

Abstract
Cholangiopathies such as primary sclerosing cholangitis (PSC) are chronic liver diseases characterized by increased cholestasis, biliary inflammation and oxidative stress. The objective of this study was to elucidate the impact of cholestatic injury on oxidative stress-related factors. Using hepatic tissue and whole cell liver extracts (LE) isolated from 11-week old C57BL/6J (WT) and Mdr2KO mice, inflammation and oxidative stress was assessed. Concurrently, specific targets of carbonylation were assessed in LE prepared from murine groups as well as from normal and human patients with end-stage PSC. Identified carbonylated proteins were further evaluated using bioinformatics analyses. Picrosirius red staining revealed extensive fibrosis in Mdr2KO liver, and fibrosis colocalized with increased periportal inflammatory cells and both acrolein and 4-HNE staining. Western blot analysis revealed elevated periportal expression of antioxidant proteins Cbr3, GSTμ, Prdx5, TrxR1 and HO-1 but not GCLC, GSTπ or catalase in the Mdr2KO group when compared to WT. From immunohistochemical analysis, increased periportal reactive aldehyde production colocalized with elevated staining of Cbr3, GSTμ and TrxR1 but surprisingly not with Nrf2. Mass spectrometric analysis revealed an increase in carbonylated proteins in the Mdr2KO and PSC groups compared to respective controls. Gene ontology and KEGG pathway analysis of carbonylated proteins revealed a propensity for increased carbonylation of proteins broadly involved in metabolic processes as well more specifically in Rab-mediated signal transduction, lysosomes and the large ribosomal subunit in human PSC. Western blot analysis of Rab-GTPase expression revealed no significant differences in Mdr2KO mice when compared to WT livers. In contrast, PSC tissue exhibited decreased levels of Rabs 4, 5 and increased abundance of Rabs 6 and 9a protein. Results herein reveal that cholestasis induces stage-dependent increases in periportal oxidative stress responses and protein carbonylation, potentially contributing to pathogenesis in Mdr2KO. Furthermore, during early stage cholestasis, there is cell-specific upregulation of some but not all, antioxidant proteins.
Keywords: Cholestasis, protein carbonylation, reactive aldehyde, liver, oxidative stress, inflammation
Graphical Abstract
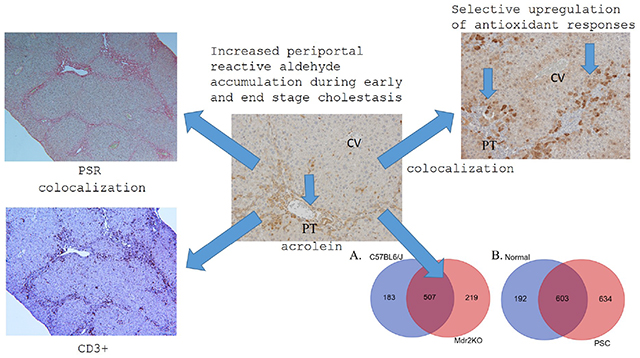
Introduction
Primary sclerosing cholangitis (PSC) is a progressive biliary disease of unknown etiology that affects about 16.2 out of 100,000 individuals and has no current therapeutic option [1, 2]. PSC can originate in both the intra and extra hepatic ducts and is characterized by cholestasis, hepatobiliary inflammation (neutrophil and T-cell infiltration), choledocholithiasis, biliary strictures and decreased biliary flow. It has been estimated that 80% of PSC patients also have other autoimmune disorders such as inflammatory bowel disease (IBD) [3]. Importantly, cholestasis induces chronic inflammation and oxidative stress which may contributes to cholestasis-induced liver injury [4–6]. Progression of PSC results in cholangitis, periportal fibrosis with progression to cirrhosis, end stage liver disease and ultimately, a requirement for liver transplantation. In general, PSC is diagnosed late in its course when fibrosis/cirrhosis are already present. If patients do not receive a liver transplant, cholangiocarcinoma (CCA) occurs in intra or extra hepatic ducts at a rate of 8–30% of adult patients with PSC and is often fatal[7]. If patients do receive a transplant, disease recurrence is not uncommon (20-50%) making understanding the mechanisms of cholestatic injury and the search for a cure for PSC evermore important [7, 8].
Multidrug resistance protein 2 (Mdr2) is a phospholipid flippase expressed in the biliary canaliculi that mediates hepatocyte secretion of phospholipids into bile [9]. Mdr2 knockout mice (Mdr2KO) develop spontaneous cholangitis and periductal liver fibrosis mirroring human PSC beginning at 3 weeks of life and progressing to florid hepatobiliary inflammation by 8-10 weeks [3, 10]. Therefore, by 10 weeks of age, Mdr2KO mice present a useful model to investigate mechanisms of cholestatic liver injury.
In the liver, an important marker of increased oxidative stress is elevated levels of lipid peroxidation and generation of electrophilic α/β unsaturated fatty acid derivatives such as 4-hydroxynonenal (4-HNE), acrolein and malondialdehyde (MDA). These reactive aldehydes can adduct critical proteins and a significant consequence of this protein carbonylation is impaired protein function. Oxidative stress clearly plays a role in cholestatic liver injury. In bile duct ligation models, supplementation of the anti-oxidant n-acetylcysteine reduces injury[11–13]. Although the source of reactive aldehydes has not been definitively identified in cholestasis, experiments in bile duct ligated mice have suggested that the influx of neutrophils may be a contributing factor[13]. As evidenced by increased periportal immunohistochemical staining for 4-HNE, Mdr2KO mice have been shown to possess elevated periportal oxidative stress. Importantly, dietary supplementation with norUrsodeoxycholic acid (norURSO) reduced 4-HNE adduct accumulation and decreased hepatocellular injury suggesting reactive aldehydes contribute to cholestatic injury[14]. Furthermore, induction of heme oxygenase 1 (HO-1) in Mdr2KO mice reduced overall hepatic damage (serum ALT levels), fibrosis (liver hydroxyproline content) and inflammation (decreased Kupffer cell infiltration and TNFα levels) further supporting the impact of oxidative stress on cholestatic liver injury[15].
Although significant data have been accumulated in a variety of other models of chronic hepatic inflammation, the direct impact of protein carbonylation during cholestasis-induced oxidative stress has not been examined in either human or murine models of cholestatic liver disease. In the present study we examined hepatocellular lipid peroxidation and antioxidant responses in hepatic tissue obtained from fibrotic 11-week-old Mdr2KO mice as well as tissue procured from human PSC patients[16, 17]. In both models, we find elevated hepatic protein carbonylation and dysregulation of anti-oxidant responses. Furthermore, in end-stage PSC, a propensity for carbonylation of proteins regulating vesicular trafficking (Rab GTPases) and proteins that compose the large ribosomal subunit was observed.
Materials and Methods
Sample procurement:
For human studies, paraffin-embedded and frozen hepatic tissues from normal and end stage PSC patients (n=8/condition; ages 38-62 years of age) procured during transplantation were provided by the University of Minnesota Liver Tissue Cell Distribution Center (NIH Contract #HHSN276201200017C) as previously described[16, 17].
To elucidate cholestatic effects on inflammation and oxidative stress, Mdr2KO mice were used. Breeding pairs were obtained from Dr. Ronald Oude-Elferink via Dr. LaRusso (Mayo, Rochester, MN)[18]. Genetic background assessment revealed that the founders possessed a highly C57BL/6J genetic background (91 ± 0.5%), supporting the use of WT C57BL6/J mice as control animals (data not shown). At 11 weeks of age, C57BL/6J and Mdr2KO mice (6/group), were injected with sodium pentobarbital (0.1mg/kg) and blood was collected from the inferior vena cava. Plasma was separated by centrifugation at 5000 rpm for 5 min at 4°C and was assayed for alanine aminotransferase (ALT) activity (Sekisui Diagnostics, P.E.I., Canada), alkaline phosphatase activity and total bilirubins (performed by the UC Denver Department of Pathology shared resource core). Whole livers were excised and weighed. Caudate and median lobes were removed, fixed in 10% neutral buffered formalin and embedded in paraffin for histological and immunohistochemical (IHC) analyses. The remaining portion of the liver was homogenized and whole cell extracts (LE) of each sample prepared by Dounce homogenization (10X) of tissue resuspended in 50mM tricine pH 8.0, 1mM NaCl and phosphatase and protease inhibitors (SIGMA ALDRICH, St Louis, MO) followed by sonication (3X15 seconds at 4°C). To remove debris, samples were centrifuged at 16,000g (4°C) for 10 minutes. Supernatants were drawn off and immediately flash frozen in liquid N2[19]. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Colorado and were performed in accordance with published National Institutes of Health guidelines.
Western blotting:
Western blotting was performed using 10-40μg of liver extract protein and primary antibodies listed in Table S1 as previously described[20, 21]. Quantification of expression of each protein was performed using ImageJ (NIH) and normalized to overall GAPDH expression. All exposures were normalized using GAPDH expression. Data are means +/− SEM, n=6 per genotype. Statistical analysis was via students t-test, *p<0.05;**p<0.01;***p<0.001.
Histological and Immunohistochemical Evaluation:
Formalin fixed slides were analyzed following hematoxylin and eosin (H&E) or picrosirius red (PSR) staining. For assessing inflammation, carbonylation and oxidative stress, the following protocol performed using the anti-bodies listed in Table S1. Heat induced antigen retrieval was performed in citrate buffer pH 7.0. Following incubation with primary antibodies overnight, slides were washed 3X5min in tris-buffered saline 1% tween and incubated in HRP-conjugated secondary antibody for 30 min (MP-7401 anti-rabbit, MP-7405 anti-goat, Vector Labs, Burlingame CA). The peroxidase substrate used was IMMPACT-DAB (SK-4105, Vector Labs). Histologic images were captured on an Olympus BX51 microscope equipped with a four-megapixel Macrofire digital camera (Optronics; Goleta, CA) using the PictureFrame Application 2.3 (Optronics). All images were cropped and assembled using Photoshop CS2 (Adobe Systems, Inc.; Mountain View, CA).
Quantitative PCR:
Using fresh hepatic tissue, RNA was isolated using a Qiagen RNeasy Plus Mini Kit (#74134), according to manufacturer’s instruction. RNA concentration was determined by optical density measurement at 260 nm on a spectrophotometer. RNA (1000 μg) was reverse-transcribed using an iScriptTM Reverse Transcription Supermix for RT-qPCR and a T100TM Thermal Cycler (Biorad). 2 μL cDNA was used for the PCR reaction. The rt-qPCR was performed using the primers listed in Table S2C., Power SYBRTM Green PCR Master Mix, and a 7900HT Fast Real-Time PCR System with 384-Well Block Module (Applied BiosystemsTM/ThermoFisher) running 40 cycles at 95°C for 15 seconds, 60°C for 60 seconds.
Biotin hydrazide purification:
Biotin hydrazide derivatization and purification of aldehyde modified proteins was performed as previously described using 500 μg of LE protein from each group (n=6 WT, 6 Mdr2KO, 8 Normal, 8 PSC [16, 17])[21].
Determination of GST and SOD2 activity:
Enzymatic activity for GST and SOD2 was determined using LE prepared from fresh frozen hepatic tissue as previously described[22, 23].
LC/MS analysis:
For LC-MS analysis, 5μl of each peptide mixture was loaded on a Bruker Maxis IMPACT LC-MS and analyzed as previously described[21, 24].
Determination of GSH, GSSG, Cys, CySS and CysSG:
At sacrifice, LE were prepared for analysis and flash frozen in liquid nitrogen. Quantification of GSH, GSSG, Cys, CySS and CysSG was performed using an Agilent model 1260 HPLC as previously described[25].
Mesoscale quantification of cytokines:
Portal blood was obtained by direct venipuncture of the portal vein and serum isolated by centrifugation at 5000 rpm for 5 min at 4°C. Portal serum cytokine concentrations were measured using a multi-plex electrochemiluminescence proinflammatory cytokine screen (Meso Scale Discovery, Rockville, MD). Assays were performed according to the manufacturer’s instructions.
Bioinformatics analysis:
The KEGG Pathway [26] and the Gene Ontology (GO) [27] databases were used to determine differential enrichment between the Mdr2KO and WT mice and between PSC patients and normal controls. For KEGG Pathways, Uniprot IDs were converted to KEGG gene identifiers using the KEGG REST API (https://www.kegg.jp/kegg/rest/keggapi.html). For GO terms, Uniprot IDs have previously been linked to GO terms in the Gene Ontology Annotation files available on the Gene Ontology Consortium website (http://www.geneontology.org/page/download-go-annotations). Initially, enrichment for pathways and GO terms was examined within group using a Fisher Exact test, e.g., proteins identified in the Mdr2KO mice compared to all proteins identified in either WT or Mdr2KO. A false discovery rate (FDR) with Benjamini post-hoc analysis was used to adjust for multiple comparisons across pathways/terms. If a pathway/term was significantly enriched in either group (FDR<0.05), differential enrichment was determined for that pathway/term by comparing the proportion of proteins associated with a pathway/term between groups, e.g., Mdr2KO vs. WT, using a Fisher Exact test. Enrichment analyses were executed in R Statistical Software (version 3.5.0).
Results:
Hepatocellular injury in 11 week old C57BL/6J Mdr2KO mice.
Previous studies have revealed that Mdr2KO mice on the FVB background develop extensive fibrosis and periportal inflammation[18]. When Mdr2KO was generated in the C57BL/6J background, serum alanine aminotransferase (ALT) were lower and inflammation was less pronounced[28]. To validate the liver injury in the colonies that we generated, serum ALT and fibrosis were quantified. As shown in Figure S1A, serum ALT increased almost 6-fold in the Mdr2KO group but alkaline phosphatase and serums bilirubins were not significantly different (Figure S1B,C). To examine the extent of fibrosis in the Mdr2KO mice, liver sections were analyzed histologically after staining with picrosirius red (PSR) or immunohistochemical analysis of alpha smooth muscle actin (μ-SMA). Fibrosis was not present in the WT animals (Figure 1). In the Mdr2KO group, dramatic periportal fibrosis was observed with some minor bridging evident (PT; Figure 1). Tissue sections stained with PSR were subsequently examined under polarized light and quantified (Figure S2, Table 1A). Quantification revealed a significant increase in accumulation of collagens I/III (PSR). Previous studies using Cytokeratin-19-staining have shown that Mdr2KO mice possess a strong ductular response, as bile ducts proliferate in response to cholestatic injury[29, 30]. To further explore the ductular response in C57BL/6J Mdr2KO mice, tissue sections were stained with another marker of the ductular reaction, cytokeratin-7 (CK7). CK7 staining was prominent in the expansive bile duct cholangiocytes or closely associated with these ducts in the Mdr2KO group (Figure 1B), verifying that these livers had a strong ductular reaction. The protein adipophilin (ADPH) is expressed on retinoic acid containing vesicles in quiescent stellate cells as well as lipid droplets making it a good marker to determine if stellate cells have been activated[31]. Surprisingly, ADPH staining was increased in the Mdr2KO group (Figure 1B arrows).
Figure 1. Basic Pathology in Mdr2KO livers.
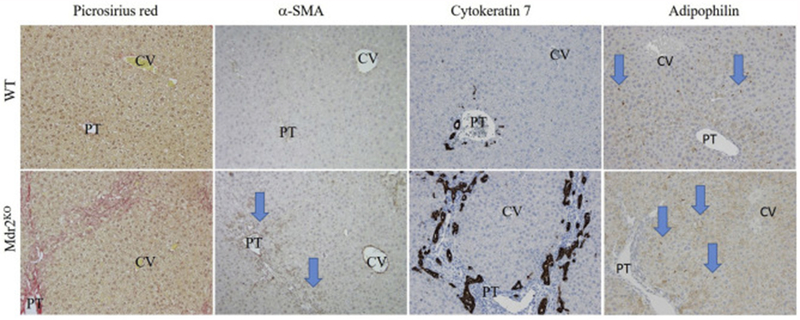
Liver sections from WT and Mdr2KO mice were stained with picrosirius red or immunostained using antibodies directed against the smooth muscle actin (αSMA), Cytokeratin 7, or adipophilin. Representative images are shown; n = at least 3 mice per genotype (CV, central vein, PT, portal triad). Blue arrows indicate increased periportal staining (aSMA) and increased ADPH staining in Mdr2KO liver sections.
Table 1. Quantification of Inflammation, proliferation and nuclear oxidative stress in Mdr2KO mice.
A. Quantitative analysis of infiltrating inflammatory cells, fibrosis, proliferation and nuclear oxidative stress. B220, F4/80, Gr1, CD3, MPO, Ki67, gH2A.x positive cells were counted per 100X field. Data are means ± SEM for n=4-6 mice/group. Significance was determined by students t-test. Groups with different letter subscripts are significant from each other. B. RT-qPCR analysis of selective inflammatory and fibrotic markers. Significance was determined by students t-test. Groups with different letter subscripts are significant from each other.
| A. Quantification of Histology≠ | Parameter | C57BL/6J | Mdr2KO | P value |
|---|---|---|---|---|
| Inflammation | MPO | 3.67+/−2.11a | 24.4+/−7.95b | p=0.0358 |
| B220 | 3.78+/−1.20a | 10.73+/−2.35b | p=0.0302 | |
| F4/80 | 57.43+/−4.73a | 80.57+/−5.08b | p=0.0103 | |
| CD3 | 22.13+/−1.23a | 47.11+/−5.78b | p=0.0029 | |
| Gr1 | 0.8+/−0.37a | 60.14+/−10.41b | p=0.0005 | |
| Fibrosis | PSR | 2.38+/−0.23a | 6.36+/−0.81b | p=0.0008 |
| αSMA | 2.81+/−0.50a | 8.03+/−2.30b | p=0.05 | |
| Proliferation | Ki67 | 2.28+/−0.69a | 38.33+/−7.77b | p=0.0018 |
| Oxidative stress | pH2A.x | 2.50+/−0.25a | 38.33+/−8.01b | p=0012 |
| B. RT-qPCR≠ | Parameter | C57BL/6J | Mdr2KO | P value |
| Inflammation | F4/80 | 1.00+/−0.10a | 1.88+/−0.67b | p=0.0126 |
| MCP-1 | 1.00+/−0.08a | 6.42+/−1.47b | p=0.0042 | |
| IL-1b | 1.00+/−0.20a | 6.30+/−1.85b | p=0.0173 | |
| IL-6 | 1.00+/−0.31a | 3.68+/−0.94b | p=0.0215 | |
| TNFα | 1.00+/−0.29 | 1.66+/−0.31 | p=0.1530 | |
| Ly6G | 1.00+/−0.10 | 1.36+/−0.27 | p=0.2461 | |
| Fibrosis | Col1A | 1.00+/−0..13a | 2.24+/−0.0.39b | p=0.0130 |
| MMP2 | 1.00+/−0.18a | 2.36+/−0.38b | p=0.0087 | |
| MMP13 | 1.00+/−0.13a | 1.98+/−0.15b | p=0.0007 | |
| TGFβ | 1.00+/−0.10a | 2.28+/−0.28b | p=0.0015 | |
| MMP9 | 1.00+/−0.11 | 1.39+/−0.18 | p=0.1005 |
Data are presented as mean± SEM. Statistical significance was determined by Students t-test Letter with similar superscripts (a, b) denote significant difference of P<0.05.
Infiltration of inflammatory cells in Mdr2KO mice.
Increased inflammation and the accumulation of reactive oxidative species (ROS) are integrally linked in cholestatic liver disease[32, 33]. To further delineate the inflammatory response in C57BL/6J Mdr2KO mice, tissue sections were probed for myeloperoxidase (MPO, a marker of activated neutrophils), Ly6G (neutrophils), F4/80 (a marker of infiltrating macrophages and resident Kupffer cells), B220 (a B cell marker), CD3 (a T- and NKT-lymphocyte marker) and Cd1d (a lipid antigen presenting ligand for NKT-cells). Few MPO-positive and Ly6G-positive neutrophils were present in control livers, whereas in Mdr2KO livers, a dramatic increase in MPO and Ly6G positive cells occurred in areas undergoing ductular reactions (Figure 2, Table 1A). A large increase in periportal F4/80-positive cells was also evident in Mdr2KO as compared to control liver (Figure 2, Table 1A). Collectively, these data reveal a strong inflammatory response in Mdr2KO livers. Recent reports have indicated that the adaptive immune response also contributes to oxidative stress in other hepatic diseases including nonalcoholic and alcoholic liver disease[34]. To examine the adaptive immune response, lymphocytes were also examined in serial sections of livers from Mdr2KO and control livers. At lower magnifications, CD3+ cells were particularly concentrated along areas of increased fibrosis (Figure S3 arrows). In control livers, B220+ plasma cells/B-lymphocytes and CD3+ T/NKT-lymphocytes were rare and randomly dispersed. In Mdr2KO livers, abundant periportal B220+ lymphocytes and CD3+ T/NKT-lymphocytes were present in regions of ductular responses (Figure 2), reaffirming that there are both innate and adaptive immune responses in areas surrounding proliferating cholangiocytes in Mdr2KO livers. Interestingly, when examining Cd1d expression by histology, there was a dramatic increase in the Mdr2KO mice. To support histological inflammation and fibrosis data, relative expression of F4/80, MCP-1, IL-6 and IL-1β , TNFα, Ly6G, Col1A, MMP2, MMP9, MMP13 and TGFβ was assessed by RT-qPCR and the primers listed in Table S2. From the data presented in Table 1B., quantitative PCR analysis revealed increased F4/80, MCP-1, IL-6 and IL-1β in Mdr2KO mice when compared to age matched controls. Additionally, in agreement with PSR quantification, Col1A, MMP2, MMP13, TGFβ were all significantly increased in the MdrKO group. To further support inflammation data, mesoscale cytokine analysis was performed using serum isolated from the portal vein of both WT control and Mdr2KO mice. As shown in Figure S4, concentrations of IFNγ, KC, IL12p70, IL-6 and IL-10 were significantly increased in Mdr2KO mice when compared to controls. Levels of IL-1β and TNFα were not significantly different.
Figure 2. Inflammatory infiltrates in Mdr2KO livers.
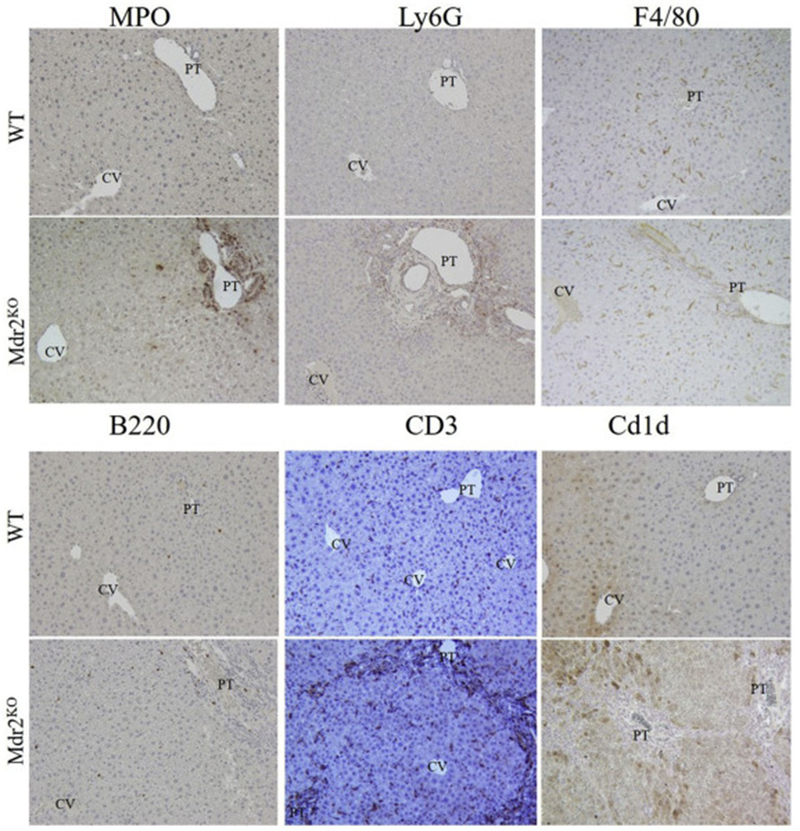
Liver sections from WT and Mdr2KO mice were immunostained using antibodies directed against the indicated markers of inflammation. Representative images are shown; n = at least 3 mice per genotype. Abbreviations as in Figure 1.
Impact of Mdr2KO on NF-κB activation.
The Nf-κB pathway is a central regulator of inflammation. Increased Nf-κB signaling results in increased macrophage production of proinflammatory cytokines including IL-1, IL-6 and TNFα. In the cell, a critical mechanism of controlling the activation of Nf-κB is by binding to the inhibitor of sequestering NF-κB in the cytosol by binding IκB. Increased phosphorylation of IκB results in its degradation and the nuclear translocation and activation of Nf-κB. We sought to determine if NF-kB activation was increased in Mdr2KO mice by Western analysis of Iκb and pIκb. Surprisingly, although there is increased cytokine production in Mdr2KO mice, quantification of IκB/pIκB revealed no significant difference between control of Mdr2KO mice (Figure S5A, B).
Impact of Mdr2KO on protein carbonylation, oxidative injury and proliferation.
Oxidative stress and protein carbonylation have been proposed to play a key role in the progression of a variety of other hepatic disorders such as alcoholic liver disease[35–37]. Previous studies have shown increased 4-HNE staining as well as MDA (TBARs) in FVB Mdr2KO mice[14, 38]. Tissue sections isolated from WT and Mdr2KO livers were probed for protein modification by the reactive aldehyde acrolein as well as 4-HNE. In healthy control liver, no significant acrolein staining was evident (Figure 3). Tissue sections from Mdr2KO livers revealed dramatically increased acrolein staining within inflammatory cells, hepatocytes and cholangiocytes within the periportal region. Furthermore, staining extended among cells along areas previously identified to exhibit fibrosis (arrows). Immunohistochemical evaluation of 4-HNE modified proteins revealed increased periportal staining that extended to surrounding hepatocytes (Figure 3)
Figure 3. Impact of Mdr2KO on protein carbonylation, oxidative injury and cellular proliferation.
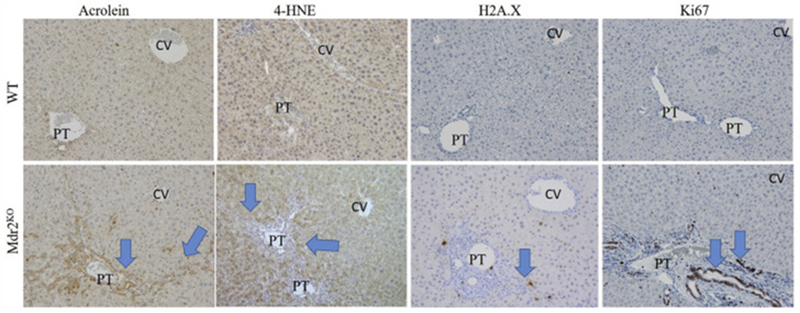
Tissue sections isolated from WT and Mdr2KO mice were immunostained using antibodies directed against acrolein, 4-HNE, γH2A.X and Ki67 as indicated, 100X magnification. Representative images are shown; n = at least 3 mice per genotype. Blue arrows indicate increased periportal staining of each marker. Abbreviations as in Figure 1.
In Mdr2KO mouse livers, there was strong acrolein staining in the cytosol and the nuclei of cholangiocytes within the periportal region as well as in cholangiocytes undergoing a ductular reaction which extended out into the hepatic parenchyma. Reactive aldehydes including 4-HNE, MDA, and acrolein can also modify DNA resulting in DNA damage and double stranded DNA breaks. Although γH2A.x has been reported to be increased in FVB Mdr2KO, it is unknown if the C57BL/6J Mdr2KO genotype exhibits a similar phenotype[38]. To assess whether this was occurring in the Mdr2KO mouse livers, hepatic tissue sections were stained for the double stranded DNA break-marker histone γH2AX followed by quantification. From the quantification, H2A.X-positive cells were significantly increased in Mdr2KO liver tissue (Figure 3, Table 1A). Oxidative stress is also associated with increased oval cell proliferation[39]. To determine if cells that display DNA damage and protein-carbonylation were also undergoing proliferation, tissue sections were stained for Ki-67 followed by quantification. As shown in Figure 3, Table 1A, increased numbers of Ki67+ cells were present in the periportal regions in Mdr2KO mice. Interestingly, Ki67+ cells were part of both the inflammatory response and within cholangiocytes undergoing ductal expansion.
Impact of Mdr2KO on expression of anti-oxidant responses
From Figure 3, increased periportal oxidative stress was evident in Mdr2KO mice. In other murine models of hepatic oxidative stress decreases in the redox capacity are observed, as evidenced by decreased GSH concentrations and increased reactive aldehyde levels[40, 41]. To further examine the impact of cholestasis-induced oxidative stress, the concentrations of GSH, GSSG, Cys, CysSS and CysSSG were measured and redox potentials calculated. Concurrently, GST and SOD2 activity assays were performed. As shown in Figure 4 (panels A–F), concentrations of cysteine were significantly increased and the Cys redox potential (Cys/CySS) significantly reduced (more negative) in the Mdr2KO mice. No significant differences in the concentrations of GSH and GSSG or the GSH/GSSG redox potential were observed between genotypes.
Figure 4.
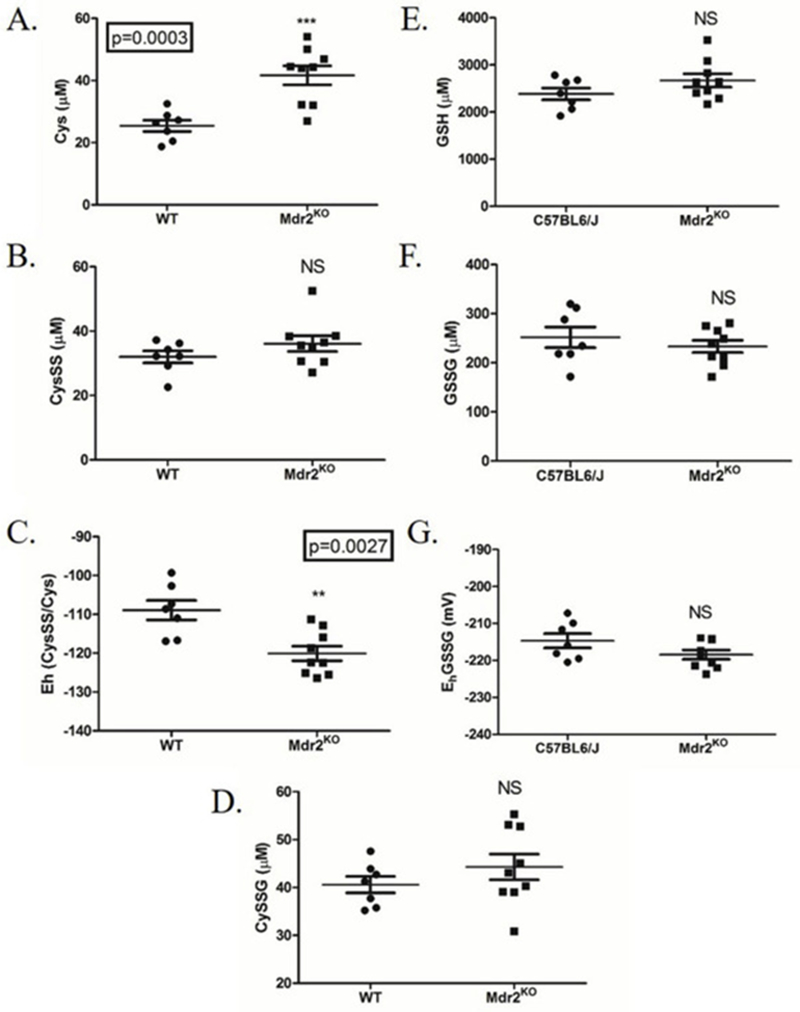
Impact of cholestasis on hepatic cysteine redox status and GSH.A. Cys, B. CysSS, C. Eh CysSS/Cys, D. CySSG, E. GSH, F. GSSG, G. Eh GSSG/GSH were determined using dansyl chloride derivatization, HPLC, and fluorescence detection. Redox potentials (panels C., G.) were calculated using the Nernst equation. N=5 per group; *P>0.05, **P>0.01.
To determine if oxidative stress induced by mild cholestasis impacted cellular antioxidant responses, the abundance of the Nrf2 response proteins glutamate-cysteine ligase catalytic subunit (GCLC), thioredoxin reductase 1 (TrxR1), heme oxygenase (HO-1), NAD(P)H quinone oxidoreductase (NQO1), glutathione peroxidase (Gpx1), glutathione S-transferase μ (GSTμ) and carbonyl reductase 3 (Cbr3) were quantified by western blot analysis (Figure 5). Additionally, the antioxidant proteins catalase, mitochondrial superoxide dismutase (SOD2), GSTπ, peroxiredoxin 5 (Prdx5), Prdx6 and Trx1 were also examined by western blot (Figure 5). A significant increase in Cbr3, GSTμ, TrxR1, SOD2, Prdx5 and Prdx6 was evident in Mdr2KO livers, but there was no change in GSTπ, Trx1, Gpx1, HO-1, catalase or GCLC levels. Surprisingly, NQO1 levels were significantly suppressed by 40% in Mdr2KO mice (Figure 5).
Figure 5. Impact of Mdr2KO on expression of antioxidant responses.
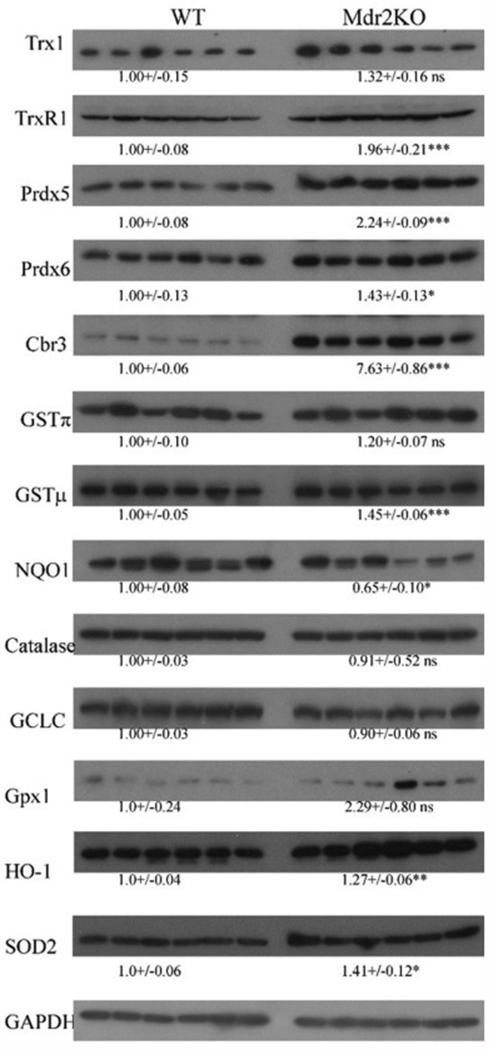
Western immunoblotting analysis of antioxidant responses in liver lysates prepared from WT and Mdr2KO mice. All exposures were normalized using GAPDH expression. Data are means +/− SEM, n=6 per genotype. Statistical analysis was via students t-test, *p<0.05;**p<0.01;***p<0.001.
Although we did not see significant differences in expression of GCLC and GSTπ, increased expression of GSTμ was evident in the Mdr2KO group. To further support western blot analysis, overall GST and SOD2 activity was determined using LE from WT and Mdr2KO mice. As shown in Figure S6, no significant differences were evident in GST activity between the two groups. We previously determined that SOD2 expression was significantly increased in human PSC[17]. To assess SOD2 activity in PSC and to determine if SOD2 activity was impacted in Mdr2KO mice, activity assays were performed. As shown in Figure S7, no significant differences were evident in SOD2 activity when comparing WT and Mdr2KO mice. In human PSC, SOD2 activity was significantly increased when compared to normal liver.
Zone-specific impact of Mdr2KO on expression of anti-oxidant response proteins.
The impact of Mdr2KO cholestatic injury on anti-oxidant responses has not been extensively examined with respect to hepatozonal changes in expression. In murine models of chemical cholestasis, pharmacological activation of Nrf2 pathways improved hepatocellular pathology and reduced injury[42, 43]. We hypothesized that the observed increase in cellular oxidative damage would result in zone-specific upregulation of oxidative detoxification enzymes. In the cell, catalase and the thioredoxin (Trx/TrxR1)/peroxiredoxin (Prdx) pathways contribute to mitigating excess ROS, specifically H2O2. The impact of oxidative injury on zone-specific expression of TrxR1, Prdx5, catalase and GSTπ, was assessed using IHC (Figure 6A). In WT livers, TrxR1 staining was moderate throughout the lobule. In Mdr2KO livers, TrxR1 staining was particularly strong within proliferating cholangiocytes and scattered periportal hepatocytes (arrows). In WT livers, weak punctate Prdx5 staining was present in hepatocytes surrounding the central vein, but stronger staining was evident in cholangiocytes and in some Kupffer cells within the sinusoids. In the Mdr2KO liver, increases in Prdx5 staining were observed and reflected increased infiltration of Kupffer cells as well as proliferation of cholangiocytes (arrows). No significant differences in catalase expression or localization were observed between WT and Mdr2KO mice. Interestingly, due to increased cholangiocyte expansion, catalase-positive cholangiocytes were increased in Mdr2KO (arrows). In WT livers, cholangiocytes exhibited very mild GSTπ staining with slightly higher GSTπ staining was evident throughout the centri-lobular hepatocytes (around the central vein). No significant differences were evident in Mdr2KO mice.
Figure 6. Impact of Mdr2KO on zonal expression of antioxidant response enzymes.
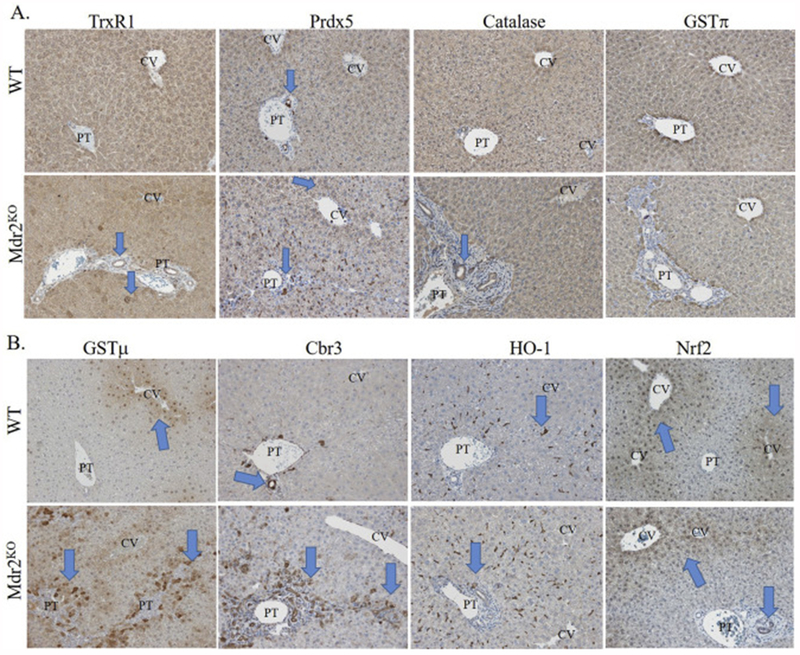
Liver sections from WT and Mdr2KO mice were immunostained using polyclonal antibodies directed against the indicated markers. Representative images are shown; n = at least 3 mice per genotype. Blue arrows indicate the hepatozonal expression for each anti-oxidant protein in WT and Mdr2KO mice as described in the results section. A. TrxR1, PRDX5, catalase and GSTπ. B. GSTμ, CBR3, HO-1 and Nrf2. Abbreviations as in Figure 1.
During conditions of increased hepatic oxidative stress, the transcription factor nuclear factor erythroid-2-like-2 (Nrf2) is activated and functions to upregulate the expression of antioxidant response genes[44]. To further explore the impact of Mdr2KO on hepatozonal expression of Nrf2 anti-oxidant responses, tissue sections were stained for the Nrf2-induced proteins, GSTμ, Cbr3, HO-1 as well as Nrf2 (Figure 6B)[45, 46]. WT livers showed mild GSTμ staining in areas surrounding the central vein and in some nuclei. In Mdr2KO livers, GSTμ staining was dramatically increased in hepatocytes (both cytoplasmic and nuclear) surrounding areas of increased biliary injury (periportal, zone 1), but not in cholangiocytes (arrows). In WT livers, high levels of Cbr3 staining was evident in cholangiocytes within the portal tract (arrow) as well as lower levels within Kupffer cells throughout the lobule. In contrast, in Mdr2KO mice, a significant elevation of Cbr3 staining was noted in cholangiocytes in the bile ducts (arrow) as well as in hepatocytes surrounding the portal triad (arrow). Hepatocyte HO-1 expression was weak and barely perceptible. Instead, robust panlobular (in all liver zones) HO-1 staining was present within Kupffer cells that resided predominantly within the sinusoids. Since the number of Kupffer cells increased on a per lobular basis, there was an overall increase in HO-1 expression within the liver lobule. In C57BL/6J mice, immunohistochemical analysis of Nrf2, revealed predominant expression in the nuclei and cytosol of hepatocytes surrounding the central vein (arrow). This expression was not increased in the Mdr2 group (arrow). Overall, Nrf2 expression was not present in tissue surrounding the portal triad. Of significance, there were a few cholangiocytes that did display Nrf2 expression in the Mdr2KO mice (arrow).
Proteomic analysis of carbonylated proteins in Mdr2KO mice
We recently reported the use of BH-derivatization followed by global LC-MS analysis to identify carbonylated proteins in subcellular fractions isolated from livers of murine models of NASH and ALD[47–49]. In the present study, BH-derivatization was performed on extracts prepared from Mdr2KO versus WT livers. Following affinity purification, carbonylated proteins were digested with trypsin, and peptides were analyzed by LC-MS. Collectively, 909 carbonylated proteins were identified in both models (Table S3).
Comparisons between groups revealed that 507 carbonylated proteins were common to both control and Mdr2KO livers, 183 carbonylated proteins were found only in the control livers, and 219 carbonylated proteins were found only in the Mdr2KO livers (Figure 7A, Table S4). Interestingly, antioxidant proteins, including GSTκ1, GSTμ3, GSTμ6 and Hsp70b, were only carbonylated in Mdr2KO livers. To identify cellular pathways preferentially impacted by carbonylation in Mdr2KO, these datasets were subjected to differential enrichment analysis using Gene Ontology and KEGG pathways [26, 50, 51]. These analyses did not reveal significant differences in any specific pathways when comparing WT and Mdr2KO, but rather, suggested that the effect of Mdr2KO deletion on protein carbonylation was protein specific when compared to other models of oxidative stress[48, 49]. Of note, GO and KEGG analysis of the subset of proteins only identified as carbonylated in Mdr2KO livers revealed a propensity for carbonylation of a variety of pathways including ribosomes, Arp2/3 complex-mediated actin nucleation, and regulation of Rho protein signal transduction (Table S5).
Figure 7. VENN analysis of carbonylated proteins during cholestatic liver disease.
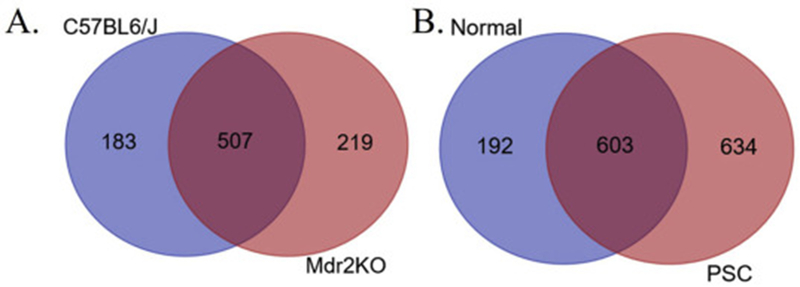
A. WT compared to Mdr2KO. B. Normal human and end-stage PSC.
The Mdr2KO murine model of cholestasis is considered an early model of cholestatic liver disease. Using hepatic tissue procured from human patients with end-stage PSC, we have recently shown that there is significant dysregulation of anti-oxidant responses and that dysregulation correlates with significant increases in protein carbonylation[16, 17]. To further delineate the impact of chronic cholestasis on protein carbonylation, global carbonylomic analysis was performed using tissue samples obtained from human livers isolated from both normal and end-stage PSC. Comparisons between the two groups revealed that 603 carbonylated proteins were common to both normal and end-stage PSC liver, 192 carbonylated proteins were found only in the normal livers, and 634 carbonylated proteins were found only in the end-stage PSC livers (Figure 7B., Table S6, S7). To identify cellular pathways preferentially impacted by carbonylation in end-stage PSC, these datasets were subjected to differential enrichment analysis using KEGG and Gene Ontology pathways [26, 50, 51]. From the KEGG analysis (Table S8) two primary pathways (KEGG Ribosome hsa03010 (Figure 8, Table S9) and KEGG Lysosome 04142 (Figure 9, Table S10)) emerged as significantly different between normal and diseased tissue. From the GO enrichment analysis (Table S11), the most consistent feature was a statistically significant enrichment of carbonylated ribosomal proteins from the large 60S ribosomal complex in the PSC group compared to normal hepatic tissue (Table S12). Just 4 ribosomal proteins were detected as carbonylated in normal tissue, compared to 24 in the PSC group. When examining the small 40S ribosomal subunits, no significant differences were evident. In addition, GTPase activity (Table S13) and Rab GTPases (Table S14) and the related GO pathway Protein transport (Table S15) were significantly enriched in patients with PSC compared to normal liver.
Figure 8. KEGG analysis of carbonylated ribosomal proteins in human end-stage PSC.
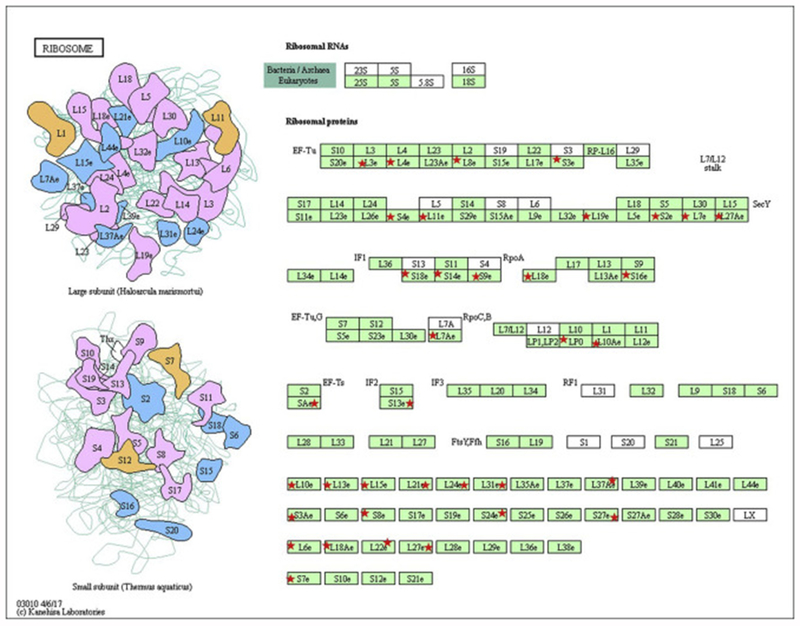
Red asterisks indicate proteins within the KEGG pathway hsa03010 (large ribosomal subunits) that were identified as carbonylated in PSC hepatic tissue.
Figure 9. KEGG analysis of carbonylated lysosomal proteins in human end-stage PSC.
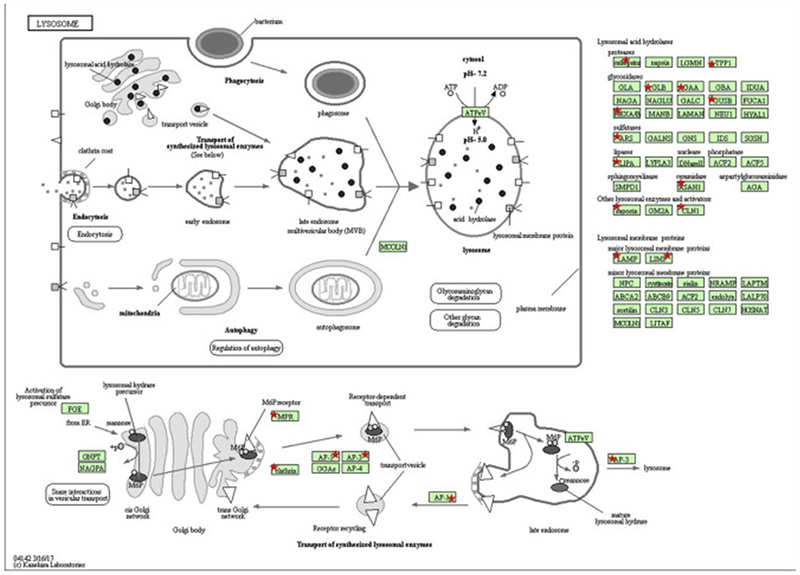
Red asterisks indicate proteins within the KEGG pathway hsa04142 (lysosomes) that were identified as carbonylated in PSC hepatic tissue.
Probing deeper into the bioinformatic analysis, several important differences emerged. Interestingly, most of these proteins also contribute to regulation of vesicular trafficking as well as actin cytoskeletal reorganization. First, in PSC tissue, a preponderance of HLA class I histocompatibility antigens were identified as carbonylated. Other carbonylated targets of interest included; ADP-ribosylation factors 1, 3, 4,and 6, Rho guanine nucleotide exchange factor 2, actin-related proteins 2, 3, 3b, and 4, actin-related protein 2/3 complex subunits 2, 3 and 4, phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 2 (SHIP-2) and type I inositol 3,4-bisphosphate 4-phosphatase (INPP4A[52]).
Bioinformatic analysis of PSC tissue strongly indicates that Rab GTPases are directly impacted by protein carbonylation. The abundance of Rab proteins has not been assessed in any murine model of cholestatic liver disease nor human cholestatic tissue. Using western blotting, the expression of Rabs 4, 5, 6, 7 and 9 was assessed for both Mdr2KO mice and human PSC. As shown in Figure 10A., no significant differences in Rab expression were evident in Mdr2KO In PSC, compared to normal tissue, expression of Rabs 4, 5 significantly decreased whereas expression of Rabs 7 and 9a significantly increased (Figure 10B.). By its ability to dephosphorylate phosphatidylinositol 3,4 bisphosphate, INPP4A is a regulator of endosomal trafficking as well as Akt activation [20, 53–55]. We sought to determine if in addition to being targeted by carbonylation, expression of INPP4A was impacted by cholestatic liver disease in both the Mdr2KO mice as well as in human PSC. In the Mdr2KO group, INPP4A abundance was slightly increased. However, in human PSC, levels of INPP4A protein was markedly suppressed.
Figure 10. Impact of cholestasis on Rab-GTPase expression.
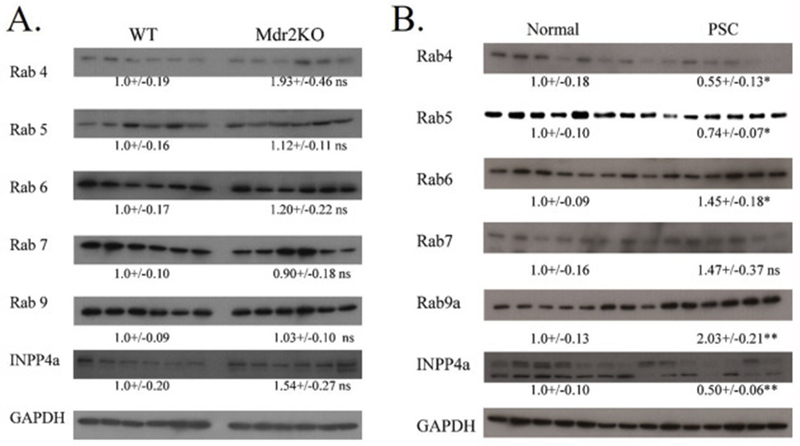
A. Western immunoblotting analysis of Rab-GTPases and INPP4A in liver lysates prepared from control and Mdr2KO mice. All exposures were normalized using GAPDH expression. Data are means +/− SEM, n=6 per genotype. Statistical analysis was via students t-test, *p<0.05;**p<0.01;***p<0.001. Data are means +/− SEM, n=6 per genotype. B. Western immunoblotting analysis of Rab-GTPases and INPP4A in liver lysates prepared from normal human and human end-stage PSC patients. Data are means +/− SEM, n=7 per condition. Statistical analysis was via students t-test, *p<0.05;**p<0.01;***p<0.001.
Discussion
Recent evidence has indicated that lowering oxidative stress and more specifically, lowering reactive aldehyde accumulation reduces hepatocellular injury during cholestatic liver disease[14, 15]. In the present study, we have used western blot and LC/MS analysis to determine the impact of early stage murine cholestasis on both the expression of anti-oxidant proteins and the carbonylation of particular proteins within the liver proteome.
Recent data has been conflicting on the role that the Nrf2 antioxidant pathway plays in cholestatic liver injury. This is partially supported by the fact that the deletion of Nrf2 promotes a cholestatic phenotype and enhances chemically induced cholestatic injury [56, 57]. Pharmacological activation of Nrf2 is protective in chemically-induced cholestatic injury but mice with obstructive cholestasis (BDL) show enhanced hepatocellular injury when treated with the Nrf2 activator Oltipraz [42, 58, 59]. By its ability to regulate expression of bile acid transporters bile salt export pump (Bsep), organic solute transporter (Ost α) as well as the multidrug resistance-associated proteins Mrp3 and Mrp4, Nrf2 also regulates bile acid homeostasis. In bile duct ligation models, constitutive induction of Nrf2 (Keap1KO) exerts a slight protective effect whereas deletion of Nrf2 by itself results in a cholestatic phenotype but exerts no effect following BDL [60, 61]. Our data using the Mdr2KO model of early cholestatic liver disease demonstrates that there is a periportal-localized antioxidant response and that this response is protein specific and includes some but not all proteins that are known to be induced by Nrf2 activation. We find that GCLC and NQO1 are not induced in the Mdr2 model whereas previous studies have demonstrated that both proteins are induced during BDL [62]. Our data is supported by the fact that we do not see a significant increase in GSH or GST activity. In cell culture models, exposure to both 4-HNE and acrolein resulted in upregulation of Nrf2 specific responses (GCLC, GSTμ, HO-1, NQO1)[63]. We find upregulation of other Nrf2 targets including Cbr3, TrxR1, HO-1 and GSTμ but not GCLC or NQO1 in the Mdr2KO mice suggesting selective induction of specific targets. This is partially explained by histological analysis of Nrf2 demonstrating predominant centrilobular Nrf2 activation with only minor staining of cholangiocytes. Although Aleksunes et al. did not examine Cbr3 or TrxR1, they did demonstrate that isoforms of GSTμ are induced in the BDL model. As both the induction and outputs of the Nrf2/Keap1 pathway are known to be highly complex [64], our observations could indicate either that cholestatic liver disease induces a unique Nrf2 response, or that another stress response pathway is driving this response. Further investigations will be needed to resolve these possibilities. In the cell, cysteine concentrations can be increased by uptake of disulfide species such as cystine or diglycinylcystine (from extracellular GSSG via Gamma glutamyl transferase) followed by reduction by the Trx1/TrxR1 system, the Trp15/TrxR1 system, or the GSH/Gsr system; it can come from increased de novo production from methionine via the methionine cycle and transsulfuration [65, 66]; or it can be acquired from protein stores by increased autophagy and protein degradation [67–69]. We find increased Trx1 and TrxR1 expression in Mdr2KO mice as well as in human PSC, which mechanistically supports at least one route for generating the observed increase in cysteine [16]. Other studies have also shown autophagy to be upregulated following BDL in the mouse as well as in human Primary Biliary Cholangitis (PBC) [70–72], potentially supporting another route of cysteine accumulation.
The increased staining of TrxR1 (hepatocytes and cholangiocytes), Cbr3 (hepatocytes and cholangiocytes) and GSTμ (periportal hepatocytes) suggests that there are cell specific responses during cholestasis. Both Cbr3 (4-oxononenal) and GSTμ (MDA, 4-HNE) possess activity towards products of lipid peroxidation and immunohistochemical localization correlates with increased acrolein and 4-HNE staining respectively in the Mdr2KO model[73, 74]. Interestingly, in human PSC which possess a more severe carbonylation phenotype, GSTμ is dramatically downregulated[17]. We hypothesize that GSTμ downregulation in PSC may be contributing to disease severity by the resulting accumulation of toxic lipid peroxides and subsequent protein carbonylation.
The relative increased staining of HO-1 in cholangiocytes when compared to hepatocytes combined with cholangiocyte Nrf2 staining also suggests that Nrf2 may play a more important role in cholangiocyte biology. In examining the Nrf2 target HO-1 using the Mdr2KO model, Barikbin et al, demonstrated that chemical upregulation of HO-1 reduced fibrosis and inflammation. We find induction of HO-1 and HO-1 localization in the Kupffer cells as well as in cholangiocytes but not in hepatocytes. This suggests that the modulation of inflammation and injury reported by Barikbin could be explained by changes in HO-1 induction in these two cell types. This is partially supported by the fact that our histology shows enhanced localization of F4/80+ Kupffer cells, CD3+ lymphocytes, plasma B cells and neutrophils in areas of fibrosis as well as surrounding proliferating bile ducts. Consistent with our findings, liver-specific Keap1 deletion, which leads to chronic Nrf2 induction, causes elevated GCLC and GSTA4 expression in the liver, but does not impact GPX levels which are not elevated in the Mdr2KO model. Overall, the data herein demonstrates that induction of anti-oxidant responses is periportally localized and based on the lack of induction of specific Nrf2 targets. Furthermore, we hypothesize that when compared to BDL, at least in the C57BL/6J background, deletion of Mdr2 produces a more mild form of oxidative injury that does not significantly impact GSH and is not sufficient to induce GCLC or NQO1. It is however, severe enough to impact cysteine metabolic pathways.
Using mass spectrometric analysis of biotin hydrazide streptavidin purified carbonylated proteins we have identified a total of 690/726 carbonylated proteins in murine (WT, Mdr2KO respectively) and 795/1237 in human (634 unique to PSC, 192 unique to normal tissue and 603 identified in both) hepatic tissue. From our Bioinformatic pathway analyses’ we did not identify specific pathways that are preferentially carbonylated in the Mdr2KO group. Interestingly, we did identify specific proteins including GSTκ1, GSTμ3, GSTμ6 and Hsp70b that are preferentially carbonylated in the Mdr2KO group. Of these, GSTμ3, GSTμ6 and Hsp70b were not identified as preferential carbonylation targets in murine models of alcoholic liver disease suggesting that these may be specific targets in cholestatic liver disease[48, 49]. At 11 weeks of age, WT mice exhibit a mild induction of oxidative stress with only a selected induction of anti-oxidant proteins. We hypothesize that the lack of identification of specific pathways in the Mdr2KO group may be due to zone specific periportal oxidative stress that is occurring given that we have identified distinct pathways in other models of chronic liver disease such as alcoholic liver disease[48, 49]. Furthermore, given the modest degree of injury in the Mdr2KO group, we anticipate that specific pathways will emerge in more severe models of cholestatic injury. Future research will be necessary using more severe models of cholestatic injury such as the BDL model of obstructive cholestasis.
In human end-stage PSC, oxidative injury is far more severe and there is significant dysregulation of anti-oxidant responses[16, 17]. Bioinformatic pathway analysis of carbonylated proteins supports the more severe phenotype by demonstrating statistical enrichment of carbonylation of both the large subunit of ribosomal protein synthesis, lysosomal trafficking and Rab-GTPase mediated vesicular trafficking. Interestingly, there are a few reports that have examined Rab-GTPases and cholestatic liver disease. Using cell culture, Rab4 and Rab11 have been demonstrated to play important roles in bile trafficking by regulating the sodium-taurocholate (TC) cotransporting polypeptide (Ntcp) and by increasing plasma membrane localization of multidrug resistance-associated protein 2 (MRP2) [75, 76]. Following BDL, Rab11a is concentrated to the crude vesicular fraction to support basolateral-to-apical transcytosis [77]. From our data, we hypothesize that decreased expression of Rab4 may be contributing to hepatocellular injury in PSC.
Notably, Rab5 regulates early endosomal trafficking and directly associates with INPP4A[78]. By its lipid phosphatase activity, combined with the 3-phosphatase PTEN, INPP4A is also a regulator of Akt and may be contributing to the aforementioned increases in phosphorylation. Importantly we have previously shown that increased carbonylation of PTEN contributes to Akt activation [19, 79]. As important, in our previous paper, we demonstrated increased Ser473Akt phosphorylation and Akt activation in human PSC [17]. We predict that carbonylation of INPP4A would also decrease lipid phosphatase activity contributing to the observed Akt activation. In addition to increased carbonylation, we demonstrate decreased expression of both Rab5 and INPP4A suggesting that vesicular and more specifically endocytic trafficking may also be dysregulated in human PSC.
In conclusion, results herein provide evidence that zone-specific induction of antioxidant proteins occurs in Mdr2KO livers and this may mitigate the accumulation of protein carbonyls. However, the induction is not sufficient to fully abrogate the accumulation of oxidative damage to proteins (as visualized by increased protein carbonylation) or DNA (as visualized by increased γH2A.x positive cells). We speculate that this may be due to only minor periportal induction of Nrf2. Interestingly, we find cell specific induction of anti-oxidant responses within the periportal region corresponded to different products of lipid peroxidation that we measured in the Mdr2KO livers. This occurred in cholangiocytes as well as in the surrounding hepatocytes suggesting cells have distinct innate abilities to defend themselves against lipid peroxide-induced damage. Histology clearly indicates that the production of ROS, the formation of products of lipid peroxidation (4-HNE, acrolein) strongly correlate with the inflammatory response as well as fibrosis. We caution however that this study only examines a single time point. We cannot conclude from this study if the source of ROS is from the inflammation itself or from cholestatic injury. Further studies that utilize a time course will be necessary to mechanistically identify the source of ROS during cholestatic injury. From our proteomic studies, protein carbonylation increased as the severity of cholestasis increased. Finally, we show that in human PSC, the dramatic accumulation of protein carbonyls target-specific pathways that also are undergoing changes in expression which would compound protein dysfunction during vesicular trafficking. Cumulatively, these results identify a novel cholestasis-associated response that implicates protein carbonylation as a potential therapeutic intervention to improve hepatocellular function.
Supplementary Material
Highlights.
Increased periportal accumulation of reactive aldehydes in cholestasis.
Atypical activation of antioxidant proteins
Colocalization of aldehydes with inflammation, fibrosis and antioxidant response
Increased carbonylation of Rab GTPases, Lysosomal proteins and ribosomes in PSC
Acknowledgements
This research was funded by a grant from the National Institutes of Health Institute of Alcohol Abuse and Alcoholism (NIAAA) 5R37 AA00930022 (To D.R.P.), the Gastrointestinal and Liver Innate Immunity Program (GALIIP) at the University of Colorado Denver Anschutz Medical Campus (To C.T.S., B.F.), and R01’s DK103712 / DK095491 to S.P.C. The funding agencies had no role in study design, data collection. The authors also wish to acknowledge the Skaggs School of Pharmacy and Pharmaceutical Sciences Mass Spectrometry Core Facility for assistance in analysis of carbonylated proteins and E. Erin Smith, HTL (ASCP)CMQIHC of the University of Colorado Denver Cancer Center Research Histology Core for assistance in preparing histology slides. The UCDCCRHC is supported in part by NIH/NCRR Colorado CTSI Grant Number UL1 RR025780 and the University of Colorado Cancer Center Grant (P30 CA046934).
Abbreviations:
- ALT
alanine aminotransferase
- BDL
bile duct ligation
- BH
biotin hydrazide
- Cbr3
carbonyl reductase isoform 3
- CID
collision-induced dissociation
- CV
central vein
- CK
cytokeratin
- ETD
electron transfer dissociation
- GO
Gene Ontology
- GST
glutathione S-transferase
- HO-1
heme oxygenase
- 4-HNE
4-hydroxy-2-nonenal
- IBD
inflammatory bowel disease
- INPP4A
inositol polyphosphate 4-phosphatase isoform A
- Keap1
Kelch-like ECH-associated protein 1
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LE
whole liver extract
- MDA
malondialdehyde
- MMP
matrix metalloproteinases
- MPO
myeloperoxidase
- NQO1
NAD(P)H quinone dehydrogenase 1
- Nrf2
nuclear factor erythroid-2-like-2
- PT
portal triad
- PSC
primary sclerosing cholangitis
- Rab-GTPase
Ras associated binding protein
- ROS
reactive oxygen species
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Boonstra K, Beuers U, Ponsioen CY, Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review, J Hepatol 56(5) (2012) 1181–8. [DOI] [PubMed] [Google Scholar]
- [2].Molodecky NA, Kareemi H, Parab R, Barkema HW, Quan H, Myers RP, Kaplan GG, Incidence of primary sclerosing cholangitis: a systematic review and meta-analysis, Hepatology 53(5) (2011) 1590–9. [DOI] [PubMed] [Google Scholar]
- [3].Pollheimer MJ, Trauner M, Fickert P, Will we ever model PSC? - “it’s hard to be a PSC model!”, Clin Res Hepatol Gastroenterol 35(12) (2011) 792–804. [DOI] [PubMed] [Google Scholar]
- [4].Vendemiale G, Grattagliano I, Lupo L, Memeo V, Altomare E, Hepatic oxidative alterations in patients with extra-hepatic cholestasis. Effect of surgical drainage, J Hepatol 37(5) (2002) 601–5. [DOI] [PubMed] [Google Scholar]
- [5].Grattagliano I, Calamita G, Cocco T, Wang DQ, Portincasa P, Pathogenic role of oxidative and nitrosative stress in primary biliary cirrhosis, World J Gastroenterol 20(19) (2014) 5746–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Aller MA, Arias JL, Prieto I, Losada M, Arias J, Bile duct ligation: step-by-step to cholangiocyte inflammatory tumorigenesis, Eur J Gastroenterol Hepatol 22(6) (2010) 651–61. [DOI] [PubMed] [Google Scholar]
- [7].Hirschfield GM, Karlsen TH, Lindor KD, Adams DH, Primary sclerosing cholangitis, Lancet 382(9904) (2013) 1587–99. [DOI] [PubMed] [Google Scholar]
- [8].Tamura S, Sugawara Y, Kaneko J, Matsui Y, Togashi J, Makuuchi M, Recurrence of primary sclerosing cholangitis after living donor liver transplantation, Liver Int 27(1) (2007) 86–94. [DOI] [PubMed] [Google Scholar]
- [9].Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, Mol CA, Ottenhoff R, van der Lugt NM, van Roon MA, et al. , Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease, Cell 75(3) (1993) 451–62. [DOI] [PubMed] [Google Scholar]
- [10].Mauad TH, van Nieuwkerk CM, Dingemans KP, Smit JJ, Schinkel AH, Notenboom RG, van den Bergh Weerman MA, Verkruisen RP, Groen AK, Oude Elferink RP, et al. , Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis, Am J Pathol 145(5) (1994) 1237–45. [PMC free article] [PubMed] [Google Scholar]
- [11].Yang YY, Lee KC, Huang YT, Wang YW, Hou MC, Lee FY, Lin HC, Lee SD, Effects of N-acetylcysteine administration in hepatic microcirculation of rats with biliary cirrhosis, J Hepatol 49(1) (2008) 25–33. [DOI] [PubMed] [Google Scholar]
- [12].Pastor A, Collado PS, Almar M, Gonzalez-Gallego J, Antioxidant enzyme status in biliary obstructed rats: effects of N-acetylcysteine, J Hepatol 27(2) (1997) 363–70. [DOI] [PubMed] [Google Scholar]
- [13].Copple BL, Jaeschke H, Klaassen CD, Oxidative stress and the pathogenesis of cholestasis, Semin Liver Dis 30(2) (2010) 195–204. [DOI] [PubMed] [Google Scholar]
- [14].Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, Zatloukal K, Liu J, Waalkes MP, Cover C, Denk H, Hofmann AF, Jaeschke H, Trauner M, 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice, Gastroenterology 130(2) (2006) 465–81. [DOI] [PubMed] [Google Scholar]
- [15].Barikbin R, Neureiter D, Wirth J, Erhardt A, Schwinge D, Kluwe J, Schramm C, Tiegs G, Sass G, Induction of heme oxygenase 1 prevents progression of liver fibrosis in Mdr2 knockout mice, Hepatology 55(2) (2011) 553–62. [DOI] [PubMed] [Google Scholar]
- [16].Petersen DR, Orlicky DJ, Roede JR, Shearn CT, Aberrant expression of redox regulatory proteins in patients with concomitant primary Sclerosing cholangitis/inflammatory bowel disease, Exp Mol Pathol 105(1) (2018) 32–36. [DOI] [PubMed] [Google Scholar]
- [17].Shearn CT, Orlicky DJ, Petersen DR, Dysregulation of antioxidant responses in patients diagnosed with concomitant Primary Sclerosing Cholangitis/Inflammatory Bowel Disease, Exp Mol Pathol 104(1) (2018) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fickert P, Pollheimer MJ, Beuers U, Lackner C, Hirschfield G, Housset C, Keitel V, Schramm C, Marschall HU, Karlsen TH, Melum E, Kaser A, Eksteen B, Strazzabosco M, Manns M, Trauner M, P.S.C.S.G. International, Characterization of animal models for primary sclerosing cholangitis (PSC), J Hepatol 60(6) (2014) 1290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shearn CT, Smathers RL, Backos DS, Reigan P, Orlicky DJ, Petersen DR, Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis, Free Radic Biol Med 65 (2013) 680–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shearn CT, Walker J, Norris FA, Identification of a novel spliceoform of inositol polyphosphate 4-phosphatase type Ialpha expressed in human platelets: structure of human inositol polyphosphate 4-phosphatase type I gene, Biochem Biophys Res Commun 286(1) (2001) 119–25. [DOI] [PubMed] [Google Scholar]
- [21].Shearn CT, Orlicky DJ, Saba LM, Shearn AH, Petersen DR, Increased hepatocellular protein carbonylation in human end-stage alcoholic cirrhosis, Free Radic Biol Med 89 (2015) 1144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shearn CT, Fritz KS, Thompson JA, Protein damage from electrophiles and oxidants in lungs of mice chronically exposed to the tumor promoter butylated hydroxytoluene, Chem Biol Interact 192(3) (2011) 278–86. [DOI] [PubMed] [Google Scholar]
- [23].Assiri MA, Roy SR, Harris PS, Ali H, Liang Y, Shearn CT, Orlicky DJ, Roede JR, Hirschey MD, Backos DS, Fritz KS, Chronic Ethanol Metabolism Inhibits Hepatic Mitochondrial Superoxide Dismutase via Lysine Acetylation, Alcohol Clin Exp Res 41(10) (2017) 1705–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shearn CT, Orlicky DJ, McCullough RL, Jiang H, Maclean KN, Mercer KE, Stiles BL, Saba LM, Ronis MJ, Petersen DR, Liver-Specific Deletion of Phosphatase and Tensin Homolog Deleted on Chromosome 10 Significantly Ameliorates Chronic EtOH-Induced Increases in Hepatocellular Damage, PLoS One 11(4) (2016) e0154152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Harris PS, Roy SR, Coughlan C, Orlicky DJ, Liang Y, Shearn CT, Roede JR, Fritz KS, Chronic ethanol consumption induces mitochondrial protein acetylation and oxidative stress in the kidney, Redox Biol 6 (2015) 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K, KEGG: new perspectives on genomes, pathways, diseases and drugs, Nucleic Acids Res 45(D1) (2017) D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gene Ontology C, Gene Ontology Consortium: going forward, Nucleic Acids Res 43(Database issue) (2015) D1049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Potikha T, Stoyanov E, Pappo O, Frolov A, Mizrahi L, Olam D, Shnitzer-Perlman T, Weiss I, Barashi N, Peled A, Sass G, Tiegs G, Poirier F, Rabinovich GA, Galun E, Goldenberg D, Interstrain differences in chronic hepatitis and tumor development in a murine model of inflammation-mediated hepatocarcinogenesis, Hepatology 58(1) (2013) 192–204. [DOI] [PubMed] [Google Scholar]
- [29].Jones H, Hargrove L, Kennedy L, Meng F, Graf-Eaton A, Owens J, Alpini G, Johnson C, Bernuzzi F, Demieville J, DeMorrow S, Invernizzi P, Francis H, Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2(−/−) mice, Hepatology 64(4) (2016) 1202–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Baghdasaryan A, Fuchs CD, Osterreicher CH, Lemberger UJ, Halilbasic E, Pahlman I, Graffner H, Krones E, Fickert P, Wahlstrom A, Stahlman M, Paumgartner G, Marschall HU, Trauner M, Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis, J Hepatol 64(3) (2016) 674–81. [DOI] [PubMed] [Google Scholar]
- [31].Orlicky DJ, Roede JR, Bales E, Greenwood C, Greenberg A, Petersen D, McManaman JL, Chronic ethanol consumption in mice alters hepatocyte lipid droplet properties, Alcohol Clin Exp Res 35(6) (2011) 1020–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hov JR, Boberg KM, Taraldsrud E, Vesterhus M, Boyadzhieva M, Solberg IC, Schrumpf E, Vatn MH, Lie BA, Molberg O, Karlsen TH, Antineutrophil antibodies define clinical and genetic subgroups in primary sclerosing cholangitis, Liver Int 37(3) (2017) 458–465. [DOI] [PubMed] [Google Scholar]
- [33].Shearn CT, Orlicky DJ, Petersen DR, Dysregulation of antioxidant responses in patients diagnosed with concomitant Primary Sclerosing Cholangitis/Inflammatory Bowel Disease, Exp Mol Pathol 104(1) (2017) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, Albano E, Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH, Hepatology 59(3) (2014) 886–97. [DOI] [PubMed] [Google Scholar]
- [35].Basaranoglu M, Basaranoglu G, Senturk H, From fatty liver to fibrosis: a tale of “second hit”, World J Gastroenterol 19(8) (2013) 1158–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rolo AP, Teodoro JS, Palmeira CM, Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis, Free Radic Biol Med 52(1) (2012) 59–69. [DOI] [PubMed] [Google Scholar]
- [37].Smathers RL, Galligan JJ, Stewart BJ, Petersen DR, Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease, Chem Biol Interact 192(1–2) (2011) 107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tebbi A, Levillayer F, Jouvion G, Fiette L, Soubigou G, Varet H, Boudjadja N, Cairo S, Hashimoto K, Suzuki AM, Carninci P, Carissimo A, di Bernardo D, Wei Y, Deficiency of multidrug resistance 2 contributes to cell transformation through oxidative stress, Carcinogenesis 37(1) (2016) 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roskams T, Yang SQ, Koteish A, Durnez A, DeVos R, Huang X, Achten R, Verslype C, Diehl AM, Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease, Am J Pathol 163(4) (2003) 1301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Galligan JJ, Smathers RL, Shearn CT, Fritz KS, Backos DS, Jiang H, Franklin CC, Orlicky DJ, Maclean KN, Petersen DR, Oxidative Stress and the ER Stress Response in a Murine Model for Early-Stage Alcoholic Liver Disease, J Toxicol 2012 (2012) 207594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shearn CT, Mercer KE, Orlicky DJ, Hennings L, Smathers-McCullough RL, Stiles BL, Ronis MJ, Petersen DR, Short Term Feeding of a High Fat Diet Exerts an Additive Effect on Hepatocellular Damage and Steatosis in Liver-Specific PTEN Knockout Mice, PLoS One 9(5) (2014) e96553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yu L, Liu X, Yuan Z, Li X, Yang H, Yuan Z, Sun L, Zhang L, Jiang Z, SRT1720 Alleviates ANIT-Induced Cholestasis in a Mouse Model, Front Pharmacol 8 (2017) 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shen K, Feng X, Pan H, Zhang F, Xie H, Zheng S, Baicalin Ameliorates Experimental Liver Cholestasis in Mice by Modulation of Oxidative Stress, Inflammation, and NRF2 Transcription Factor, Oxid Med Cell Longev 2017 (2017) 6169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wu KC, Liu J, Klaassen CD, Role of Nrf2 in preventing ethanol-induced oxidative stress and lipid accumulation, Toxicol Appl Pharmacol 262(3) (2012) 321–9. [DOI] [PubMed] [Google Scholar]
- [45].Schaupp CM, White CC, Merrill GF, Kavanagh TJ, Metabolism of doxorubicin to the cardiotoxic metabolite doxorubicinol is increased in a mouse model of chronic glutathione deficiency: A potential role for carbonyl reductase 3, Chem Biol Interact 234 (2015) 154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ebert B, Kisiela M, Malatkova P, El-Hawari Y, Maser E, Regulation of human carbonyl reductase 3 (CBR3; SDR21C2) expression by Nrf2 in cultured cancer cells, Biochemistry 49(39) (2010) 8499–511. [DOI] [PubMed] [Google Scholar]
- [47].Petersen DR, Saba LM, Sayin VI, Papagiannakopoulos T, Schmidt EE, Merrill GF, Orlicky DJ, Shearn CT, Elevated Nrf-2 responses are insufficient to mitigate protein carbonylation in hepatospecific PTEN deletion mice, PLoS One 13(5) (2018) e0198139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shearn CT, Pulliam CF, Pedersen K, Meredith K, Mercer KE, Saba LM, Orlicky DJ, Ronis MJ, Petersen DR, Knockout of the Gsta4 Gene in Male Mice Leads to an Altered Pattern of Hepatic Protein Carbonylation and Enhanced Inflammation Following Chronic Consumption of an Ethanol Diet, Alcohol Clin Exp Res 42(7) (2018) 1192–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shearn CT, Fritz KS, Shearn AH, Saba LM, Mercer KE, Engi B, Galligan JJ, Zimniak P, Orlicky DJ, Ronis MJ, Petersen DR, Deletion of GSTA4-4 results in increased mitochondrial post-translational modification of proteins by reactive aldehydes following chronic ethanol consumption in mice, Redox Biol 7 (2016) 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Saba LM, Flink SC, Vanderlinden LA, Israel Y, Tampier L, Colombo G, Kiianmaa K, Bell RL, Printz MP, Flodman P, Koob G, Richardson HN, Lombardo J, Hoffman PL, Tabakoff B, The sequenced rat brain transcriptome - its use in identifying networks predisposing alcohol consumption, FEBS J (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, Gene ontology: tool for the unification of biology. The Gene Ontology Consortium, Nat Genet 25(1) (2000) 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shearn CT, Norris FA, Biochemical characterization of the type I inositol polyphosphate 4-phosphatase C2 domain, Biochem Biophys Res Commun 356(1) (2007) 255–9. [DOI] [PubMed] [Google Scholar]
- [53].Norris FA, Majerus PW, Hydrolysis of phosphatidylinositol 3,4-bisphosphate by inositol polyphosphate 4-phosphatase isolated by affinity elution chromatography, J Biol Chem 269(12) (1994) 8716–20. [PubMed] [Google Scholar]
- [54].Ivetac I, Munday AD, Kisseleva MV, Zhang XM, Luff S, Tiganis T, Whisstock JC, Rowe T, Majerus PW, Mitchell CA, The type Ialpha inositol polyphosphate 4-phosphatase generates and terminates phosphoinositide 3-kinase signals on endosomes and the plasma membrane, Mol Biol Cell 16(5) (2005) 2218–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li H, Marshall AJ, Phosphatidylinositol (3,4) bisphosphate-specific phosphatases and effector proteins: A distinct branch of PI3K signaling, Cell Signal 27(9) (2015) 1789–98. [DOI] [PubMed] [Google Scholar]
- [56].Weerachayaphorn J, Mennone A, Soroka CJ, Harry K, Hagey LR, Kensler TW, Boyer JL, Nuclear factor-E2-related factor 2 is a major determinant of bile acid homeostasis in the liver and intestine, Am J Physiol Gastrointest Liver Physiol 302(9) (2012) G925–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tan KP, Wood GA, Yang M, Ito S, Participation of nuclear factor (erythroid 2-related), factor 2 in ameliorating lithocholic acid-induced cholestatic liver injury in mice, Br J Pharmacol 161(5) (2010) 1111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tanaka Y, Aleksunes LM, Cui YJ, Klaassen CD, ANIT-induced intrahepatic cholestasis alters hepatobiliary transporter expression via Nrf2-dependent and independent signaling, Toxicol Sci 108(2) (2009) 247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ma X, Zhao YL, Zhu Y, Chen Z, Wang JB, Li RY, Chen C, Wei SZ, Li JY, Liu B, Wang RL, Li YG, Wang LF, Xiao XH, Paeonia lactiflora Pall. protects against ANIT-induced cholestasis by activating Nrf2 via PI3K/Akt signaling pathway, Drug Des Devel Ther 9 (2015) 5061–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Okada K, Shoda J, Taguchi K, Maher JM, Ishizaki K, Inoue Y, Ohtsuki M, Goto N, Sugimoto H, Utsunomiya H, Oda K, Warabi E, Ishii T, Yamamoto M, Nrf2 counteracts cholestatic liver injury via stimulation of hepatic defense systems, Biochem Biophys Res Commun 389(3) (2009) 431–6. [DOI] [PubMed] [Google Scholar]
- [61].Weerachayaphorn J, Cai SY, Soroka CJ, Boyer JL, Nuclear factor erythroid 2-related factor 2 is a positive regulator of human bile salt export pump expression, Hepatology 50(5) (2009) 1588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Aleksunes LM, Slitt AL, Maher JM, Dieter MZ, Knight TR, Goedken M, Cherrington NJ, Chan JY, Klaassen CD, Manautou JE, Nuclear factor-E2-related factor 2 expression in liver is critical for induction of NAD(P)H:quinone oxidoreductase 1 during cholestasis, Cell Stress Chaperones 11(4) (2006) 356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang H, Forman HJ, Signaling pathways involved in phase II gene induction by alpha, beta-unsaturated aldehydes, Toxicol Ind Health 25(4–5) (2009) 269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Suzuki T, Yamamoto M, Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress, J Biol Chem 292(41) (2017) 16817–16824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Prigge JR, Coppo L, Martin SS, Ogata F, Miller CG, Bruschwein MD, Orlicky DJ, Shearn CT, Kundert JA, Lytchier J, Herr AE, Mattsson A, Taylor MP, Gustafsson TN, Arner ESJ, Holmgren A, Schmidt EE, Hepatocyte Hyperproliferation upon Liver-Specific Co-disruption of Thioredoxin-1, Thioredoxin Reductase-1, and Glutathione Reductase, Cell Rep 19(13) (2017) 2771–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mosharov E, Cranford MR, Banerjee R, The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes, Biochemistry 39(42) (2000) 13005–11. [DOI] [PubMed] [Google Scholar]
- [67].Pader I, Sengupta R, Cebula M, Xu J, Lundberg JO, Holmgren A, Johansson K, Arner ES, Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase, Proc Natl Acad Sci U S A 111(19) (2014) 6964–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Holmgren A, Bovine thioredoxin system. Purification of thioredoxin reductase from calf liver and thymus and studies of its function in disulfide reduction, J Biol Chem 252(13) (1977) 4600–6. [PubMed] [Google Scholar]
- [69].Mandal PK, Seiler A, Perisic T, Kolle P, Banjac Canak A, Forster H, Weiss N, Kremmer E, Lieberman MW, Bannai S, Kuhlencordt P, Sato H, Bornkamm GW, Conrad M, System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency, J Biol Chem 285(29) (2010) 22244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nakanuma Y, Sasaki M, Harada K, Autophagy and senescence in fibrosing cholangiopathies, J Hepatol 62(4) (2015) 934–45. [DOI] [PubMed] [Google Scholar]
- [71].Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y, Increased expression of mitochondrial proteins associated with autophagy in biliary epithelial lesions in primary biliary cirrhosis, Liver Int 33(2) (2013) 312–20. [DOI] [PubMed] [Google Scholar]
- [72].Kim S, Han SY, Yu KS, Han D, Ahn HJ, Jo JE, Kim JH, Shin J, Park HW, Impaired autophagy promotes bile acid-induced hepatic injury and accumulation of ubiquitinated proteins, Biochem Biophys Res Commun 495(1) (2018) 1541–1547. [DOI] [PubMed] [Google Scholar]
- [73].Berhane K, Widersten M, Engstrom A, Kozarich JW, Mannervik B, Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases, Proc Natl Acad Sci U S A 91(4) (1994) 1480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Doorn JA, Maser E, Blum A, Claffey DJ, Petersen DR, Human carbonyl reductase catalyzes reduction of 4-oxonon-2-enal, Biochemistry 43(41) (2004) 13106–14. [DOI] [PubMed] [Google Scholar]
- [75].Park SW, Schonhoff CM, Webster CR, Anwer MS, Rab11, but not Rab4, facilitates cyclic AMP- and tauroursodeoxycholate-induced MRP2 translocation to the plasma membrane, Am J Physiol Gastrointest Liver Physiol 307(8) (2014) G863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Schonhoff CM, Thankey K, Webster CR, Wakabayashi Y, Wolkoff AW, Anwer MS, Rab4 facilitates cyclic adenosine monophosphate-stimulated bile acid uptake and Na+-taurocholate cotransporting polypeptide translocation, Hepatology 48(5) (2008) 1665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Larkin JM, Coleman H, Espinosa A, Levenson A, Park MS, Woo B, Zervoudakis A, Tinh V, Intracellular accumulation of pIgA-R and regulators of transcytotic trafficking in cholestatic rat hepatocytes, Hepatology 38(5) (2003) 1199–209. [DOI] [PubMed] [Google Scholar]
- [78].Shin HW, Hayashi M, Christoforidis S, Lacas-Gervais S, Hoepfner S, Wenk MR, Modregger J, Uttenweiler-Joseph S, Wilm M, Nystuen A, Frankel WN, Solimena M, De Camilli P, Zerial M, An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway, J Cell Biol 170(4) (2005) 607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Shearn CT, Smathers RL, Stewart BJ, Fritz KS, Galligan JJ, Hail N Jr., Petersen DR, Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibition by 4-hydroxynonenal leads to increased Akt activation in hepatocytes, Mol Pharmacol 79(6) (2011) 941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.