

Abstract
The RNA-guided Cas9 endonuclease from Streptococcus pyogenes is a single turnover enzyme that displays a stable product state after double-stranded DNA cleavage. Here, we present cryo-EM structures of pre-catalytic, post-catalytic, and product states of the active Cas9-sgRNA-DNA complex in the presence of Mg2+. In the pre-catalytic state, Cas9 adopts the “checkpoint” conformation with the HNH nuclease domain positioned far away from the DNA. Transition to the post-catalytic state involves a dramatic ~34 Å swing of the HNH domain and disorder of the REC2 recognition domain. The post-catalytic state captures the cleaved substrate bound to the catalytically competent HNH active site. In the product state, the HNH domain is disordered, REC2 returns to the pre-catalytic conformation, and additional interactions of REC3 and RuvC with nucleic acids are formed. The coupled domain motions and interactions between the enzyme and nucleic acids provide new insights into the mechanism of genome editing by Cas9.
The Cas9 endonuclease from Streptococcus pyogenes is a powerful tool for genome editing in mammalian cells 1,2. Cas9 has been ubiquitously adopted for genome editing due to the minimal requirements for nuclease activity, ease of targeting to desired genomic sites, and sequence specificity for DNA hydrolysis 3-5. Understanding the Cas9 reaction cycle at biophysical and structural levels is important for overcoming barriers to the safe and effective usage of this bacterial ribonucleoprotein for research and therapeutic applications.
Previous studies on the Cas9 reaction sequence have converged on a model with several conformational checkpoints that are thought to ensure proper RNA-DNA hybridization, and facilitate concerted, high-fidelity DNA cleavage 4,6-8. Cas9 binds the single guide RNA (sgRNA) with high affinity, forming a Cas9-sgRNA binary complex 9. The interactions between the enzyme-RNA complex and DNA are short-lived until a protospacer adjacent motif (PAM) sequence on the DNA is recognized 4. Upon PAM recognition, the sgRNA forms Watson-Crick base pairs with the target strand (TS), leading to unwinding of the DNA in a direction away from the PAM sequence 4 and formation of the Cas9-sgRNA-DNA R-loop structure 10. Subsequently, double-stranded DNA (dsDNA) hydrolysis is initiated by docking of the HNH nuclease domain to the TS. This is followed by concerted cleavage of the TS and non-target strand (NTS) by the HNH and RuvC subdomains, respectively 3, forming a stable enzyme-product complex 4,9. However, all current models for the Cas9 reaction sequence rely primarily on structures preceding DNA cleavage, obtained either in the absence of added Mg2+ or from catalytically inactive Cas9 mutants 9-12. High-resolution structures of catalytically competent Cas9 complexes typifying reaction steps during and after DNA cleavage are not yet available. Knowledge of these structures will add to our basic understanding of the Cas9 reaction sequence model and provide new opportunities for rational engineering of enzyme activity.
To elucidate the structural mechanisms underlying the Cas9 reaction cycle during and after DNA cleavage, we performed cryo-EM analysis of catalytically active Cas9 bound to sgRNA and a 40 base-pair (bp) dsDNA substrate (Figs. 1a, b) in the presence of Mg2+. Our studies elucidate three concurrent and distinct conformational states (I-III) (Figs. 1c-h, Supplementary Fig. 1, Supplementary Notes 1 and 2, Table 1). Density maps for each of these states, determined at a resolution of ~3.3 Å, show that each state displays the previously reported two-lobed architecture of Cas9 with the DNA substrate in an unwound state (PDB IDs 4OO8, 4UN3, 5F9R) 10-12. In state I, the intact TS is hybridized to the 20 nucleotide (nt) guide segment of sgRNA. In states II and III, the bound TS DNA is cleaved between the third and fourth nucleotide upstream of the PAM sequence, but with substantial differences in the ordering and positioning of HNH and REC2 domains.
Fig. 1. Cryo-EM structures of three states of Cas9-sgRNA-dsDNA complex.
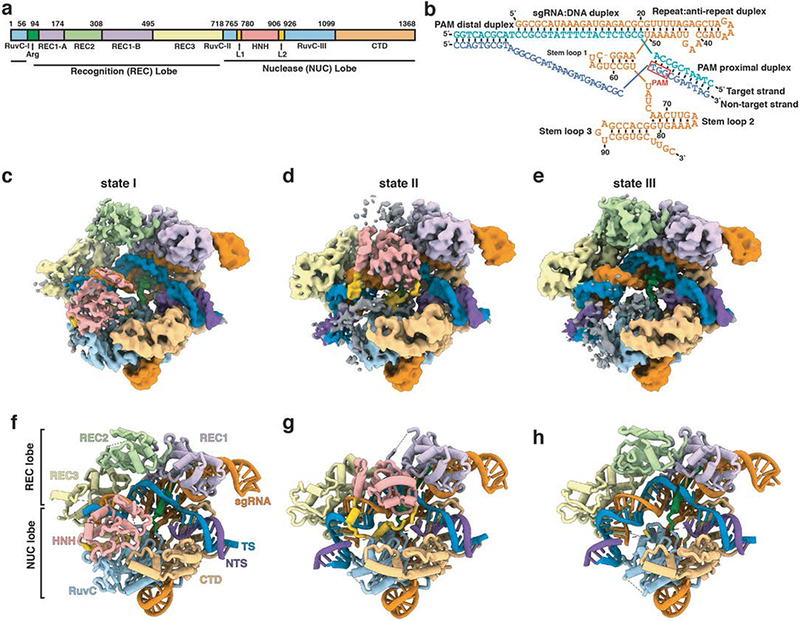
(a) Domain organization of S. pyogenes Cas9. (b) Schematic diagram of the nucleic acids used in the study. The sgRNA is orange, and the target (TS) and non-target strands (NTS) of the dsDNA are blue and purple, respectively. Cryo-EM density maps (c-e) and schematic representations (f-h) of states I, II and III of the Cas9-sgRNA-dsDNA ternary complex. Protein domains and nucleic acids are colored as in (a) and (b). Some regions of weak density, shown in grey in panels c-e, are not included in the atomic models shown in panels f-h.
Table 1.
Cryo-EM data collection, refinement and validation statistics
| State I (EMD-0585, PDB 6O0Z) |
State II (EMD-0584, PDB 6O0Y) |
State III (EMD-0583, PDB 6O0X) |
|
|---|---|---|---|
| Data collection and processing | |||
| Magnification | 165,000 | 165,000 | 165,000 |
| Voltage (kV) | 300 | 300 | 300 |
| Electron exposure (e−/Å2) | 73 | 73 | 73 |
| Defocus range (μm) | −0.7 to −3.0 | −0.7 to −3.0 | −0.7 to −3.0 |
| Pixel size (Å) | 0.836 | 0.836 | 0.836 |
| Symmetry imposed | C1 | C1 | C1 |
| Initial particle images (no.) | 1,301,136 | 1,301,136 | 1,301,136 |
| Final particle images (no.) | 40,328 | 27,433 | 30,513 |
| Map resolution (Å) | 3.30 | 3.37 | 3.28 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 2.8-6.8 | 2.8-6.8 | 2.8-6.8 |
| Refinement | |||
| Initial model used (PDB code) | 5B2R | 5B2R | 5B2R |
| Model resolution (Å) | 3.30 | 3.37 | 3.28 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Model resolution range (Å) | 2.8-6.8 | 2.8-6.8 | 2.8-6.8 |
| Map sharpening B factor (Å2) | −75.6 | −74.7 | −68.9 |
| Model composition | |||
| Non-hydrogen atoms | 11,912 | 11,828 | 11,257 |
| Protein residues | 1,288 | 1,146 | 1,112 |
| Nucleotides | 135 | 150 | 150 |
| B factors (Å2) | |||
| Protein | 132.8 | 123.0 | 118.4 |
| Nucleic acid | 131.3 | 141.5 | 141.9 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.006 | 0.006 | 0.006 |
| Bond angles (°) | 0.99 | 0.98 | 0.99 |
| Validation | |||
| MolProbity score | 1.46 | 1.42 | 1.47 |
| Clashscore | 2.58 | 2.52 | 2.70 |
| Poor rotamers (%) | 0 | 0.37 | 0.14 |
| Ramachandran plot | |||
| Favored (%) | 93.67 | 94.45 | 93.81 |
| Allowed (%) | 6.33 | 5.55 | 6.19 |
| Disallowed (%) | 0 | 0 | 0 |
Results
The “checkpoint” conformation of Cas9 is captured in State I
Detailed inspection of state I shows that DNA binding to the Cas9-sgRNA binary complex results in a conformational change of the REC2 and REC3 domains (Fig. 2a), which leads to an opening of the central channel in Cas9 4,7,10. The opening facilitates accommodation of the newly formed DNA-RNA hybrid within the channel. In the crystal structure of the “Mg2+-free” Cas9-sgRNA ternary complex (PDB ID 5F9R) 10, the NTS threads into a tunnel buttressed by the HNH and RuvC domains. This tunnel is sterically blocked in state I, as revealed by superimposition of the two structures (Figs. 2b, c, and Supplementary Fig. 2), and although the NTS is only poorly resolved, our density map suggests that the NTS runs parallel to the RNA-DNA heteroduplex following the trajectory that is closer to the REC2 domain (Figs. 2b, c).
Fig. 2. The central channel of Cas9 accommodates the R-loop structure in state I.
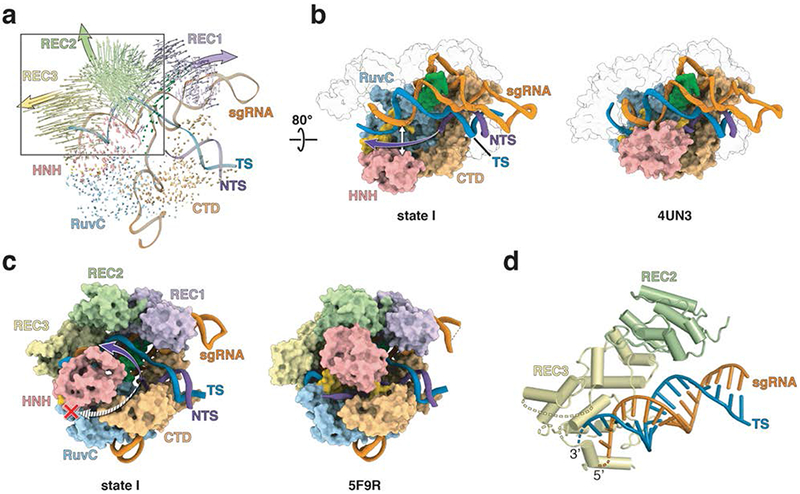
(a) Superposition of Cas9-sgRNA (PDB ID 4ZT0) onto state I, with RuvC as the common reference, reveals the extent of Cas9 domain movements upon dsDNA and Mg2+ binding. Arrow lengths correspond to the magnitude of Cα atom movements; arrowheads show domain movement directions and large solid arrows point to the general direction of the domain movement. (b) The central channel in state I (left panel) and Cas9 bound to the partial duplex (right panel; PDB ID 4UN3). The purple arrow (left panel) indicates the trajectory for the disordered NTS. The helical recognition lobes are shown as transparent surfaces for clarity. (c) The central channel in state I (left panel) and the “Mg2+-free” complex (right panel; PDB ID 5F9R). The purple and striped arrows (left panel) indicate two possible trajectories for the disordered NTS. The red cross indicates that the tunnel between HNH and RuvC is closed in state I. (d) A global view showing interactions between REC3 domain and the sgRNA-TS duplex in the PAM-distal. Dashed lines indicate unmodeled regions of L1 and REC3. The domains and nucleic acids are colored as in Figs. 1a, b.
The state I structure is the first reported visualization of the “checkpoint” conformation of Cas9 at high resolution, confirming in part earlier models resulting from of bulk and single-molecule fluorescence energy transfer (smFRET) studies 4,5,8 while revealing the true location of the HNH and recognition lobes in this state. Importantly, state I reveals that the HNH nuclease domain is in an inactive conformation and positioned >30 Å away from the cleavage site in the TS, a significantly larger distance (by ~20 Å) than previously observed 10. Also, in state I, the REC2 domain sterically occludes the HNH domain from accessing the scissile phosphodiester bond, which is consistent with earlier proposals 10 and smFRET data 7. The residues previously examined by FRET (Asp435 and Glu945) were located in state I at a distance of 83 Å, which is consistent with both previous ternary complex crystal structures 11,12 and measured EFRET values. The most frequently used pair in FRET, Ser355 and Ser867, is separated by 73 Å in state I, which is halfway between values derived from crystal structures of the apo- and sgRNA-bound enzyme (~80 Å) and the ternary complex (~60 Å) 4. These distances are consistent with measured EFRET values. The distance between Ser867 and Asn1054 is 5.8 Å in state I, which is also comparable to that in the apo- and sgRNA-bound Cas9 (~6.5 Å) crystal structures (see Extended Data Table 1 in Sternberg et al 4). Because of the disorder in the RuvC segment in the ternary complex crystal structures, only estimated Ser867-Asn1054 distances of ~30 Å were reported (see Extended Data Table 1 in Sternberg et al 4), so it is difficult to make a precise correlation of these values with our structure. Furthermore, the reconstruction shows that the REC3 domain is mostly ordered except for the three solvent-exposed loops in REC3 (residues 530-537, 574-588, 686-689). Considering that interactions between the REC3 and PAM distal end of TS have been reported to function as a conformational checkpoint for proper sgRNA-DNA base pairing 7, the disordered loops in REC3 domain may be actively sampling different regions of the sgRNA-DNA hybrid prior to DNA cleavage (Fig. 2d). Taken together, these observations suggest that state I likely represents the “checkpoint” conformation of Cas9.
HNH domain remains bound to the cleaved DNA and REC2 becomes disordered immediately after cleavage
The structure of state I reveals rearrangements of the Cas9-sgRNA upon DNA binding and in the presence of Mg2+ in solution; however, the HNH domain is in an inactive conformation, and the DNA is intact. By contrast, state II captures the catalytically competent conformation of the HNH domain that has already cleaved its substrate (Figs. 1d-g, 3). Notably, the HNH domain is resolved in complex with the TS, which is cleaved between the third and fourth nucleotide upstream of the PAM sequence. Persistent binding of HNH to the cleaved on-target DNA is in agreement with smFRET data 5. Superposition of states I and II reveals that the HNH domain must undergo a large conformational change to reach the site of its catalytic activity (Fig. 3a). In the transition from state I to state II, the displacement of the HNH domain corresponds to a ~34 Å translation accompanied with a rotation of either ~220° (clockwise) or ~140° (anti-clockwise), relative to the axis perpendicular to the sgRNA-DNA hybrid (Fig. 3a, Supplementary Video 1). The proposed model of the HNH domain docked onto DNA 4 is consistent with the conformation of HNH in state II. For example, the docked model predicted that Ser355-Ser867 and Ser867-Asn1054 distances should be 21 Å and 57 Å, respectively 4. Remarkably, these distances are 20.6 Å and 60 Å in state II, supporting the assignment of state II as the catalytic conformation of Cas9.
Fig. 3. HNH domain adopts catalytic conformation in state II.

(a) A conformational change of the HNH domain during transition from state I to state II. The HNH domain in state I (beige cartoon) rotates around a central axis (grey rod) and translates ~34 Å to reach the scissile bond in TS (shown with dashed circle, and either arrow or asterisk) where it adopts an active conformation (pink cartoon). The nucleic acid backbone is colored as in Fig. 1 with the rest of Cas9 shown in grey transparent surface. (b) Close-up view of the catalytically-competent active site of HNH in state II with ball-and-stick representation of the catalytic site. The newly formed 5’- and 3’-ends of the cleaved TS are shown in blue, with hydrogen bonds shown as dashed lines. (c) Cryo-EM density maps and models spanning the cleavage site demonstrate that the TS is intact in state I and cleaved in states II and III. Arrow designates the scissile bond in state I, while the asterisk marks the cleaved phosphodiester bond in states II and III.
Importantly, because the orientation of the REC2 domain in state I prevents the HNH domain from adopting its catalytic conformation, the HNH movement is coupled to a REC2 domain motion away from the TS and towards the solvent (Fig. 3a). Interestingly, the outward REC2 motion during HNH docking was previously suggested based on reaction sequence simulations and other biophysical studies10,13. To this end, smFRET studies reported reciprocal changes in bulk FRET values, implying that the catalytic activity of Cas9 may be tightly regulated by coupling the movements of the REC2 and HNH domains 7. The loss of tight contacts with the sgRNA-TS hybrid may enable REC2 to be more dynamic, as evidenced by the weaker density for this domain (Figs. 1d, g). To rule out the possibility that the loss of domain density was a result of Cas9 degradation, the ternary complex was assessed for activity and stability over time (Supplementary Fig. 3, Supplementary Data Set 1). Time-course analysis established that Cas9 exhibits full activity and expected single-turnover kinetics, and that there is no detectable degradation of the Cas9 protein (Supplementary Fig. 3, Supplementary Data Set 1).
The transition to state II, the solvent-exposed loops of REC3 that were disordered in state I become ordered and establish new interactions with the PAM-distal hybrid reminiscent of the type of movements observed in other Cas9 complex structures (PDB IDs 4OO8 and 5F9R) 10,12. This is contrast to the disordering of REC2 in state II as compared to state I. The observed large-scale movements of the HNH and REC2 domains, the accompanied disorder in REC2, the coordinated nature of the HNH, REC2 and REC3 domain motions during the transition from the “checkpoint” conformation to DNA hydrolysis add new elements to our understanding of the Cas9 reaction sequence.
Detailed analysis of the state II complex structure reveals significant additional changes and interactions between the RuvC domain and the extended DNA duplex that could be of significance. The solvent exposed loop carrying positively charged residues (Lys948, Arg951 and Lys954) in the RuvC domain interacts with the extended DNA duplex (Supplementary Fig. 4a). Moreover, the region in RuvC encompassing residues 1000-1076 is disordered in state II. Structural comparison with the 30 bp dsDNA-bound Cas9 (PDB ID 5F9R) reveals that the longer distal DNA duplex used in our study would be expected to clash sterically with this particular segment of RuvC in the “Mg2+-free” ternary complex. Interestingly, the TS DNA products are bound to the HNH active site while the NTS is positioned slightly farther away from the RuvC catalytic center as compared to its location in the absence of Mg2+ 10. Considering that TS DNA hydrolysis by the HNH domain results in simultaneous cleavage of NTS by the RuvC domain 4, it is likely that the NTS has already been cleaved by the RuvC and has dissociated from the active site. This suggestion is consistent with our observation of weak density covering the region where the distal NTS may be located (Supplementary Figs. 1b, 4b). Taken together, we conclude that state II represents the structure of the “post-catalytic” state in which the HNH domain has not yet dissociated from the cleaved TS.
The HNH active site adopts catalytically competent conformation in State II
Previous studies demonstrated Cas9-sgRNA-DNA is trapped in an inactive conformation in the absence of Mg2+, and that catalysis can be rapidly activated by addition of the divalent cation 5,10,14,15. Although DNA hydrolysis has been completed in state II, the orientation and proximity of the catalytic residues and reaction product groups indicates that the HNH active-site crevice adopts a conformation that closely resembles the catalytic conformation (Fig. 4). Notably, side chains of Asp839 and Asn863, and leaving groups, 5'-PO3 of T14 of and 3'-OH of G13, point towards one another and the putative Mg2+-binding pocket (Figs. 3b, 4a). Although the overall similarity of the conformation of the HNH active site with that of other homologous enzymes implies that Mg2+ is bound in this location (Fig. 4a), we cannot establish this definitively at the present resolution of this map.
Fig. 4. The HNH active site conformation in state II.
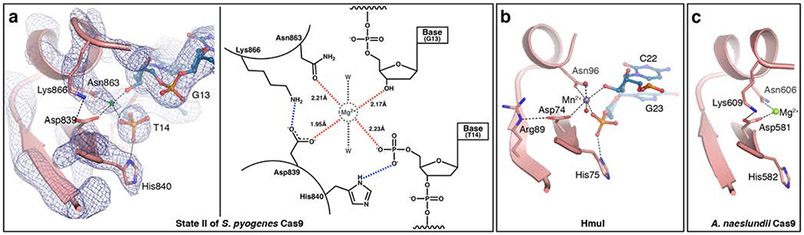
(a) Left: Close-up view of the active site of the HNH domain in state II (pink) with the corresponding cryo-EM map (blue mesh) superposed. A putative metal ion position is marked with green asterisk. Right: Schematic representation of the state II HNH active site. Putative Mg2+ ion, water molecules and their interactions are colored grey. Observed H-bonds are blue dashed lines, while red dashed lines designate putative interactions between Cas9 side chains and unmodeled Mg2+. Distances between coordinating Cas9 residues and the proposed Mg2+ion location are shown in Å. (b) The active site of the homing endonuclease I, HmuI (PDB ID 1U3E) 26. (c) The active site of the HNH domain from A. naeslundii Cas9 (PDB ID 4OGE) 9. In (a, left), (b) and (c), catalytic residues and reaction products are shown as pink and blue sticks, respectively, and hydrogen bonds, both observed and putative, are shown as dashed lines. In panels b and c, purple, green and red spheres are Mn2+, Mg2+ and water molecules, respectively.
Our results explain that the Asn863Ala mutation abolishes HNH catalytic activity by hindering formation of the Mg2+-binding pocket 12. Further, analogous to His75 in the homing endonuclease HmuI (Fig. 4b), the side chain of His840 forms a hydrogen bond with the newly formed 5’-end of the cleaved TS (Figs. 3b, 4a). Because of its proximity, His840 is a likely general base that, together with Mg2+ ion, orients and activates a water molecule for nucleophilic attack onto the TS scissile phosphodiester bond. Additionally, the amino group of Lys866 is within hydrogen-bonding distance from the side-chain carboxyl of Asp839 (Figs. 3b, 4a). Given its significance in HmuI and Actinomyces naeslundii Cas9 (Fig. 4c), we therefore propose that the interaction between Asp839 and Lys866 is significant for Mg2+ coordination. Concurrently, the side chain of Arg864 points away from the active site and interacts with the minor groove of the DNA-RNA hybrid (Figs. 3b, 4a). Interestingly, the importance of Asn863 for catalysis and interactions between Arg864 and the hybrid were not predicted in earlier molecular dynamics experiments 16,17. Altogether, the conformation of the HNH active site captured in state II is consistent with a state immediately following DNA hydrolysis, and best described as the “post-catalytic” conformation of the HNH active site.
In the “product” complex, the HNH domain is disordered and REC2 is ordered and bound to nucleic acids
Substantial Cas9 domain rearrangements are observed during transition from state II to state III (Figs. 1d-h, Supplementary Fig. 5, Supplementary Video 2). Most notably, the HNH domain dissociates from the cleaved product and is completely disordered in state III. By contrast, the REC2 domain is now ordered and occupies the same position as in state I, re-establishing interactions with the nucleic acid. In addition, the TS retains base-pairing to both sgRNA and the non-spacer NTS as in state II, and the single-stranded NTS product remains dissociated from RuvC. These changes are accompanied by disorder in loops L1 and L2 (residues 766-779 and 907-925). Unexpectedly, interactions between REC3 and the RNA-DNA hybrid as well as between RuvC and the extended DNA duplex (Supplementary Fig. 4a) are nearly the same in states II and III, indicating that these interactions persist during transition from the “post-catalytic” state to state III. Given its structural features and previously published observations 6,18,19, we suggest that state III represents a stable “product” complex that precedes release of the cleaved DNA. The persistent interactions between REC3 and RNA-DNA hybrid, and RuvC and the DNA duplex likely contribute to the unusual stability of the Cas9 product complex.
Discussion
In this study, we present novel structures of the wild-type Cas9 in complex with sgRNA and DNA and in the presence of Mg2+. Three distinct states were captured in solution, providing a glimpse into the mechanism by which Cas9 cleaves its DNA target. Based on the new structural findings and their implication, we propose a revised model for the Cas9-catalyzed RNA-guided DNA cleavage (Fig. 5). States I-III are assigned to distinct steps of the Cas9 catalytic pathway that closely match those observed in previous FRET experiments 4,5,7,8. Transitions between these states involves structural rearrangements within the Cas9 protein. Binding of sgRNA to apo-Cas9 9 induces major rearrangements of the Cas9 protein domains 20. Subsequent binding of Mg2+ and DNA to the binary complex promotes formation of an intermediate state I or the “checkpoint” conformation, which is a proof-reading state that assesses whether sufficient Watson-Crick base pairing has been established between the sgRNA and TS. Following the RNA-DNA hybridization, the newly displaced NTS runs parallel to the RNA-DNA hybrid within the central channel, while REC3 scans for proper base-pairing between TS and sgRNA at the PAM-distal end. The enzyme may sample a ‘pre-catalytic’ conformation that was observed under Mg2+-free conditions before final HNH docking 10. In state II, the HNH and REC2 domains move simultaneously; the HNH domain docks onto the TS DNA, while REC2 becomes disordered as it moves outward, towards the solvent. Solvent-exposed loops of REC3 become structured and interact with the PAM-distal of the RNA-DNA hybrid. With the NTS now displaced and the R-loop formed, the HNH and RuvC simultaneously cleave the TS and NTS in a Mg2+-dependent manner. In the “product” complex (state III), the HNH domain dissociates from and moves away from the DNA, becoming disordered, while REC2 reorients and adopts the same conformation as in state I. Persistent interactions of REC3 and RuvC with distal duplex presumably stabilize the “product” complex. As noted earlier, under the conditions used for sample storage and grid preparation, Cas9 is fully stable, catalytically active and exhibits expected single-turnover kinetics (Supplementary Fig. 3, Supplementary Data Set 1). The observation of multiple ternary complex states, perhaps fortuitous to the specific experimental conditions we used, nevertheless suggest that understanding reaction conditions that slow or delay Cas9 activity could be useful for manipulation of the enzyme in vitro.
Fig. 5. Proposed mechanism for the concerted series of domain movements involved in Cas9-mediated DNA cleavage.
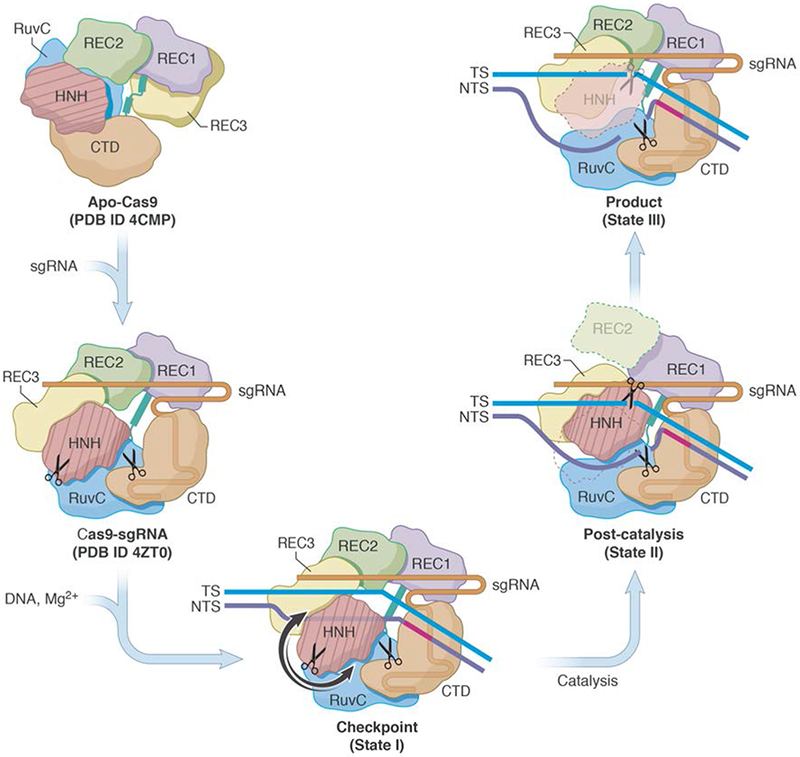
The binding of sgRNA to apo-Cas9 induces major domain rearrangements and formation of the binary complex. In the presence of dsDNA and Mg2+, the “checkpoint” conformation (State I) is formed. Target strand base-pairs with NTS and runs parallel to sgRNA, HNH adopts an inactive conformation, and the distal DNA duplex is short. Upon activation, HNH rotates and swings (arrows indicate possible routes) towards the cleavage site in TS. This is accompanied by REC2 disorder, ordering of REC3 loops that bind to the distal PAM, and interactions between RuvC and the longer distal DNA duplex. During catalysis, the HNH and RuvC active sites (scissors) cleave the TS and NTS, respectively. After cleavage, HNH remains bound to products, whereas the RuvC active site is near the cleaved NTS. This arrangement is captured in the “post-catalytic” complex (State II). In the “product” complex (State III), HNH dissociates from the cleaved TS and becomes disordered, REC2 is ordered and adopts its State I conformation, while interactions between REC3, RuvC and nucleic acids persist. Apo-Cas9 and Cas9-sgRNA are based on PDB ID 4CMP 9 and 4ZT0 20, respectively.
Taken together, it appears that the mechanism of DNA hydrolysis by Cas9 involves a remarkable and unexpected set of coupled conformational transitions, with alternating sequences of order-disorder transitions in key protein domains. Moreover, dramatic quaternary rearrangements are required to enable the HNH and REC2 to exchange their positions around the DNA during the course of a single catalytic cycle. These coupled domain movements indicate that Cas9 catalysis is choreographed with both large-scale and subtle motions to enable the reaction to take place. Traditionally, analyses of available Cas9-sgRNA-DNA ternary complex structures have aided targeted and functional mutagenesis of the enzyme. Though the structures utilized for previous engineering visualized Cas9 before DNA cleavage 9-12, they nevertheless enabled substantial improvement of genome editing outcomes and capabilities. For example, mutation of the PAM interaction domain can broaden targeting range 21,22, while mutations of the REC3 domain can enhance the fidelity of DNA cleavage 7.
The studies we present here bring our understanding of the Cas9 reaction sequence closer to completion by contributing the first structures of Cas9 immediately after DNA hydrolysis and in a persistent enzyme-product complex. Considering our structures were resolved with catalytically active Cas9-sgRNA-DNA product complexes, it is reasonable to expect that these molecular and structural insights are highly relevant for enzyme engineering and that will offer new opportunities for modulation and manipulation of the catalytic functions of Cas9 under native conditions. Recent efforts converged to Cas9 mutants with altered PAM recognition specificities 21 and those that displayed enhanced fidelity 23, among many others (reviewed by Adli 24,25 and in Wilkinson et al 24,25). Going forward, mutations of the HNH active-site and its surrounding residues or modulation of coupled HNH-REC2 domain motions may yield Cas9 variants with altered DNA catalysis rates. Manipulation of the newly identified enzyme-nucleic acid interfaces in the product complex could affect the dissociation rate of Cas9 from its DNA products, potentially enhancing its processivity. Solvent-exposed loops of REC3 (residues 530-537, 574-588, 686-689) that interact with the distal PAM are obvious targets and the limited evolution of this segment could yield enzyme mutants with improved characteristics. Another area of interest is a segment of RuvC (residues 1,000-1,076) that moves out of the way to allow binding of the distal duplex to REC3 after DNA cleavage. In addition, the RuvC loop that carries a patch of positively charged residues (Lys948, Arg951 and Lys954) which interact with the distal duplex could be another target for mutational studies. Modulation of any, or a combination, of these elements could produce Cas9 mutants with altered properties that could be of immediate practical value by expanding applications of Cas9 throughout the genome, manipulating the rate of genome editing and its efficiency.
On-line Methods
Substrate preparation.
The 40 base-pair double-stranded DNA (dsDNA) substrate was constructed from two single-stranded DNA (ssDNA) oligonucleotides ordered from IDT:
| Target strand | 5’-CCAGTGCGTAGGCGCATAAAGATGAGACGCTGGCGATTAG-3’ |
| Non-target strand | 5’-CTAATCGCCAGCGTCTCATCTTTATGCGCCTACGCACTGG-3’ |
Oligonucleotides were hydrated in water to a final concentration of 100 mM and mixed at a 1:1 ratio. After heating to +95 °C, the dsDNA sample was cooled to +25 °C in 1 °C/min intervals.
RNA preparation.
The sgRNA spacer sequence containing BsmBI compatible ends (lower case nucleotide letters) was cloned into the DR274 (Addgene #57048) plasmid upstream of the 80 nucleotide sgRNA scaffold using the following primers ordered from IDT:
| Forward | 5’-taggGGCGCATAAAGATGAGACGC-3’ |
| Reverse | 5’-aaacGCGTCTCATCTTTATGCGCC-3’ |
To generate the template DNA for in vitro transcription, a large-scale PCR amplification starting from the T7 promoter to the end of the sgRNA scaffold was performed on the sgRNA containing plasmid using the following set of PCR primers:
| Forward | 5’-TAATACGACTCACTATAGG-3’ |
| Reverse | 5’-GCAAGCACCGACTCGGT-3’ |
PCR reactions were completed using Phusion DNA polymerase, Phusion HF buffer (NEB) and the following thermocycling conditions: initial denaturation at +98 °C for 30 s; 30 cycles of denaturation at +98 °C for 5 s, annealing at +64 °C for 10 s and elongation at +72 °C for 15 s; the final elongation at +72 °C for 5 min. The PCR products were column purified (Zymo DNA clean and concentrator) and eluted to a final volume of 100 μL. To generate the RNA transcript, a 20 mL transcription reaction was performed in the presence of 100 μL of purified PCR product, 5.0 μg/mL purified recombinant T7 RNA Polymerase, and 1x transcription buffer (40mM Tris-HCl pH 8.0, 2mM spermidine, 10mM MgCl2, 5mM dithiothreitol (DTT), 2.5mM rNTPs). Following incubation at +37 °C for 4 h, reactions were spun at 5,000 × g for 20 min to pellet and remove the pyrophosphate precipitate. The filtered transcription reaction was loaded onto a Resource-Q column (HiLoad 16/60 Superdex 200, GE Healthcare) and purified over a linear NaCl gradient (0.4-0.7 M) in 20 mM Tris-HCl, pH 8.0. The sgRNA was then separated by gel filtration on a S200 Superdex size-exclusion column (GE Healthcare) equilibrated with 20 mM Tris-HCl, pH 8.0, and 150 mM NaCl. The eluted sgRNA was concentrated to ~2 mg/mL, flash-frozen, and stored at −80 °C until use.
Cas9 expression and purification.
Recombinant wild-type Streptococcus pyogenes Cas9 (Cas9) possessing an N-terminal His6-MBP tag (Addgene #39312) was expressed in Escherichia coli strain Rosetta2 (DE3) (Novagen) and purified as described previously 27. Briefly, when cells reached an OD600 of ~0.6, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.2 mM to induce protein expression. Cells were then grown for an additional 18-20 h at +18 °C. Harvested cells were resuspended in 5 mL/g pellet of lysis buffer (20 mM Tris pH 8.0, 250 mM NaCl, 10% (v/v) glycerol, and 3 mM β-mercaptoethanol) containing an EDTA-free protease inhibitor tablet (Roche). The cell suspension was sonicated on ice and clarified by centrifugation at 27,000 × g for 45 min. The soluble lysate fraction was loaded onto a nickel-charged His-Trap FF crude column (GE Healthcare). The His6-MBP-Cas9 eluted from the column with lysis buffer supplemented with 250 mM imidazole. The His6-MBP tag was removed by overnight TEV cleavage (1 mg of TEV protease was added per 50 mg of protein). For cleavage, the Ni2+ eluate was diluted to ~1 mg/mL in dialysis buffer (20 mM HEPES-KOH, pH 7.5, 150 mM KCl, 10% (v/v) glycerol, and 1 mM DTT) and dialyzed against dialysis buffer overnight at +4 °C. Following the His6-tag removal, Cas9 was loaded onto a SP Sepharose High Performance cation exchange column (HiTrap SP HP, GE Healthcare) and eluted in 100-500 mM gradient of KCl. Finally, the Cas9 protein storage buffer was exchanged by dialysis into 30 mM Tris-HCl pH 8.0, 200 mM NaCl, 20 mM EDTA, 10% (v/v) glycerol and 5 mM DTT. The final Cas9 sample was concentrated to ~10 mg/mL, filtered through 0.22 μm filter, aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C.
Reconstitution of the Cas9-sgRNA-DNA ternary complex.
Reconstitution of the Cas9-sgRNA-DNA was carried out by adding substrate dsDNA to a pre-formed the Cas9-sgRNA binary complex. Purified sgRNA was heated to +94 °C for 4 min and then cooled at room temperature to promote proper secondary structure formation. Cas9 and sgRNA were mixed in a 1:1.5 molar ratio and incubated for 10 min at room temperature before adding substrate dsDNA to bring the final molar ratio of ternary complex components Cas9, sgRNA, and dsDNA to 1:1.5:2.0. The ternary complex was diluted in Cas9 complex ‘storage’ buffer (30 mM Tris-HCl pH 7.5, 200 mM NaCl, 20 mM EDTA, 10% glycerol and 5 mM DTT) to final concentration of ~0.5 mg/mL. To initiate nuclease activity, MgCl2 was added to the ternary complex to a final concentration of 40 mM. Following incubation at +37 °C for 30 min, samples were flash-frozen in liquid nitrogen.
Electromobility shift assays, SDS-PAGE, and TBE-urea analyses of Cas9 activity and stability.
To assess Cas9 activity and the stability of the ternary complex in the complex storage buffer over time, the ternary complex was formed using 1:1.5:1.5 molar ratio of Cas9-sgRNA-DNA. Aliquots of the ternary complex were taken at 0, 1, 5, 30, 240 and 960 min after DNA addition and subjected to several analyses. Briefly, 2.6 mL of the master mix (i.e. the complex storage buffer containing 40mM MgCl2) was prepared and kept on ice. The sgRNA was heated at +94 °C for 3 min, allowed to cool to room temperature, and then added to the master mix to a final concentration of 600 nM. Cas9 was then added to a final concentration of 400 nM and incubated for 5 min before the addition of the substrate DNA to a final concentration of 600 nM. Subsequently, 12 150 μl aliquots (i.e. 6 duplicate timepoints) were distributed into a 96-well plate that was pre-chilled on ice. 25 μl of 0.5 M EDTA was added to the first 2 reactions to inhibit DNA cleavage for the 0 min timepoint. The plate was then transferred to a +37 °C heated block and EDTA was simultaneously added to two samples corresponding to each timepoint.
To assess stability of the Cas9 protein over time, 22.5 μL of the ternary complex from each timepoint was mixed with 7.5 μL of 4x SDS-polyacrylamide gel electrophoresis (PAGE) loading buffer, heated for 5 min at +95 °C, and loaded onto a 4-20% precast SDS-PA gel (Bio-Rad). The gel was stained with Coomassie Brilliant Blue R-250 to visualize Cas9.
After the addition of EDTA, one of the samples was subjected to RNase A (Invitrogen) and proteinase K (Life Technologies) treatments for 60 and for 30 min, respectively, at +37 °C. After treatments, samples were flash-frozen and kept at −80 °C until further use. RNase A and proteinase K treatments allow for analysis of free and Cas9-bound DNA at each timepoint and ensure the visualization of cleaved DNA products.
To visualize the ternary complex stability and DNA cleavage levels, samples were subjected to electromobility shift assays (EMSA) on a native TBE-PA gel. Briefly, 18 μl of both untreated and RNaseA/proteinase K-treated samples corresponding to each time point was mixed with 2 μl of 5x Novex Hi-Density TBE sample buffer (Thermo Fisher) and then immediately loaded onto an 8% 0.5x TBE-PA gel. To prevent cleaved DNA from running off the gel, electrophoresis was run until the bromophenol dye front reached 3/4 of the gel. The gel was first stained in ethidium bromide (0.05 μg/mL) and then in Coomassie Brilliant Blue R-250 to visualize nucleic acids (i.e. DNA and sgRNA) and Cas9 on the same gel.
To assess kinetics of DNA cleavage, 20 μL of untreated and RNaseA/proteinase K-treated sample corresponding to each time point was mixed with 20 μL of 2x Novex TBE-urea sample buffer (Thermo Fisher), heated at +95 °C for 3 min, and loaded onto a 15% TBE-urea PA gel. To visualize the nucleic acid species, the gels were stained with 0.05 μg/mL ethidium bromide solution for 10 min.
Control samples contained 400 nM Cas9, 600 nM sgRNA, or 600 nM DNA only in the complex storage buffer. All gels were imaged on the GL212 Pro Imager (Carestream) using UV transillumination for ethidium bromide staining and white light trans-illumination for Coomassie staining.
Cryo-EM grid preparation and data collection.
The Cas9-sgRNA-DNA ternary complex sample (0.5 mg/mL) was concentrated to a volume of ~ 1.8 mL and then vitrified. A volume of 2.8 μl of the sample was applied to Cu R1.2/1.3, 300 mesh holey carbon grids (Quantifoil) plasma cleaned using H2/O2 gas mixture for 5 s in a Solarus plasma cleaner (Gatan Inc.) operating at 50 W. The sample was allowed to adsorb for 2 s prior to blotting for 6 s, followed by plunge freezing into liquid ethane cooled at liquid N2 temperature with a Leica EM GP (Leica Microsystems Inc.) and operating at +20 °C and 90% humidity. Cryo-EM grids were imaged using a Titan Krios transmission electron microscope (FEI Company), operated at 300 kV and aligned for parallel illumination, with the specimen maintained at liquid nitrogen temperatures (Table 1). Images were recorded on a K2 Summit camera equipped with the XP sensor (Gatan Inc., Pleasanton, CA) operated in super-resolution counting mode, placed at the end of a GIF Quantum Energy Filter (Gatan Inc., Pleasanton, CA), operating in zero-energy-loss mode with a slit width of 20 eV. Images were typically collected with a defocus range between −0.7 to −3.0 μm at a nominal magnification of 165,000x corresponding to a super-resolution pixel size of 0.418 Å. A total of 2405 micrographs were collected using automated data acquisition in Latitude software (Gatan Inc., Pleasanton, CA) as a 70-frame movie with intermediate frames recorded every 0.2 s. The dose rate used was ~5.2 e−/Å2·s (at the specimen plane) with a total exposure time of 14 s, giving an accumulated dose of ~73 e−/Å2·s per micrograph.
Image processing.
All image processing was performed within RELION 3.0-beta-2 28 unless stated otherwise. Two batches of movies were first motion-corrected and dose-weighted separately using MotionCor2 29. Then all aligned micrographs were merged and processed together. About 1.3 million particles were picked by Gautomatch and cleaned by 2D classification. An initial model was generated from cleaned particle stack and used as reference in 3D classification. Particles corresponding to the best class showing high-resolution features were then re-extracted with a box size of 608 and a binning factor of 2. Local 3D refinement was performed, followed by CTF refinement and Bayesian polishing. The refined particles were further classified into 6 classes with mask. For the final reconstruction, particles of each class were subjected to homogeneous refinement in cryoSPARC 30. The overall resolutions were estimated based on the gold-standard criterion of Fourier shell correlation (FSC) = 0.143. Local resolutions were estimated from two half maps in cryoSPARC. Fourier shell correlation was calculated with Mtriage in Phenix 31.
Model building and refinement.
Initial models for all three states were derived from the high-resolution crystal structures of Cas9-sgRNA-DNA complexes (PDB ID 5B2R, 5F9R and 4UN3). Each domain was fitted into electron density maps and adjusted manually in Coot 32. For the active site of state II, corresponding residues from the homing endonuclease I-HmuI (PDB ID 1U3E) were used as starting point. For the additional PAM-distal DNA duplex in state II and III, an ideal B-form DNA duplex was docked into the density. Final structures were refined using crystal structures as reference in Phenix 31. Final models were validated with statistics from Ramachandran plots, MolProbity scores, and EMRinger scores (Table 1).
Reporting Summary Statement:
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data Availability Statement:
All data needed to assess and evaluate the conclusions in the paper are available in the main text and Supplementary Information. The coordinates and electron density maps are deposited in the Protein Data Bank and EMDB with the following accession numbers: 6O0Z and 0585 for pre-catalytic complex (state I), 6O0Y and 0584 for post-catalytic complex (state II), and 6O0X and 0583 for product complex (state III). Source data for Supplementary Fig. 3 are available with the paper on-line. All other data are available upon request.
Supplementary Material
Acknowledgments:
This work was supported by the intramural research program of the National Cancer Institute (X.Z, S.C. and S.S.), NIH grants (GM097042 to M.S. and HD081534 to B.J.M.), and UIC Center for Clinical and Translational Sciences (R.C. and A.K.P.).
Footnotes
Competing Interests Statement: The authors declare no competing interests.
References:
- 1.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823, doi: 10.1126/science.1231143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826, doi: 10.1126/science.1232033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyle EA et al. High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding. Proc Natl Acad Sci U S A 114, 5461–5466, doi: 10.1073/pnas.1700557114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sternberg SH, LaFrance B, Kaplan M & Doudna JA Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113, doi: 10.1038/nature15544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dagdas YS, Chen JS, Sternberg SH, Doudna JA & Yildiz A A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci Adv 3, eaao0027, doi: 10.1126/sciadv.aao0027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sternberg SH, Redding S, Jinek M, Greene EC & Doudna JA DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67, doi: 10.1038/nature13011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen JS et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410, doi: 10.1038/nature24268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sung K, Park J, Kim Y, Lee NK & Kim SK Target Specificity of Cas9 Nuclease via DNA Rearrangement Regulated by the REC2 Domain. J Am Chem Soc 140, 7778–7781, doi: 10.1021/jacs.8b03102 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Jinek M et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997, doi: 10.1126/science.1247997 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang F et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871, doi: 10.1126/science.aad8282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anders C, Niewoehner O, Duerst A & Jinek M Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573, doi: 10.1038/nature13579 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishimasu H et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949, doi: 10.1016/j.cell.2014.02.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palermo G, Miao Y, Walker RC, Jinek M & McCammon JA Striking Plasticity of CRISPR-Cas9 and Key Role of Non-target DNA, as Revealed by Molecular Simulations. ACS Central Science 2, 756–763, doi: 10.1021/acscentsci.6b00218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong S, Yu HH, Johnson KA & Taylor DW DNA Unwinding Is the Primary Determinant of CRISPR-Cas9 Activity. Cell Rep 22, 359–371, doi: 10.1016/j.celrep.2017.12.041 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raper AT, Stephenson AA & Suo Z Functional Insights Revealed by the Kinetic Mechanism of CRISPR/Cas9. J Am Chem Soc 140, 2971–2984, doi: 10.1021/jacs.7b13047 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Palermo G, Miao Y, Walker RC, Jinek M & McCammon JA CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc Natl Acad Sci U S A 114, 7260–7265, doi: 10.1073/pnas.1707645114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuo Z & Liu J Structure and Dynamics of Cas9 HNH Domain Catalytic State. Sci Rep 7, 17271, doi: 10.1038/s41598-017-17578-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brinkman EK et al. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol Cell 70, 801–813 e806, doi: 10.1016/j.molcel.2018.04.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke R et al. Enhanced Bacterial Immunity and Mammalian Genome Editing via RNA-Polymerase-Mediated Dislodging of Cas9 from Double-Strand DNA Breaks. Mol Cell 71, 42–55 e48, doi: 10.1016/j.molcel.2018.06.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang F, Zhou K, Ma L, Gressel S & Doudna JA A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481, doi: 10.1126/science.aab1452 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Kleinstiver BP et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485, doi: 10.1038/nature14592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63, doi: 10.1038/nature26155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleinstiver BP et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495, doi: 10.1038/nature16526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adli M The CRISPR tool kit for genome editing and beyond. Nat Commun 9, 1911, doi: 10.1038/s41467-018-04252-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilkinson RA, Martin C, Nemudryi AA & Wiedenheft B CRISPR RNA-guided autonomous delivery of Cas9. Nat Struct Mol Biol 26, 14–24, doi: 10.1038/s41594-018-0173-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen BW, Landthaler M, Shub DA & Stoddard BL DNA binding and cleavage by the HNH homing endonuclease I-HmuI. J Mol Biol 342, 43–56, doi: 10.1016/j.jmb.2004.07.032 (2004). [DOI] [PubMed] [Google Scholar]
Methods-only References:
- 27.Anders C & Jinek M In vitro enzymology of Cas9. Methods Enzymol 546, 1–20, doi: 10.1016/B978-0-12-801185-0.00001-5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530, doi: 10.1016/j.jsb.2012.09.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332, doi: 10.1038/nmeth.4193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296, doi: 10.1038/nmeth.4169 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501, doi: 10.1107/S0907444910007493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to assess and evaluate the conclusions in the paper are available in the main text and Supplementary Information. The coordinates and electron density maps are deposited in the Protein Data Bank and EMDB with the following accession numbers: 6O0Z and 0585 for pre-catalytic complex (state I), 6O0Y and 0584 for post-catalytic complex (state II), and 6O0X and 0583 for product complex (state III). Source data for Supplementary Fig. 3 are available with the paper on-line. All other data are available upon request.