

Abstract
Tumor heterogeneity provides a complex challenge to cancer treatment and is a critical component of therapeutic response, disease recurrence, and patient survival. Single-cell RNA-sequencing (scRNA-seq) technologies have revealed the prevalence of intra- and inter-tumor heterogeneity. Computational techniques are essential to quantify the differences in variation of these profiles between distinct cell types, tumor subtypes, and patients to fully characterize intra- and inter-tumor molecular heterogeneity. In this study, we adapted our algorithm for pathway dysregulation, Expression Variation Analysis (EVA), to perform multivariate statistical analyses of differential variation of expression in gene sets for scRNA-seq. EVA has high sensitivity and specificity to detect pathways with true differential heterogeneity in simulated data. EVA was applied to several public domain scRNA-seq tumor datasets to quantify the landscape of tumor heterogeneity in several key applications in cancer genomics such as immunogenicity, metastasis, and cancer subtypes. Immune pathway heterogeneity of hematopoietic cell populations in breast tumors corresponded to the amount of diversity present in the T-cell repertoire of each individual. Cells from head and neck squamous cell carcinoma (HNSCC) primary tumors had significantly more heterogeneity across pathways than cells from metastases, consistent with a model of clonal outgrowth. Moreover, there were dramatic differences in pathway dysregulation across HNSCC basal primary tumors. Within the basal primary tumors there was increased immune dysregulation in individuals with a high proportion of fibroblasts present in the tumor microenvironment. These results demonstrate the broad utility of EVA to quantify inter- and intra-tumor heterogeneity from scRNA-seq data without reliance on low dimensional visualization.
Keywords: tumor heterogeneity, single-cell RNA-seq, T-cell receptor, genomics, cancer systems biology
1. Introduction
Tumor heterogeneity poses significant challenges in the clinical diagnosis and treatment of cancer. Variation can occur among tumors of the same histological subtype, giving rise to variability in therapeutic responses among patients. Cellular heterogeneity can also occur within tumors, allowing cancer to evolve over the course of disease progression, resulting in drug resistance, treatment failure, and disease recurrence(1-3). An important source of tumor heterogeneity is the molecular variation among subclones and even individual cells within a tumor. This variation drives tumor progression through dysregulation of key cancer pathways and contributes to the evolutionary fitness of tumors(3,4). Differential variability analysis of bulk transcriptional data from microarrays and RNA-sequencing have also demonstrated that tumors with worse prognosis have a corresponding increase in transcriptional variation(5-8). Single-cell RNA-sequencing (scRNA-seq) technologies provide an unprecedented ability to measure gene expression from individual cells, enabling in-depth exploration of tumor heterogeneity(9,10).
Accurate characterization of inter-sample variation from scRNA-seq data of tumors is critical to quantify tumor heterogeneity. Molecular heterogeneity of scRNA-seq data is often analyzed visually, using computational methods for dimensionality reduction that enable qualitative interpretations based upon the dissimilarity in transcriptional profiles between cells(11-19). These techniques provide visual representations of the cellular composition within high-dimensional data. However, stochasticity, overplotting, and nonlinearity can challenge biological interpretation from visual analysis of scRNA-seq data. Moreover, the embeddings produced by these algorithms often rely on pre-selection of highly variable genes for clustering that may bias analyses based on variation. Highly variable genes are often identified based upon the coefficient of variation(20,21) or dispersion(22,23) of each gene across a cell population. Further gene set analysis of these statistics can be applied to quantify pathways or biological processes that contribute to cell-to-cell differences within a group of cells. Multivariate methods for analyzing transcriptional heterogeneity provide alternatives to quantify transcriptional heterogeneity from scRNA-seq data, such as PAGODA which quantifies overdispersion of annotated gene sets(24). Similarly, phenotypic volume was introduced to quantify the variation between cells in a single sample(25). These methods are all tailored to identify highly variable gene sets or samples across a population of cells from a single phenotype. Differences in variation within cells from a diseased population relative to variation within cells from a normal population may drive critical phenotypes, such as carcinogenesis or metastasis. Additional analysis techniques are essential to capture relevant pathway level heterogeneity that drives the observed deviations between groups of cells from distinct phenotypes.
In this paper, we extend our algorithm to quantify relative pathway dysregulation between experimental conditions from bulk transcriptional data(26) called Expression Variation Analysis (EVA) to scRNA-seq. Briefly, EVA provides a robust statistical test to compare the heterogeneity of transcriptional profiles of genes in a gene set between groups of cells from two phenotypes. We benchmark EVA using simulated data and compare its performance to other methods to demonstrate the accuracy, robustness, and interpretability of the algorithm. With the recent outpouring of large scale scRNA-seq studies in cancer, publicly available datasets provide a breadth of transcriptional data to explore the role of heterogeneity in a variety of contexts. We utilize datasets from head and neck(27) and breast(25) cancers, which contain thousands of cells comprising dozens of cell types from different tissues, subtypes, and individuals. These datasets were selected to benchmark the performance of our algorithm to characterize cases with known differences in heterogeneity, such as between tumor and normal cells. Pathways found to be statistically significant from EVA are called differentially variable or heterogeneous between cells from distinct sample groups. These analyses enable novel characterization of the role of tumor heterogeneity in complex processes in cancer. For example, these analyses enable quantification of pervasive, differentially variable pathways between primary tumors and metastases consistent with the hypothesis of clonal outgrowth(28). They also enable us for the first time to define the relationship between variation in immune pathways and TCR clonality. Finally, they quantify inter-tumor heterogeneity between primary tumors of a single subtype and identify immune dysregulation related to the degree of fibroblasts present in the tumor microenvironment (TME). Together, these results suggest that EVA provides an important tool to quantify inter-cellular heterogeneity directly from scRNA-seq data to yield novel biological insights.
2. Methods
EVA analysis
We use EVA from the R/Bioconductor package GSReg(26) version 1.17.0 to quantify pathway dysregulation in sets of cells from one group relative to the set of cells in another. Kendall-tau dissimilarities are computed with the function in the GSReg package and other dissimilarity measures using the R package philentropy version 0.2.0. Imputed scRNA-seq data are input to this algorithm, with imputation method described for each dataset below. P-values obtained from EVA analysis are FDR adjusted with the Benjamini-Hochberg correction and FDR adjusted p-values below 0.05 are called statistically significant. Additional details of our methods, including all code and datasets used to generate our results are available online.
Simulated datasets
We generate several simulated datasets to benchmark the performance of EVA, with varying degrees of complexity to balance controlled testing of the algorithm with the complex properties of scRNA-seq data. To examine the sensitivity of distance metrics to missing data, we simulate count data with different amounts of missing data using the squamous cell carcinoma bulk transcriptional dataset with a binary phenotype from the R/Bioconductor package GSBenchmark version 0.112.0. We randomly replace expression values with specified percentages of zeros to generate multiple datasets with varying degrees of missingness. We also generate a dataset with no signal by duplicating the transcriptional profiles for one phenotype. Again, we randomize zeros to determine the effect on the false positive rate in data without signal. We perform 100 iterations of all randomizations and test the performance against 35 distance measures.
While random zeros can be used to examine the general effect of missing data on dissimilarity, this does not accurately capture the nature of zeros in scRNA-seq data. To explore this, we simulate scRNA-seq data generated using the R/Bioconductor package Splatter version 1.0.3(29). We first generate a simulated dataset with no signal to assess the dependence of EVA to missing data from scRNA-seq data. Count data was simulated for a single group of 100 cells and 10,000 genes using default parameters. A second group was simulated under the same conditions, with the parameter for dropout = TRUE. Merging these outputs resulted in a single dataset with a population of cells equally distributed between two groups with identical transcriptomes and a varying number of zeros in one group. After imputation of this dataset, we randomize pathway expression profiles for each cell in one group to simulate data with differential heterogeneity in specified pathways.
For comparison of EVA to pre-existing methods we use PAGODA(24) from the R/Bioconductor package scde version 3.8. To generate simulated scRNA-seq data consisting of two groups with a high degree of differential variability, we modified Splatter so that the mean gene expression could follow a different distribution for each gene pathway and we added an additional variance parameter that allowed the variance to be specified for each cell group and pathway. We simulate count data for two groups containing 500 cells and 340 genes representing 10 synthetic pathways. We use default Splatter parameters with no added pathway variance for one group and set the added variance to 1 for each pathway in the second group to simulate differential variation between groups. We impute the simulated datasets described above for EVA analyses with the R package Rmagic version 1.3.0(30).
Public domain scRNA-seq datasets
We use 45,000 immune cells from 8 primary breast carcinomas with matched normal breast tissue, blood, and lymph nodes along with 27,000 T-cells with paired single-cell RNA and single-cell TCR sequencing previously described in Azizi et al.(25). The scRNA-seq dataset from Azizi et al.(25) was previously imputed from their study using BISCUIT(31). These datasets are available under GEO: GSE114727, GSE114724, and GSE114725.
We also use scRNA-seq datasets of 6,000 cells from 18 head and neck squamous cell carcinoma (HNSCC) patients containing 5 sets of matched primary tumors and lymph node metastases as previously described in Puram et al.(27) In our study, we impute the scRNA-seq data from Puram et al.(27) with MAGIC version 0.1.0 (Python) prior to analysis(30). HNSCC subtypes present in the data were called using The Cancer Genome Atlas (TCGA) classification profiles on bulk data from primary cancer cells(32). For inter-subtype comparisons batch effect correction was performed using the function ComBat from R/Bioconductor package sva version 3.26.0(33), considering each patient as a batch to isolate differences between cells from distinct HNSCC subtypes. This dataset is available under GEO: GSE103322.
TCR repertoire analysis
TCR repertoire clonality, richness, and Morisita-Horn similarity index between samples were computed on the TCR sequencing data from Azizi et al.(25) using the tcrSeqR(34) R package version 1.0.6 available from https://github.com/ahopki14/tcrSeqR.
Differential expression and gene set enrichment analysis
Differential expression analyses were performed across all expressed genes using the Monocle R/Bioconductor package version 2.6.1(35). In all tests, the number of genes detected in each cell was included in both the full and reduced models as a nuisance parameter. Gene set enrichment was performed on differentially expressed genes with FDR adjusted p-values below 0.05 using the wilcoxGST function from the limma package version 3.32.10(36). The alternative hypotheses of “up” and “down” were used to determine if genes were generally upregulated or downregulated, respectively.
Pathways and gene sets used in EVA and enrichment analyses
EVA and gene set enrichment analyses are performed for distinct sets of pathways appropriate for each analysis. Molecular signaling pathways are determined from the Hallmark gene sets in MSigDB version 6.1(37), meta-signatures defined from NMF analysis of the scRNA-seq data in Puram et al.(27), and the Myeloid Innate Immunity Panel pathways from NanoString (NanoString Technologies).
Code Availability
All code for the EVA analyses is available from https://github.com/edavis71/scEVA.
3. Results
The EVA algorithm provides a multivariate statistical framework to quantify differences in transcriptional heterogeneity between sets of cells from two phenotypes
EVA is a statistical algorithm designed to compare the relative heterogeneity of expression profiles within pathways between two phenotypes. It does this by computing the expected dissimilarity of expression profiles between any pair of samples from one phenotype relative to the expected dissimilarity of expression profiles between any pair of samples from another. When applied to the set of genes in a pathway, the expected dissimilarity between any pair of samples from one phenotype provides a measure of pathway dispersion or dysregulation. The difference of empirical estimates of the dispersion from a phenotype to another phenotype is called EVA statistics. EVA statistics test the null hypothesis that pathway dysregulation is equal between the phenotypes. Previously, we derived a computationally efficient approximation of p-values for these EVA statistics from U-theory statistics(26,38). Briefly, let xi denote the expression profile for sample i for the set of genes annotated to a specific pathway and d(xi,xj) the dissimilarity between the profiles for sample i and j for any dissimilarity metric d. EVA tests the null hypothesis that the E[d(xi,xj)] = E[d(xk,xl)], where E[] denotes the expectation and i and j index a pair of i.i.d. samples from one phenotype while k and l index a pair of i.i.d. samples from another. U-theory statistics provide an asymptotic approximation for the standard deviation of the dissimilarity measures for each phenotype as described in Asfari et al.(26,39,40), resulting in an analytic framework to test the null hypothesis. The resulting EVA algorithm provides a robust, non-competitive gene set measure to quantify the relative inter-phenotype heterogeneity of pathway usage. In our previous applications, we based our comparisons on the Kendall-tau dissimilarity measure in bulk transcriptional data. This measure was selected both because its rank-based nature reduces sensitivity to data preprocessing and models discordance between the expression of genes in a profile, which is indicative of pathway dysregulation. Bulk data lacks the resolution to quantify cellular heterogeneity because it is inherently an aggregate. EVA is poised to perform variation analysis based upon the measures of cellular heterogeneity in scRNA-seq data. If we treat each individual cell as a sample, we can adapt EVA to compare transcriptional heterogeneity scRNA-seq data between specified sets of cells (Figure 1A-1B).
Figure 1. Overview of EVA algorithm to compare pathway-level transcriptional heterogeneity between groups of cells from two phenotypes.
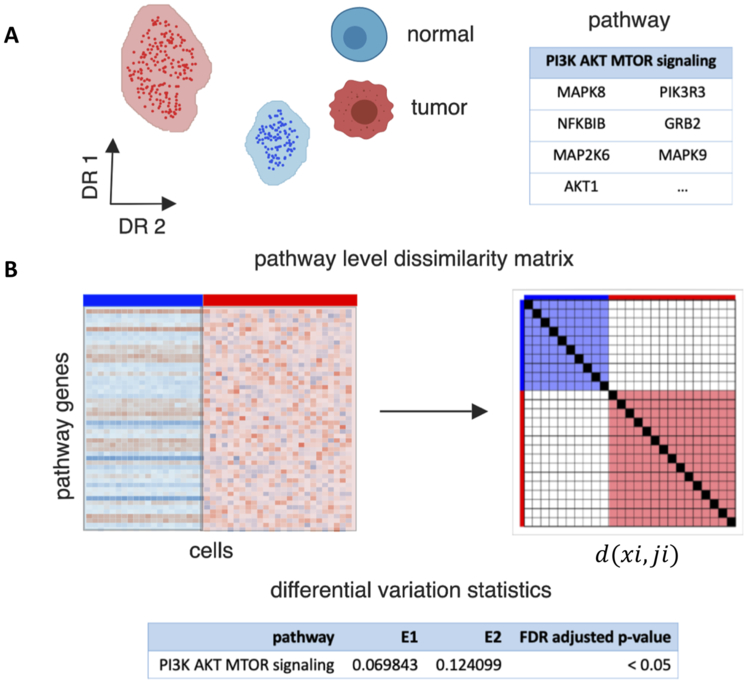
A. EVA inputs a single-cell gene expression matrix for cells from two phenotypes, such as tumor and normal cells, and a list of genes annotated to a single pathway. B. EVA extracts the expression profiles for pathway specific genes. It then computes the dissimilarity between the expression profiles for each pair of cells from the same phenotype using a user specified dissimilarity metric. Finally, EVA computes the expected dissimilarity between pairs of cells of each phenotype and U-theory statistics are applied to test the null hypothesis that the expected dissimilarity between pairs of cells from one phenotype is equal to the expected dissimilarity between paris of cells in the other. The expected dissimilarity between pairs of cells from one phenotype is called the EVA statistic, which quantifies the inter-cellular heterogeneity for a given pathway. The U-theory statistics provide a robust estimate to quantify p-values that compare this relative heterogeneity between phenotypes.
Given that Kendall-tau dissimilarity is rank-based, it is robust to normalization and read depth. However, the abundance of zero counts from scRNA-seq data would lead to an increase of ties in the ranking. Moreover, dropout events in scRNA-seq data occur when an mRNA transcript is not captured by the library preparation reaction prior to sequencing and this generally happens more frequently in genes expressed at low levels. This, combined with the general bursting nature of the transcription machinery, leads to “false” zero counts, indistinguishable from biological zeros of truly unexpressed transcripts, and inappropriate rank assignments in the Kendall-tau dissimilarity.
Simulated data studies reveal varying sensitivities of distance metrics to missing data
The EVA algorithm defaults to comparisons based upon the Kendall-tau dissimilarity metric. Because this metric quantifies the number of gene pairs which switch ranks between two conditions, it directly quantifies how tightly a set of genes in a pathway are regulated(5,26). Previous work in simulated data studies for bulk RNA-seq data have shown that this algorithm performs optimally at detecting differences between phenotypes whose expression profiles vary in rank between within samples from a single phenotype, but does not detect mean shifts in expression profiles between the phenotypes(38). In the case of single-cell data, it is critical to quantify the sensitivity of the algorithm to the dissimilarity metric used for analysis, missing data from dropout, and relative to other algorithms for pathway variation in scRNA-seq data. We have devised a series of simulated data experiments described in the Methods to evaluate the performance of EVA for each of these contexts. Briefly, we use the simulated data to examine the sensitivity of EVA to various distance metrics, the dependence of EVA to missing data from scRNA-seq, and to compare EVA to other available techniques.
First, the U-theory statistics to compare the expected dissimilarity between groups of cells from distinct phenotypes in EVA are general and can be applied to any dissimilarity measure. In order to compare the results of EVA using various dissimilarity measures, we use a dataset in GSBenchmark containing expression profiles of 22 matched samples from HNSCC tumor and normal tissues(41). It was previously observed that pathways between tumor and normal samples are significantly dysregulated in this dataset(26). We apply EVA using 35 dissimilarity metrics from Philentropy(42) and found that the number of significant pathways between tumor and normal vary widely across metrics (Supplemental Figure 1A). Several metrics including cosine and Ruzicka found no significant differentially variable pathways between normal and tumor samples. Kendall-tau detected the highest number of significant pathways, followed by Euclidean which is a commonly used distance to compare transcriptomes between single cells in visualization methods such as tSNE(11-17).
We next examined the sensitivity of different dissimilarity measures to variable sparsity by randomly replacing transcription values with specified percentages of zeros. For each metric, the significant pathways calculated on the previously described data with no sparsity are used as our true positives to benchmark the performance. The number of significant pathways varies greatly depending on the amount of missing data, with an overall loss of signal when the amount of zeros is the highest (Supplemental Figure 1B-1D). While cosine initially found no significant differentially variable pathways, this metric detected the most false positives when count data was replaced with 80% zeros. Of note, Kendall-tau had the most consistency of the results without dropout in these simulations.
Altogether, these simulations demonstrate that each dissimilarity metric has varying degrees of sensitivity to missing data. We select Kendall-tau for the remainder of the analyses in this paper based on the observed accuracy in the two simulated datasets without additional normalization. We note the rank-based nature of the Kendall-tau dissimilarity renders the EVA statistics performed on Kendall-tau dissimilarity independent of common normalization procedures, such as log transformation.
EVA captures differential variation in imputed scRNAseq simulations
The previous simulated datasets were designed with random zeros to test the performance of EVA to missing data. Yet, dropout in scRNA-seq may not be missing at random. To determine the effect of dropout and imputation on EVA’s robustness to detect pathway variability, we conducted an additional simulation study using synthetic scRNA-seq datasets generated using the Splatter pipeline(29). We first examined the performance of EVA on a dataset with no signal and a bias in zeros. We used Splatter to generate a simulated dataset from two identical groups, one containing only biological zeros, and one where dropout was also present (Figure 2A). Due to the abundance of zeros in the group with dropout and the sensitivity of Kendall-tau to missing data, EVA failed to recognize that the groups were otherwise identical and detected differential heterogeneity across 62% (31 out of 50) MSigDB Hallmark gene set pathway comparisons. We then imputed the missing values in the simulated dataset using MAGIC(30). EVA analysis of this imputed data had no pathways with statistically significant differential heterogeneity between the two groups (Figure 2B).
Figure 2. Performance of EVA with Kendall-tau dissimilarity on simulated data.
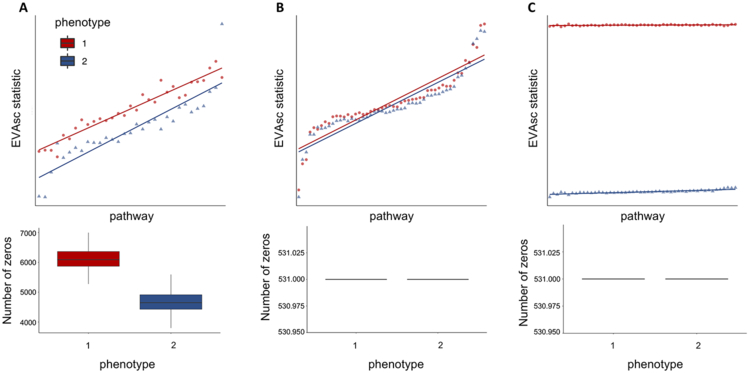
A. We apply EVA to a simulated dataset containing 50 pathways with no differential variation between cells from two phenotypes, but differential bias in their respective dropout rates. EVA statistics using a Kendall-tau dissimilarity have differential heterogeneity consistent with the simulated dropout rates. B. After MAGIC imputation of the data from A, EVA finds no significant differentially variable pathways and EVA statistics overlap for the two groups. C. We generate an additional simulated dataset by adding randomized signal to one group from the imputed data. The EVA statistics for significant pathways reflects the true heterogeneity in the simulated dataset.
We next examined the performance of EVA to detect known differential variation in imputed scRNA-seq data. Using the previously described imputed dataset, pathway expression profiles for each cell in one group were randomized to simulate heterogeneity. EVA detected dramatic differences in variation between the two groups across all randomized hallmark pathways. 100% (50 out of 50) of the comparisons were statistically significant (Figure 2C). These simulations demonstrate that EVA is able to assess the degree of pathway dysregulation between conditions in imputed scRNA-seq data.
EVA detects differential variation not observed by other methods
To benchmark EVA against previously published methods that are commonly used for variation analysis, namely coefficient of variation(20,21) and PAGODA(24), we compared the ability of these methods to recognize highly variable pathways in simulated data. To generate simulated scRNA-seq data containing heterogeneity between groups, we modified the Splatter pipeline to include an additional variance parameter. This allowed for increased variance in the simulated gene expression between cells, specifically with the option to specify the amount of variance between groups. We then generated 10 pathway expression profiles for 500 normal cells and 500 tumor cells with different values for this variance. Each of the pathway-level comparisons were statistically significant when analyzed with EVA. In comparison, gene set enrichment performed on the coefficient of variation statistics identified no significantly variable pathways and no significantly overdispersed pathways were identified by PAGODA with adjusted z-scores greater than 1.96 (Supplemental table 1). While previously existing methods quantify the overall variation across transcriptional profiles of all cells, EVA is unique in determining differential variation between cells from two distinct phenotypes.
EVA detects greater variation in tumor than normal in scRNA-seq data from breast cancer samples
We next evaluated the ability of EVA to compare heterogeneity between normal and tumor samples in scRNA-seq data from breast tumors for distinct immune cell types(25). Azizi et al.(25) reported an increase in the variance of tumor cell-intrinsic gene expression compared to normal breast tissue. Genes with the largest differential variance were enriched in signaling pathways important to the TME. To demonstrate that EVA enables robust statistical comparison of this heterogeneity in pathways, we compared tumor to normal immune cells across multiple cell types, which included T-cells, myeloid, and NK cells. EVA analysis detected greater variation in breast tumor than normal breast tissue across each immune cell type tested. All 50 pathways tested were statistically significant in each comparison (FDR adjusted p-value < 0.05) (Figure 3, Supplemental table 2). This suggests that increased pathway heterogeneity within tumor-associated immune cell types may be driven by distinct TMEs present within a single tumor.
Figure 3. All pathways are significantly dysregulated in immune cell types from breast tumors relative to normal breast tissue.
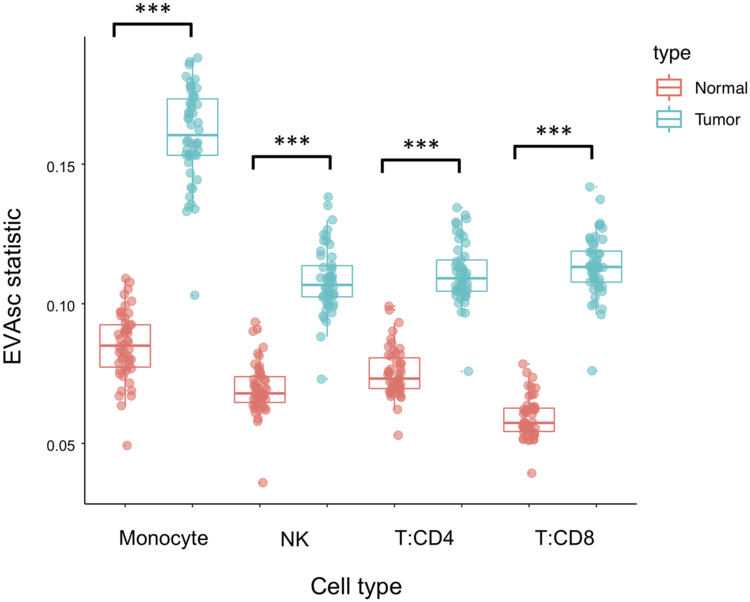
Boxplot of EVA statistics of inter-cellular heterogeneity for all 50 hallmark pathways in major immune cell types from both tumor (blue) and normal (red) breast tissue.
EVA finds increased immune pathway heterogeneity in tumors with high T-cell clonality
With the rapid increase of interest in the field of immunotherapy, T-cell receptor (TCR) sequencing is becoming a valuable tool for assessing immune response. Accordingly, we used T-cells from breast cancer data(25) to explore the relationship between the TCR repertoire and heterogeneity in immune signaling pathways using 27,000 T-cells with paired single-cell RNA and V(D)J sequencing from three breast cancer tumors. For each individual tumor, we computed Shannon entropy for TCR clonality and richness as a measure of TCR diversity based on the single-cell TCR sequencing data (Figure 4A). A Morisita-Horn similarity matrix was generated to compare the similarity of TCR repertoires across tumor replicates (Figure 4B). We then applied EVA to the scRNA-seq data using the Myeloid Innate Immunity Panel pathways from NanoString (NanoString Technologies) to compare each T-cell subtype between individuals. Hierarchical clustering of the EVA statistics revealed a gradient of pathway dysregulation directly correlated with the degree of TCR clonality (Figure 4C, Supplemental table 3). We further applied GSEA to differentially expressed genes to compare the overlap between the enrichment of upregulated and downregulated immune pathways and the immune pathway dysregulation found with EVA (Supplemental table 4). The majority of the significantly dysregulated pathways from EVA overlapped with pathways that were enriched for upregulation in higher clonality compared to lower clonality individuals, with seven additional pathway comparisons that are significantly downregulated. We note that clonal expansion of T-cells is generally associated with a mounting immune response after antigen recognition. Our EVA results suggest that increased clonality of the TCR repertoire leads to increased heterogeneity in immune pathway expression as well as upregulated immune pathway expression.
Figure 4. TCR clonality is associated with immune pathway dysregulation in breast tumors.

A. TCR clonality and richness for individual breast tumors with matched scRNA-seq and TCR-seq data. B. Heatmap of Morisita-Horn similarity index to quantify agreement of CDR3 clonotypes from duplicate TCR-seq data for the same breast tumor and between individual breast tumors. C. Hierarchical heatmap of EVA statistics of inter-cellular heterogeneity for immune pathways in each breast tumor T cell subtype.
EVA finds increased variation in primary tumors relative to metastases and subtype-specific pathway dysregulation
After demonstrating the ability of EVA to detect heterogeneity between tumors, we sought to characterize intra-tumor heterogeneity within primary tumors and associated metastases. Further, we aimed to identify differences in pathway heterogeneity between cancer subtypes, within subtypes, and within the TME. In order to make these comparisons, we applied EVA to scRNA-seq data for 18 HNSCC patients, including five matched primary tumors and lymph node metastases(27).
We first applied EVA to matched primary and metastatic cancer cells within five individual HNSCC patients to examine intra-tumor pathway dysregulation. 98% (55 out of 56) of the pathways are statistically significant for patient HN25, with 100% (56 out of 56) statistically significant for patient HN26 (FDR adjusted p-value < 0.05) (Supplemental table 5). In both cases, all significant hits have greater variation in the primary tumor than the metastasis (Figure 5A). For the remaining three patients, no significant pathway dysregulation was observed. Puram et al.(27) previously observed that the expression profiles of lymph node metastases overlapped with the corresponding primary tumors. While this indicates that there appears to be no mean differences between the paired samples, our method is able to capture significant differential variation between these phenotypes which was previously unrecognized.
Figure 5. Inter- and intra-tumor heterogeneity distinguish HNSCC subtypes and metastases.

A. Boxplot of EVA statistics in primary and metastatic HNSCC cancer cells for each patient demonstrate higher inter-cellular heterogeneity in primary cancer cells than metastatic cells for two patients. B. A heatmap of EVA statistics reveals that inter-cellular heterogeneity varies between primary cancer cells of the basal tumor type for all hallmark pathways, although no differences in mean expression were observed previously with tSNE(27). C. EVA analysis observes significant increases in inter-cellular variation of immune pathways for fibroblasts that are associated with the total fibroblast content in each basal HNSCC tumor. Previous observations of TCGA subtypes noted that tumors with high fibroblast content (red) were classified as mesenchymal and low fibroblast content (blue) as basal, suggestive of fibroblast mediated differences between immune pathway activity in these subtypes. D. Heatmap of EVA statistics of inter-cellular heterogeneity in hallmark pathways for cancer cells from patients in distinct HNSCC subtypes.
To determine the degree of inter-tumor heterogeneity between patients within a single subtype we compared primary cancer cells between seven individuals with basal primary tumors. 78% (923 out of 1176) of the comparisons are statistically significant when all pairwise combinations of patients were considered (FDR adjusted p-value < 0.05) (Supplemental table 6). EVA analysis revealed dramatic differences in pathway dysregulation across patients (Figure 5B). Additionally, we explored heterogeneity within cells of the primary TME across individuals with basal primary tumors. Previously, Puram et al.(27) observed that the proportion of cell types within the TME vary for each patient. Notably, they found that the differences in the basal and mesenchymal subtypes of HPV-negative head and neck cancer can be attributed to a larger proportion of fibroblasts in the TME. Thus, we stratified these basal samples into a binary classification of high (>40%) or low-fibroblast (<40%). To determine the transcriptional status of immune-pathways within patient-specific fibroblast populations we applied EVA using the Myeloid Innate Immunity Panel pathways from NanoString (NanoString Technologies). Hierarchical clustering of the EVA statistics demonstrated increased immune dysregulation in individuals with a high proportion of fibroblasts present in the TME (Figure 5C). 69% (348 out of 504) of the comparisons are statistically significant when all pairwise combinations of patients were considered (FDR adjusted p-value < 0.05) (Supplemental table 7). We note that the fibroblast composition in each basal tumor is independent of the pathway dysregulation observed across cancer cells from distinct patients.
We next applied EVA to primary cancer cells of HNSCC subtypes to examine the differences between inter-tumor heterogeneity. Subtypes were previously called by TCGA classification and ComBat(43) was performed to remove the impact of patient identity on transcriptional profiles. This batch correction enables EVA to compare cells from several patients to isolate only subtype-specific differences(33). We include all MSigDB Hallmark gene set pathways and six meta-signatures derived from non-negative matrix factorization programs that represent common expression programs variable within multiple tumor forms(27) in our comparisons. 46% (77 out of 168) of the comparisons are statistically significant when all pairwise combinations of subtypes were considered (FDR adjusted p-value < 0.05) (Supplemental Table 7). Hierarchical clustering of the EVA statistics demonstrated patterns of subtype-specific pathway dysregulation (Figure 5D).
4. Discussion
We develop EVA to quantify heterogeneity in pathway level gene expression from imputed scRNA-seq data to quantify differential variability between conditions. We demonstrate the suitability of EVA for identifying differential variability of pathway gene expression by applying it to simulated and real scRNA-seq data. Simulated data generated with Splatter(29) was used to demonstrate the ability of EVA to detect known variability between conditions. Validation was performed by comparing immune cell types between normal breast tissue and breast tumors from Azizi et al.(25). As expected, EVA detected increased variability in the tumor cells for all cell type comparisons relative to normal cells (Figure 3).
We then applied EVA to perform novel analyses of differential heterogeneity on two publicly available cancer scRNA-seq datasets. We used paired single-cell RNA and single-cell TCR sequencing data(25) to compare inter-patient T-cell subtype heterogeneity in relation to TCR clonality. TCR repertoire analysis showed differences in the level of TCR clonality for each individual (Figure 4A). EVA analysis revealed significant differences in immune pathway heterogeneity between individuals, consistent with the degree of TCR clonality: increased TCR clonality, increased heterogeneity (Figure 4C). We then performed differential expression analysis between individuals to explore the direction of gene set enrichment. There was a large amount of overlap in differentially variable and differentially upregulated pathways, indicating increased heterogeneity as well as increased gene expression in higher clonality individuals.
Ikeda et al.(44) examined the relationship between intra-tumor expression levels of immune-related genes and TCR repertoire in endometrial cancer. They found increased mRNA expression levels in cases with high T-cell clonality, which was associated with a better prognosis. These results were obtained using total RNA and quantitative real-time PCR in relatively few genes and are consistent with our findings at a comprehensive single-cell RNA level. Recent data has also shown that increased clonal expansion of T-cells and low baseline clonality are associated with longer survival after being treated with anti-CTLA4 inhibitors in pancreatic ductal adenocarcinoma(34). Thus, characterizing the immune microenvironment by expression of immune pathways, immune pathway heterogeneity, and the clonality of infiltrated T-cell receptors may be an important biomarker for clinical response to immunotherapy. With the advent of paired single-cell RNA and TCR profiling methods, studying the transcriptional effect of TCR repertoire changes across cancer cells may provide further insight into the mechanisms of immunotherapy.
Further, an HNSCC scRNA-seq dataset from Puram et al.(27) was used to examine differences in heterogeneity between HNSCC tumor subtypes. Previously, bulk studies have classified HNSCC tumors into four distinct molecular subtypes based on their expression profiles(32): atypical, basal, classical, and mesenchymal. EVA analysis revealed unique patterns of pathway dysregulation in each of the subtypes detected by TCGA classification (Figure 5D). Overall, immune pathways are enriched in the atypical subtype. It has been reported that mesenchymal and atypical subtypes have the highest degree of immune infiltration, making them attractive targets for immunotherapy(45). Our results suggest a key immune component specific to the atypical subtype.
Previous analyses of the HNSCC scRNA-seq data found that the cancer cells in the mesenchymal and basal subtypes have similar expression profiles when stromal contribution was removed(27) and refined the classification of mesenchymal to basal subtype. We speculated that the cellular compositions of the TME within individual basal tumors could contribute to the molecular heterogeneity. Importantly, fibroblasts have opposing roles in the TME and showed a wide-range of inter-tumor proportional variability. Normal fibroblasts exert anti-tumorigenic effects to suppress tumor growth but can be reprogrammed to a cancer-associated phenotype supportive of tumor evolution. EVA analysis comparing fibroblast populations between individuals with basal primary tumors demonstrated that TMEs with a large proportion of fibroblasts have a high degree of immune pathway dysregulation. This indicated immune pathway heterogeneity within the fibroblast expression states, likely due to the immunomodulatory role of cancer-associated fibroblasts within the TME(46).
Beyond immunology, the intra-patient comparison with EVA enables evaluation of the role of intra-tumor heterogeneity in metastasis. Specifically, we compared cancer cells from primary tumors to metastases from individual patients in HNSCC single-cell data. This analysis revealed a clear pattern: either uniform dysregulation or no significant differences between the primary tumor and metastasis. For the two patients that had differential variability, the heterogeneity within the primary tumor was significantly higher than the metastatic cancer cells (Figure 5A). This observation agrees with Nowell’s theory of clonal evolution, which states that cancer originates from a single cell, accumulates genetic alterations, and during the process of metastasis there is an enrichment for the most aggressive clones(28). This theory would indicate that clonal metastases are more homogeneous, as very few cells gain invasive and metastatic potential. Such intra-tumor discrepancies that may evolve as the disease progresses between the primary tumor and disseminated metastasis can result in incorrect biomarkers being used to make clinical decisions and lead to therapeutic failure(1). The differences in molecular heterogeneity may also give rise to different therapeutic responses in primary tumors than metastases. We note that the analyses performed in this study used current landmark cohorts of breast and head and neck tumors, which were limited in sample size. Future work with EVA analysis on larger sample cohorts is essential to establish the role of heterogeneity in complex dynamic processes such as cancer progression and therapeutic response.
Together, the results of these analyses show that EVA is a robust algorithm for detecting inter- and intra-tumor heterogeneity in scRNA-seq data. EVA is applicable to imputed scRNA-seq datasets, which we demonstrate using MAGIC and BISCUIT imputed data. In the applications to some of the cancer datasets in this study, such as tumor versus normal and primary tumor versus metastasis, we observe widespread changes across a majority of pathways between phenotypes. We attribute these changes to the pervasive transcriptional reprogramming in cancer. While the pathways examined in this study are in no way exhaustive, this is suggestive of global disruption of gene expression and makes for broad interpretations. Comparisons within immune cells and primary cancer subtypes show phenotype specific patterns of dysregulation, allowing more specific interpretation of the molecular mechanisms in tumor heterogeneity. We note that EVA can be widely applied beyond cancer, for example to evaluate the role of transcriptional variation on cell fate specification in development(47). In this context, heterogeneity is more constrained than in cancer and EVA finds different patterns of inter-cellular heterogeneity for distinct pathways, with some pathways increasing over developmental time and others decreasing. Because EVA statistics compare cells from two phenotypes, this time course analysis was performed by applying EVA to compare cells from pairs of consecutive developmental time points. Extensions to EVA to quantify dysregulation relative to continuous phenotypes(48) or adaptation of alternative kernel based methods to scRNA-seq data(49) will be essential to evaluate as extensions to more complex statistical comparisons in future work.
In addition, EVA is broadly applicable to any dissimilarity metric and is not limited to Kendall-tau (Supplemental fig 1). This flexibility in the algorithm allows users to specify appropriate distance metrics for datasets and enables the direct comparison of performance across various metrics. We have demonstrated that different dissimilarity metrics have different sensitivities to missing data and note that these metrics may also have different sensitivities to the preprocessing used for the scRNA-seq datasets. The rank-based Kendall-tau dissimilarity metric used for the majority of this study is independent of many sample-specific normalization procedures, such as log transformation or quantile normalization. Other dissimilarity measures may be sensitive to these transformations, and this effect must be evaluated before applying EVA to compare dissimilarity based upon these metrics. Emerging variance stabilization methods to account for the pervasive heteroscedastic mean variance relationship of scRNA-seq data may impact the results obtained with this algorithm and are essential to evaluate in future studies. Thus, EVA is a robust multivariate statistical method to quantify differential variation of pathway gene expression and provides the ability to explore transcriptional variation in numerous disease and normal contexts at a single-cell resolution. Future work to improve the EVA algorithm will involve integrating mathematical models to compute comparisons on scRNA-seq data without the need for imputation.
Supplementary Material
{kind=link}
Significance: This study presents a robust statistical algorithm for evaluating gene expression heterogeneity within pathways or gene sets in single-cell RNA-seq data
Acknowledgements
This work was supported by grants from the NIH (R01CA177669 and U01CA212007 to EJF), the Chan-Zuckerberg Initiative DAF (2018-183445 to LAG and 2018-183444 to EJF) an advised fund of Silicon Valley Community Foundation, the Johns Hopkins University Catalyst (EF & LAG) and Discovery awards (EJF), and the Johns Hopkins University School of Medicine Synergy Award (LAG, & EJF). EMJ and ERT acknowledge funding from the Broccoli Foundation, The Bloomberg~Kimmel Institute for Cancer Immunotherapy, and The Skip Viragh Center for Pancreas Cancer Clinical Research and Patient Care, and The Commonwealth Foundation for Cancer Research. ERT is funded through the MacMillan Pathway to Independence Fellowship. EMJ, ERT, and AH are also supported through NIH R01CA184926, and through a Stand Up To Cancer-Lustgarten Foundation Pancreatic Cancer Convergence Dream Team Name Translational Research Grant (SU2C-AACR-DT14-14). Stand Up to Cancer is a division of the Entertainment Industry Foundation administered by the American Association for Cancer Research, the Scientific Partner of SU2C. Additional support is provided through NIH P30CA006973, P50CA062924, U01CA196390, RFBR 17-00-00208 and Russian Academic project 0112-2019-0001. The authors thank L. Cope, A. Ewald, K. Schuebel, R. Scharpf, V. Yegnasubramanian, R. Riggins, L. Kagohara, D. Gaykalova, T. Triche, and W. H. Jin for feedback on the algorithm and manuscript.
Footnotes
Disclosure of potential conflicts of interest: The authors declare no potential conflicts of interest.
References
- 1.Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501:355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pogrebniak KL, Curtis C. Harnessing Tumor Evolution to Circumvent Resistance. Trends Genet. 2018;34:639–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gatenby RA, Gillies RJ, Brown JS. Of cancer and cave fish. Nat Rev Cancer. 2011;11:237–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338. [DOI] [PubMed] [Google Scholar]
- 5.Eddy JA, Hood L, Price ND, Geman D. Identifying tightly regulated and variably expressed networks by Differential Rank Conservation (DIRAC). PLoS Comput Biol. 2010;6:e1000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bravo HC, Pihur V, McCall M, Irizarry RA, Leek JT. Gene expression anti-profiles as a basis for accurate universal cancer signatures. BMC Bioinformatics. 2012;13:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dinalankara W, Bravo HC. Gene Expression Signatures Based on Variability can Robustly Predict Tumor Progression and Prognosis. Cancer Inform. 2015;14:CIN.S23862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dinalankara W, Ke Q, Xu Y, Ji L, Pagane N, Lien A, et al. Digitizing omics profiles by divergence from a baseline. Proc Natl Acad Sci U S A. 2018;115:4545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levitin HM, Yuan J, Sims PA. Single-Cell Transcriptomic Analysis of Tumor Heterogeneity. Trends Cancer Res. Elsevier; 2018;4:264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saadatpour A, Lai S, Guo G, Yuan G-C. Single-Cell Analysis in Cancer Genomics. Trends Genet. 2015;31:576–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Maaten L, Hinton G. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–605. [Google Scholar]
- 12.McInnes L, Healy J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv. 2018. http://arxiv.org/abs/1802.03426 [Google Scholar]
- 13.Moon KR, van Dijk D, Wang Z, Chen W, Hirn MJ, Coifman RR, et al. PHATE: A Dimensionality Reduction Method for Visualizing Trajectory Structures in High-Dimensional Biological Data. bioRxiv. 2017. https://www.biorxiv.org/content/early/2017/03/24/120378 [Google Scholar]
- 14.Angerer P, Haghverdi L, Büttner M, Theis FJ, Marr C, Buettner F. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics. 2016;32:1241–3. [DOI] [PubMed] [Google Scholar]
- 15.Pierson E, Yau C. ZIFA: Dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 2015;16:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeTomaso D, Yosef N. FastProject: a tool for low-dimensional analysis of single-cell RNA-Seq data. BMC Bioinformatics. 2016;17:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amir E-AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cleary B, Cong L, Cheung A, Lander ES, Regev A. Efficient Generation of Transcriptomic Profiles by Random Composite Measurements. Cell. 2017;171:1424–36.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DiGiuseppe JA, Cardinali JL, Rezuke WN, Pe’er D. PhenoGraph and viSNE facilitate the identification of abnormal T-cell populations in routine clinical flow cytometric data. Cytometry B Clin Cytom. 2018;94:588–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantsoki A, Devailly G, Joshi A. Gene expression variability in mammalian embryonic stem cells using single cell RNA-seq data. Comput Biol Chem. 2016;63:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brennecke P, Anders S, Kim JK, Kołodziejczyk AA, Zhang X, Proserpio V, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods. 2013;10:1093–5. [DOI] [PubMed] [Google Scholar]
- 22.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat Methods. 2016;13:241–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174:1293–1308.e36. 10.1016/j.cell.2018.05.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afsari B, Geman D, Fertig EJ. Learning dysregulated pathways in cancers from differential variability analysis. Cancer Inform. 2014;13:61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017;171:1611–24.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zappia L, Phipson B, Oshlack A. Splatter: simulation of single-cell RNA sequencing data. Genome Biol. 2017;18:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell. 2018;174:716–29.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azizi E, Prabhakaran S, Carr A, Pe’er D. Bayesian Inference for Single-cell Clustering and Imputing. Genomics and Computational Biology. 2017;3:46. [Google Scholar]
- 32.Walter V, Yin X, Wilkerson MD, Cabanski CR, Zhao N, Du Y, et al. Molecular subtypes in head and neck cancer exhibit distinct patterns of chromosomal gain and loss of canonical cancer genes. PLoS One. 2013;8:e56823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopkins AC, Yarchoan M, Durham JN, Yusko EC, Rytlewski JA, Robins HS, et al. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight. 2018;3 10.1172/jci.insight.122092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Afsari B, Guo T, Considine M, Florea L, Kagohara LT, Stein-O’Brien GL, et al. Splice Expression Variation Analysis (SEVA) for inter-tumor heterogeneity of gene isoform usage in cancer. Bioinformatics. 2018;34:1859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Afsari B, Guo T, Considine M, Florea L, Kagohara LT, Stein-O’Brien GL, et al. Splice Expression Variation Analysis (SEVA) for inter-tumor heterogeneity of gene isoform usage in cancer. Bioinformatics. 2018;34:1859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afsari B Modeling cancer phenotypes with order statistics of transcript data. 2013. http://search.proquest.com/openview/5feaee27e99f17d27b10470db987c6ca/1?pq-origsite=gscholar&cbl=18750&diss=y
- 41.Kuriakose MA, Chen WT, He ZM, Sikora AG, Zhang P, Zhang ZY, et al. Selection and validation of differentially expressed genes in head and neck cancer. Cell Mol Life Sci. 2004;61:1372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drost H-G. Philentropy: Information Theory and Distance Quantification with R. JOSS. 2018;3:765. [Google Scholar]
- 43.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27. [DOI] [PubMed] [Google Scholar]
- 44.Ikeda Y, Kiyotani K, Yew PY, Sato S, Imai Y, Yamaguchi R, et al. Clinical significance of T cell clonality and expression levels of immune-related genes in endometrial cancer. Oncol Rep. 2017;37:2603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mandal R, Şenbabaoğlu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1:e89829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alkasalias T, Moyano-Galceran L, Arsenian-Henriksson M, Lehti K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int J Mol Sci. 2018;19 10.3390/ijms19051532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clark B, Stein-O’Brien G, Shiau F, Cannon G, Davis E, Sherman T, et al. Comprehensive analysis of retinal development at single cell resolution identifies NFI factors as essential for mitotic exit and specification of late-born cells. 2018. 10.1101/378950 [DOI] [PMC free article] [PubMed]
- 48.Afsari B, Favorov A, Fertig EJ, Cope L. REVA: a rank-based multi-dimensional measure of correlation. bioRxiv. 2018. [Google Scholar]
- 49.Zhao N, Chen J, Carroll IM, Ringel-Kulka T, Epstein MP, Zhou H, et al. Testing in Microbiome-Profiling Studies with MiRKAT, the Microbiome Regression-Based Kernel Association Test. Am J Hum Genet. 2015;96:797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.