

Abstract
Randomized cardiovascular trials aimed to reduce the excessive residual risk in high‐risk patients through a more aggressive low‐density lipoprotein‐cholesterol control or targeting triglycerides or high‐density lipoprotein‐cholesterol levels have shown a null or, at best, limited incremental benefit. In some cases, the treatment produced meaningful effects only in study subgroups. As a consequence, some compounds were withdrawn (e.g., nicotinic acid derivatives and cholesteryl ester transfer protein inhibitors), whereas others (fibrates) are utilized with reluctance due to the low level of evidence‐based data. By reviewing these trials analytically, we identified a common feature that might explain their meager results: most of them involved patients generically at high cardiovascular risk with normal or near normal lipid levels and not patients with “true” dyslipidemia, who would receive the treatment if it were part of usual care. These observations may warrant re‐examining a central criterion of pragmatism, eligibility, in the outline of forthcoming cardiovascular trials with novel lipid‐modifying drugs.
Morbidity and mortality in patients at high cardiovascular risk remain high, even in those receiving the best current standards of care. This “residual risk”1 may be attributed to: (i) nonlipid risk factors, including inflammatory factors, (ii) persistently high low‐density lipoprotein‐cholesterol (LDL‐C) in patients with very high baseline levels or statin intolerance, or (iii) concomitant lipid abnormalities, such as high triglycerides (TGs), low high‐density lipoprotein‐cholesterol (HDL‐C), or high lipoprotein(a). The advent of proprotein convertase subtilisin/kexin type 9 (PCSK9)‐inhibitor therapy represents a medical breakthrough for LDL‐C lowering and residual risk reduction in patients with “hard‐to‐treat” hypercholesterolemia. On the other hand, negative results of cardiovascular outcome trials with triglyceride‐lowering compounds (fibrates) or HDL‐C–raising agents (nicotinic acid derivatives and cholesteryl ester transfer protein (CETP) inhibitors) in patients on statins suggest that these treatments do not reduce residual risk. Yet, one possible explanation for these negative results is that most of the patients included in the trials did not have the lipid abnormality that the tested interventions actually correct (Figure 1 and Figures S1–S3 ). Using an everyday analogy, you cannot straighten something that is already straight, even if the tool used may successfully straighten something that is bent. Indeed, as the relationship between the level of a risk factor and the cardiovascular risk is not linear, targeting patients with relatively normal lipid values to achieve “super‐normal” values (e.g., super‐low LDL‐C or TGs or super‐high HDL‐C) may dilute the efficacy of the tested intervention toward an effect smaller than expected or even yield an overall null result.
Figure 1.
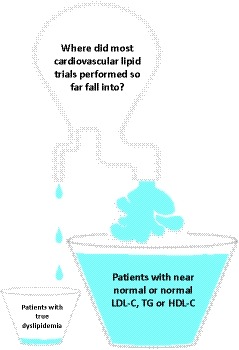
Allegorical sketch representing the distribution of lipid trials with cardiovascular outcomes performed so far, according to the presence of “true” dyslipidemia at baseline. HDL‐C, high‐density lipoprotein‐cholesterol; LDL‐C, low‐density lipoprotein‐cholesterol; TG, triglycerides.
We herein describe key features of published explanatory lipid trials, not specifically targeted at patients with “true” dyslipidemia, who might have undermined the chance to provide definite data about the effect of a comprehensive lipid management on residual risk, appealing for pragmatic studies.
LDL trials
Early epidemiological studies and evidence from monogenic disorders affecting LDL metabolism have shown that increased concentrations of LDL‐C are linked with an increased risk of myocardial infarction and cardiovascular death. Mendelian randomization studies have documented that this association is causal2 and that a lifelong exposure even to a few‐mg/dL lower LDL‐C determines a significantly reduced risk of cardiovascular events.3 Relevantly, the relationship between LDL‐C levels and cardiovascular risk is not linear.4 As a consequence, for a given absolute LDL‐C reduction, patients with high LDL‐C levels before treatment should rationally get from the intervention a larger risk reduction than patients with lower baseline levels. The most common form of severe hypercholesterolemia encountered in clinical practice is observed in patients with familial hypercholesterolemia (FH), a disease caused by pathogenic mutations in genes involved in LDL metabolism.5 Heterozygous FH is a relatively frequent condition (1:200 to 1:500 in a general population), which determines, if untreated, lifelong LDL‐C levels usually between 190 and 500 mg/dL and an at least 10‐fold higher risk of premature coronary events than that of unaffected individuals.5, 6, 7 Accordingly, in current prevention guidelines, patients with heterozygous FH are automatically assigned a high‐risk category (without the need for risk score tools) and recommended precocious and intensive drug treatment.8, 9 What may not be so patent is that this treatment recommendation, although undeniably warranted, is based on the lowest level of evidence (level C, expert opinion), inasmuch as, weirdly enough, not a single controlled LDL‐C trial with cardiovascular end points has been carried out so far in patients with heterozygous FH. Consequently, no empirical data on the cardiovascular effects of LDL‐C lowering in patients that fall within the steeper part of the LDL‐C‐related cardiovascular risk are available.
Instead, early randomized controlled trials (e.g., 4S, CARE)10, 11 demonstrated that lowering LDL‐C with statins in high‐risk patients with moderate hypercholesterolemia (i.e. LDL‐C 140–190 mg/dL), largely of polygenic plus dietary basis, reduces events and mortality. In the studies where an analysis of subgroups according to baseline LDL‐C was performed, such as in the CARE study, baseline LDL‐C had a clear influence on the impact on clinical outcomes. Later studies showed that a more intensive LDL‐C lowering through statins further reduces cardiovascular morbidity, but not mortality, compared with less intensive schemes.12 The addition of ezetimibe to a background of statin therapy was also effective in reducing the residual risk.13 Thereafter, the concept “the lower, the better” pervaded the scientific and clinical communities.
Anti–PCSK‐9 monoclonal antibodies show an exceptionally potent LDL‐C lowering action and good tolerability. Evolocumab and alirocumab, the two compounds that reached the market, were tested in full programs of phase III studies (PROFICIO and ODYSSEY, respectively) focusing on changes in the lipid profile and tolerability in candidates suitable for this type of medicine (i.e., high‐risk statin‐intolerant patients with primary hypercholesterolemia and patients with heterozygous FH inadequately controlled by standard therapy) (Table 1 ). However, the two main cardiovascular outcome trials using anti–PCSK‐9 antibodies (FOURIER and ODYSSEY OUTCOMES, respectively)14, 15 did not target these “challenging” patients but rather broad categories of high‐risk patients with LDL‐C levels perhaps not “optimal” but acceptably controlled through statins (baseline LDL‐C 92 mg/dL in FOURIER and 87 mg/dL in ODYSSEY OUTCOMES). These values are substantially lower than those of most studies with surrogate outcomes that provided backing to regulatory filing (Table 1 ). This might account, at least in part, for the discrepancy between the impressive results of exploratory analyses of extension studies with evolocumab (OSLER 1‐2)16 and alirocumab (ODYSSEY LONG TERM)17 in patients with “true” hypercholesterolemia and those of the FOURIER and ODYSSEY OUTCOMES studies, respectively.
Table 1.
Summary of clinical studies with evolocumab and alirocumab showing that the only two RCTs with cardiovascular outcomes (FOURIER and ODYSSEY OUTCOMES) were carried out in patients with fairly well‐controlled LDL‐C at baseline
| Program | Trial name | N a | PubMed ID | Baseline LDL‐C (mg/dL)a | Aim of the study |
|---|---|---|---|---|---|
| ODYSSEY (Alirocumab) | Odyssey FH I | 322 | 26330422 | 144.7 ± 52.0
|
Lipid changes and safety |
| Odyssey FH II | 166 | 26330422 | 134.6 ± 41.2 | ||
| Odyssey High FH | 72 | 27618825 | 196.3 ± 57.9 | ||
| Odyssey Combo I | 209 | 26027630 | 94.8 ± 29.3 | ||
| Odyssey Combo II | 479 | 25687353 | 108 ± 35 | ||
| Odyssey Mono | 52 | 25037695 | 141 ± 27 | ||
| Odyssey Alternative | 126 | 26687696 | 191.1 ± 72.7 | ||
| Odyssey Options I | 57–47 | 26030325 | 103.9 ± 34.9–116.4 ± 37.4 | ||
| Odyssey Options II | 48–53 | 26638010 | 94.7 ± 33.6–115.2 ± 48.4a | ||
| Odyssey Choice I | 312c–37a | 27639753 | 115.4 ± 30.6c–154.7 ± 39.2a | ||
| Odyssey Choice II | 116–59 | 27625344 | 154.1 ± 42.4–167.5 ± 69.0a | ||
| Odyssey Long Term | 1,553 | 25773378 | 122.7 ± 42.6
|
Lipid changes, safety, CV outcomes | |
| Odyssey Outcomes | 9,462 | 30403574 | 87 (73–104) | ||
| PROFICIO (Evolocumab) | Rutherford‐2 | 110–110 | 25282519 | 155 ± 43–163 ± 50a
|
Lipid changes and safety |
| Taussig | 106 | 28215937 | 325.9 ± 134.4 | ||
| Tesla | 33 | 25282520 | 356 ± 135 | ||
| Yukawa | 205 | 24662398 | 143 ± 19 | ||
| Descartes | 145–126 | 24678979 | 94.6 ± 12.9–116.8 ± 35.3a | ||
| Laplace‐2 | 1,117 | 24825642 | 109.7 ± 42.3 | ||
| Mendel‐2 | 153–153 | 24691094 | 142 ± 22–144 ± 23a | ||
| Gauss‐2 | 103–102 | 24694531 | 192 ± 57–192 ± 61a | ||
| Gauss‐3 | 145 | 27039291 | 218.8 ± 73.1 | ||
| Osler 1‐2 | 2,976 | 25773607 | 120 (97–148)
|
Lipid changes, safety, CV outcomes | |
| Fourier | 13,784 | 28304224 | 92 (80–109) |
CV, cardiovascular; FH, familial hypercholesterolemia; LDL‐C, low‐density lipoprotein‐cholesterol; RCTs, randomized controlled trials.
Number and values in the active group/s. bDifferent groups or assigned doses of proprotein convertase subtilisin/kexin type 9 monoclonal antibody; In Osler 1‐2, Fourier, and Odyssey Outcomes LDL‐C is expressed as mean (interquartile range); cOn statins; dOff statins.
The results of a non‐prespecified post hoc subgroup analysis of ODYSSEY OUTCOMES give further support to the relevance of baseline LDL‐C in residual risk reduction with PCSK‐9 inhibitors: the number of patients needed to treat to prevent one primary event in 2.8 years (the median follow‐up) is 29 for those with LDL‐C ≥ 100 mg/dL (median LDL‐C 118 mg/dL) and 125 (4.3‐fold larger) for those with LDL‐C < 100 mg/dL. Regrettably, these data arise from subgroup analyses and therefore they do not provide definite conclusions.
In line with the former data is the comparison of the results of cardiovascular trials with bococizumab,18 another monoclonal antibody against PCSK‐9: a significant reduction of the primary cardiovascular event (hazard ratio: 0.79; 95% confidence interval (CI): 0.65−0.97; P = 0.02) was observed in SPIRE‐2 (mean baseline LDL‐C = 134 mg/dL, 7% with heterozygous FH) but not in SPIRE‐1 (mean baseline LDL‐C = 94 mg/dL, 1.7% with heterozygous FH).
To comprehensively address the impact of baseline LDL‐C levels on cardiovascular risk reduction, we performed a meta‐regression analysis of LDL‐C cardiovascular trials, stratifying the clinical response according to baseline LDL‐C levels above or below 100 mg/dL. The meta‐regression scatter plot was constructed using the same data on statin trials published by Silverman et al.19 in 2016 (eFigure S3 in the Supplemental Material of the quoted paper) with the addition of seven more recent studies with ezetimibe and PCSK‐9 inhibitors.13, 14, 15, 16, 17, 18 Data of individual trials included in the analysis are shown in Table S1 . Figure 2 clearly shows that the relative risk reduction per 1 mmol/L (1 mmol/L = 38.6 mg/dL) of LDL‐C reduction is −23% (95% CI: −32% to −13%; P = 0.0002) for trials with mean baseline LDL‐C levels ≥ 100 mg/dL and −8% (95% CI: −22% to +9%; P = 0.22) for those with mean baseline LDL‐C levels <100 mg/dL. The interaction was statistically significant (P = 0.015). These observations are in line with a recent meta‐analysis20 showing that more intensive—as compared with a less intensive—LDL‐C lowering produces a greater reduction in the risk of total and cardiovascular mortality in patients with baseline levels of LDL‐C above but not below 100 mg/dL. Therefore, although a certain benefit may still be obtained from LDL‐C lowering in high‐risk patients without “true” dyslipidemia to LDL‐C levels typical of healthy neonates (about 30–50 mg/dL), the return is dramatically lower than in patients with true dyslipidemia.
Figure 2.
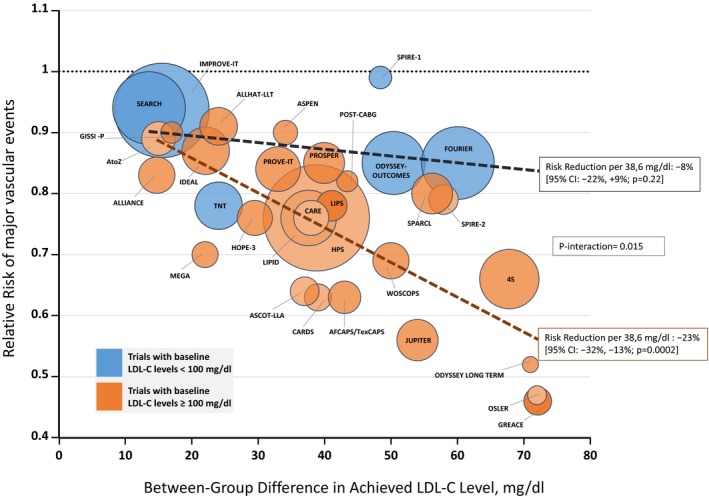
Relative risk (RR) of major vascular events plotted against the between‐group difference in achieved low‐density lipoprotein‐cholesterol (LDL‐C) level, stratified by baseline LDL‐C level in the experimental group < 100 mg/dL (blue bubbles) or ≥ 100 mg/dL (orange bubbles). The tags indicate the acronyms of the randomized LDL‐C cardiovascular trials included (25 with statins, 1 with statins plus ezetimibe, 6 with inhibitors of proprotein convertase subtilisin/kexin type 9). Meta‐regression analysis was performed by weighting each study according to the inverse of the variance of the RR. Size of the bubbles is proportional to the weight in the meta‐regression. The dashed lines represent the meta‐regression lines for each strata, and the slopes indicate the risk reduction per 38.6 mg/dL (1 mmol/L) of between‐group difference in achieved LDL‐C level. The difference between the two slopes was assessed by testing the interaction term in the meta‐regression. CI, confidence interval.
Looking behind, we can say that the large amount of data produced in the last 2 decades of prevailing explanatory LDL‐C cardiovascular trials substantiate the pioneering view of Grundy,21 based at that time just on subgroup analyses of 4S, CARE, and WOSCOPS, about the “….need to use clinical judgment as to whether to intensify therapy in patients whose LDL cholesterol levels are already nearing (these) goals,” which at that time were ≤ 130 mg/dL in high‐risk primary prevention and ≤ 100 mg/dL in secondary prevention.
TG trials
The role of blood TG in cardiovascular disease has been under scrutiny for decades. A main claim against the atherogenicity of high TG is that patients with extremely high TG levels (5,000–10,000 mg/dL or higher)—due to the so‐called familial hyperchylomicronemia—do not have a substantially increased risk of cardiovascular events.22 Paradoxically, several epidemiological studies (reviewed in refs. 22, 23) showed that mildly‐to‐moderately increased TG levels (200–800 mg/dL) are associated with coronary heart disease and, in some of these studies, also with ischemic stroke and total mortality. This association, however, was attenuated or even abrogated after adjustment for standard risk factors, casting doubts on whether mildly‐to‐moderately high TG levels are directly deleterious to arteries (i.e., a true risk factor) or merely an innocuous bystander or marker of other underlying pathogenic conditions or factors (e.g., diabetes, insulin resistance, low HDL‐C, high cholesterol in remnant lipoproteins, etc.). This uncertainty has been fueled by the inconsistency of the results of explanatory TG trials with fibrates. Concisely, compared with placebo, fibrates reduced major cardiovascular events in primary prevention (Helsinki Heart Study (HHS))24 and in a large secondary prevention trial with gemfibrozil (VA‐HIT trial)25 but not in another one with bezafibrate (Bezafibrate Infarction Prevention (BIP) study).26 In these studies, mean baseline TG values were 176, 160, and 145 mg/dL, respectively, and most patients did not have “true” hypertriglyceridemia. Yet, post hoc subgroup analyses of HHS and BIP26, 27 showed a much more remarkable risk reduction in patients with high baseline TGs (Table 2 ). Unfortunately, in the design of later trials with fibrates, this piece of information was overlooked. In FIELD28 and ACCORD LIPIDS29 indeed, a fibrate in addition to statins was not significantly superior to placebo in the whole cohorts (which had mean baseline triglycerides of 150 and 162 mg/dL, respectively), but, again, subgroup analyses showed a reduction of major events in patients with TGs >204 mg/dL at baseline29, 30 (Table 2 ). A meta‐analysis shows that fibrates reduce cardiovascular risk by 30% (95% CI: 19−40%; P < 0.0001) in patients with high TGs and only by 6% (95% CI: −2 to 13%; P = 0.13) in those without, with a significant heterogeneity between groups (P = 0.0002).31 Thus, fibrates only work in the patients under‐represented in the trials (Table 2 ) who might customarily have a fibrate prescribed to normalize TGs in clinical practice. Yet, because subgroup analyses and meta‐analyses are the best available experimental evidence, current guidelines assign treatment with fibrates in addition to statins as a low class of recommendation.8, 9
Table 2.
Summary of clinical trials with fibrates and cardiovascular outcomes showing that the treatment effect is markedly different in the entire cohorts and in subgroups of patients with true hypertriglyceridemia
| TRIAL | Drug | Primary end point: entire cohort (P value) | Lipid subgroup criterion | Primary end point: subgroup (P value) | Prevalence of the lipid subgroup in the whole cohort | Type of subgroup analysis | Heterogeneity in treatment effect |
|---|---|---|---|---|---|---|---|
| HHS | Gemfibrozil | −34% (0.02) |
TG > 200 mg/dL LDL‐C/HDL‐C > 5.0 |
−71% (0.005) | 7.3% | Post hoc | Not assessed |
| BIP | Bezafibrate | −7.3% (0.24) | TG ≥ 200 mg/dL | −39.5% (0.02) | 14.8% | Post hoc | Not assessed |
| FIELD | Fenofibrate | −11% (0.16) |
TG ≥ 204 mg/dL HDL‐C < 42 mg/dL |
−27% (0.005) | 20.6% | Prespecified | P = 0.053 for interaction |
| ACCORD | Fenofibrate | −8% (0.32) |
TG ≥ 204 mg/dL HDL‐C < 34 mg/dL |
−31% | 17.0% | Prespecified | P = 0.057 for interaction |
HDL‐C, high‐density lipoprotein‐cholesterol; LDL‐C, low‐density lipoprotein‐cholesterol; TG, triglyceride.
In the last years, the interest in TG was reignited by the results of Mendelian randomization studies, which strongly support a causal relationship between genes involved in TG metabolism and coronary heart disease.32 Conveniently, it seems that, in this period of renewed scientific attention to blood levels of TGs, more pragmatism is being adopted in the design of trials aimed to assess the efficacy of TG‐lowering treatments in reducing residual cardiovascular risk (see below REDUCE‐IT, STRENGTH, and PROMINENT).
HDL trials
Epidemiological studies consistently demonstrated that blood levels of HDL‐C and risk of coronary heart disease events are linearly and inversely correlated with HDL‐C values up to about 60 mg/dL.33, 34, 35 However, certain genetic variants associated with high concentrations of HDL‐C (e.g., in the CETP gene, in the hepatic lipase promoter and in the SCARB1 gene) have been associated with a high risk of cardiovascular disease,36, 37, 38 whereas other genetic variants associated with low concentrations of HDL‐C (e.g., in the apoA1 gene, in the ABCA1 gene, and in the LCAT gene) do not seem to convey an increased cardiovascular risk.39, 40, 41, 42 Further elements against the cardioprotective role of HDL‐C arise from the results of Mendelian randomization studies showing no evident link between genetic variants associated with HDL‐C levels and cardiovascular risk.32, 43, 44 Thus, the available data demonstrating a first criterion of causality (i.e., low HDL‐C correlates with increased risk) are not fully consistent. Nevertheless, low HDL‐C is the most common lipid abnormality found in patients with premature coronary heart disease,45 and there was strong expectation that increasing HDL‐C through therapy could reduce cardiovascular events. The compounds tested in HDL trials were nicotinic acid derivatives and CETP inhibitors (briefly “trapibs”). Although nicotinic acid had earlier demonstrated efficacy in reducing recurrent myocardial infarction in the CORONARY DRUG PROJECT,46 it was not superior to placebo in the statin‐treated patients of AIM‐HIGH47 and HPS2‐THRIVE.48 Yet, did the patients enrolled in these trials actually have a low HDL‐C? The available data indicate that most of them did not (Table 3 ).
Table 3.
Summary of cardiovascular trials with nicotinic acid derivatives or CETP inhibitors showing that most patients included in these trials had normal HDL‐C levels
| Study compound | Study name | Baseline HDL‐C (mg/dL)a |
|---|---|---|
| Nicotinic acid | ||
| Extended‐release niacin | AIM‐HIGH | 34.5 ± 5.6 |
| Extended‐release niacin + laropiprant | HPS‐2 THRIVE | 43.9 ± 11.2 |
| CETP inhibitors | ||
| Torcetrapib | ILLUMINATE | 48.6 ± 12.0 |
| Dalcetrapib | DAL‐OUTCOMES | 42.5 ± 11.7 |
| Evacetrapib | ACCELERATE | 45.3 ± 11.7 |
| Anacetrapib | REVEAL | 40 ± 10 |
CETP, cholesteryl ester transfer protein; HDL‐C, high‐density lipoprotein‐cholesterol.
Values in the active group.
CETP inhibitors induce a much greater HDL‐C raising effect than nicotinic acid, and early studies focusing on lipid changes in patients with low HDL‐C levels49 had nurtured great expectation. However, in the cardiovascular outcome trials ILLUMINATE (torcetrapib),50 DAL‐OUTCOMES (dalcetrapib),51 and ACCELERATE (evacetrapib),52 the trapibs performed not better, or even worse, than placebo. The last trapib trial, REVEAL (anacetrapib),53 showed a meager 9% relative reduction in the risk of events among patients receiving anacetrapib, and the producer decided not to submit applications for regulatory approval. It is worth noting that baseline HDL‐C was in the normal range in all these trials (Table 3 ). Thus, the short life of trapibs was closed without a single hard outcome trial in patients at additional risk due to low HDL‐C. Interestingly, a recent large epidemiological study54 demonstrates a U‐shaped relationship between HDL‐C and risk of total and cardiovascular mortality and cardiovascular events, with a flat low risk at HDL‐C between 40 and 80 mg/dL, a sharp rise below 40 mg/dL, and a gradual rise above 80 mg/dL. This might explain, at least in part, beyond the theoretical pros and cons of CETP inhibition and/or adverse effects of trapibs unrelated to lipid changes, the futility, or even harmfulness of these interventions.
Thus, although up‐to‐date knowledge from observational studies raises serious doubts about low HDL‐C as a causal factor in vascular disease and as a target for therapy, it is also fair to recognize that the second criterion of causality (i.e., reversing low HDL‐C reduces residual cardiovascular risk) has never been truly tested in the explanatory HDL trials performed so far.
Critical aspects of pragmatism needed in cardiovascular outcome lipid trials
Ford and Norrie55 recently stated: “Explanatory trials investigate a physiological hypothesis, whereas pragmatic trials inform a clinical or policy decision by providing evidence for adoption of the intervention into real‐world clinical practice.” The first aspect to estimate a trial's pragmatism is eligibility,56 which is assessed through the question: “To what extent are the participants in the trial similar to patients who would receive this intervention if it was part of usual care?” We believe that for the vast majority of the trials mentioned above, the most factual answer is to a minimal extent, if any.
Hopefully, pragmatism in trial design will get its place. Potential participants in pragmatic trials with emerging LDL‐C–lowering compounds are patients with LDL‐C levels plainly above the goals (i.e., with a clinically meaningful gap between the LDL‐C goal and the LDL‐C inclusion criteria) despite using the highest tolerated doses of consolidated therapy (e.g., statins and/or statins plus ezetimibe). This insufficient response may be due to very high baseline lipid levels (such as in some patients with FH), poor individual response,57 or intolerance to treatment, which affects about 10% of patients on statins58 and rarely patients on ezetimibe. Potential participants in pragmatic TG and HDL‐C trials are patients with TG and HDL‐C levels, respectively, frankly within the range associated with an increased risk of cardiovascular events: TG between 200 and 800 mg/dL for TG trials and HDL‐C ≤ 39 mg/dL (1 mmol/L) for HDL‐C trials. Of note, these HDL‐C values are not unusual, as they were observed in 16.2% of men and 3.7% of women in the general population of a recent large Danish epidemiological study.54 Analogously, we hope that cardiovascular trials aimed to evaluate the effect of forthcoming lipoprotein(a)‐lowering compounds59 will focus on patients at high cardiovascular risk with hyperlipoprotein(a) and not on patients generically at high risk.
Why did so many clinical outcome lipid trials not focus on patients with overtly abnormal lipid values?
One claim is the “proof of concept,” which investigates hypotheses, such as “the‐lower‐the‐better” (for LDL‐C or TGs) or “the‐higher‐the‐better” (for HDL‐C). This argument seems timely for LDL‐C but not for TGs or HDL‐C, where there is still a “black hole” about the impact of normalization of “true” abnormal levels on residual risk. Another possibility is the righteous hope of clinical researchers of reducing residual risk in a broad category of patients at high risk, independently of their lipid levels. In this case, even a small positive result allows claiming a favorable effect on the whole category, although many patients (and sometimes most) actually get a marginal benefit (see ODYSSEY OUTCOMES above). Conversely, a negative overall result may lead to consider that the intervention is outright futile, often leading to the withdrawal of the drug (see HDL trials above), although it might have been useful in the right patient. In other cases, positive results in subgroup analyses of negative explanatory trials (see FIELD and ACCORD LIPIDS above) provide backing to low‐class guideline recommendations, erratically followed in clinical practice. Last but not least, given the high cost of drug development, preclinical and clinical testing, and registration, pharmaceutical companies may be more interested in endorsing trials whose results (even quantitatively small) are potentially applicable to a market large enough to cover investments and yield a profit, rather than obtaining larger effects in a more specific but smaller group of treatment candidates.
Who should promote and perform pragmatic cardiovascular outcome lipid trials?
The purpose of healthcare stakeholders of evolving toward personalized evidence‐based medicine calls for pragmatism in future lipid trials, in order to inform physicians and patients about the real cost‐effectiveness of a treatment in certain candidates for the treatment. The design of some new studies suggests that things are hopefully starting to move in this direction.
REDUCE‐IT60 was aimed to evaluate the efficacy in reducing residual cardiovascular risk of a 4 g daily dose of eicosapentaenoic acid ethyl ester in patients at elevated cardiovascular risk on LDL‐C–lowering drugs with fairly well‐controlled LDL‐C levels (median 74 mg/dL) and baseline fasting TG levels between 150 and 500 mg/dL (median 216 mg/dL). Even though the effect of the therapy on TG levels was mild (18.3%; −39.0 mg per deciliter), the treatment produced, after 5 years, a 25% reduction of the primary cardiovascular end point. To what extent these effects are the result of TG lowering is not clear, as omega‐3 have many other putative cardioprotective pleiotropic effects. Yet, the trial pragmatically tested a coherent hypothesis in candidates suitable for the intervention in the real‐world clinical practice, responding to a frequent question of physicians: can we reduce residual risk in patients with moderate hypertriglyceridemia through omega‐3 fatty acids?
Propitiously, STRENGTH,61 another pragmatic randomized placebo‐controlled trial with omega‐3 fatty acids in statin‐treated patients at high risk of developing cardiovascular events, with well‐controlled LDL‐C, hypertriglyceridemia (≥ 180 to < 500 mg/dL), and low levels of HDL‐C (< 42 mg/dL in men or < 47 mg/dL in women) is ongoing, and the results are awaited by the end of 2019.
Another example of a pragmatic TG‐lowering trial in high‐risk patients is the ongoing PROMINENT trial,62 a randomized, double‐blind, placebo‐controlled cardiovascular outcome trial with the novel fibrate compound Pemafibrate. The PROMINENT trial plans to recruit high‐risk patients with diabetes with and without cardiovascular disease, and, for the first time in fibrate outcome trials, all the patients will have TGs >200 mg/dL and low HDL‐C despite best evidence‐based treatment, including high‐intensity statin therapy. The results of the PROMINENT trial are awaited by 2022.
Plausibly, the logical sequence of lipid trials with new compounds should recapitulate the history of those performed with statins: first, pragmatic studies in patients with definite dyslipidemia, then explanatory studies to assess usefulness in broader groups. Quoting once again Ford and Norrie55: “…who should pay for these [pragmatic] trials… Perhaps the best solution would be joint industry–governmental funding.” Definitely highly desirable, such a joint venture might be encouraged by regulatory changes intended to compensate the industry for taking on the cost and the business risk of pragmatic research.
Key messages
Performing only explanatory cardiovascular outcome lipid trials has important negative consequences: (i) some drugs are withdrawn prematurely without a specific assessment of efficacy in appropriate patients, (ii) physicians prescribe some drugs inconsistently and without certainty due to the low level of evidence‐based data, and (iii) the cost‐effectiveness of the treatment (number needed to treat (NNT), number needed to harm (NNH), etc.) in certain candidates for the medicine often remains indefinitely unknown. Pragmatic trials in individuals with “true” dyslipidemia are urged.
Funding
This work was funded by the Italian Ministry of Health, Ricerca Corrente, to Centro Cardiologico Monzino, IRCCS.
Conflict of Interest
The authors declared no competing interests for this work.
Supporting information
Figure S1. Mismatch between baseline LDL‐C levels at trial entry and range of lipid‐related high cardiovascular risk (red zone).
Figure S2. Mismatch between baseline triglyceride levels at trial entry and range of lipid‐related high cardiovascular risk (red zone).
Figure S3. Mismatch between baseline HDL‐C levels at trial entry and range of lipid‐related high cardiovascular risk (red zone).
Table S1. Data of cardiovascular LDL‐C trials included in Figure 2 (meta‐regression analysis) of the main manuscript.
Acknowledgments
The authors thank Dr Amitava Banerjee and Michela Palmieri for their valuable suggestions to refine English writing.
References
- 1. Sampson, U.K. , Fazio, S. & Linton, M.F. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr. Atheroscler. Rep. 14, 1–10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burgess, S. & Harshfield, E. Mendelian randomization to assess causal effects of blood lipids on coronary heart disease: lessons from the past and applications to the future. Curr. Opin. Endocrinol. Diabetes Obes. 23, 124–130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ference, B.A. et al Effect of long‐term exposure to lower low‐density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J. Am. Coll. Cardiol. 60, 2631–2639 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Martin, M.J. , Hulley, S.B. , Browner, W.S. , Kuller, L.H. & Wentworth, D. Serum cholesterol, blood pressure, and mortality: implications from a cohort of 361,662 men. Lancet 2, 933–936 (1986). [DOI] [PubMed] [Google Scholar]
- 5. Sniderman, A.D. , Tsimikas, S. & Fazio, S. The severe hypercholesterolemia phenotype: clinical diagnosis, management, and emerging therapies. J. Am. Coll. Cardiol. 63, 1935–1947 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Cuchel, M. et al Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 35, 2146–2157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wong, B. , Kruse, G. , Kutikova, L. , Ray, K.K. , Mata, P. & Bruckert, E. Cardiovascular disease risk associated with familial hypercholesterolemia: a systematic review of the literature. Clin. Ther. 38, 1696–1709 (2016). [DOI] [PubMed] [Google Scholar]
- 8. Catapano, A.L. et al 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur. Heart J. 37, 2999–3058 (2016). [DOI] [PubMed] [Google Scholar]
- 9. Piepoli, M.F. et al 2016 European Guidelines on cardiovascular disease prevention in clinical practice: the Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 37, 2315–2381 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 344, 1383–1389 (1994). [PubMed] [Google Scholar]
- 11. Sacks, F.M. et al The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med. 335, 1001–1009 (1996). [DOI] [PubMed] [Google Scholar]
- 12. Cannon, C.P. , Steinberg, B.A. , Murphy, S.A. , Mega, J.L. & Braunwald, E. Meta‐analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J. Am. Coll. Cardiol. 48, 438–445 (2006). [DOI] [PubMed] [Google Scholar]
- 13. Cannon, C.P. et al Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 372, 2387–2397 (2015). [DOI] [PubMed] [Google Scholar]
- 14. Sabatine, M.S. et al Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 376, 1713–1722 (2017). [DOI] [PubMed] [Google Scholar]
- 15. Schwartz, G.G. et al Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 379, 2097–2107 (2018). [DOI] [PubMed] [Google Scholar]
- 16. Sabatine, M.S. et al Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 372, 1500–1509 (2015). [DOI] [PubMed] [Google Scholar]
- 17. Robinson, J.G. et al Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 372, 1489–1499 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Ridker, P.M. et al Cardiovascular efficacy and safety of bococizumab in high‐risk patients. N. Engl. J. Med. 376, 1527–1539 (2017). [DOI] [PubMed] [Google Scholar]
- 19. Silverman, M.G. et al Association between lowering LDL‐C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta‐analysis. JAMA 316, 1289–1297 (2016). [DOI] [PubMed] [Google Scholar]
- 20. Navarese, E.P. et al Association between baseline LDL‐C level and total and cardiovascular mortality after LDL‐C lowering: a systematic review and meta‐analysis. JAMA 319, 1566–1579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grundy, S.M. Statin trials and goals of cholesterol‐lowering therapy. Circulation 97, 1436–1439 (1998). [DOI] [PubMed] [Google Scholar]
- 22. Nordestgaard, B.G. & Varbo, A. Triglycerides and cardiovascular disease. Lancet 384, 626–635 (2014). [DOI] [PubMed] [Google Scholar]
- 23. Miller, M. et al Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 123, 2292–2333 (2011). [DOI] [PubMed] [Google Scholar]
- 24. Frick, M.H. et al Helsinki Heart Study: primary‐prevention trial with gemfibrozil in middle‐aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N. Engl. J. Med. 317, 1237–1245 (1987). [DOI] [PubMed] [Google Scholar]
- 25. Rubins, H.B. et al Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high‐density lipoprotein cholesterol. Veterans Affairs High‐Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 341, 410–418 (1999). [DOI] [PubMed] [Google Scholar]
- 26. Bezafibrate Infarction Prevention (BIP) . Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation 102, 21–27 (2000). [DOI] [PubMed] [Google Scholar]
- 27. Manninen, V. et al Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation 85, 37–45 (1992). [DOI] [PubMed] [Google Scholar]
- 28. Keech, A. et al Effects of long‐term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 366, 1849–1861 (2005). [DOI] [PubMed] [Google Scholar]
- 29. ACCORD Study Group et al Effects of combination lipid therapy in type 2 diabetes mellitus. N. Engl. J. Med. 362, 1563–1574 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scott, R. et al Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care 32, 493–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruckert, E. , Labreuche, J. , Deplanque, D. , Touboul, P.J. & Amarenco, P. Fibrates effect on cardiovascular risk is greater in patients with high triglyceride levels or atherogenic dyslipidemia profile: a systematic review and meta‐analysis. J. Cardiovasc. Pharmacol. 57, 267–272 (2011). [DOI] [PubMed] [Google Scholar]
- 32. Holmes, M.V. et al Mendelian randomization of blood lipids for coronary heart disease. Eur. Heart J. 36, 539–550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Castelli, W.P. et al HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation 55, 767–772 (1977). [DOI] [PubMed] [Google Scholar]
- 34. Gordon, D.J. et al High‐density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 79, 8–15 (1989). [DOI] [PubMed] [Google Scholar]
- 35. Wilson, P.W. , Abbott, R.D. & Castelli, W.P. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis 8, 737–741 (1988). [DOI] [PubMed] [Google Scholar]
- 36. Agerholm‐Larsen, B. , Nordestgaard, B.G. , Steffensen, R. , Jensen, G. & Tybjaerg‐Hansen, A. Elevated HDL cholesterol is a risk factor for ischemic heart disease in white women when caused by a common mutation in the cholesteryl ester transfer protein gene. Circulation 101, 1907–1912 (2000). [DOI] [PubMed] [Google Scholar]
- 37. Andersen, R.V. , Wittrup, H.H. , Tybjaerg‐Hansen, A. , Steffensen, R. , Schnohr, P. & Nordestgaard, B.G. Hepatic lipase mutations, elevated high‐density lipoprotein cholesterol, and increased risk of ischemic heart disease: the Copenhagen City Heart Study. J. Am. Coll. Cardiol. 41, 1972–1982 (2003). [DOI] [PubMed] [Google Scholar]
- 38. Zanoni, P. et al Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 351, 1166–1171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Calabresi, L. et al Functional lecithin: cholesterol acyltransferase is not required for efficient atheroprotection in humans. Circulation 120, 628–635 (2009). [DOI] [PubMed] [Google Scholar]
- 40. Franceschini, G. , Sirtori, C.R. , Capurso, A. 2nd , Weisgraber, K.H. & Mahley, R.W. A‐IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J. Clin. Invest. 66, 892–900 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Frikke‐Schmidt, R. et al Association of loss‐of‐function mutations in the ABCA1 gene with high‐density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 299, 2524–2532 (2008). [DOI] [PubMed] [Google Scholar]
- 42. Kuivenhoven, J.A. , Pritchard, H. , Hill, J. , Frohlich, J. , Assmann, G. & Kastelein, J. The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J. Lipid Res. 38, 191–205 (1997). [PubMed] [Google Scholar]
- 43. Johannsen, T.H. et al Hepatic lipase, genetically elevated high‐density lipoprotein, and risk of ischemic cardiovascular disease. J. Clin. Endocrinol. Metab. 94, 1264–1273 (2009). [DOI] [PubMed] [Google Scholar]
- 44. Voight, B.F. et al Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet 380, 572–580 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Genest, J.J. Jr et al Familial lipoprotein disorders in patients with premature coronary artery disease. Circulation 85, 2025–2033 (1992). [DOI] [PubMed] [Google Scholar]
- 46. Clofibrate and niacin in coronary heart disease. JAMA 231, 360–381 (1975). [PubMed] [Google Scholar]
- 47. AIM‐HIGH Investigators et al Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 365, 2255–2267 (2011). [DOI] [PubMed] [Google Scholar]
- 48. HPS2‐THRIVE Collaborative Group et al Effects of extended‐release niacin with laropiprant in high‐risk patients. N. Engl. J. Med. 371, 203–212 (2014). [DOI] [PubMed] [Google Scholar]
- 49. Brousseau, M.E. et al Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N. Engl. J. Med. 350, 1505–1515 (2004). [DOI] [PubMed] [Google Scholar]
- 50. Barter, P.J. et al Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 357, 2109–2122 (2007). [DOI] [PubMed] [Google Scholar]
- 51. Schwartz, G.G. et al Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 367, 2089–2099 (2012). [DOI] [PubMed] [Google Scholar]
- 52. Lincoff, A.M. et al Evacetrapib and cardiovascular outcomes in high‐risk vascular disease. N. Engl. J. Med. 376, 1933–1942 (2017). [DOI] [PubMed] [Google Scholar]
- 53. HPS3/TIMI55‐REVEAL Collaborative Group et al Effects of anacetrapib in patients with atherosclerotic vascular disease. N. Engl. J. Med. 377, 1217–1227 (2017). [DOI] [PubMed] [Google Scholar]
- 54. Madsen, C.M. , Varbo, A. & Nordestgaard, B.G. Extreme high high‐density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: two prospective cohort studies. Eur. Heart J. 38, 2478–2486 (2017). [DOI] [PubMed] [Google Scholar]
- 55. Ford, I. & Norrie, J. Pragmatic trials. N. Engl. J. Med. 375, 454–463 (2016). [DOI] [PubMed] [Google Scholar]
- 56. Loudon, K. , Treweek, S. , Sullivan, F. , Donnan, P. , Thorpe, K.E. & Zwarenstein, M. The PRECIS‐2 tool: designing trials that are fit for purpose. BMJ 350, h2147 (2015). [DOI] [PubMed] [Google Scholar]
- 57. Karlson, B.W. , Wiklund, O. , Palmer, M.K. , Nicholls, S.J. , Lundman, P. & Barter, P.J. Variability of low‐density lipoprotein cholesterol response with different doses of atorvastatin, rosuvastatin, and simvastatin: results from VOYAGER. Eur. Heart J. Cardiovasc. Pharmacother. 2, 212–217 (2016). [DOI] [PubMed] [Google Scholar]
- 58. Newman, C.B. et al Statin safety and associated adverse events: a scientific statement from the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 39, e38–e81 (2019). [DOI] [PubMed] [Google Scholar]
- 59. Viney, N.J. et al Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double‐blind, placebo‐controlled, dose‐ranging trials. Lancet 388, 2239–2253 (2016). [DOI] [PubMed] [Google Scholar]
- 60. Bhatt, D.L. et al Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N. Engl. J. Med. 380, 11–22 (2019). [DOI] [PubMed] [Google Scholar]
- 61. Nicholls, S.J. et al Assessment of omega‐3 carboxylic acids in statin‐treated patients with high levels of triglycerides and low levels of high‐density lipoprotein cholesterol: rationale and design of the STRENGTH trial. Clin. Cardiol. 41, 1281–1288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fruchart, J.C. Pemafibrate (K‐877), a novel selective peroxisome proliferator‐activated receptor alpha modulator for management of atherogenic dyslipidaemia. Cardiovasc. Diabetol. 16, 124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mismatch between baseline LDL‐C levels at trial entry and range of lipid‐related high cardiovascular risk (red zone).
Figure S2. Mismatch between baseline triglyceride levels at trial entry and range of lipid‐related high cardiovascular risk (red zone).
Figure S3. Mismatch between baseline HDL‐C levels at trial entry and range of lipid‐related high cardiovascular risk (red zone).
Table S1. Data of cardiovascular LDL‐C trials included in Figure 2 (meta‐regression analysis) of the main manuscript.