cancer patients in Taiwan and defined several highly mutated genes. Furthermore, we compared the public datasets of these highly mutated genes from TCGA (The Cancer Genome Atlas)13, METABRIC (Molecular Taxonomy of Breast Cancer International Consortium)14, COSMIC (Catalogue of Somatic Mutations in Cancer)15, and NCBI GEO (National Center for Biotechnology Information, Gene Expression Omnibus)16. The findings were correlated with clinical outcomes and recurrence-free survival of breast cancer patients.
Results
Mutation burden versus the fraction of the genome altered in breast cancer patients
A total of 22 patients with cancer recurrence within 24 months postoperatively were collected (recurrence group). Twenty-two matched patients without cancer recurrence were selected as the control group. Breast cancer tissue was analyzed by targeted deep sequencing for mutations in 143 cancer-related genes (Supplementary Table S1). Patient characteristics are shown in Table 1. The patients with cancer recurrence had a larger tumor size than the matched controls (P = 0.044, Table 1). Age, operative method, TNM staging (tumor-node-metastasis), and intrinsic subtypes were similar between recurrence and matched control groups. CLC Genomics Workbench and Gene Ontology (GO) were used to analyze our sequencing data of 44 patients. Enrichment of GO terms among the mutated genes was clustered with top ten of the significant GO terms in the REViGO algorithm (Reduce + Visualize Gene Ontology, Fig. 1A and Table 2). GO terms from the categories “biological processes”, “cellular components” and “molecular function” with an adjusted p-value of <0.05 were considered overrepresentative in a subset of analyzed genes. These highly mutated genes had a high correlation with protein phosphorylation, such as peptidyl-tyrosine phosphorylation, protein phosphorylation and autophosphorylation (Fig. 1A). The highly mutated genes also correlated with cell adhesion, cell migration, extracellular matrix organization, axon guidance, actin cytoskeleton organization, heart development, and the regulation of small GTPase-mediated signal transduction (Table 2). Supplementary Table S2 provides all detailed information about the correlation between the novel GO terms and mutation in our breast cancer patients.
Table 1.
Characteristics of patients with breast cancer (n = 44). Comparison between recurrence and control patients was done.
| Characteristic | Recurrence* | Control** | p-value |
|---|---|---|---|
| Patients, n (%) | 22 (50%) | 22 (50%) | |
| Age at surgery (years)*** | 47 (29–63) | 50 (35–75) | 0.289 |
| Tumor size (cm)*** | 3.3 (1.4–8.0) | 2.6 (1.5–5.5) | 0.044 |
| Operation methods | 0.203 | ||
| Partial mastectomy and sentinel lymph node biopsy | 1 (33%) | 2 (67%) | |
| Partial mastectomy and axillary lymph node dissection | 1 (100%) | 0 | |
| Total mastectomy and sentinel lymph node biopsy | 2 (100%) | 0 | |
| Modified radical mastectomy | 18 (47%) | 20 (53%) | |
| Tumor-infiltrating lymphocytes | |||
| Low | |||
| High | |||
| Histology grade | >0.999 | ||
| Nuclear grade II | 4 (44%) | 5 (56%) | |
| Nuclear grade III | 18 (51%) | 17 (49%) | |
| Extensive intraductal components | 10 (67%) | 5 (33%) | 0.203 |
| Lymphatic tumor emboli | 13 (52%) | 12 (48%) | >0.999 |
| Extranodal extension | 6 (67%) | 3 (33%) | 0.460 |
| Tumor stage | 0.493 | ||
| T1 | 1 (33%) | 2 (67%) | |
| T2 | 18 (49%) | 19 (51%) | |
| T3 | 3 (75%) | 1 (25%) | |
| Nodal stage | 0.103 | ||
| N0 | 9 (47%) | 10 (53%) | |
| N1 | 6 (46%) | 7 (54%) | |
| N2 | 3 (38%) | 5 (62%) | |
| N3 | 4 (100%) | 0 | |
| AJCC TNM stage | 0.168 | ||
| Stage IA | 1 (33%) | 2 (9%) | |
| Stage IIA | 7 (47%) | 8 (53%) | |
| Stage IIB | 7 (50%) | 7 (50%) | |
| Stage IIIA | 3 (37%) | 5 (63%) | |
| Stage IIIC | 4 (100%) | 0 | |
| Intrinsic subtypes | 0.716 | ||
| Luminal A | 3 (37%) | 5 (63%) | |
| Luminal B without Her-2/Neu overexpression | 3 (50%) | 3 (50%) | |
| Luminal B with Her-2/Neu overexpression | 6 (67%) | 3 (33%) | |
| Her-2/Neu overexpression | 3 (37%) | 5 (63%) | |
| Triple-negative breast cancer | 7 (54%) | 6 (46%) |
AJCC TNM stage, American Joint Committee on Cancer tumor-node-metastases (TNM) staging system, 7th ed.
*Cancer recurrence within 24 months postoperatively. **No recurrence after 24 months postoperatively. ***Values are expressed as median (range).
Figure 1.

Overview of frequently mutated genes in the present study and comparison with the COSMIC database of breast cancer. (A) The significantly enriched Gene Ontology (GO) terms were calculated by CLC Genomics Workbench according to the mutated genes from sequencing data, and the graph was plotted with the REViGO package for the top ten significant GO terms. Each of the GO terms represents a node in the graph. The scatterplot showing functional clusters according to biological processes and the bubble color refer to the p-values from the GO result, whereas the bubble size indicates the frequency of the biology process from the GO database. The larger bubbles have more general terms, and these bubbles were highly correlated with protein phosphorylation, such as peptidyl-tyrosine phosphorylation, protein phosphorylation, and protein autophosphorylation. (B) Mutation prevalence in our cohort and comparison with the COSMIC database. The X-axis is the total number of mutations in the particular gene. The percentage close to the bar is the number of mutations in the particular gene divided by sum of all mutations in these 15 genes.
Table 2.
Predicting Gene Ontology (GO) biological process according to the mutation data from 44 sequenced breast cancer patients.
| GO term | Description | p-values |
|---|---|---|
| GO:0007155 | cell adhesion | 1.53E-14 |
| GO:0016477 | cell migration | 8.63E-11 |
| GO:0030198 | extracellular matrix organization | 1.01E-10 |
| GO:0018108 | peptidyl-tyrosine phosphorylation | 1.21E-08 |
| GO:0006468 | protein phosphorylation | 2.38E-08 |
| GO:0007411 | axon guidance | 3.83E-08 |
| GO:0030036 | actin cytoskeleton organization | 3.99E-08 |
| GO:0046777 | protein autophosphorylation | 5.64E-08 |
| GO:0007507 | heart development | 6.76E-07 |
| GO:0051056 | regulation of small GTPase mediated signal transduction | 6.99E-07 |
| GO:0048010 | vascular endothelial growth factor receptor signaling pathway | 7.79E-07 |
| GO:2000463 | positive regulation of excitatory postsynaptic potential | 2.34E-06 |
| GO:0009968 | negative regulation of signal transduction | 2.69E-06 |
| GO:0071300 | cellular response to retinoic acid | 4.30E-06 |
| GO:0007165 | signal transduction | 8.22E-06 |
| GO:0043410 | positive regulation of MAPK cascade | 1.21E-05 |
| GO:0007399 | nervous system development | 1.34E-05 |
| GO:0010595 | positive regulation of endothelial cell migration | 1.76E-05 |
| GO:0006024 | glycosaminoglycan biosynthetic process | 1.98E-05 |
| GO:0060997 | dendritic spine morphogenesis | 2.64E-05 |
Enrichment of functionally related GO terms and clustering of significant GO terms was analyzed by CLC Genomics Workbench. The first 20 GO terms are listed.
Selected alterations that appeared in target genes were compared with those in the COSMIC database (containing results from TCGA, METABRIC, and NCBI GEO, Fig. 1B). The results from TCGA was compared separately and listed in Supplementary Fig. S1. Previous studies reported that BRCA2, APC, TP53, and PIK3CA were highly mutated genes in breast cancer patients, and these genes were served as positive controls for our analysis13,14. Remarkably, we found a high mutation rate of PTCH1 in Taiwanese breast cancer patients (3.70% in our series vs. 0.91% in COSMIC, Fig. 1B, or 1.39% in TCGA, Supplementary Fig. S1). Among the 15 most often mutated genes, some mutated genes were also correlated with good prognosis, such as APC, ATM, NF1, TSC2, NOTCH1, and FBXW7. Others were correlated with poor prognosis, such as BRCA1, TET2, TSC1, MSH2, PTCH1, and PIK3CA. Mutation of PTCH1 predicted poor recurrence-free survival with statistical significance (P = 0.0275, Fig. 2). The incidence of PTCH1 mutation was higher in patients with recurrence compared with nonrecurrence control (68% vs. 34%, Table 3). Since only a few studies have focused on this gene in breast cancer, we selected PTCH1 for further analyses.
Figure 2.

Kaplan-Meier analysis of recurrence-free survival based on the mutation status of each gene. Mutation of PTCH1 was correlated with poor prognosis.
Table 3.
Correlation of genetic mutations with the two group of patients (n = 44).
| Characteristic | Recurrence* | Control** | p-value |
|---|---|---|---|
| Patient, n (%) | 22 (50%) | 22 (50%) | |
| BRCA2 mutation | 22 (50%) | 22 (50%) | *** |
| APC mutation | 20 (49%) | 21 (51%) | >0.999 |
| ATM mutation | 19 (46%) | 22 (53%) | >0.999 |
| BRCA1 mutation | 18 (51%) | 17 (49%) | >0.999 |
| NF1 mutation | 12 (41%) | 17 (59%) | 0.203 |
| TET2 mutation | 16 (52%) | 15 (48%) | >0.999 |
| TSC1 mutation | 12 (63%) | 7 (37%) | 0.223 |
| TSC2 mutation | 12 (44%) | 15 (56%) | 0.537 |
| NOTCH1 mutation | 15 (45%) | 18 (55%) | 0.488 |
| MSH2 mutation | 10 (59%) | 7 (41%) | 0.537 |
| PTCH1 mutation | 13 (68%) | 6 (34%) | 0.067 |
| TP53 mutation | 22 (50%) | 22 (50%) | *** |
| PIK3CA mutation | 10 (56%) | 8 (44%) | 0.760 |
| FBXW7 mutation | 3 (37%) | 5 (63%) | 0.698 |
| RB1 mutation | 6 (55%) | 5 (45%) | >0.999 |
*Cancer recurrence within 24 months postoperatively. **No recurrence after 24 months postoperatively. ***p-values could not be calculated because every patient had BRCA2 and TP53 mutations.
Mutation allele frequency and phosphorylation analysis
The frequency of mutation alleles was compared with the copy number of each gene. Amplification of the ERBB2 gene was detected in Her-2 (human epidermal growth factor receptor 2) enriched patients, and anti-Her-2 therapy was successfully applied to these patients. We used the ERBB2 gene as a control. The ERBB2 gene had a high rate of copy-number variations (CNV) and a particularly low mutation rate. However, PTCH1 had particularly low CNVs but a surprisingly high mutation rate in these breast cancer patients (Fig. 3A). The details of PTCH1 mutations are presented in Supplementary Fig. S2, and the most common 5 mutant sites on PTCH1 were analyzed (Fig. 3B). The mutation frequency was the product of allele frequency (AF) and read depth (DP). PTCH1 c.154C > T and c.158C > T (exon 1) were detected at a low frequency. We presumed that the number of cancer cells with PTCH1 c.154C > T or c.158C > T was too small and the small number of mutated cells could not represent the whole heterogeneous cancer scenario. There were only two patients with PTCH1 c.3907C > T (exon 23) and another two patients with c.3538C > T (exon 21) mutations. The patient number was insufficient to perform further analysis. We identified that c.3583A > T was the most frequent mutation in the present study and this mutation site was located in exon 22. Validation with Sanger sequencing was listed in Supplementary Table S3.
Figure 3.

Mutation allele frequency and copy-number variation of PTCH1 in breast cancer patients. (A) Comparison across 44 breast cancer samples by mutation allele frequency and copy-number variations (CNVs). ERBB2 had a high CNV rate but low mutant rate, whereas PTCH1 had a high mutation rate but an extremely low CNV rate in our study. (B) The upper graph shows the mutation frequencies of the top 5 mutation sites in PTCH1, including c.3907C > T, c.3583A > T, C.3538C > T, c.158C > T, and c.154C > T. Each dot represents an individual patient with a mutated gene. The mutation frequency was the product of allele frequency (AF) and read depth (DP). c.3583A > T had the highest mutation frequency and more patient number. The lower table shows detailed information. The mutation frequency is presented as median (range) and mean ± standard deviation. PTCH1 mutation sites (Alt) were compared with the reference genome (Ref). The biological function and patient number of each mutated site in the present study and in the TCGA database are also listed.
The highly mutated genes were correlated with protein phosphorylation through GO analysis (Fig. 1A). CLC Genomics Workbench was used to predict the impact of the PTCH1 mutants on posttranslational modification (PTM). Our results were analyzed with a comprehensive resource of known phosphorylation sites, the PhosphoSite database17. More than half of the mutation sites showed novelty (Fig. 4). c.3583A > T occurred at the intracellular C-terminus and changed the amino acid from threonine to serine (mutation of amino acid: p.T1195S, Fig. 5). In the prediction model, c.3583A > T (p.T1195S) directly interrupted the protein kinase C phosphorylation (Fig. 4) and changed the protein structure (Supplementary Fig. S3). Multiple mutation sites located in exon 22 and exon 23 were predicted to affect phosphorylation, from c.3583A (p.T1195) to c.3992C (p.S1331) (Fig. 4). Based on the aforementioned results, we presumed that exons 22 and 23 were the potentially crucial segments of PTCH1.
Figure 4.
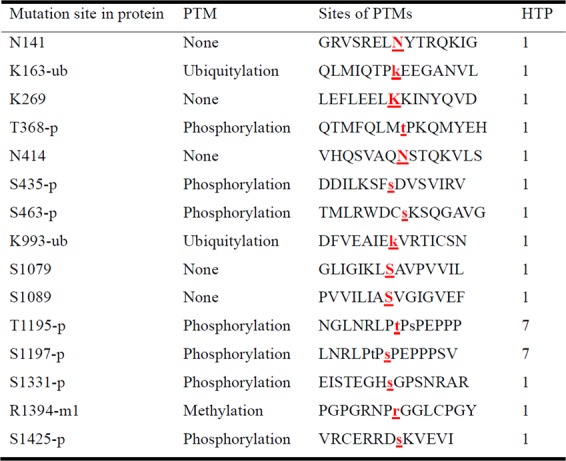
Posttranslational modification (PTM) sites of PTCH1 protein were identified by PhosphoSite. The PTMs in the proteins were located, and the below list focuses on the surrounding amino acid sequences. The targeted amino acid is marked with highlighted, red-colored, and bold. HTP, high-throughput publication.
Figure 5.

The alignment of the PTCH1 amino acid sequence of reference and the biology effect of the mutated sites for posttranslational modification. The 1st line is the UniProt Knowledgebase (UniProtKB) sequence of human PTCH1 protein, entry number Q13635. The 2nd line is the full sequence of PTCH1, patched homolog 1. The structural motif of PTCH1 protein is drawn in line 3. The transmembrane helices are shown as red boxes, and the extracellular and intracellular domains are shown as blue lines. The 4th line is the sterol-sensing domain (SSD) for cholesterol signaling. The 5th line is the binding site for ligand proteins (patched and patched family proteins). The 6th line is the known sites of posttranslational modification. The green nodes are sites of phosphorylation, and the blue nodes are sites of methylation. c.3591C (phosphorylation of p.S1197 protein) is expressed as a green node. The mutation site c.3583A > T (p.T1195S in the protein) is marked with a red line and is located in the intracellular domain at the C-terminus of PTCH1 protein.
Mutations of the PTCH1 gene in breast cancer are correlated with recurrence
Mutations of PTCH1 were correlated with demographics, histopathological findings and intrinsic subtypes (Table 4). Decreased amounts of tumor-infiltrating lymphocytes (TILs) were detected in breast cancer samples with mutated PTCH1 (21% low TILs in mutated PTCH1 vs. 79% low TILs in wild-type). A higher ratio of PTCH1 mutations was found in luminal B breast cancer without Her-2/neu overexpression (67%) and triple-negative breast cancer (TNBC, 62%) compared with other subtypes (Table 4). The PTCH1 mutation burden was correlated with TILs, intrinsic subtypes, prognostic predictors, and cancer recurrence by PheWAS analysis (Fig. 6A). The patients with mutated PTCH1 had a higher ratio of metastasis in the lung, liver, and distant lymph nodes (Table 5). The patients with PTCH1 mutations had poor recurrence-free survival (P = 0.0275, Figs 2 and 6B). Multivariate analysis of recurrence-free survival showed that the N3 nodal stage and PTCH1 mutation strongly predicted recurrence (Table 6). PTCH1 is a long gene with a total of 23 exons. To better understand the effect of PTCH1 mutation on the clinical outcome of patients, we analyzed patients according to the exon harboring the mutation or to the specific mutation site (Supplementary Table S4 and Supplementary Fig. S4). Only one patient had a PTCH1 mutation in exon 10 or 12, and no solid conclusion was obtained. PTCH1 mutations in exons 3, 7, 15, and 21 were not correlated with survival. PTCH1 mutations in exons 1, 6, 8, 14, 17, 19, 20, and 22 led to poor prognosis, though without statistical significance. The patients with mutated exon 23 of PTCH1 had poor survival (P = 0.0179, Supplementary Fig. S4O). Mutation in the union of mutations in exons 22 and 23 predicted worse survival (P = 0.0273, Fig. 6C). PTCH1 c.3583A > T (exon 22) mutations were correlated with poor patient survival, though the p-value was not statistically significant due to the small patient number (number of mutated patients = 6, Fig. 6D). Meanwhile, multiple mutation sites, from c.3583A (p.T1195) to c.3992C (p.S1331), located on exon 22 and exon 23, were predicted to affect phosphorylation (Fig. 4). All patients with lung or liver metastasis had mutations in exon 22, or 23 of PTCH1, which is in the C-terminus of the intracellular domain of PTCH1 protein (Supplementary Table S5). This result confirmed that exons 22 and 23 were potentially crucial segments of PTCH1.
Table 4.
Correlation of wild-type or mutated PTCH1 with demographics, histological findings, and intrinsic subtype in 44 patients with breast cancer who underwent radical resection and standard adjuvant therapy.
| Wild-type PTCH1 | Mutated PTCH1 | p-value | |
|---|---|---|---|
| Patients, n (%) | 25 (57%) | 19 (43%) | |
| Age at surgery (years)* | 47 (31–75) | 50 (29–63) | 0.454 |
| Tumor size (cm)* | 2.8 (1.5–6.0) | 3.1 (1.4–8.0) | 0.648 |
| Nuclear grade | 0.710 | ||
| Grade II | 6 (67%) | 3 (33%) | |
| Grade III | 19 (54%) | 16 (46%) | |
| Extensive intraductal components | 8 (53%) | 7 (47%) | 0.759 |
| Lymphatic tumor emboli | 13 (52%) | 12 (48%) | 0.547 |
| Tumor-infiltrating lymphocytes | 0.058 | ||
| Low | 11 (79%) | 3 (21%) | |
| High | 14 (47%) | 16 (53%) | |
| Skin invasion | 2 (67%) | 1 (33%) | >0.999 |
| Nipple invasion | 3 (43%) | 4 (57%) | 0.432 |
| Axillary lymph node metastasis | 0.761 | ||
| Negative | 10 (53%) | 9 (47%) | |
| Positive | 15 (60%) | 10 (40%) | |
| Positive lymph node numbers | 1 (0–17) | 1 (0–43) | 0.853 |
| Total resected lymph node numbers | 19 (1–38) | 21 (5–45) | 0.297 |
| Extranodal extension | 3 (33%) | 6 (67%) | 0.257 |
| Nodal staging | 0.933 | ||
| N0 | 10 (53%) | 9 (47%) | |
| N1 | 8 (62%) | 5 (38%) | |
| N2 | 5 (63%) | 3 (37%) | |
| N3 | 2 (50%) | 2 (50%) | |
| Tumor stage | 0.906 | ||
| T1 | 2 (67%) | 1 (33%) | |
| T2 | 21 (57%) | 16 (43%) | |
| T3 | 2 (50%) | 2 (50%) | |
| AJCC TNM stage | 0.921 | ||
| Stage I | 2 (67%) | 1 (33%) | |
| Stage II | 16 (55%) | 13 (45%) | |
| Stage III | 7 (58%) | 5 (42%) | |
| Estrogen receptor | 0.361 | ||
| Negative (<10%) | 10 (48%) | 11 (52%) | |
| Positive (≥ 10%) | 15 (65%) | 8 (35%) | |
| Progesterone receptor | 0.474 | ||
| Negative (<10%) | 18 (53%) | 16 (47%) | |
| Positive (≥ 10%) | 7 (70%) | 3 (30%) | |
| Her-2/Neu | 0.124 | ||
| Negative | 12 (46%) | 14 (54%) | |
| Positive | 13 (72%) | 5 (28%) | |
| Intrinsic subtype | 0.192 | ||
| Luminal A | 6 (75%) | 2 (25%) | |
| Luminal B without Her-2/Neu overexpression | 2 (33%) | 4 (67%) | |
| Luminal B with Her-2/Neu overexpression | 7 (78%) | 2 (22%) | |
| Her-2/Neu overexpression | 5 (63%) | 3 (37%) | |
| Triple-negative breast cancer | 5 (38%) | 8 (62%) |
AJCC TNM stage, American Joint Committee on Cancer tumor-node-metastases (TNM) staging system, 7th ed.
*Values are expressed as median (range).
Figure 6.

Mutations of PTCH1 in breast cancer patients were correlated with poor survival and a high recurrence rate. (A) The upper graph shows the mutation burden of PTCH1 from the current study; each bar represents an individual patient. The lower one represents the PheWAS analysis of the correlation between mutation of PTCH1 and clinical outcomes. Each bar represents a single patient. A high mutation burden of PTCH1 was correlated with a high rate of recurrence. (B–D) Kaplan-Meier analysis of recurrence-free survival according to the different mutation sites. (B) The patients were stratified into no mutation and all PTCH1 mutations. (C) The patients were stratified into those without and with mutations in exons 22 & 23 of PTCH1. (D) The patients were stratified into without and with the PTCH1 c.3583A > T mutation.
Table 5.
Correlation between cancer recurrence and wild-type or mutated PTCH1 in patients with breast cancer who underwent radical resection and standard adjuvant therapy.
| Wild-type PTCH1 | Mutated PTCH1 | p-value | |
|---|---|---|---|
| Patients, n (%) | 25 (57%) | 19 (43%) | |
| Recurrence | 9 (41%) | 13 (59%) | 0.067 |
| Lung metastasis | 5 (31%) | 11 (69%) | 0.013* |
| Liver metastasis | 2 (20%) | 8 (80%) | 0.011* |
| Bone metastasis | 6 (40%) | 9 (60%) | 0.123 |
| Brain metastasis | 3 (50%) | 3 (50%) | >0.999 |
| Regional lymph node recurrence | 2 (40%) | 3 (60%) | 0.638 |
| Distant lymph node metastasis | 4 (31%) | 9 (69%) | 0.044* |
| Local recurrence | 0 | 2 (100%) | 0.181 |
| Skin metastasis | 1 (50%) | 1 (50%) | >0.999 |
| Adrenal gland metastasis | 1 (50%) | 1 (50%) | >0.999 |
| Splenic metastasis | 1 (33%) | 2 (67%) | 0.570 |
*p-value less than 0.05.
Table 6.
Multivariate analysis of hazard ratio according to clinicopathological factors and PTCH1 mutation in patients with breast cancer (n = 44).
| Hazard radio | 95% Confidence interval | p-value | |
|---|---|---|---|
| Age at surgery (years) | 0.973 | 0.925–1.023 | 0.280 |
| Nodal stage* | 0.007** | ||
| N0 | 1 | ||
| N1 | 0.793 | 0.274–2.295 | 0.669 |
| N2 | 0.589 | 0.147–2.360 | 0.455 |
| N3 | 7.799 | 1.940–31.353 | 0.004** |
| PTCH1 gene | 0.010** | ||
| Wild-type PTCH1 | 1 | ||
| Mutated PTCH1 | 3.453 | 1.344–8.870 |
*According to American Joint Committee on Cancer tumor-node-metastasis (TNM) staging system, 7th ed. **p-value less than 0.05.
Discussion
The breakthroughs of the Human Genomic Project have accelerated the search for somatic mutations that drive the initiation and progression of human cancers. A combination of public databases of online information and targeted deep sequencing of 44 breast cancer patients in National Cheng Kung University Hospital identified PTCH1 mutations in the present study. Mutation of PTCH1 was predicted to correlate with protein phosphorylation. The cancer samples with mutated PTCH1 had a lower number of tumor-infiltrating lymphocytes. The patients with mutated PTCH1 had more metastasis in the lung, liver or distant lymph nodes. The patients with mutated PTCH1, especially exons 22 and 23, had a worse recurrence-free survival. These findings suggest that mutation of PTCH1 is a powerful predictor for breast cancer patients and the exons 22 and 23 are potentially vital regions.
Previous studies emphasize that the few mutations occur at a high frequency within or across cancer types; however, most mutations in cancer genomes occur infrequently. These rare mutations collectively play a defining role in one-fourth of all human cancers. High-frequency mutations drive critical molecular, biological and clinical phenotypes, while rare mutations promote the development of precision medicine10. Somatic mutations spontaneously occur in the body’s cells and accumulate throughout a lifetime, which may contribute to cancer and other diseases18,19. In contrast, germline mutations are present in gametes and passed to the next generation by inheritance20. Advancements in genome sequencing techniques and large-scale databases have revolutionized our knowledge of the mutations in cancer. When genetic mutation yields a clone of cells that circumvent the restrictions of normal cells, it will further promote cancer development and progression21,22.
The present study identified mutations in 44 breast cancer patients in Taiwan. A total of 143 cancer-related genes were sequenced. We also used bioinformatics approaches to identify potential mutations in breast cancer and predict their effects. Identifying genomic signatures in breast cancer patients will provide a better understanding of further clinical applications. Based on our results, the most frequent mutations were identified in BRCA2, APC, ATM, BRCA1, NF1, TET2, TSC1, TSC2, NOTCH1, MSH2, PTCH1, TP53, PIK3CA, FBXW7, and RB1 (Fig. 1B and Supplementary Fig. S1). Our data identified different incidence of mutation in these genes. All patients had mutations in the BRCA2 and TP53 genes. The clinical significance of these mutations requires further study. Other patients with gene mutations had similar recurrence-free survival to those without mutations. The patients with the PTCH1 mutation had a worse recurrence-free survival than those with wild-type PTCH1 (Fig. 2). High mutated allele frequency and low copy-number variation of PTCH1 was a unique presentation (Fig. 3A). According to our data, the function of PTCH1 in breast cancer was presumed to depend on mutation frequency, not the expression level. Based on the aforementioned results, PTCH1 was chosen for further study.
PTCH1 gene encodes the patched homolog 1 (PTCH1) protein. PTCH1, a 12-pass transmembrane protein, contains two large extracellular loops and two large intracellular loops. PTCH1 protein is one of the membranous receptors in Hedgehog signaling23,24. The sterol-sensing domain (SSD) is the segment involved in cholesterol synthesis and transport. Hedgehog signaling is central to the embryonic development of various tissues and organs. An increasing number of tumors arise due to mutations in this pathway, such as basal cell carcinoma (BCC) of the skin25. Many studies have focused on the function of PTCH1 protein in human disorders and tumorigenesis25–27. The extracellular loop of PTCH1 protein interacts with sonic hedgehog (SHH) or desert hedgehog (DHH) protein. Without ligand binding, PTCH1 protein inhibits Smoothened (SMO) receptors and inactivates downstream pathways. Binding of SHH/DHH protein with PTCH1 receptor activates the SMO coreceptor and downstream GLI1 signaling. Without the presence of hedgehog ligand, PTCH1 receptor has other roles in the cell, such as restricting the range of signaling, inhibiting cell cycle progression by sequestering cyclin B1, or mediating cell apoptosis via caspase 3- and caspase 9-related signaling pathways28,29. The PTCH1 receptor is supposed to be a tumor suppressor. The Hedgehog signaling pathway potentially activates breast carcinogenesis by increasing the expression of SHH, SMO and GLI1 proteins and reducing the expression of PTCH130. However, overexpression of PTCH1 protein is detected in breast cancer, especially in luminal B and TNBC subtypes31. While overexpression of PTCH1 protein is detected in many kinds of cancer, the function of PTCH1 changes from tumor suppressor to drug transporter for chemotherapeutic agents. PTCH1 protein uses the proton-motive force and establishes a reversed pH gradient to efflux drugs, such as doxorubicin. The GXXXD motif of putative transmembrane segment 4 is highly conserved in humans and drosophila. The glycine residue at position 509 and the aspartic acid residue at position 513 are important in drug efflux32. In cancer cell lines from adrenocortical carcinoma or melanoma, doxorubicin efflux is handled by overexpressing the PTCH1 protein33,34. PTCH1 is a potential therapeutic target for PTCH1-overexpressing lung, breast, prostate, ovary, colon, brain, adrenocortical carcinoma and melanoma35. However, these studies focus on overexpression of PTCH1 protein, not sequencing of the PTCH1 gene. Overexpression of a past-identified tumor suppressor in cancer tissue potentially implicates an alteration of protein structure and function. Mutation of the PTCH1 gene may function as the turning point from tumor suppressor to drug transporter.
Mutations of the PTCH1 gene are believed to be responsible for the development of both sporadic and familial BCCs24,26. However, there is a lack of research on PTCH1 mutations in breast cancer. Based on the results of next-generation sequencing, the polymorphism of PTCH1 c. 3944C > T (p.P1315L) is associated with the incidence of breast cancer after the use of oral contraceptives36. We excluded PTCH1 c. 3944C > T because of the high prevalence of this polymorphism and the low usage of oral contraceptives in Taiwan. We conducted deep targeted sequencing of the PTCH1 gene from 44 breast cancer patient samples. The samples with mutated PTCH1 contained a lower number of TILs (Table 4). The patients with PTCH1 mutations had a higher ratio of lung, liver, or distant lymph node metastasis (Table 5) and worse recurrence-free survival (Figs 2 and 6B). After analysis of all mutation sites on PTCH1, c.3583 A > T was the highest mutation point (Fig. 3B). The patients with mutated exon 22 and exon 23 of PTCH1 or mutated exon 23 had poor prognosis (Fig. 6C and Supplementary Fig. S4O). The clinical importance of PTCH1 mutations in breast cancer is implicated.
We found that all highly mutated genes were correlated with protein phosphorylation, such as peptidyl-tyrosine phosphorylation, protein phosphorylation, and protein autophosphorylation (Fig. 1A). We tried to analyze PTCH1 mutations in protein structure and function. The PhosphoSite database showed that half of the mutation sites in PTCH1 genes can alter posttranslational modifications of the protein (Fig. 4). CLC Genomics Workbench was used to predict the protein structure of the PTCH1 c.3583A > T mutant (Supplementary Fig. S3). PTCH1 c.3583A (p.T1195) mutation interrupted protein phosphorylation (Fig. 5). Multiple mutation sites, from c.3583A to c.3992C (p.S1331), located in exon 22 and exon 23, were predicted to affect phosphorylation (Fig. 4). Exons 22 and 23 encode parts of the intracellular C-terminus of the PTCH1 protein and interact with the SMO receptor. Blockage of the interaction between PTCH1 and SMO will retrain signal transduction and suppress GLI1 protein expression. Exons 22 and 23 encode potentially crucial segments for PTCH1 protein in breast cancer. Although further efforts should be applied to understand the role of PTCH1 in breast cancer, we believe that the present study suggests unprecedented value in assessing PTCH1 in breast cancer patients. Future research can be based on these cancer susceptibility genes in this study and tease out their roles in breast cancer.
Our study also had some limitations because of the low sample numbers, with only 44 patients, and the risk of DNA damage in formalin-fixed paraffin-embedded (FFPE) samples. Most breast cancer patients have a better survival after standard treatment and seldom develop recurrence within 24 months postoperatively. The patients with the worst outcome were rare, and this was the reason that the present study had to run from 2006 to 2017 to let us collect 22 recurrent patients. A previous study indicated that the sensitivity and specificity to detect mutations in FFPE samples were reliable when comparing next-generation sequencing assays with Oncomine Cancer Panel assays37. The damage of the DNA template in FFPE samples was a possible source of sequencing artifacts38. We tried to eliminate artifacts with improved DNA extraction and PCR amplification techniques. Sanger sequencing was also used to confirm the mutation sites. However, the possibility of artifacts could not be completely excluded, and further study of PTCH1 mutations in breast cancer patients is necessary.
Conclusions
We performed targeted deep sequencing of 143 cancer-related genes for 44 breast cancer samples. Genomic mutation and copy-number variations were analyzed and compared with the data in public databases. The functions of mutated genes were predicted by GO analysis, the PhosphoSite database, and CLC Genomics Workbench. We detected the PTCH1 mutation in 18 of 44 patients, and c.3583A > T had the highest mutation frequency. Multiple mutation sites from c.3583A to c.3992C located in exon 22 and exon 23 of PTCH1 were predicted to affect phosphorylation. The patients with mutated PTCH1 had a high risk of metastasis in the lung, liver, or distant lymph nodes and a decreased number of TILs in breast cancer samples. The patients with mutated exon 23 or mutated exons 22 and 23 of PTCH1 had poor survival. These data suggest that exons 22 and 23 are crucial for PTCH1 and that PTCH1 can be a powerful predictor for the recurrence of breast cancer patients.
Materials and Methods
Patients and targeted deep sequencing for mutation detection
Formalin-fixed, paraffin-embedded (FFPE) tissue specimens from breast cancer patients were collected from the Department of Pathology in the National Cheng Kung University Hospital (NCKUH). Written informed consent was collected for all patients, and the Institutional Review Board of NCKUH approved this study under NCKUH IRB number A-ER-105-233. The study also complied with the formal guidelines of the Institutional Review Board. The collection period was from April 2006 to January 2017. A total of 22 patients were defined as the recurrence group, having cancer recurrence within 24 months postoperatively. Another 22 matched patients without cancer recurrence after surgery and standard treatment were selected as the control group. All these patients received radical operation and standard adjuvant therapy. Patient information was collected from a retrospective chart reviewing demographics, histopathologic findings, and clinical results as we previously described39,40. The TNM (tumor, node, metastasis) stage was defined according to the American Joint Committee on Cancer (AJCC) classification of 2010, 7th edition (corrected printing 2015)41.
DNA and RNA were extracted from these cancer tissues. Sufficient amount of tissue was used to extract DNA/RNA after removal of formaldehyde-induced crosslinks by heat and removal of protein-DNA crosslinks by proteinase K. Fluorometry was used to assess quality of double-stranded DNA. Short amplicons of DNA was generated to increase number of templates for PCR and specific primers for each strand was applied in amplicon-based target enrichment. RNA was reverse-transcribed to make cDNA, and target regions from DNA or cDNA were amplified by the DNA/RNA Oncomine Cancer Research Panel version 1 (Thermo Fisher Scientific, Waltham, USA). High-fidelity DNA polymerase was used to reduce polymerase errors and bypass uracil/abasic sites. An Ion 318 chip (Thermo Fisher Scientific) was prepared and loaded according to the manufacturer’s recommendation. The Ion PGM Sequencing 200 Kit v.2 (Thermo Fisher Scientific) was used with the Ion PGM sequencer (Thermo Fisher Scientific) as described in the Ion PGM Sequencing Kit User Guide. The sequencing reads were aligned to the reference genome (hg19) and variants called by Torrent Suite 5.0.442,43. The sequencing data of 44 breast cancer samples were available for analysis of 143 genes (Supplementary Table S1). Variant calling was conducted using a locked data analysis pipeline, Torrent Suite version 5.2.2 (Thermo Fisher Scientific) and Oncomine Comprehensive w2.3 - DNA and Fusions - Single Sample v5.6 in Ion Reporter version 5.6 (Thermo Fisher Scientific). The minimal depth of single-nucleotide variants (SNVs) and small insertions/deletions (indels) was 15. The minimal allele frequency (AF) was 7% for indels. Some SNV hotspots were designed in the Oncomine system, and the minimal AF was 3% for SNVs in hotspots. For SNVs not in hotspots, the minimal AF was 4%.
Whole-genome sequencing data of 499 normal Taiwanese were provided by Taiwan Biobank to compare the distribution and frequency of different genes between cancer patients and the normal population as previously described44. Publicly available data were used to compare and validate the distribution of the PTCH1 genetic variant worldwide. Phenome-wide association scans (PheWAS) were used to link the characteristics of cancer and mutations of PTCH1.
Sanger sequencing for validation of mutation
To confirm the variant detection analysis, we used Sanger sequencing for the high-frequency variants. PCR amplification and DNA sequencing primers were designed and synthesized by Mission Biotech, Taipei, Taiwan. The primer sequences for the PTCH1 gene were as follows: PTCH1-F: TGA CAC TGT CGT CTG GGA AC; PTCH1-R: AAC AGA GGC CCC TGA AAA AT. For the PCR amplification, the target regions were amplified using the KAPA HiFi HotStart PCR kit (KAPA Biosystems, Wilmington, USA) in a total reaction volume of 50 μL. Reactions were run in a 9700-thermal cycler (Applied Biosystems, Waltham, USA) using the following cycling parameters: 3 min holding at 95 °C, followed by 25 cycles of 20 sec at 95 °C, 20 sec at 66 °C, and 30 sec at 72 °C. The presence of amplicons was confirmed by gel electrophoresis on a 1.5% agarose gel. The PCR Fragment Extraction Kit (Geneaid, New Taipei, Taiwan) was used for the PCR amplicon purification process. DNA sequencing was done by using the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit v3.1 (Applied Biosystems) on the ABI Prism 3730XL DNA Analyzer. Sanger chromatograms were analyzed using CLC Genomics Workbench, Aarhus, Denmark.
Gene ontology cluster and analysis
Clustering of Gene Ontology (GO) terms furnishes groups of genes sharing similar functions or characteristics. Based on standard GO terms, comparative genomic studies in different diseases are made possible45,46. GO term analysis begins with three main features: biological process, molecular function, and cellular component47. The GO list was generated from the CLC Genomics Workbench by analyzing mutation patterns in 44 breast cancer patients. Afterward, sequences from these patients were grouped with GO terms using BLAST2GO PRO plugin 1.1.0 within CLC Genomics Workbench. A hypergeometric distribution test was used to link the association between 143 genes and GO class. The Benjamini-Hochberg procedure was used to avoid type I errors and decrease the false discovery rate. A corrected p-value cutoff <0.05 was set. Finally, the output results were processed on the REViGO web-server tool to remove redundant GO terms (http://revigo.irb.hr/). REViGO has many features for postanalysis of GO terms that connect to third-party plug-ins, such as Cytoscape Scatterplots and Tree Mapping48.
Statistical analysis
All statistical results were conducted using SPSS version 17.0 (SPSS, New York, USA). The continuous variables are reported as median (range) and were compared using the nonparametric Kruskal-Wallis H test. Fisher’s exact test and the chi-square test were used to compare the differences in categorical variables. Kaplan-Meier survival analysis and the log-rank test were used to estimate patient survival and quantify the differences between groups. Those factors with P < 0.2 in the univariate analysis were included in the multivariate analysis. A Cox proportional hazards regression model was used to analyze significant factors in recurrence-free survival in multivariate analysis. The results are expressed as the hazard ratio (HR) with 95% confidence interval (95% C.I.). The p-value cut-off was set at 0.05 for statistically significant differences.
Supplementary information
Acknowledgements
Bioinformatics analyses and data mining were conducted in the Bioinformatics Core at the National Cheng Kung University (Tainan, Taiwan). The authors are grateful for the support from the Human Biobank, Research Center of Clinical Medicine, National Cheng Kung University Hospital. We were blessed with support from the late superintendent, Professor Pin-wen Lin. Furthermore, we thank Mrs. Ya-Li Hsiao, Dr. Wei-Pang Chung, Dr. Wen-Chung Chen, and Prof. Yi-Ling Chen for their support; Dr. Dao-Peng Chen and Dr. I-Hui Chen of Kim Forest Enterprise for technical consultation; and Dr. Zhengda Sun from the University of California, San Francisco, for the editing of this manuscript. The study was supported by the Ministry of Science and Technology (MOST) of Taiwan (grant MOST105-2314-B-006-028, MOST108-2314-B-006-082 and MOST108-2634-F-006-011), the National Cheng Kung University Hospital (grant NCKUH-106-01002), the Ministry of Health and Welfare (grant MOHW106-TDU-B-211-113003, MOHW108-TDU-B- 211-124018, and MOHW108-TDU-B-211-133003), and Chi Mei Medical Center (grant B106-F058E).
Author contributions
C.Y.W. was responsible for bioinformatics analysis and wrote the first draft. Y.C.C. and C.H.A.C. predicted the structure and posttranslational modification of the mutated protein. Y.L.K., K.T.L. and H.P.H. managed patients and performed surgery. P.S.C., C.P.C. and M.R.S. were involved in the study concept and design. N.N.P. was responsible for English review. H.P.H. performed the statistical analysis and revised the manuscript. All authors edited the manuscript and approved the final version for publication.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-52617-4.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Registry, T. C. Cancer Incidence and Mortality Rates in Taiwan (2014).
- 3.Pleasance ED, et al. A comprehensive catalog of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–1489. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- 6.Robinson D, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;162:454. doi: 10.1016/j.cell.2015.06.053. [DOI] [PubMed] [Google Scholar]
- 7.Taniguchi K, Okami J, Kodama K, Higashiyama M, Kato K. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci. 2008;99:929–935. doi: 10.1111/j.1349-7006.2008.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 9.Low SK, Zembutsu H, Nakamura Y. Breast cancer: The translation of big genomic data to cancer precision medicine. Cancer Sci. 2018;109:497–506. doi: 10.1111/cas.13463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohno T. Implementation of “clinical sequencing” in cancer genome medicine in Japan. Cancer Sci. 2018;109:507–512. doi: 10.1111/cas.13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisman PS, et al. Genetic alterations of triple negative breast cancer by targeted next-generation sequencing and correlation with tumor morphology. Mod. Pathol. 2016;29:476–488. doi: 10.1038/modpathol.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu X, et al. Genetic alterations and oncogenic pathways associated with breast cancer subtypes. Mol. Cancer Res. 2009;7:511–522. doi: 10.1158/1541-7786.MCR-08-0107. [DOI] [PubMed] [Google Scholar]
- 13.Koboldt DC, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: A bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- 18.Pennington KP, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014;20:764–775. doi: 10.1158/1078-0432.CCR-13-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janoueix-Lerosey I, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 20.Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007;98:1817–1824. doi: 10.1111/j.1349-7006.2007.00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodis E, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peifer M, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harvey MC, Fleet A, Okolowsky N, Hamel PA. Distinct effects of the mesenchymal dysplasia gene variant of murine Patched-1 protein on canonical and non-canonical Hedgehog signaling pathways. J. Biol. Chem. 2014;289:10939–10949. doi: 10.1074/jbc.M113.514844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boutet N, et al. Spectrum of PTCH1 mutations in French patients with Gorlin syndrome. J. Invest. Dermatol. 2003;121:478–481. doi: 10.1046/j.1523-1747.2003.12423.x. [DOI] [PubMed] [Google Scholar]
- 25.Kimonis VE, Mehta SG, Digiovanna JJ, Bale SJ, Pastakia B. Radiological features in 82 patients with nevoid basal cell carcinoma (NBCC or Gorlin) syndrome. Genet. Med. 2004;6:495–502. doi: 10.1097/01.GIM.0000145045.17711.1C. [DOI] [PubMed] [Google Scholar]
- 26.Pan S, Dong Q, Sun LS, Li TJ. Mechanisms of inactivation of PTCH1 gene in nevoid basal cell carcinoma syndrome: modification of the two-hit hypothesis. Clin. Cancer Res. 2010;16:442–450. doi: 10.1158/1078-0432.CCR-09-2574. [DOI] [PubMed] [Google Scholar]
- 27.Mancuso M, et al. Basal cell carcinoma and its development: insights from radiation-induced tumors in Ptch1-deficient mice. Cancer Res. 2004;64:934–941. doi: 10.1158/0008-5472.CAN-03-2460. [DOI] [PubMed] [Google Scholar]
- 28.Adolphe C, Hetherington R, Ellis T, Wainwright B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006;66:2081–2088. doi: 10.1158/0008-5472.CAN-05-2146. [DOI] [PubMed] [Google Scholar]
- 29.Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009;9:873–886. doi: 10.2174/156652409789105570. [DOI] [PubMed] [Google Scholar]
- 30.Monkkonen T, Lewis MT. New paradigms for the Hedgehog signaling network in mammary gland development and breast Cancer. Biochim. Biophys. Acta Rev. Cancer. 2017;1868:315–332. doi: 10.1016/j.bbcan.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riaz SK, et al. Involvement of hedgehog pathway in early onset, aggressive molecular subtypes and metastatic potential of breast cancer. Cell Commun. Signal. 2018;16:3. doi: 10.1186/s12964-017-0213-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bidet M, et al. The Hedgehog receptor Patched functions in multidrug transport and chemotherapy resistance. Mol. Cancer Res. 2012;10:1496–1508. doi: 10.1158/1541-7786.MCR-11-0578. [DOI] [PubMed] [Google Scholar]
- 33.Hasanovic A, et al. Targeting the multidrug transporter Patched potentiates chemotherapy efficiency on adrenocortical carcinoma in vitro and in vivo. Int. J. Cancer. 2018;143:199–211. doi: 10.1002/ijc.31296. [DOI] [PubMed] [Google Scholar]
- 34.Fiorini L, et al. Natural paniceins from mediterranean sponge inhibit the multidrug resistance activity of Patched and increase chemotherapy efficiency on melanoma cells. Oncotarget. 2015;6:22282–22297. doi: 10.18632/oncotarget.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasanovic A, Mus-Veteau I. Targeting the multidrug transporter Ptch1 potentiates chemotherapy efficiency. Cells. 2018;7:E107. doi: 10.3390/cells7080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang-Claude J, et al. The patched polymorphism PRO1315LEU (C3944T) may modulate the association between use of oral contraceptives and breast cancer risk. Int. J. Cancer. 2003;103:779–783. doi: 10.1002/ijc.10889. [DOI] [PubMed] [Google Scholar]
- 37.Lih CJ, et al. Analytical validation of the next-generation sequencing assay for a nationwide signal-finding clinical trial - molecular analysis for therapy choice clinical trial. J. Mol. Diagn. 2017;19:e313–e327. doi: 10.1016/j.jmoldx.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Do H, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin. Chem. 2015;61:64–71. doi: 10.1373/clinchem.2014.223040. [DOI] [PubMed] [Google Scholar]
- 39.Hsu HP, Shan YS, Jin YT, Lai MD, Lin PW. Loss of E-cadherin and beta-catenin is correlated with poor prognosis of ampullary neoplasms. J. Surg. Oncol. 2010;101:356–362. doi: 10.1002/jso.21493. [DOI] [PubMed] [Google Scholar]
- 40.Wu CL, et al. Dual role of CD44 isoforms in ampullary adenocarcinoma: CD44s predicts poor prognosis in early cancer and CD44v is an indicator for recurrence in advanced cancer. BMC Cancer. 2015;15:903. doi: 10.1186/s12885-015-1924-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fleming I. D. et al. Of referencing in AJCC Cancer Staging Manual (7th ed. Edge, S. B.) 345–376 (New York: Springer, 2010) (Corrected at 7th printing 2015).
- 42.Casper J, et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018;46:D762–D769. doi: 10.1093/nar/gkx1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeh YM, et al. The c.1085A > G genetic variant of CSF1R gene regulates tumor immunity by altering the proliferation, polarization, and function of macrophages. Clin. Cancer Res. 2017;23:6021–6030. doi: 10.1158/1078-0432.CCR-17-1007. [DOI] [PubMed] [Google Scholar]
- 45.Sun Z, et al. Single-cell RNA sequencing reveals gene expression signatures of breast cancer-associated endothelial cells. Oncotarget. 2018;9:10945–10961. doi: 10.18632/oncotarget.23760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang CY, et al. PSMB5 plays a dual role in cancer development and immunosuppression. Am. J. Cancer Res. 2017;7:2103–2120. [PMC free article] [PubMed] [Google Scholar]
- 47.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.