

Abstract
The rapidly‐advancing field of pharmaceutical and clinical research calls for systematic, molecular‐level characterization of complex biological systems. To this end, quantitative proteomics represents a powerful tool but an optimal solution for reliable large‐cohort proteomics analysis, as frequently involved in pharmaceutical/clinical investigations, is urgently needed. Large‐cohort analysis remains challenging owing to the deteriorating quantitative quality and snowballing missing data and false‐positive discovery of altered proteins when sample size increases. MS1 ion current‐based methods, which have become an important class of label‐free quantification techniques during the past decade, show considerable potential to achieve reproducible protein measurements in large cohorts with high quantitative accuracy/precision. Nonetheless, in order to fully unleash this potential, several critical prerequisites should be met. Here we provide an overview of the rationale of MS1‐based strategies and then important considerations for experimental and data processing techniques, with the emphasis on (i) efficient and reproducible sample preparation and LC separation; (ii) sensitive, selective and high‐resolution MS detection; iii)accurate chromatographic alignment; (iv) sensitive and selective generation of quantitative features; and (v) optimal post‐feature‐generation data quality control. Prominent technical developments in these aspects are discussed. Finally, we reviewed applications of MS1‐based strategy in disease mechanism studies, biomarker discovery, and pharmaceutical investigations.
Keywords: MS1 quantification, ion current‐based proteomics, LC‐MS, reproducible protein measurement, large cohorts
Abbreviations
- AMRT
accurate mass retention time
- AUC
area under curve
- BAL
bronchoalveolar lavage
- COW
correlation‐optimized warping
- CPM
continuous profile model
- CLL
chronic lymphocytic leukemia
- DTW
dynamic time warping
- DTT
dithiothreitol
- DIA
data‐independent acquisition
- DDA
data‐dependent acquisition
- DICE
direct ion current extraction
- FFId
feature finder Identification
- FC
fold changes
- FDR
false discovery rate
- FASP
filter‐assisted sample preparation
- HGSOC
high‐grade serous ovarian cancer
- iTRAQ
isobaric tagging for relative and absolute quantification
- iBAQ
intensity‐based absolute quantification
- iST
in‐StageTip
- LC‐MS
liquid chromatography‐mass spectrometry
- LIMMA
linear models for microarray data
- MPs
membrane proteins
- PTW
parametric time warping
- PCT
pressure cycling technology
- PBMC
peripheral blood mononuclear cells
- PLOT
porous layer open tubular
- PTW
parametric time warping
- PCA
principal component analysis
- RT
retention time
- SILAC
stable isotope labelling by amino acids in cell culture
- SpC
Spectral counting
- SWATH
sequential window acquisition of all theoretical fragment‐ion spectra
- SDS
sodium dodecyl sulfate
- SDC
sodium deoxycholate
- SPE
solid‐phase extraction
- SEPOD
surfactant cocktail‐aided extraction/precipitation/on‐pellet digestion
- SCAD
surfactant and chaotropic agent assisted sequential extraction/on pellet digestion
- SC
surfactant cocktail
- SAM
significance analysis of microarray
- TPP
trans‐proteomic pipeline
- TDA
target‐decoy approach
- TIC
total ion current
- TMT
tandem mass tag
- TBI
traumatic brain injuries
- UHF
ultra‐high‐field
INTRODUCTION
Quantitative proteomics based on liquid chromatography‐mass spectrometry (LC‐MS) represents a powerful tool for biomedical research. For pharmaceutical and clinical investigations, it is often necessary to analyze large numbers of biological samples (e.g., tissues, body fluids, or cellular conditions) to warrant quantitative reliability and statistical power, and more importantly, to minimize the false‐positive discovery of altered proteins arising from the typically high inter‐individual variability in these studies. Nonetheless, large‐scale proteomics quantification has been quite challenging owing to the technical difficulties in achieving high‐quality quantification of large cohorts, including but not limited to suboptimal quantitative accuracy, precision, and robustness when measuring proteins in many samples, as well as high missing data and false‐positive discoveries (Domon & Aebersold, 2010; Webb‐Robertson et al., 2015). Isotope‐labeling‐based methods such as stable isotope labeling by amino acids in cell culture (SILAC) (Ong et al., 2002), isobaric tagging for relative and absolute quantification (iTRAQ) (Ross et al., 2004), stable isotope dimethyl labeling (Boersema et al., 2009), tandem mass tag (TMT) (Thompson et al., 2003), rely on incorporation of isotopic tags (either chemically or biologically) for relative quantification. Despite recent advances that have substantially improved quantitative depth, accuracy, and precision (Mallick & Kuster, 2010; McAlister et al., 2014; Sonnett et al., 2018), labeling methods fall short in achieving robust large‐cohort analysis, as the replicate capacity of labeling methods is limited by the number of isotopic tags (usually ≤10 for commercially‐available reagents) (Wasinger et al., 2013). Moreover, selectivity for low‐abundant peptides quantification might be compromised by some labeling methods because of co‐isolation and co‐fragmentation of interfering ions (Ow et al., 2009; Erickson et al., 2017). By comparison, label‐free quantification methods are theoretically unlimited in sample numbers, and have more flexible samples preparation options at lower costs, thus appearing to be a logical choice for analyzing large biological cohorts (Higgs et al., 2008; Merl et al., 2012). Nevertheless, achieving reliable, high‐quality qualification of many biological samples (e.g., ≥20) is also challenging for label‐free strategies (Cox et al., 2014; Shen et al., 2015). On one hand, it is difficult to attain high quantitative accuracy and precision when analyzing a large cohort of samples by label‐free methods, largely because the absence of an internal standard to correct quantitative variations arising from sample preparation and analytical process. Such technical variations could lead to the inaccurate measurement especially for low‐abundance proteins, as well as high false‐positives in discovering altered‐proteins (or biomarkers in certain contexts) (Nahnsen et al., 2013). To alleviate this issue, highly robust, reproducible, and well‐controlled experimental procedure for sample preparation, LC separation, and MS analysis across large cohorts is critical. On the other hand, missing data remains a prominent issue for both labeling and label‐free methods even though the recent advancements in informatics and LC‐MS instruments have greatly enhanced the proteome coverage, sensitivity and selectivity of quantitative proteomics. This problem becomes much more pronounced when the number of samples increases (Old et al., 2005; Zhang et al., 2009). The high missing data critically compromises the quality of quantification, and may lead to incorrect biological interpretation owing to the suboptimal characterization of biological functions, pathways, and networks (Domon & Aebersold, 2010). One primary reason of missing data is the use of data‐dependent acquisition (DDA) in the quantitative process (Karpievitch et al., 2012). In DDA, a survey scan of precursors is performed followed by sequential MS2 events based on the observed precursors, where the most intensive precursors are usually prioritized (Mann et al., 2001; Xie et al., 2011). Spectral counting (SpC) and other DDA‐MS2‐based methods (e.g., SpC‐Normalized Spectral Abundance Factor (NSAF) (Paoletti et al., 2006), Exponentially Modified Protein Abundance Index (emPAI) (Ishihama et al., 2005), MS2 Total‐Ion‐Current (TIC) (Tu et al., 2014b), normalized Spectral Index (SIn) (Griffin et al., 2010)), constitute a prevalently‐employed type of approach which measure a protein based on the total number of tandem mass spectra matching to peptides of the protein (Paoletti et al., 2006). The stochastic nature of DDA in precursor selection among different runs leads to considerable under‐sampling of low‐abundance, regulatory proteins (Zhou et al., 2012b; Geib et al., 2016). Furthermore, dynamic exclusion which is devised to improve the depth of identification, substantially decreases the reproducibility of MS2 spectra acquisition among runs and thereby increasing the stochasticity and missing data of MS2‐based quantification. The acquisition of MS2 spectra usually occurs outside the elution peak apex, which may compromise the sensitivity and quality of MS2 spectra (Michalski et al., 2011). Because of these issues, MS2‐based methods show low consistency in quantification, especially for low abundance ones; for example, as high as 20–50% identified proteins with missing quantitative values in 6–20 samples even higher when sample size increases (Bruderer et al., 2015; Zhang et al., 2016b). To improve the reproducibility of protein measurement across a large number of biological replicates, the MS2‐based “data‐independent acquisition (DIA)” was developed, and the most prominent example is SWATH (sequential window acquisition of all theoretical fragment‐ion spectra), which triggers MS2 scans in a window‐based, independent and unbiased manner, and therefore alleviates the missing data to <10% at protein level for relatively large cohorts (Geib et al., 2016; Hu et al., 2016 Collins et al., 2017). Although MS2‐DIA represents an enormous advance in reproducible protein measurement, some limitations are also noted: (i) as DIA uses multiplexed fragmentation of many precursors, it may be difficult to interpret these MS2 spectra containing multiple co‐fragmented precursors while maintaining low false‐positives and (ii) the depth of identification using spectral library matching is often limited (Rost et al., 2014). More recently, a number of new pipelines were developed to address these issues, such as PECAN (Ting et al., 2017), DIA‐Umpire (Tsou et al., 2015), and DirectDIA in Spectronaut™ Pulsar (Bruderer et al., 2016). These methods still suffer from the above problems intrinsic to MS2‐DIA, and their performance in large‐cohort analysis remains to be comprehensively evaluated.
Recently, MS1‐based methods showed considerable promise in high‐quality quantification of large cohorts. In this review, we will introduce the rationale and technicality of MS1‐based quantification, and discuss its potential for large‐cohort analysis, important considerations (e.g., experimental procedures, MS resolution, data processing strategies, etc.) affecting the quantitative data quality, and applications in molecular mechanism exploration of diseases, biomarker discovery, and drug discovery/therapeutics.
RATIONALE OF MS1‐BASED QUANTIFICATION AND ITS HIGH PROMISE IN LARGE‐COHORT ANALYSIS
A generic scheme for MS1‐based quantification is shown in Figure 1. As with all label‐free methods, samples are analyzed sequentially by LC‐MS. The MS is usually operated in scan cycles each containing one high‐resolution MS1 full scan followed by dependent fragmentation events of precursors. The high‐resolution precursor (i.e., MS1) ion current peaks are extracted as quantitative features (Shen et al., 2017a) while the accompanying MS2‐DDA is often utilized merely to assign peptide ID to quantitative features but not involved in determining quantitative values.
Figure 1.
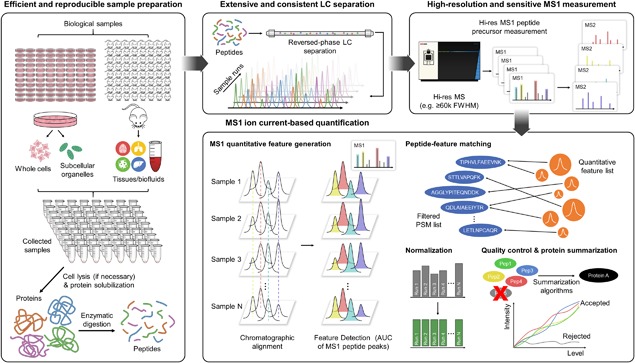
The general workflow for MS1 ion current‐based quantitative strategy.
The MS1‐based method exhibits remarkable potential for large‐cohort analysis for several reasons. First, MS1 quantification is performed in a strictly MS2‐independent manner, which opens the possibility for reproducible protein measurement among many samples. Additionally, this approach allowed inference of peptide ID across all sample runs, which is tremendously useful for reliable and consistent quantification of low‐abundance species. For example, even if a peptide was identified successfully only once by MS2 in the entire dataset, it could be successfully quantified across all biological samples (i.e., without missing data) as long as well‐defined MS1 ion current peaks of this peptide are acquired in all samples. An illustration of this point is shown Figure 2, where quantification of a low‐abundance peptide by a DDA method (spectra count) exhibited severe missing data (in 10 out of 12 samples) while quantification by MS1‐based method is missing‐data‐free with ID inferred from the few runs with successful identification. Therefore, MS1 strategies can be used to substantially alleviate the missing data problem that plagues DDA‐based proteomics quantitation in large sample sets.
Figure 2.
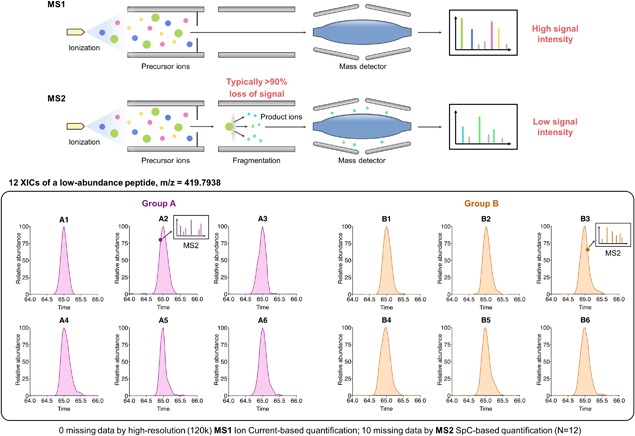
Comparison of MS1 and MS2‐based method. Upper: an illustration showing MS1 based methods resulted in much higher signal intensity of a peptide comparing to MS2‐based methods. Therefore, MS1‐based methods can achieve high sensitivity if a high selectivity is realized, for example, by high MS resolution. Lower: an example for quantifying a low‐abundance protein in human tissue samples (group A vs. B, n = 6 per group). MS1 ion current‐based method was able to quantify the protein in all samples without missing data while MS2 spectral counting (SpC) method is not useful owing to the very high missing data, that is, only two MS2 identifications in all runs.
Second, MS1‐based methods have the potential for highly sensitive and selective protein quantification, which permits both in‐depth proteomics analysis and high‐quality quantification of low‐abundance proteins in large‐cohort analysis. This feature greatly reduces the need for sample fractionation prior to LC‐MS analysis, as is often used in identification experiments to enhance the depth of proteomic analysis but not practical for analyzing a large sample set (Pernemalm et al., 2009; Zhang et al., 2011; Choi, 2012). Therefore, the MS1‐based strategy can employ extensive one‐dimensional separation (e.g., using long columns with small particles) to substitute the need of sample fractionation and to achieve extensive separation, enabling a practical, high‐throughput large‐cohort analysis. Moreover, comparing to MS2 product ions, MS1 signal intensity of a peptide is significantly higher (often by >10‐fold) as the fragmentation process can markedly decrease the signal strength (Qu & Straubinger, 2005). However, it has long been well recognized that MS2‐based methods (e.g., SRM, product ion scan, PRM, etc. (Ronsein et al., 2015; Meyer & Schilling, 2017)) almost always achieve higher sensitivity than MS1 approaches, because of its much higher selectivity and thereby lower chemical noises, which markedly improves signal‐to‐noise ratio despite the lower signal strength. Consequently, if MS1‐based strategy could achieve high selectivity with substantially lowered chemical noises, then a highly sensitive analysis can be attained by taking advantage of the high MS1 signal intensity. This rationale is illustrated in Figure 2. The selectivity for MS1‐based methods can be achieved by (i) sufficient chromatographic separation and (ii) high‐resolution MS1 detection AND narrow m/z window for XIC extraction. Detailed discussions are in the following sections. As exemplified in Figure 3, the increase of resolutions resulted in drastically improved selectivity and S/N when analyzing a highly complex proteomics sample.
Figure 3.
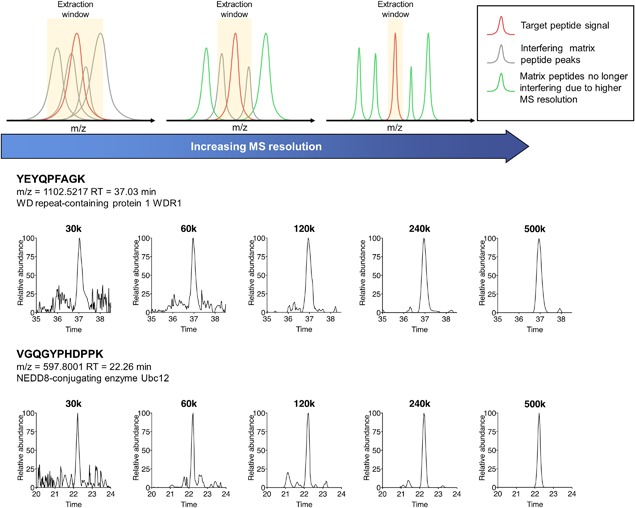
High‐resolution MS measurement of peptide precursor substantially improves selectivity and therefore sensitivity. Upper: the effect of MS resolution on the selectivity for MS1‐based quantification, illustrated via simulated MS spectra. Lower: examples showing higher MS resolution drastically lowered chemical noises for MS1‐based analysis of low‐abundance protein in tissue samples.
IMPORTANT CONSIDERATIONS FOR MS1‐BASED QUANTIFICATION IN LARGE COHORTS
To fully realize the potential of MS1‐based quantification in large‐cohort analysis with high quantitative accuracy, precision, low missing‐data, and false‐positives, it is essential to meet some critical requirements in terms of experimental strategies, data processing methods as well as stringent control of false‐positives. These requirements and related techniques are reviewed in this section.
A. Experimental Strategies
Experimental procedures should enable in‐depth proteomics analysis (i.e., quantify as many proteins as possible), as well as reliable, consistent quantification in large‐cohorts, especially for low‐abundance proteins. To achieve this, highly robust, reproducible, and well‐controlled experimental strategies including (i) efficient and reproducible sample preparation across large cohorts; (ii) extensive and consistent LC separation in many samples; and (iii) high‐resolution, sensitive MS1 measurements are all essential to warrant high‐quality MS1‐based quantification. Nonetheless, these prerequisites have long been underappreciated while most efforts have been focused on the informatics approaches to correct bias and variability, which is difficult to achieve in the event of large errors and variations rooting from suboptimal experimental practices (Shen et al., 2017a).
1. Efficient and Reproducible Sample Preparation Across Large Cohorts
Efficient and well‐controlled sample preparation is the key to successful large‐cohort proteomics analysis. Specifically, the sample preparation strategy should fulfill the following important requirements across a large sample set: (a) exhaustive and consistent protein extraction; (b) robust and reproducible cleanup of detrimental non‐protein matrix components which may potentially undermine digestion and LC‐MS analysis; (c) extensive protein denaturation to ensure efficient and reproducible proteolytic cleavage; (d) reasonable throughput for large‐cohort analysis. These requirements also universally apply to all quantitative proteomics techniques.
Protein extraction profoundly affects both proteomic coverage and quality of quantification. Extraction buffers with denaturing agents such as surfactants coupled with mechanical disruptions (e.g., sonication, mechanical homogenization, French pressing, pressure cycling technology (PCT) (Olszowy et al., 2013)) were shown to achieve effective protein extraction (Raynie, 2010). It is important to ensure high and consistent protein yields for large‐cohort analysis, especially for membrane proteins (MPs), which consist a substantial fraction of the total proteome and are usually more versatile and critical in biological functions (Savas et al., 2011). However, extraction of MPs remains challenging because of their heterogeneous and hydrophobic characteristics (Duan et al., 2009). Various detergents are often utilized to solubilize MPs and to avoid the formation of hydrophobic aggregates when the MPs are extracted from the lipid bilayer. Two commonly used surfactants are sodium dodecyl sulfate (SDS) and sodium deoxycholate (SDC). SDC has shown the ability to improve protein solubility while retaining trypsin digestion efficiency even at high concentrations (Lin et al., 2013, 2008), for example, 77.4% trypsin activity can be retained with 10% SDC (Lin et al., 2008; Masuda et al., 2008). SDC can be removed via acidification or ethyl acetate phase transfer before MS analysis (Lin et al., 2010; Masuda et al., 2008). SDS is another common surfactant that performs well in solubilizing MPs, yet it compromises enzyme activity and therefore should be removed prior to digestion. One popular method utilizing SDS is Filter‐Assisted Sample Preparation (FASP) (Wisniewski et al., 2009), which dissolves the proteins in SDT buffer (contains SDS and dithiothreitol (DTT)) followed by a molecular weight‐based cutoff centrifugal filter or spin plate to effectively remove small‐molecule components (e.g., SDS, salts, lipids, etc.), while proteins are retained and then digested on the filter unit (Coleman et al., 2017). Consequently, FASP is quite versatile, for example, can be applied to process samples extracted with high concentrations of compatible surfactants (e.g., 4% SDS). One reported drawback is irreproducible and low peptide yields likely arising from in‐filter adsorption (Choksawangkarn et al., 2012), which may considerably compromise quantitative quality in large sample cohorts. Furthermore, the capacity of sample loading on the filter is limited. Recently, a number of modified FASP procedures have been reported. For instance, an enhanced FASP (eFASP) strategy (Erde et al., 2014) was found to improve proteome coverage and sample recovery by adding 0.2% deoxycholic acid (DCA) in digestion buffer, though a later study observed eFASP showed no significant differences over the original procedure other than slightly more recovery of basic peptides (Nel et al., 2015); the multi‐enzyme digestion FASP (MED‐FASP) through consecutive use of LysC and trypsin in digestion showed significantly more identified proteins and phosphorylation sites with improved sequence coverage than FASP (Wisniewski & Mann, 2012; Wisniewski, 2016); a high‐throughput FASP (Potriquet et al., 2017) using 96‐well plate with polyethersulfone molecular weight cutoff membrane instead of the cellulose membrane was found to enable efficient, high‐throughput processing of protein samples (Potriquet et al., 2017). To overcome sample loss via filter‐adsorption which is an intrinsic issue of FASP, a “single vessel” in‐StageTip (iST) approach was introduced. The procedure performs all steps (e.g., protein extraction, digestion, cleanup) in a vial containing a C18 disk, which serves as a barrier for macromolecules, and enables sample cleanup using solid‐phase extraction (SPE) (Kulak et al., 2014). Although representing a remarkable advancement, the method falls short in the limited use of reagents, for example, SDS and other surfactants cannot be utilized as these cannot be removed by the C18 material, which limits its application in proteomic studies (Sielaff et al., 2017). Another single‐tube sample preparation approach is the single‐pot solid‐phase‐enhanced sample preparation (SP3). In SP3, surface‐functionalized (e.g., carboxylate‐coated) paramagnetic beads are used to trap proteins in the hydrophilic layers around the beads using increased organic composition with properly buffered pH. The contaminants and detergents can be removed by washing with different organic solvents (e.g., ethanol, acetonitrile), while the bound proteins can be eluted from the beads with an aqueous solution. The applicability of SP3 for sensitive proteome analysis has been demonstrated by the identifying >15,000 unique peptides from as little as 1000 HeLa cells (Hughes et al., 2014). In a recent study, both SP3 and iST were found to have provided higher proteome coverage in the low sample amount (<10 µg) than FASP, and the reproducibility of SP3 was higher than iST (Sielaff et al., 2017). The performance of these device‐dependent single‐vessel approaches on large‐cohort samples has yet been demonstrated.
The in‐solution digestion is an alternative method of single‐vessel sample preparation performing both protein extraction and digestion in single tube, which has been popular owing to the minimized sample loss and variability introduced by sample transfer/manipulation. However, strong denaturing agents such as surfactants are often not permitted in the protocol and therefore urea is most often employed as the only denaturant, which results in suboptimal extraction efficiency; moreover, matrix components (e.g., salts, phospholipids, fatty acids, DNA/RNA, etc.) remaining in the solution may severely compromise digestion efficiency, reverse‐phase LC separation and MS detection (Zhou et al., 2012a). Thus, extra cleanup procedures are often necessary, though at the cost of introducing considerable quantitative variability and biases. To address these issues, a number of acid‐cleavable or degradable surfactants (i.e., MS‐compatible surfactants) have been developed such as PPS Silent Surfactant (Norris et al., 2003), RapiGest SF and ProteaseMAX™, which help to solubilize and denature proteins with minimized negative impact on both digestion and LC‐MS analysis, as shown in both in‐gel and in‐solution digestion applications (Norris et al., 2003; Chen et al., 2007; Winter & Steen, 2011). Though the above studies showed these surfactants allowing improved protein extraction and in‐solution digestion, a recent report found that a traditional, less expensive surfactant, SDC, enabled superior proteomics coverage especially for membrane proteins compared with RapidGest SF in Saccharomyces cerevisiae (Moore et al., 2016).
Another type of approaches compatible with strong surfactant extraction/denaturation is precipitation/on‐pellet‐digestion, which was firstly described in 2009 (Duan et al., 2009). After detergent extraction, the detergent as well as most matrix components are removed via an organic solvent precipitation; without re‐suspension of the pellet, digestion buffer containing enzymes is added followed by incubation with agitation, which rapidly dissolves the pellet by continuously cleaving pelleted proteins into solution. A number of similar procedures were later reported for global proteomics quantification (Ouyang et al., 2012; Gong et al., 2015; Ma et al., 2018), as well as reproducible, robust, and rapid sample preparation target protein quantification (An et al., 2015). The most recently developed methods were surfactant cocktail‐aided extraction/precipitation/on‐pellet digestion (SEPOD), and surfactant and chaotropic agent assisted sequential extraction/on pellet digestion (SCAD). SEPOD utilizes a high‐concentration surfactant cocktail (SC) buffer containing multiple non‐ionic/anionic surfactants (e.g., SDS, SDC, IGEPAL CA‐630), which are then removed by precipitation with organic solvent (e.g., acetone). The detergent cocktail achieves three important goals: (i) exhaustive/reproducible protein extraction, including MPs from cells and tissues; for example, the SC buffer significantly outcompeted SDT buffer in protein extraction from lung and brain, suggesting the use of multiple surfactants may afford more efficient disruption of cellular compartments and thereby enhancing protein extraction from tissues (Shen et al., 2018a); (ii) effective removal of detrimental non‐protein matrix components (e.g., fatty acids, phospholipids, etc.) which would otherwise compromise the robustness of digestion and LC‐MS analysis; (iii) highly effective proteolytic digestion owing to the through, dual‐action (surfactants + precipitation) denaturation. Compared with FASP and in‐solution digestion, SEPOD showed substantially higher peptide/protein (including MPs) recovery and ∼20–40% more peptide identifications, as well as improved quantification of peptides with extreme physicochemical properties in large sample cohorts (Shen et al., 2018a). More importantly, the procedure enabled highly efficient, reproducible and robust preparation of large sample cohorts, as exemplified by analysis of 44 lung tissue samples in a time‐course investigation post virus infection (Shen et al., 2018a). SCAD utilizes extraction buffer containing SDS, which is removed afterwards by a two‐round acetone‐precipitation (Ma et al., 2018). This protocol provides high protein yields and peptide recovery with minimal sample loss. Compared with FASP and in‐solution digestion with urea, SCAD showed superior protein extraction efficiency and peptide yield (three‐fold and 20% more peptide yields than FASP and in‐solution with urea method, respectively) (Ma et al., 2018). Moreover, precipitation/on‐pellet digestion methods are not subjected to limited sample loading amount, a prominent issue for FASP.
One common issue of most digestion approaches is the long digestion time (e.g., overnight) needed to achieve complete and reproducible cleavage, not only limiting the throughput of large‐scale quantitative proteomics studies, but more importantly, affecting the reproducibility and sensitivity arising from unstable proteolytic peptides under digestion conditions that appears to be quite common albeit overlooked (Duan et al., 2012; Nouri‐Nigjeh et al., 2014; Shen et al., 2017a). Despite various harsh denaturation approaches such as high pressure or temperature, ultrasound, microwave, and infrared radiation have been used to accelerate digestion speed (Wang et al., 2008; Ye & Li, 2012), the suboptimal digestion efficiency and reproducibility owing to the compromised enzymatic activity under these harsh conditions, have been reported (Havlis et al., 2003; Canas et al., 2007; An et al., 2015). It was observed that extensive denaturation by a high concentration of detergents followed by organic solvent precipitation in SEPOD protocol allowed highly efficient and consistent on‐pellet digestion of tissue proteome across a large number of samples, within 6 hr at mild conditions that maximize trypsin efficiency (i.e., 37°C, without pressurization or radiation) (Shen et al., 2018a), which permits reproducible same‐day sample preparation for large cohorts.
2. Extensive and Consistent LC Separation Across a Large Numbers of Samples
In order to achieve in‐depth, high quality MS1 analysis of large numbers of samples, three prerequisites in chromatographic separation are indispensable: first, extensive chromatographic separation to enable in‐depth identification and quantification, as well as selective procurement of MS1 peptide signals in complex matrices; second, high separation reproducibility among many sample runs to permit reliable peak alignment, accurate ID inference among runs as well as precise quantification of large‐cohorts; finally, high analytical sensitivity to warrant reliable quantification of low‐abundance proteins.
Though two‐dimensional (2D) chromatographic fractionation has been used to increase sensitivity and the depth of analysis by some quantitative proteomics pipelines (Dowell et al., 2008), this time‐consuming procedure is apparently not practical for analysis of a large cohort. To realize extensive separation of the highly complex proteomics samples without fractionation, single‐dimension LC separation (usually reversed‐phase LC) with increased column lengths and smaller particle sizes is employed. The early developed capillary columns were usually short (≤20 cm) with low peak capacity (e.g., ∼100 (Shen et al., 2005a,2005b)). Shen et al. firstly demonstrated peak capacity of ∼1000 with long capillary column (80 cm × 150 µm i.d, 3‐µm porous particles) over a 3‐hr gradient (Shen et al., 2002). Since then, various lengths of LC columns and particle sizes (≤3 µm) were examined (Hsieh et al., 2013; Shen et al., 2017b, 2005b; Lan et al., 2018). Using an Orbitrap XL, it was demonstrated the 100‐cm column with 2‐µm particles and a 7‐hr gradient resulted in 3.2‐fold greater numbers of quantifiable proteins than a 25 cm columns packed with same materials with a 1.5‐hr gradient (Nouri‐Nigjeh et al., 2014). While the quickly expanding availability of the fast‐scanning ultra‐high‐field Orbitrap seems to have decreased the need for long gradient, using long column remains beneficial in terms of sensitivity and selectivity of analysis (Shen et al., 2017a). However, it remained difficult to achieve BOTH extensive separation with a long column and excellent run‐to‐run reproducibility across a large number of biological samples, largely because of the difficulties in stabilizing the long‐column‐LC‐MS system for an extended period of time and the buildup of matrix components in the LC‐MS system, which significantly deteriorate chromatographic performance over time. Another issue is the very high pressure needed to drive such columns. Though recent developments showed feasibility of attaining 45 k psi by isolated pneumatic amplifier pump with a storage loop (Grinias et al., 2016), the access to such instruments is limited to proteomics communities. One commonly‐utilized method is elevated separation temperature (e.g., up to 60°C), which not only substantially reduces back pressure but also improves peptide separation, especially for hydrophilic ones (Yan et al., 2000). Furthermore, it has been demonstrated that highly homogeneous heating of long nano‐column in heat‐conductive silicon markedly improved run‐to‐run repeatability compared to a standard column oven (Nouri‐Nigjeh et al., 2014; Shen et al., 2017a). One practical and easy‐to‐implement approach to achieve highly reproducible and robust separation across a large sample cohort is to use a large‐ID trap, which permits high separation reproducibility for two reasons (Nouri‐Nigjeh et al., 2014; Shen et al., 2017a): first, the trap enables reproducible gradient delivery to the downstream column by providing homogeneous mobile phase mixing and dampened pump noise, as confirm by real‐time conductivity profiles; second, a optimized, selective peptide trapping/delivery strategy on the trap prevents detrimental hydrophobic and hydrophilic matrix components from entering the nano‐LC − MS system, which permits highly reproducible and robust separation of many biological samples without appreciable loss of chromatographic resolution. The use of large‐ID trap also eliminates the need of offline sample cleaning, one major source of compromised reproducibility of label‐free quantification in large‐cohorts (Nouri‐Nigjeh et al., 2014; Tu et al., 2014a; Shen et al., 2017a). Moreover, the large‐ID trap enables rapid sample loading and 5–10 folds higher loading of peptide digests without compromising the chromatographic resolutions and quantitative linearity, which substantially improved the sensitivity for MS1‐based quantification (Shen et al., 2017a).
Another approach to enhance analytical sensitivity is to use smaller‐ID columns, based on the notion that ESI‐MS is a concentration‐dependent detector (Zhang et al., 2018). For example, Shen et al. (2002) showed drastically improved protein identification in 100 ng yeast digest when column I.D. decreased from 75 to 15 µm. A more recent work demonstrated a 30‐µm I.D. column increased signal intensity by >3‐fold and 32% more peptides identifications than using a 75 µm I.D. column (Zhu et al., 2018b). Although the increased signal strength by smaller‐I.D. columns is highly valuable in the event of very small sample size, these columns fall short because of difficulties in high‐quality column packing, compromised separation efficiency and robustness, as well as low loading capacity (Horie et al., 2014), which markedly limits their application in large‐scale quantitative analysis. The advent of monolithic columns with small‐sized skeletons, high permeability (i.e., low backpressure) and relatively large pores are considered promising alternatives to packed columns. These techniques allowed practical utilization of columns with very small I.D. (e.g., 10 µm) and extended lengths (up to 8 m), which markedly improves sensitivity (Yi et al., 2017). A number of works relating to this technique are reported (Luo et al., 2007; Iwasaki et al., 2010). Furthermore, more recently developed polymer monolithic porous layer open tubular (PLOT) columns showed improved reproducibility (Rogeberg et al., 2013). The performance of these techniques in the analysis of large‐cohorts of biological samples have not been evaluated yet.
3. High‐Resolution, Sensitive, and Consistent MS1 Measurement
Selecting an MS instrument with the following characteristics is critical for MS1‐based quantification: (a) high‐resolution MS measurement to achieve selective procurement of MS1 signal; (b) high sensitivity to analyze low‐abundance proteins; and (c) stable, robust signal intensity with a wide quantitative linear range. Resolution or resolving power, the ability of a MS analyzer to separate adjacent mass peaks, is one of the most important considerations as it profoundly affects the quality of MS1 quantification. A MS analyzer with higher resolution enables extraction of peptide ion currents with narrower m/z windows, which substantially reduces the chemical noises and matrix interference and thereby improving sensitivity, accuracy and specificity of MS1‐based quantification (Tu et al., 2014b; Shen et al., 2017a). Moreover, higher MS resolution also enables more precise peptide matching among samples and improves the confidence of peptide identification (May et al., 2008; Tu et al., 2017a,2017b). A growing record of studies utilize high‐resolution MS analyzers to achieve sensitive and specific protein quantification (Henry et al., 2012; Krey et al., 2014; Geib et al., 2016). Historically, when low‐resolution MS (e.g., an ion trap) was used, the performance of MS1‐based quantification was found far inferior to MS2‐based methods (e.g., spectral counting) owing to the poor selectivity (Tu et al., 2014b). Nonetheless, the advent of high‐resolution MS techniques shifted the balance drastically and greatly encouraged the development/optimization of MS1‐based strategies (Zhang et al., 2006; Gautier et al., 2012). For example, more recently, the Orbitrap MS analyzer rapidly gained popularity (Kelstrup et al., 2014; Rose et al., 2012), as reflected by ∼3000 peer‐reviewed publications to date. Figure 3 demonstrates that the higher resolutions afforded by Orbitrap enables higher selectivity, lower chemical noises and thereby higher sensitivity for MS1 ion current‐based quantification of low‐abundance species in biological samples. At the resolution of 60 k and 120 k (FWHM@m/z = 200, same below) which is typical for the first‐generation Orbitrap (e.g., Orbitrap XL, Q‐Exactive, etc.), substantially higher signal‐to‐noise ratios were achieved than 30 k resolution (i.e., the resolution by a typical Q‐TOF). Higher resolutions by the ultra‐high‐field (UHF) Orbitrap (240 k and 500 k, respectively by a Q‐Exactive HF and a Fusion LUMOS) enabled extraction of peptide ion currents at extremely‐narrow m/z windows, which further enhanced selectivity and sensitivity. Higher MS resolution was also found to improve quantitative accuracy and precision for MS1‐based analysis in large cohorts, as benchmarked by a recent study (Shen et al., 2017a). Moreover, UHF‐Orbitrap enables faster scan speed with higher sensitivity than the first‐generation Orbitrap, which reduces the need of long‐gradient LC separation: for example, it was reported that a 2.5‐hr gradient on a 100‐cm column achieved optimal proteomic depth and throughput with UHF‐Orbitrap, while a 7‐hr gradient was deemed optimal for the same column on a first‐generation Orbitrap (Nouri‐Nigjeh et al., 2014; Shen et al., 2017a). Finally, besides high MS resolution, Orbitrap also provides excellent signal stability, which renders it an instrument‐of‐choice for MS1‐based quantification.
In order to obtain consistent, stable MS signal across many samples, it is important to attain efficient and robust ionization and ion transmission. Recent techniques such as Ion Funnel (Kelly et al., 2010) and new front‐end designs greatly contribute to this regard by affording resistant to contamination and highly efficient ion transmissions. Furthermore, it is essential to remove detrimental matrix components by both sample preparation and chromatography in order for stable ionization. The abovementioned IonStar experimental pipeline (Shen et al., 2017a; Shen et al., 2018a) significantly cleaned biological samples, which showed extraordinary reproducibility in MS signal and excellent robustness: no appreciable signal decrease across 100 samples, which laid a solid foundation for reliable large‐cohort analysis (Shen et al., 2017a).
B. Optimal Data Processing Methods
For MS1‐based quantification methods, a generic workflow for data processing encompasses several essential steps: (1) peptide and protein identification; (2) sensitive and selective generation of quantitative features; (3) data integration and quality control. Among these, approaches for quantitative feature generation and subsequent data processing steps vary substantially among different techniques in terms of the rationales and algorithms used. To achieve high‐quality quantification in large‐cohort analysis, rigorous evaluation, and optimization of these procedures are essential.
1. Peptide and Protein Identification
This section provides a brief overview of peptide and protein identification as a common procedure in most quantitative proteomics pipelines. Generally speaking, peptide, and protein identification is performed by three steps: (i) conversion of MS2 spectra to putative peptide sequences; (ii) protein inference (i.e., mapping peptide sequences to the corresponding protein entries); (iii) filtering and validation of peptide/protein identifications. As the quantification process of MS1‐based methods is independent of the identification process, theoretically an MS1 quantitative pipeline can be conjugated with any identification strategy chosen by the users.
Peptide and protein identification is usually accomplished by either sequence database searching or spectral library matching. Because of its ease to use, sequence database searching is by far the most prevalent approach for MS1 quantitative proteomics, which compares and matches the observed versus theoretical MS2 spectra. To date, a number of database search engines have been developed with varied descriptive models, searching speed, matching accuracy, and sensitivity/specificity, for example, SEQUEST (Eng et al., 1994), Mascot (Perkins et al., 1999), MS Amanda (Dorfer et al., 2014), Andromeda (Cox et al., 2011), OMSSA (Geer et al., 2004), X!Tandem (Craig & Beavis, 2004), MS‐GF + (Kim & Pevzner, 2014), Phenyx (Allet et al., 2004), MyriMatch (Tabb et al., 2007), Morpheus (Wenger & Coon, 2013), and MSFragger (Kong et al., 2017). By comparison, spectral library matching, though less practiced and requiring significant efforts to build spectral libraries, may confer several advantages over sequence database searching by including non‐canonical product ions and the measured distribution of product ion intensities under the collision energy set (Griss, 2016) as well as shrinking the searching space by eliminating a large population of sequences either nonexistent or undetectable. Currently, this strategy is mainly adopted by MS2‐DIA strategies but may represent a promising alternative to enhance the depth of analysis for MS1‐based quantification.
The assignment of identified peptides (i.e., peptide‐spectrum matches; PSMs) to protein entries (i.e., protein inference) is another crucial step, though to some extents has been plagued by several problems intrinsic to the peptide‐centric nature of bottom‐up proteomics. First, due to sequence homology among different protein isoforms, an identified peptide may be assigned to multiple protein entries (Zhang et al., 2010). These so‐called “shared peptides” often confounds the assembly process. A simple solution is to remove all shared peptides at the cost of losing these valid peptide IDs. The second problem is the so‐called “one‐hit wonder,” referring to compromised confidence in identification of proteins with only one valid peptide (Huang et al., 2012). Therefore, >2 peptides/protein for protein identification is widely accepted, although objection to this practice has also been documented (Gupta & Pevzner, 2009). A variety of computational methods have been developed to assemble proteins from peptides and cluster proteins into protein groups, and these methods can be either dependent or independent of search engines. Pro and cons of these methods are reviewed extensively by Huang et al. (2012).
As the identification process could be susceptible to random errors where an MS2 spectrum is assigned to a spurious peptide sequence (i.e., false discovery) (Deutsch et al., 2010), it is necessary to confirm the validity of peptide identification and control false discoveries. PSM validity are often evaluated by two classes of statistical and computational strategies, recalibration of search engine scores and global assessment of false discovery rate (FDR) (Nesvizhskii, 2010). The former interrogates single‐spectrum confidence by recalculating confidence scores for individual PSMs and setting cutoff thresholds to distinguish genuine versus spurious PSMs, for example, methods calculating P‐value based on null distribution of search engine scores, or E‐value calculation of PSM (Fenyo & Beavis, 2003). In practice, a number of proteomics software suites also provide their own functional module for PSM score recalibration, for example, Percolator in Proteome Discoverer (Kall et al., 2008), PeptideProphet in Trans‐Proteomic Pipeline (TPP) (Deutsch et al., 2015), MaxQuant (Cox et al., 2014), and OpenMS (Lange et al., 2007). Though recalibration of single‐spectrum confidence scores is a straightforward (Choi & Nesvizhskii, 2008), the large number of PSMs in a dataset also brings about the multiple testing problem and the lack of the overall statistical characteristics. Therefore, global FDR assessment methods have been devised.
The most prevalently practiced global FDR assessment method is target‐decoy approach (TDA) (Elias & Gygi, 2007, 2010), which appends a decoy database generated by reversal, shuffling, or randomization of all sequences in the original sequence database (Wang et al., 2009)), and estimates global FDR based on the assumption that the search engine scores from incorrect and decoy matches follow the same distribution. FDR can then be controlled by adopting an optimized score cutoff, above which the PSMs are accepted. TDA is quite straightforward in both concept and application, and thereby is universally applied in proteomics. Nonetheless, users need to be cautious as the performance of TDA can vary tremendously depending on a number of factors, including the way the decoy sequence database is created (Blanco et al., 2009), the scheme of searching (Keich et al., 2015), and the formula used for FDR calculation (Elias & Gygi, 2007; Jeong et al., 2012). Moreover, TDA also bear certain limitations and pitfalls, such as incompatibility with multi‐stage searching strategies (Zhang et al., 2012), overrepresentation of decoy peptide sequences due to single nucleotide polymorphisms (Bessant, 2016), suboptimal performance with scoring systems using protein inference, and varied reliability when selecting a subset of whole PSM population (Chalkley, 2013).
Finally, the estimation of protein‐level FDR is another important topic in sequence identification. Conventionally, protein‐level FDR is directly calculated based on the fixed PSM‐level FDR obtained from methods such as TDA; while such a scheme works pretty well in smaller sample sets, it may result in misleading outcomes in larger sample sets where overestimation of protein‐level FDR could occur frequently due to the faster increase of spurious PSM/proteins than that of the target ones (Savitski et al., 2015). A number of approaches, such as MAYU (Reiter et al., 2009) and a “picked” TDA method (Savitski et al., 2015) have been proposed to mitigate this problem (Hather et al., 2010), however in general, accurate and confident estimation of protein‐level FDR still remains elusive and challenging.
2. Sensitive and Selective Generation of Quantitative Features
For data processing pipeline of MS1‐based strategy, two fundamental components are (i) chromatographic alignment, which corrects retention time (RT) deviation among different sample runs and clusters chromatographic peaks from the same peptide and (ii) feature detection, which extracts the intensities or peak areas of clustered peaks from the same peptide. Currently, there are a variety of ready‐to‐use software packages to execute these functions, and in this section, we will compare the rationales and algorithms of some prevalently used methods.
Chromatographic Alignment
MS1‐based quantification requires retention time (RT) match of the same peptide among all runs. However, perfectly reproducible LC separation is not achievable especially for large‐cohort analysis, due to a series of factors such as random variations of separation conditions, fluctuation of environmental temperature, column aging, and systematic RT shifts over time (Smith et al., 2015), which can be controlled but never completely eliminated. As a result, an effective chromatographic alignment procedure is critical to accurately associate corresponding peaks among runs and thereby set solid foundation for reliable feature generation. Warping is the most widely used method so far, which reversely shift/stretch/squeeze peaks to minimize variation based on a reference chromatogram selected. A number of warping‐based alignment algorithms have been developed either as stand‐alone tools or built‐in modules in software platforms, which can be categorized into profile‐based or feature‐based workflows (Sandin et al., 2014). Prominent examples of profile‐based methods, where alignment is conducted prior to feature detection or peptide identification, include dynamic time warping (DTW) (Charfi & Zrida, 2011), correlation‐optimized warping (COW) (Frusawa et al., 2003), ChromAlign (Sadygov et al., 2006), parametric time warping (PTW) (Eilers, 2004) and parallel factor analysis (PARAFAC) (Smith et al., 2015). DTW models the variation of RT between runs by equal weighing of all ion peaks from the total ion current (TIC), with later modification adding the m/z dimension to further facilitate RT alignment. COW emphasizes the alignment of correlating ion peaks, and warps user‐defined RT segments by piecewise linear alignment to the reference chromatogram (Frusawa et al., 2003). The selection of an optimal reference and segment width therefore plays an important role on the performance of COW as well as its derivatives. ChromAlign is a two‐step derivative of COW, which conducts a quick Fourier transform‐based pairwise correlation based on RT and intensity, and then adopts a three‐dimensional algorithm using m/z, RT and intensity for more precise alignment. The crude alignment as the first step significantly shortens the overall processing time, and offers much improved alignment performance compared to two‐dimensional algorithms (Sadygov et al., 2006). Recently, the use of an improved ChromAlign method in the IonStar quantitative pipeline provided superior performance in decreasing RT deviations (∼97%) compared with the feature‐based alignment algorithm used in MaxQuant (∼50%) as benchmarked with a N = 20 sample set, setting a solid foundation for high‐quality MS1 ion‐current based quantification (Shen et al., 2018b). PTW employs a polynomial warping function to minimize differences in abundance among sample runs (Eilers, 2004), yet its performance could be suboptimal for ion peaks with low intensities, as PTW largely relies on the intensity dimension (Yao et al., 2007). PARAFAC is an extension of principal component analysis (PCA) based on the assumption of trilinear data structure in LC‐MS chromatograms, similar to COW (Bylund et al., 2002). Other examples of profile‐based methods encompass continuous profile model (CPM) (Listgarten et al., 2007), MZmine (Katajamaa & Oresic, 2005), MapAligner in OpenMS (Lange et al., 2007), SuperHirn (Mueller et al., 2007), XCMS (Callister et al., 2006), SpecArray (Li et al., 2005), and XAlign (Li et al., 2005).
Feature‐based algorithms conduct alignment after the feature detection process (discussed in the following section), which only pick true peptide signals and then condense the raw data into m/z‐RT coordinates of these features. Alignment is only performed on detected features, enabling fast data processing speed data albeit at the cost of possible compromise to quantitative accuracy and sensitivity. Canonical examples of feature‐based algorithms include MaxQuant (MaxLFQ) (Cox et al., 2014), OpenMS (Lange et al., 2007), MZmine (Katajamaa & Oresic, 2005), XCMS (Smith et al., 2006), SuperHirn (Mueller et al., 2007), SpecArray (Li et al., 2005), and XAlign (Li et al., 2005). As one of the most popular software suites for MS1‐based quantitative proteomics, MaxQuant exerts a “match‐between‐runs” function to infer the peptide ID of quantitative features runs with valid identification to those without, which demands aligned peak RTs among different sample runs (Cox et al., 2014). Upon the completion of feature detection, a hierarchical clustering is performed to determine the similarities among all chromatographic profiles of sample runs; then the runs with the highest similarities are aligned first, followed by those with lower similarities. A two‐dimensional Gaussian kernel smoothing algorithm is employed for pair‐wise RT calibration. OpenMS offers a “MapAligner” module for identification‐based RT adjustment, which calibrates the RT of peptides in a run with either RT from a user‐defined reference or the median RT across all runs based on a double Gaussian distribution fitting model (Lange et al., 2007). MZMine assigns an ion current feature in each sample to the closest one on the master feature list within a tolerance window (Katajamaa & Oresic, 2005). SuperHirn employs a modified accurate mass retention time (AMRT) method to cluster common features between two runs and generate a model reflecting RT fluctuations. A robust smoothing method LOWESS is then applied to obtain a nonlinear fitting model subsequently used for the RT prediction of features with no valid identification (Mueller et al., 2007). XAlign utilizes a piecewise alignment algorithm for defined m/z‐RT window segments after feature detection, and pinpoints features with the highest intensity in each window as a landmark, which is then employed for alignment via linear warping function (Li et al., 2005). One potential pitfall for XAlign, though, is that ion peaks from the same peptide may vary significantly across different sample/groups, which may lead to inaccurate landmark correspondence (Li et al., 2005). Besides warping algorithms, several non‐warping algorithms for RT alignment have also been developed, such as MassUntangler (Ballardini et al., 2011), MSInspect (Bellew et al., 2006), and Peakmatch (Li et al., 2005). To sum up, a wide range of LC‐MS alignment approaches have been developed; since the quality of chromatographic alignment is of extraordinary importance to sensitivity and accuracy of MS1‐based quantification in large sample cohorts, an appropriate approach should be identified based on extensive evaluation.
Quantitative Feature Detection
For MS1 ion current‐based quantitative proteomics, feature detection refers to the process of discovering and extracting the set of MS1 ion chromatographic currents of the same peptide throughout all sample runs. As the basic unit for MS1‐based quantification, a quantitative feature often encompasses a set of data including m/z of the peptide, apex RT, charge state, and peptide peak intensity in all sample runs. Under most circumstances, the area under curve (AUC) of the peptide ion peaks is calculated as peptide signal intensity, though apex intensity may sometimes be used for LC‐MS run with high noise levels (Sandin et al., 2014). The process also assigns peptide sequences to quantitative data. For MS1‐based quantification, it is of key importance to achieve sensitive, accurate, and robust feature detection in order to warrant high‐quality quantification, but remains challenging for large‐cohort analysis. The consistency of peak selection often deteriorates in large sample cohorts, which is a major factor contributing to missing data. This problem is further confounded by the difficulty in robust detection of features for low‐abundance proteins with high sensitivity while eliminating interfering signals and noises.
Generally speaking, there are two major types of feature detection algorithms, and the Peak Property‐Based (PPB) methods (Sandin et al., 2014) is the one more frequently used. PPB methods differentiate peptide signals from noises by wavelet‐based techniques and then filters the signals based on a number of peak properties, for example, peak shape, peak intensity, isotopic envelope, and peak length (Zhang et al., 2009). For example, approaches such as VIPER (Monroe et al., 2007), SuperHirn (Mueller et al., 2007), OpenMS (Sturm et al., 2008), and PepList (in SpecArray) (Li et al., 2005) detects features primarily based on isotopic envelope matching in the m/z dimension. Other packages such as MaxQuant (Tyanova et al., 2016), MEND (Andreev et al., 2003), Vectorized peak detection (Hastings et al., 2002) and MZMine (Katajamaa et al., 2006) rely more on chromatographic peak shape. Some alternative peak detection algorithms (e.g., MapQuand (Leptos et al., 2006) and msInspect (Bellew et al., 2006)) are based on the two‐dimensional nature of LC‐MS, which process the data in the LC dimension or on the LC‐MS plane before performing isotopic envelope matching. Theoretically, PPB methods should enable stringent quality control for feature generation. In practice, quantitative features for peptides of relatively high abundance can be confidently generated by these methods; however, it is very difficult to accurately modulate the peak properties of low‐abundance peptides against a complex background of co‐eluted matrix interferences, which is typical for biological samples. Such a scenario could markedly compromise sensitivity and reproducibility of feature generation. Therefore, PPB methods are prone to missing ion current peaks of low abundance peptides which often do not conform to property. The elevated missing data for low‐abundance species and thereby compromised quantitative reproducibility are further manifested in large‐cohort analysis.
The other type of feature detection algorithms is Direct Ion Current Extraction (DICE) (Sandin et al., 2014). Comparing with PPB, DICE methods generate quantitative features by directly extracting the peptide ion currents within a pre‐defined m/z‐RT window after chromatographic alignment. Prominent advantages of DICE includes straightforward extraction algorithms and more importantly, highly sensitive, and comprehensive generation of quantitative features because no match of peak properties is required. Nonetheless, inclusion of low‐quality quantitative data by DICE is inevitable, which could compromise quantitative quality. This problem can be effectively minimized by two measures. First, MS1 measurement at very high resolution allows reliable ion current extraction within a very narrow m/z window, which permits high‐quality procurement of AUC data for low‐abundance peptides. Second, a stringent, effective post‐feature‐generation quality control is essential to remove low‐quality quantitative data, as described below. Currently, DICE has been employed in MS1‐based quantitative packages such as Skyline (Schilling et al., 2012), DeMix‐Q (Zhang et al., 2016a; FeatureFinderIdentification (FFId) plugin in OpenMS (Weisser & Choudhary, 2017), and IonStar (Shen et al., 2018b). For example, IonStar uses a combination of ChromAlign and a unique DICE method optimized for very high‐resolution MS (e.g., FWHM = 120 k or higher) to achieve sensitive and reproducible generation of quantitative feature sets termed as “frames.” Combined with a series of stringent post‐feature generation quality control measures, IonStar showed more efficient and consistent feature generation compared with a number of PPB‐based quantitative packages such as MaxQuant and OpenMS, as well as superior quantitative performance in large sample cohorts (e.g., lower missing data level, improved quantitative reproducibility, and accuracy/precision, better sensitivity/specificity in detecting protein changes) (Shen et al., 2018b). Some representative data are shown in Figure 4.
Figure 4.
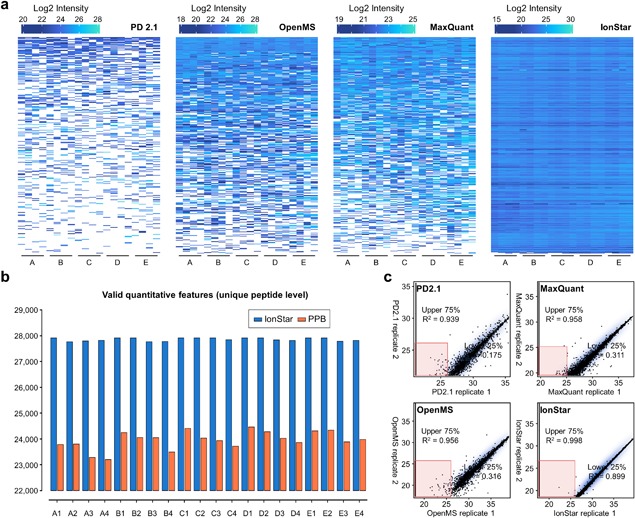
Comparison of representative MS1‐based strategies on (a) levels of missing data, as showed by the abundance heat maps of proteins with the lowest 10% abundances in the benchmark sample set (n = 20 in total). White areas indicate missing data. (b) Comparison of valid quantitative feature numbers generated by IonStar vs. a representative PPB method in the same sample set; (c) Reproducibility of the methods as examined by Pearson correlation of two replicate runs from the same sample. The R 2 values were separately calculated for proteins in the upper 75% and lower 25% abundance percentiles. (Reprinted with permission from (Shen et al., 2018b), copyright 2018, Proc Natl Acad Sci USA).
C. Optimal Quantitative Data Integration and Quality Control
1. Normalization
Due to the inevitable experimental variability among a large‐cohort analysis, an optimal normalization method to alleviate biases and variations and improve quantitative accuracy/precision, is indispensable. The performance of several normalization approaches for MS1‐based quantification has been investigated in the past decade. For example, Tu et al. evaluated the performance of six normalization methods (i.e., LOESS, quantile, upper‐quantile, maximum intensity, median intensity, and total intensity) for peptide‐level normalization in one spike‐in BSA dataset and two rat brain digest datasets, and found that the LOESS approach obtained the best results, followed by quantile normalization by a small margin (Tu et al., 2014b). Valikangas et al. (2018) compared eleven popular normalization strategies using three spike‐in datasets and one mouse experimental dataset, and also examined the outcomes respectively by global and segmental normalization. It was found that variance stabilization normalization (Vsn) was the most effective measure to mitigate technical variability in all datasets involved. Kultima et al. attempted ten types of normalization methods on three sets of neuropeptidomics data and found that RegrRun method achieved the best results by combining linear regression and analysis order normalization (Kultima et al., 2009). This agrees with finding by Callister et al., who also identified linear regression as the best among four prevalent methods (i.e., central tendency, linear regression, locally weighted regression, and quantile techniques). The poor consensus from these aforementioned studies likely indicates that the performance of normalization approaches is largely dependent on the experimental setup and data analysis pipeline, and thus the selection of an appropriate approach should be based on an extensive evaluation. Besides normalization based on the quantitative data in each run, an alternative is to normalize based on proteins of constant levels, which can be either endogenous, “housekeeping” proteins, or spiked‐in proteins with known concentrations. For example, Wisniewski and Mann (2016) reported the ubiquitous and stable expression of deglycase DJ‐1 in a diversity of cell types and tissues, and therefore may serve as standard for proteomics normalization.
2. Missing Data Handling
Despite the high potential for reproducible protein measurement in large sample cohorts, missing data remains a major concern for the majority of MS1‐based methods, where a substantially large proportion (10–20%) of proteins bear missing data and this scenario continues to exacerbate with expanded sample sizes (Bruderer et al., 2015; Chen et al., [Link]). Imputation of missing data are frequently practiced in quantitative proteomics, which can be generally classified into three types of methods: (i) single‐value replacement, which assigns constant or randomly generated values to replace missing data, for example, half of the global minimum (LOD1) (Polpitiya et al., 2008; Clough et al., 2012), half of the peptide minimum (LOD2) (Clough et al., 2012; Polpitiya et al., 2008), and random tail imputation (RTI) (Deeb et al., 2012) and (ii) local similarity‐based methods, which refers to quantitative patterns of other peptides with similar intensity levels in the dataset to estimate missing data, example is, K nearest neighbors (KNN) (Webb‐Robertson et al., 2015), local least‐squares (LLS) (Kim et al., 2005), least‐squares adaptive (LSA) (Bo et al., 2004), regularized expectation maximization (REM) (Schneider, 2001), and model‐based imputation (MBI) (Webb‐Robertson et al., 2015); and (iii) global structure‐based methods, which utilizes PCA‐based dimension reduction to break down the data matrix and reconstruct missing data by iteration, for example, probabilistic principal component analysis (PPCA) (Tipping & Bishop, 1999), and Bayesian principal component analysis (BPCA) (Nounou et al., 2002). Though imputation facilitates downstream informatics analysis (Webb‐Robertson et al., 2015), researchers should use extreme cautions when practicing imputation because there is not one imputation method capable of addressing the numerous sources of missing data in quantitative proteomics and that imputation of biological replicates may result in substantial distortion of the conclusion (Lazar et al., 2016).
The best strategy to tackle the miss data problem, of course, is to improve reproducibility of measurement and thus minimizing miss data on experimental level. To this end, a number of low‐missing data quantitative approaches have been devised (e.g., IonStar (Shen et al., 2018b), DeMix‐Q (Zhang et al., 2016a), and FFId (Weisser & Choudhary, 2017)). For example, in IonStar, extremely low levels of missing data (e.g., typically <0.5% proteins with missing data) can be achieved in large‐cohort analysis, which eliminates the need of imputation (Figure 4) (Shen et al., 2018b; Wang et al., 2018).
3. Quantitative Data Quality Control and Peptide‐to‐Protein Summarization
A proper strategy to summarize peptide quantitative data to protein level is another crucial prerequisite for quantitative proteomics. Peptide‐to‐protein summarization can be accomplished by simple approaches such as arithmetic mean or sum of intensities. The main concern for summing intensity is the underrepresentation of peptides with lower digestion or ionization efficiencies, where relative quantification is driven by the most intensive peptides. To cope with this concern, more sophisticated ones such as linear regression models and identification of temporal patterns are developed (Suomi et al., 2015). Using both simulated and experimental datasets, Carrillo et al. (2010) evaluated several commonly used protein summarization methods in terms of accuracy in estimating protein abundance. The authors found that sum of intensities and total least squares returned the best results, followed by average of ratios. The performance of PCA turned out to be mediocre, and surprisingly linear regression consistently gave the poorest outcomes (Carrillo et al., 2010). Tu et al. (2014b) also evaluated several prevalently used protein summarization methods, and concluded that sum of intensities plus Grubbs’ test achieved the superior outcomes over the variance‐weighted method, average ratio, TOP3, and linear regression.
As mentioned above, post‐feature‐generation quality control is essential to attain high‐quality quantification, especially for DICE‐type methods. This step excludes peptides with aberrant quantitative behaviors to ensure accuracy, precision, and robustness of quantification, and is often performed along with quantitative data summarization process from peptide‐ to protein‐level. Rejection of “outlier” peptides is a straightforward albeit effective method in this regard. When one protein is quantified by multiple, unique peptides, in an ideal situation, each of these peptides would give the same quantitative results (i.e., inter‐group ratio, variance, etc.) because they derive from the same protein. Nonetheless in reality, peptides from the same protein frequently show incoherent quantitative data, owing to a number of factors such as inclusion of low‐quality data, biotransformation, and incorrect peptide/protein identification. Modulation of inter‐group ratio and variance among multiple peptides from the same protein can be employed to identify and eliminate “outliers” carrying false or low‐quality quantitative information and thus achieving reliable quantification.
Traditional outlier detection methods, such as Grubbs’ test, have been frequently applied in quantitative proteomics and incorporated in several software packages (Polpitiya et al., 2008; Park & Yates, 2010). Yet Grubbs’ test only works well for the simplest two‐group case‐control comparison and can be overwhelmed by high‐dimension proteomics data obtained from large‐cohort studies (Shen et al., 2018b). To address this issue, a handful of more advanced outlier detection methods have been adapted for quantitative proteomics. For example, Cho et al. reported an R‐based package, OutlierD, to pinpoint outlier peptides by linear, non‐linear, and non‐parametric quantile regression (Cho et al., 2008). Forshed (2013) described Protein Quantification by Peptide Quality control (PQPQ), which performs clustering analysis for peptides inferred to the same protein. Peptides contained in the largest cluster will be retained for calculation of protein intensities, while others will be deemed as outliers and removed. Webb‐Robertson et al. (2014) devised a Bayesian model (BP‐Quant) to delineate the statistical signature of peptides inferred to a protein by hypothesis testing, and then exclude peptides outside of the dominant patterns from quantification. Zhang et al. proposed a factor analysis‐based approach termed as Diffacto, which utilizes the covariation of peptide intensities in individual samples for outlier peptide detection and weighted peptide‐to‐protein summarization. It was demonstrated that Diffacto provided sensitive and specific recognition of peptides with uninformative or contradictory intensity profiles, as well as reliable summarization of protein intensities (Zhang et al., 2017). Shen et al. employed PCOut, a PCA‐based multivariate mean‐variation modeling algorithm, in the IonStar quantitative package to interrogate inter‐group ratios of peptides assigned to the same proteins (Shen et al., 2018b). It was found PCOut offered excellent performance on removing peptides with poor data quality from high‐dimensional proteomics data set and afforded high quantitative accuracy and precision for large‐cohort quantification.
4. Discovery of Altered Proteins
One of the ultimate goals of quantitative proteomics is to detect proteins with differential abundances between case and control groups. In most cases, significantly altered proteins (alternatively known as differentially‐expressed proteins, dysregulated proteins, or biomarker candidates) are determined using experience‐based cutoff thresholds for protein fold changes (FC) and/or significance score (e.g., P‐value) from hypothesis testing (e.g., Student's t‐test, one‐way ANOVA) (Ting et al., 2009). However, there is an increasing awareness that such conventional schemes are often plagued by surprisingly high false‐positive rates (Gillet et al., 2012). False‐positive discovery of altered proteins may arise from different sources: (i) the FC‐based cutoff method assumes uniform variance levels of all variables, which may not apply to all proteins or experimental settings (Ting et al., 2009; Gillet et al., 2012); (ii) the hypothesis testing procedure on the large number of quantified proteins could easily result in multiple testing problem (Diz et al., 2011), which can be further confounded by peptides shared among different proteins (Serang et al., 2013); (iii) the use of an inadequate number of biological replicates in any group renders the quantification liable to bias and variation due to biological variability, which is especially problematic for clinical and pharmaceutical applications where inter‐individual variability is often substantial. In general, there are three types of methods introduced to improve the reliability in discovery of significantly altered proteins, including multiple testing correction, moderated testing statistics, and experimental‐based methods. Multiple testing correction addresses random errors during the testing procedure by either readjusting significance score threshold or predicting the FDR of the discovered “significant” proteins. A few classical examples are Bonferroni correction (Noble, 2009), Benjamini‐Hochberg method (Benjamini et al., 2001), and Storey‐Tibshirani method (Storey & Tibshirani, 2003). Nevertheless, the application of multiple testing correction in quantitative proteomics is quite limited owing to several unique natures of proteomics data (Pascovici et al., 2016). The second class of methods is moderated testing statistics. Permutation is one prominent example which requires no assumption of data normality or independence of the data (as normal hypothesis testing approaches do), which has been proved to offer robust and sensitive detection of protein changes (Nguyen et al., 2017). Significance Analysis of Microarray (SAM) determines whether a protein is significantly changed by calculating a score contrasting the measured level of changes via repeated measurement of the given protein (Tusher et al., 2001), which proves to perform better than FC or conventional t‐test methods (Roxas & Li, 2008). Linear Models for Microarray Data (LIMMA) adopts an empirical Bayes principle to reduce sample variance and establish a linear model for individual proteins quantified (Smyth, 2005), which works best for proteomics data with low replicate number or high missing data rate (Smyth, 2005; Kammers et al., 2015). Other testing statistics used for proteomics data encompass rank product (RP) method (Schwammle et al., 2013), Mixture Model Method (MMM) (Booth et al., 2011), Power Law Global Error Model (PLGEM) (Pavelka et al., 2008), and Reproducibility Optimized Test Statistics (ROTS) (Elo et al., 2009). It should be emphasized that the selection of testing statistics and optimization of parameters based on the characteristics of the data set is of utmost importance.
Finally, experimental strategies for estimation/control of false‐positives in discovering protein changes have been gaining popularity. Such strategies measure null distribution in a project by experimentally comparing groups of samples without biological difference (e.g., control vs. control or case vs. case groups) using the same experimental design and sample size per group as the case‐vs.‐control study. Such methods can accurately assess false‐positive discoveries by accounting for the collective effects of technical variability (e.g., variation and bias in sample preparation, LC/MS analysis and data processing), biological variability and project‐specific issues on false‐positive discovery (Shen et al., 2015). Experimental‐based methods have been initially employed in microarray quantification (Tusher et al., 2001) and gel‐based proteomics (Karp et al., 2007) as a validation approach, yet its application in MS1‐based quantitative proteomics studies has not been explored until recently. Shen et al. devised an Experimental Null (EN) method measuring null distribution of a given proteomics project, by random interspacing of additional control‐group samples in the same LC‐MS sequence for case‐vs.‐control analysis (Shen et al., 2015). Null distribution of protein ratios and P‐values can then be constructed by comparing the two sets of control‐group data (i.e., the EN dataset), and false‐positive rate for discovering changed proteins can be estimated by dividing the number of significantly changed proteins in the EN dataset (i.e., false‐positives) to that in the case‐control dataset. Cutoff thresholds for detecting significantly changed proteins could also be adjusted accordingly to achieve an optimal balance of discovery sensitivity versus false positives. As demonstrated by an extensive evaluation in large‐cohort sample sets, this EN method appears to be markedly more accurate in estimating false‐positives in discovery of changed proteins, compared with a number of statistical approaches including Student's t‐test, LIMMA, and Fisher's exact test plus Benjamini‐Hochberg method.
Overall, confident detection of changed proteins is one cardinal component of quality control for quantitative proteomics but remains challenging and underrepresented. If the false‐positive discovery issue was not well addressed, it could severely undermine the credibility of proteomics quantification in large‐cohort. The existing statistical and experimental methods offer a range of tools in this regard, yet we would have to give serious consideration to choose the right method owing to their varying performance and characteristics.
APPLICATIONS OF MS1‐BASED QUANTIFICATION IN RELATIVELY LARGE‐COHORT ANALYSIS
With the rapidly improving experimental and data processing strategies in the last decades, MS1‐based quantitative proteomics has been applied in a number of preclinical, clinical, and pharmaceutical studies where excellent quantitative quality and analysis of relatively large biological cohorts are often prerequisites, such as molecular mechanism investigation of human diseases, biomarker discovery, and investigation of the mechanism of actions by therapeutic agents.
A. Molecular Mechanism Investigation of Diseases
The survey of protein changes in human diseases resulting from genetic and environmental factors is highly valuable in elucidating the underlying molecular mechanisms. One of the most important examples is cancer, the second leading cause of death worldwide (Siegel et al., 2018). The complexity of cancer is reflected by the signaling network alterations accumulating at each stage of the multiple‐step carcinogenesis. To understand the complicated molecular mechanisms of carcinogenesis, it is essential to analyze the corresponding protein changes, and to procure reliable information of individual proteins in a cellular context so as to determine their roles in signaling networks and disease stages. However, protein profiling of cancer samples (e.g., cells, tissues, body fluids) has always been a prominent challenge because of the biological complexity and the difficulties in reliable analysis of a large number of individuals, which is indispensable owing to high inter‐individual variability. During last decade, MS1‐based proteomic analyses have contributed to molecular studies of cancer biology. Recently, the proteomics and metabolomics analysis of primary human B cell chronic lymphocytic leukemia (B‐CLL) cells and healthy B cells (from younger and elderly donors) (n = 32) was performed to investigate the role of aging in CLL development (Mayer et al., 2018). With MaxQuant, 6954 proteins in total were quantified, and it was found the proteome signature for immune senescence (e.g., ROS formation, DNA damage repair, inflammatory response, mitochondrial dysfunction) in elderly B cells could be related to tumorigenesis. Similarly, a large‐cohort proteomic study of cancerous (CEC) and normal (NEC) cells isolated from five different tumor and normal locations in resected colon tissue from each of the 12 patients has been reported (N = 120) (Tu et al., 2017b). With an Ion current‐based quantitative pipeline, much better run‐to‐run reproducibility (∼0.98%) and lower intra‐group CV (9.2%) compared to other quantitative methods (e.g., MS2‐TIC, emPAI, and NASF) were attained. A total of 458 altered proteins were found to be involved in deregulation of mitochondrial function, RNA post‐transcriptional modification and infection by RNA viruses in CEC, which fostered an improved understanding of CRC development and provides potential biomarkers for CRC diagnosis or therapeutic targets. A study with ovarian cancer cell lines (n = 30), high‐grade serous ovarian cancer (HGSOC) tissues (n = 8) and primary fallopian tube epithelia cells (n = 3) was able to quantify >10,000 proteins (<1% protein and peptide FDR) totally using MaxQuant iBAQ and 77% were reproducibly quantified. A 67‐protein signature was identified to separate the entire proteomic data set into epithelia and mesenchymal HGSOC tumor cluster, which provides proteomics‐based epithelial/mesenchymal stratification of cell lines and human tumors (Coscia et al., 2016). Similarly, a colorectal cancer study using 32 samples (paraffin embedded normal (N), adenoma (A), and cancer tissue (C)) quantified a combined collection of 10,900 proteins (1% FDR) with label free module of MaxQuant software. A total of 8237 proteins were quantified in at least 75% of samples. Between N and A, 23% of all proteins changed significantly, 17.8% from A to C and 21.6% from N to C. This study further allowed the evaluations in basic biological processes including the energy metabolism, plasma membrane transport, DNA replication, and transcription (Wisniewski et al., 2015).
MS1‐based quantitative proteomics has also been applied to extend our knowledge on the molecular mechanisms of several other diseases. To investigate how the mitochondria‐associated ER membrane (MAM) is affected under diabetic condition, Ma et al. (2017) performed a comprehensive proteome profiling of the isolated brain MAM from long‐term type 2 diabetic mice with non‐diabetic controls and totally quantified 1,313 MAM proteins (0.19% peptide FDR) using MS1 intensity‐based quantification (IonStar), and confident quantification of ∼95% of total proteins (1,239 out of 1,313) was achieved across all samples (i.e., no missing data). The in‐depth analysis of altered proteins uncovered the reduced MAM tethering protein, GRP75, and activated unfolded protein response molecules in diabetes. To elucidate the mechanism of HIV virus control in the long‐term‐nonprogressors (LTNP), a comparative proteome analysis of peripheral blood mononuclear cells (PBMC) from LTNP (n = 10) and normal‐progressors (NP) (n = 10) was performed using an ion‐current‐based quantitative strategy. A total of 87 altered proteins between are implicated in key processes such as cytoskeleton organization, defense response, and apoptosis regulation (Shen et al., 2015b). With a similar quantification strategy, a study of rat striatal proteomic profiling with short‐ (WD1) and long‐term (WD22) cocaine withdrawal (n = 40) identifies several key biological processes of drug‐induced neuroplasticity by altered proteins (Shen et al., 2016). Another example is the ion‐current‐based proteomics investigation of bronchoalveolar lavage fluid in chronic obstructive pulmonary disease (COPD) patients (n = 20) observes gluconeogenesis/glycolysis, inflammatory response, proteolysis as well as a novel alcohol metabolic process through 76 altered proteins (Tu et al., 2014c).
B. Biomarker Discovery
Biomarker, which usually refers to a measurable molecule found in cells, tissues, or body fluids that can serve as an indicator of a physiological condition or disease state, plays a critical role in prediction of disease aggressiveness, early diagnosis, accurate assessment of prognosis, and response evaluation to therapies. With recent advances in genomics and proteomics techniques, a mass of candidate DNA, RNA and protein biomarkers have been identified. Compared to nucleic acid biomarkers, protein biomarkers are believed to be more associated with functional information and more precisely reflecting physiopathological states (Wang et al., 2016). Discovering protein biomarker candidates is urgently needed but remains challenging because most protein biomarkers are likely present at low abundances, which are difficult to measure owing to the high sensitivity needed and the wide range of protein abundances in typical clinical samples (Wang et al., 2016). LC‐MS has greatly accelerated the identification of protein biomarker candidates through combining high‐resolution LC separations with sensitive MS detection. Minimization of false‐positives is a critical factor for successful LC‐MS‐based biomarker discovery. MS1‐based quantification strategies, when strictly controlled, have shown the ability to uncover biomarker candidates with good reliability and low missing data in large‐cohort biological samples.
To date the most popular biomarker studies are directed toward cardiac diseases or cancer, the top two most lethal diseases worldwide. Investigations in these categories have been on the rise. For example, protein biomarkers of invasive breast cancer is valuable for the early diagnosis and progression monitoring. Beretov et al. (2015) performed an unbiased proteomic analysis in urine from breast cancer patients (n = 20) and healthy women (n = 20) by ion current relative quantification using Progensis QI. Among 59 urinary proteins with significant difference in breast cancer patients, novel stage‐specific markers respectively associated with pre‐, early, and metastatic breast cancer, were identified. Lung cancer represents one of the deadliest types of cancer. As some conventional early screening methods (e.g., exhaled breath condensate analysis, low‐dose computed tomography) lead to an increased burden on bronchoscopy units, new diagnostic approaches are essential. A proteome analysis of acellular bronchoalveolar lavage (BAL) fluid samples from 90 suspected lung cancer cases was reported (Carvalho et al., 2017). MaxQuant and VEMS iBAQ are used to compare lung cancer versus non‐cancer controls, which identified 130 potential “biomarkers.” These candidates showed a large overlap with biomarkers detected in tissue samples. Using LC‐MS using MaxQuant, Liu et al. analyzed 126 triple‐negative breast cancer samples and quantified >3,500 proteins. The work identified a signature pattern encompassing 11 proteins with potential prognostic value (Liu et al., 2014).
A number of studies were performed to seek protein biomarker candidates for cardiovascular diseases. For instance, Addona et al. developed a proteomic pipeline to identify early plasma biomarkers of cardiac injury before, during, and after controlled myocardial injury (N = 24). The MS1 peak areas were calculated by Spectrum Mill, and ∼100 altered proteins were identified from 1,105 totally quantified proteins (FDR <1.5%) (Addona et al., 2011).
Using MS1‐based method, biomarkers discovery for other diseases in sizable cohorts are reported as well. For one example, proteomics study of schistosomiasis, which is found in tropical regions and associated with the risk of bladder cancer, was conducted using MaxQuant. The authors found a total of 1,306 proteins in 49 human urine samples from Eggua region and were screened for the presence of Schistosoma haematobium infection. Out of these proteins, 54 human proteins and 36 schistosoma proteins were found to be potential biomarker candidates for this disease (Onile et al., 2017).
C. Pharmaceutical Investigations
As proteins are the primary effectors responsible for drug efficacy and safety, quantitative proteomics represent a powerful tool in pharmaceutical investigations, such as target identification, candidate selection, toxicological characterization, validation of drug candidates, and clinical evaluation of products. To this end, a number of studies has been conducted on identification of potential drug targets, investigation of the mechanistic basis of drug action, toxicity, and drug resistance. Identification of therapeutic targets is of the utmost importance for drug discovery, which can be greatly facilitated by global protein profiling of diseased versus normal or treated versus non‐treated samples in a high‐throughput manner. Beside the need to profile a large biological cohort, one major challenge is that a large portion of drug targets are of low‐abundance and membrane‐associated, such as membrane receptors and ion channels (Savas et al., 2011). To address these challenges, sample preparation strategies (e.g., using strong surfactants) have been developed to increase the recovery of membrane proteins (An et al., 2015; Ma et al., 2018; Shen et al., 2018a), and advanced MS1‐based quantitative LC‐MS approaches have been explored for target discovery in large cohorts. For example, a proteomic profiling of detached versus attached cancer cells with drug treatment using MS1 intensity‐based quantification strategy (MaxQuant) quantified 4,885 proteins in 90 cell samples. Despite high missing data level (only 1,950 commonly quantified in all samples), the study found six proteins consistently altered in the detached versus attached cells regardless of the drug treatment status and cell type, which highlighted the importance of detached cells for investigation of anticancer drugs (Saei et al., 2018).
Molecular mechanisms underlying drug resistance is another critical issue to be addressed. For instance, most cancer cells utilize multiple redundant intracellular signaling pathways to maintain functions critical to their survival, especially in cells with drug‐resistance (Bermudez‐Crespo & Lopez, 2007). The signaling pathways that are necessary to cancer cell survival, proliferation, and receptor expression could be potential targets for therapeutic intervention. Moreover, the proteomics profile associated with drug actions of an individual could be highly valuable to inform therapy (Schirle et al., 2012; Shruthi et al., 2016). For example, overcoming the resistance to trastuzumab, an antibody drug targeting HER2‐overexpressing tumor, is crucial for the development of a proper therapeutic strategy. The comparison of trastuzumab‐sensitive versus resistant gastric cancer cells using MS1 intensity‐based absolute quantification (iBAQ) strategy has been described (n = 12) (Liu et al., 2017). The activated mTOR signaling uncovered in this study could mediate the resistance of trastuzumab, which provided new candidate targets for treating trastuzumab‐resistant tumors. A similar study was performed using MaxQuant on 112 tumors from breast cancer patients who manifested either good or poor outcomes to tamoxifen treatment upon recurrence. Totally >3000 proteins were quantified by LFQ and 1,960 proteins were without missing data. They found a four‐protein signature capable of predicting tamoxifen treatment outcome in recurrent breast cancer, and two proteins (ANXA1, CALD1) were associated with tamoxifen resistance (De Marchi et al., 2016). Using a MS1‐based method (IonStar), our recent study of molecular mechanisms underlying the synergism of combined birinapant/paclitaxel in treating pancreatic cancer cells quantified 4,069 proteins (99.8% without missing data, >2 unique peptides/protein) across 48 samples, and 541 proteins were significantly altered in treatment groups. Most of these proteins were altered only by combined birinapant/paclitaxel, which indicated suppressed Warburg effect, enhanced cell apoptotic regulation and cell cycle arrest were associated with drug combination but not any single drug alone (Wang et al., 2018). This temporal and large‐scale proteomic study provides novel insights contributing to the synergistic inhibitory effect of drug combination on cancer cell growth. A similar time‐series proteome analysis of pancreatic cancer cells treated with the combination of gemcitabine/birinapant was performed to characterize the synergistic mechanisms of this combined therapy (Zhu et al., 2018a).
Studies for non‐cancer‐related therapeutics emerged as well. Germany et al. recently studied the mechanism of ineffectiveness of Acamprosate, an FDA‐approved agent for alcoholism treatment (Germany et al., 2018). Using MS1 intensity‐based quantification method, a total of 3,634 proteins were quantified in 16 patient samples. The 1,040 proteins with expression changes indicated the neuroimmune restoration could be a potential efficacy mechanism in the Acamprosate treatment of certain sub‐populations of alcohol‐dependent subjects. Another example is the temporal proteomic profiling for drug‐responsive proteins after treatment with a corticosteroid drug, which enabled the first‐ever pharmacodynamics measurement on proteome level. With IonStar quantification pipeline, the time‐series study quantified 323 drug‐responsive proteins induced by methylprednisolone administration on 60 rats, and revealed diverse temporal changes of biological processes associated with hepatic metabolism, response to hormone stimuli, gluconeogenesis, and inflammatory responses. The high‐quality time course data provided by IonStar greatly facilitated pharmacodynamics modeling (Nouri‐Nigjeh et al., 2014).
Even though drug side effects are quite common problem, the mechanisms of drug toxicity in human organs (e.g., kidney, liver, heart) are often poorly understood. In recent years, proteomics has been used in the evaluation of drug toxicity, termed as toxicoproteomics (Le & Wang, 2014; Titz et al., 2014). Toxicity‐related proteins from such studies are valuable in drug screening and understanding the mechanism of toxic effects. Collins et al. developed a panel of potential pharmaceutical hepatotoxicity biomarkers for hepatotoxic compound EMD 335823 in a rat model (Collins et al., 2012). Totally 48 putative toxicity biomarkers were discovered and confirmed with the selected reaction monitoring assay. A recent work demonstrated the use of IonStar on a large‐scale investigation of 100 rat brains with traumatic brain injuries (TBI) and pharmaceutical treatments. In total >7,000 unique proteins were quantified with ≥2 peptides/protein, and >99.8% of proteins without missing data in any of the 100 samples. Low false‐positive rates (<5%) in identifying altered proteins were achieved across all groups. This study provides unprecedented high‐quality, reproducible proteomics analysis of large cohorts, and sets a new paradigm for reliable large‐cohort analysis (Shen et al., 2018b).
CONCLUSION AND FUTURE PERSPECTIVES
Quantitative proteomics capable of reliable large‐cohort analysis is highly valuable for pharmaceutical/clinical investigations but remains challenging. MS1 ion current‐based strategies show high potential owing to the following features: (i) reproducible protein measurement with low missing data since MS1‐based quantification are data‐independent; (ii) highly sensitive quantification with excellent selectivity when a high‐resolution MS is used; and (iii) high potential for accurate and precise quantification in complex matrices. In the past decade, a number of MS1‐based proteomics studies emerged including technical development in experimental and data processing approaches, as well as applications in fields such as disease mechanism study, biomarker discovery, and pharmaceutical investigations.
Despite the tremendous technical advances of MS1‐based proteomics in recent years, there is still much room for improvement. First, it is important to achieve highly reproducible and robust experimental procedures but this critical need has long been overlooked by most. Second, currently MS1 strategies rely on DDA MS2 fragmentation to assign peptide IDs to quantitative features, which considerably limits the depth of proteomics coverage. For example, in our studies (unpublished data), generally >60% quantified features were lacking peptide ID owing to the limited sensitivity of the accompanying MS2 DDA identification. Third, development of new data processing pipelines is desirable to take full advantage of the very‐high‐resolution MS (e.g., >250k FWHM). Finally, while the field has put in strenuous efforts to maximize the number of identifiable proteins (i.e., depth of analysis), the importance of achieving high‐quality quantitative data are often neglected.
Consequently, we anticipate that new techniques emerging in near future will be directed toward unleashing the full potential of MS1‐based quantification. These may include but not limited to: (i) streamlined, efficient and robust sample preparation and LC‐MS procedures that are standardized for large‐cohort quantification; (ii) methods to markedly improve proteomics coverage of MS1‐based strategies, for example, extensive peptide identification via spectral library matching; and (iii) new algorithms optimized for very‐high‐resolution MS to greatly improve sensitivity and selectivity for low‐abundance proteins, as well as informatics approaches enabling more accurate and precise quantification with lower false discovery rate of altered proteins.
REFERENCES
- Addona TA, Shi X, Keshishian H, et al. 2011. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nature Biotechnology 29(7):635‐U119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allet N, Barrillat N, Baussant T, et al. 2004. In vitro and in silico processes to identify differentially expressed proteins. Proteomics 4(8):2333–2351. [DOI] [PubMed] [Google Scholar]
- An B, Zhang M, Johnson RW, Qu J. 2015. Surfactant‐aided precipitation/on‐pellet‐digestion (SOD) procedure provides robust and rapid sample preparation for reproducible, accurate and sensitive LC/MS quantification of therapeutic protein in plasma and tissues. Anal Chem 87(7):4023–4029. [DOI] [PubMed] [Google Scholar]
- Andreev VP, Rejtar T, Chen HS, Moskovets EV, Ivanov AR, Karger BL. 2003. A universal denoising and peak picking algorithm for LC‐MS based on matched filtration in the chromatographic time domain. Anal Chem 75(22):6314–6326. [DOI] [PubMed] [Google Scholar]
- Ballardini R, Benevento M, Arrigoni G, Pattini L, Roda A. 2011. Mass untangler: A novel alignment tool for label‐free liquid chromatography‐mass spectrometry proteomic data. J Chromatogr A 1218(49):8859–8868. [DOI] [PubMed] [Google Scholar]
- Bellew M, Coram M, Fitzgibbon M, Igra M, Randolph T, Wang P, May D, Eng J, Fang R, Lin C, Chen J, Goodlett D, Whiteaker J, Paulovich A, McIntosh M. 2006. A suite of algorithms for the comprehensive analysis of complex protein mixtures using high‐resolution LC‐MS. Bioinformatics. 22(15):1902–1909. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. 2001. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125(1–2):279–284. [DOI] [PubMed] [Google Scholar]
- Beretov J, Wasinger VC, Millar EK, Schwartz P, Graham PH, Li Y. 2015. Proteomic analysis of urine to identify breast cancer biomarker candidates using a label‐free LC‐MS/MS approach. PLoS ONE 10(11):e0141876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez‐Crespo J, Lopez JL. 2007. A better understanding of molecular mechanisms underlying human disease. Proteomics Clin Appl 1(9):983–1003. [DOI] [PubMed] [Google Scholar]
- Bessant C. Proteome informatics. London, UK: Royal Society of Chemistry; 2016: 412 p. [Google Scholar]
- Blanco L, Mead JA, Bessant C. 2009. Comparison of novel decoy database designs for optimizing protein identification searches using ABRF sPRG2006 standard MS/MS data sets. J Proteome Res 8(4):1782–1791. [DOI] [PubMed] [Google Scholar]
- Bo TH, Dysvik B, Jonassen I. 2004. LSimpute: accurate estimation of missing values in microarray data with least squares methods. Nucleic Acids Res 32(3):e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. 2009. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4(4):484–494. [DOI] [PubMed] [Google Scholar]
- Booth JG, Eilertson KE, Olinares PD, Yu H. 2011. A bayesian mixture model for comparative spectral count data in shotgun proteomics. Mol Cell Proteomics 10(8):M110 007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer R, Bernhardt OM, Gandhi T, et al. 2015. Extending the limits of quantitative proteome profiling with data‐independent acquisition and application to acetaminophen‐treated three‐dimensional liver microtissues. Mol Cell Proteomics 14(5):1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer R, Bernhardt OM, Gandhi T, Reiter L. 2016. High‐precision iRT prediction in the targeted analysis of data‐independent acquisition and its impact on identification and quantitation. Proteomics 16(15‐16):2246–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylund D, Danielsson R, Malmquist G, Markides KE. 2002. Chromatographic alignment by warping and dynamic programming as a pre‐processing tool for PARAFAC modelling of liquid chromatography‐mass spectrometry data. J Chromatogr A 961(2):237–244. [DOI] [PubMed] [Google Scholar]
- Callister SJ, Barry RC, Adkins JN, et al. 2006. Normalization approaches for removing systematic biases associated with mass spectrometry and label‐free proteomics. J Proteome Res 5(2):277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canas B, Pineiro C, Calvo E, Lopez‐Ferrer D, Gallardo JM. 2007. Trends in sample preparation for classical and second generation proteomics. J Chromatogr A 1153(1‐2):235–258. [DOI] [PubMed] [Google Scholar]
- Carrillo B, Yanofsky C, Laboissiere S, Nadon R, Kearney RE. 2010. Methods for combining peptide intensities to estimate relative protein abundance. Bioinformatics. 26(1):98–103. [DOI] [PubMed] [Google Scholar]
- Carvalho AS, Cuco CM, Lavareda C, et al. 2017. Bronchoalveolar lavage proteomics in patients with suspected lung cancer. Sci Rep 7:42190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley RJ. 2013. When target: Decoy false discovery rate estimations are inaccurate and how to spot instances. J Proteome Res 12(2):1062–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charfi M, Zrida J. 2011. Speed improvement of B‐snake algorithm using dynamic programming optimization. IEEE Trans Image Process. 20(10):2848–2855. [DOI] [PubMed] [Google Scholar]
- Chen EI, Cociorva D, Norris JL, Yates JR, 3rd . 2007. Optimization of mass spectrometry‐compatible surfactants for shotgun proteomics. J Proteome Res 6(7):2529–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YT, Parker CE, Chen HW, et al. Discovery and validation case studies, recommendations: A pipeline that integrates the discovery and verification studies of urinary protein biomarkers reveals candidate markers for bladder cancer. Comprehensive Biomarker Discovery and Validation for Clinical Application 2013(33):271–314. [Google Scholar]
- Cho H, Kim YJ, Jung HJ, Lee SW, Lee JW. 2008. OutlierD: An R package for outlier detection using quantile regression on mass spectrometry data. Bioinformatics 24(6):882–884. [DOI] [PubMed] [Google Scholar]
- Choi H, Nesvizhskii AI. 2008. False discovery rates and related statistical concepts in mass spectrometry‐based proteomics. J Proteome Res 7(1):47–50. [DOI] [PubMed] [Google Scholar]
- Choi YS. 2012. Reaching for the deep proteome: Recent nano liquid chromatography coupled with tandem mass spectrometry‐based studies on the deep proteome. Arch Pharm Res 35(11):1861–1870. [DOI] [PubMed] [Google Scholar]
- Choksawangkarn W, Edwards N, Wang Y, Gutierrez P, Fenselau C. 2012. Comparative study of workflows optimized for in‐gel, in‐solution, and on‐filter proteolysis in the analysis of plasma membrane proteins. J Proteome Res 11(5):3030–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clough T, Thaminy S, Ragg S, Aebersold R, Vitek O. 2012. Statistical protein quantification and significance analysis in label‐free LC‐MS experiments with complex designs. BMC Bioinformatics 13(Suppl 16):S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman O, Henry M, Clynes M, Meleady P. 2017. Filter‐aided sample preparation (FASP) for improved proteome analysis of recombinant chinese hamster ovary cells. Methods Mol Biol 1603:187–194. [DOI] [PubMed] [Google Scholar]
- Collins BC, Hunter CL, Liu Y, et al. 2017. Multi‐laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH‐mass spectrometry. Nat Commun 8(1):291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BC, Miller CA, Sposny A, et al. 2012. Development of a pharmaceutical hepatotoxicity biomarker panel using a discovery to targeted proteomics approach. Mol Cell Proteomics 11(8):394–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscia F, Watters KM, Curtis M, et al. 2016. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat Commun 7:12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. 2014. Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13(9):2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. 2011. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10(4):1794–1805. [DOI] [PubMed] [Google Scholar]
- Craig R, Beavis RC. 2004. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 20(9):1466–1467. [DOI] [PubMed] [Google Scholar]
- De Marchi T, Liu NQ, Stingl C, et al. 2016. 4‐protein signature predicting tamoxifen treatment outcome in recurrent breast cancer. Mol Oncol 10(1):24–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeb SJ, D'Souza RC, Cox J, Schmidt‐Supprian M, Mann M. 2012. Super‐SILAC allows classification of diffuse large B‐cell lymphoma subtypes by their protein expression profiles. Mol Cell Proteomics 11(5):77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Mendoza L, Shteynberg D, et al. 2010. A guided tour of the trans‐proteomic Pipeline. Proteomics 10(6):1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Mendoza L, Shteynberg D, Slagel J, Sun Z, Moritz RL. 2015. trans‐proteomic pipeline, a standardized data processing pipeline for large‐scale reproducible proteomics informatics. Proteomics Clin Appl 9(7‐8):745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diz AP, Carvajal‐Rodriguez A, Skibinski DO. 2011. Multiple hypothesis testing in proteomics: a strategy for experimental work. Mol Cell Proteomics 10(3):M110 004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon B, Aebersold R. 2010. Options and considerations when selecting a quantitative proteomics strategy. Nature Biotechnol 28(7):710–721. [DOI] [PubMed] [Google Scholar]
- Dorfer V, Pichler P, Stranzl T, et al. 2014. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J Proteome Res 13(8):3679–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell JA, Frost DC, Zhang J, Li L. 2008. Comparison of two‐dimensional fractionation techniques for shotgun proteomics. Anal Chem 80(17):6715–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Dai L, Chen SC, Balthasar JP, Qu J. 2012. Nano‐scale liquid chromatography/mass spectrometry and on‐the‐fly orthogonal array optimization for quantification of therapeutic monoclonal antibodies and the application in preclinical analysis. J Chromatogr A 1251:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Young R, Straubinger RM, et al. 2009. A straightforward and highly efficient precipitation/on‐pellet digestion procedure coupled with a long gradient nano‐LC separation and orbitrap mass spectrometry for label‐free expression profiling of the swine heart mitochondrial proteome. J Proteome Res 8(6):2838–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers PH. 2004. Parametric time warping. Anal Chem 76(2):404–411. [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. 2007. Target‐decoy search strategy for increased confidence in large‐scale protein identifications by mass spectrometry. Nat Methods 4(3):207–214. [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. 2010. Target‐decoy search strategy for mass spectrometry‐based proteomics. Methods Mol Biol 604:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elo LL, Hiissa J, Tuimala J, Kallio A, Korpelainen E, Aittokallio T. 2009. Optimized detection of differential expression in global profiling experiments: Case studies in clinical transcriptomic and quantitative proteomic datasets. Brief Bioinform 10(5):547–555. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5(11):976–989. [DOI] [PubMed] [Google Scholar]
- Erde J, Loo RR, Loo JA. 2014. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J Proteome Res 13(4):1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson BK, Rose CM, Braun CR, et al. 2017. A strategy to combine sample multiplexing with targeted proteomics assays for high‐Throughput protein signature characterization. Mol Cell 65(2):361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenyo D, Beavis RC. 2003. A method for assessing the statistical significance of mass spectrometry‐based protein identifications using general scoring schemes. Anal Chem 75(4):768–774. [DOI] [PubMed] [Google Scholar]
- Forshed J. 2013. Protein quantification by peptide quality control (PQPQ) of shotgun proteomics data. Methods Mol Biol 1023:149–158. [DOI] [PubMed] [Google Scholar]
- Frusawa H, Fukagawa A, Ikeda Y, et al. 2003. Aligning a single‐lipid nanotube with moderate stiffness. Angew Chem Int Ed Engl 42(1):72–74. [DOI] [PubMed] [Google Scholar]
- Gautier V, Mouton‐Barbosa E, Bouyssie D, et al. 2012. Label‐free quantification and shotgun analysis of complex proteomes by one‐dimensional SDS‐PAGE/NanoLC‐MS: Evaluation for the large scale analysis of inflammatory human endothelial cells. Mol Cell Proteomics 11(8):527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, Markey SP, Kowalak JA, et al. 2004. Open mass spectrometry search algorithm. J Proteome Res 3(5):958–964. [DOI] [PubMed] [Google Scholar]
- Geib T, Sleno L, Hall RA, Stokes CS, Volmer DA. 2016. Triple quadrupole versus high resolution quadrupole‐time‐of‐flight mass spectrometry for quantitative LC‐MS/MS analysis of 25‐Hydroxyvitamin d in human serum. J Am Soc Mass Spectrom 27(8):1404–1410. [DOI] [PubMed] [Google Scholar]
- Germany CE, Reker AN, Hinton DJ, et al. 2018. Pharmacoproteomics profile in response to acamprosate treatment of an alcoholism animal model. Proteomics 18(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, et al. 2012. Targeted data extraction of the MS/MS spectra generated by data‐independent acquisition: A new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11(6):O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Zheng N, Zeng J, Aubry AF, Arnold ME. 2015. Post‐pellet‐digestion precipitation and solid phase extraction: A practical and efficient workflow to extract surrogate peptides for ultra‐high performance liquid chromatography–tandem mass spectrometry bioanalysis of a therapeutic antibody in the low ng/mL range. J Chromatogr A 1424:27–36. [DOI] [PubMed] [Google Scholar]
- Griffin NM, Yu JY, Long F, et al. 2010. Label‐free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nature Biotechnol 28(1):83‐U116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinias KM, Godinho JM, Franklin EG, Stobaugh JT, Jorgenson JW. 2016. Development of a 45kpsi ultrahigh pressure liquid chromatography instrument for gradient separations of peptides using long microcapillary columns and sub‐2mum particles. J Chromatogr A 1469:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss J. 2016. Spectral library searching in proteomics. Proteomics 16(5):729–740. [DOI] [PubMed] [Google Scholar]
- Gupta N, Pevzner PA. 2009. False discovery rates of protein identifications: a strike against the two‐peptide rule. J Proteome Res 8(9):4173–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings CA, Norton SM, Roy S. 2002. New algorithms for processing and peak detection in liquid chromatography/mass spectrometry data. Rapid Commun Mass Spectrom 16(5):462–467. [DOI] [PubMed] [Google Scholar]
- Hather G, Higdon R, Bauman A, von Haller PD, Kolker E. 2010. Estimating false discovery rates for peptide and protein identification using randomized databases. Proteomics 10(12):2369–2376. [DOI] [PubMed] [Google Scholar]
- Havlis J, Thomas H, Sebela M, Shevchenko A. 2003. Fast‐response proteomics by accelerated in‐gel digestion of proteins. Anal Chem 75(6):1300–1306. [DOI] [PubMed] [Google Scholar]
- Henry H, Sobhi HR, Scheibner O, Bromirski M, Nimkar SB, Rochat B. 2012. Comparison between a high‐resolution single‐stage orbitrap and a triple quadrupole mass spectrometer for quantitative analyses of drugs. Rapid Commun Mass Spectrom 26(5):499–509. [DOI] [PubMed] [Google Scholar]
- Higgs RE, Knierman MD, Gelfanova V, Butler JP, Hale JE. 2008. Label‐free LC‐MS method for the identification of biomarkers. Methods Mol Biol 428:209–230. [DOI] [PubMed] [Google Scholar]
- Horie K, Kamakura T, Ikegami T, et al. 2014. Hydrophilic interaction chromatography using a meter‐scale monolithic silica capillary column for proteomics LC‐MS. Anal Chem 86(8):3817–3824. [DOI] [PubMed] [Google Scholar]
- Hsieh EJ, Bereman MS, Durand S, Valaskovic GA, MacCoss MJ. 2013. Effects of column and gradient lengths on peak capacity and peptide identification in nanoflow LC‐MS/MS of complex proteomic samples. J Am Soc Mass Spectrom 24(1):148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu A, Noble WS, Wolf‐Yadlin A. 2016. Technical advances in proteomics: new developments in data‐independent acquisition. F1000Res 5:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Wang J, Yu W, He Z. 2012. Protein inference: A review. Brief Bioinform 13(5):586–614. [DOI] [PubMed] [Google Scholar]
- Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, Krijgsveld J. 2014. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol 10:757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama Y, Oda Y, Tabata T, et al. 2005. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics 4(9):1265–1272. [DOI] [PubMed] [Google Scholar]
- Iwasaki M, Miwa S, Ikegami T, Tomita M, Tanaka N, Ishihama Y. 2010. One‐dimensional capillary liquid chromatographic separation coupled with tandem mass spectrometry unveils the escherichia coli proteome on a microarray scale. Analyt Chem. 82(7):2616–2620. [DOI] [PubMed] [Google Scholar]
- Jeong K, Kim S, Bandeira N. 2012. False discovery rates in spectral identification. BMC Bioinformatics 13(Suppl 16):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L, Storey JD, MacCoss MJ, Noble WS. 2008. Posterior error probabilities and false discovery rates: Two sides of the same coin. J Proteome Res 7(1):40–44. [DOI] [PubMed] [Google Scholar]
- Kammers K, Cole RN, Tiengwe C, Ruczinski I. 2015. Detecting significant changes in protein abundance. EuPA Open Proteom 7:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp NA, McCormick PS, Russell MR, Lilley KS. 2007. Experimental and statistical considerations to avoid false conclusions in proteomics studies using differential in‐gel electrophoresis. Mol Cell Proteomics 6(8):1354–1364. [DOI] [PubMed] [Google Scholar]
- Karpievitch YV, Dabney AR, Smith RD. 2012. Normalization and missing value imputation for label‐free LC‐MS analysis. BMC Bioinformatics. 13(Suppl 16):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katajamaa M, Miettinen J, Oresic M. 2006. MZmine: Toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics. 22(5):634–636. [DOI] [PubMed] [Google Scholar]
- Katajamaa M, Oresic M. 2005. Processing methods for differential analysis of LC/MS profile data. BMC Bioinformatics. 6:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keich U, Kertesz‐Farkas A, Noble WS. 2015. Improved false discovery rate estimation procedure for shotgun proteomics. J Proteome Res. 14(8):3148–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RT, Tolmachev AV, Page JS, Tang K, Smith RD. 2010. The ion funnel: theory, implementations, and applications. Mass Spectrom Rev. 29(2):294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelstrup CD, Jersie‐Christensen RR, Batth TS, et al. 2014. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra‐high‐field Orbitrap mass spectrometer. J Proteome Res. 13(12):6187–6195. [DOI] [PubMed] [Google Scholar]
- Kim H, Golub GH, Park H. 2005. Missing value estimation for DNA microarray gene expression data: Local least squares imputation. Bioinformatics. 21(2):187–198. [DOI] [PubMed] [Google Scholar]
- Kim S, Pevzner PA. 2014. MS‐GF+ makes progress towards a universal database search tool for proteomics. Nat Commun. 5:5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI. 2017. MSFragger: ultrafast and comprehensive peptide identification in shotgun proteomics. Nature methods. 14(5):513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey JF, Wilmarth PA, Shin JB, et al. 2014. Accurate label‐free protein quantitation with high‐ and low‐resolution mass spectrometers. J Proteome Res. 13(2):1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. 2014. Minimal, encapsulated proteomic‐sample processing applied to copy‐number estimation in eukaryotic cells. Nat Methods 11(3):319–324. [DOI] [PubMed] [Google Scholar]
- Kultima K, Nilsson A, Scholz B, Rossbach UL, Falth M, Andren PE. 2009. Development and evaluation of normalization methods for label‐free relative quantification of endogenous peptides. Mol Cell Proteomics. 8(10):2285–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J, Nunez Galindo A, Doecke J, et al. 2018. Systematic evaluation of the use of human plasma and serum for mass‐spectrometry‐based shotgun proteomics. J Proteome Res 17(4):1426–1435. [DOI] [PubMed] [Google Scholar]
- Lange E, Gropl C, Schulz‐Trieglaff O, Leinenbach A, Huber C, Reinert K. 2007. A geometric approach for the alignment of liquid chromatography‐mass spectrometry data. Bioinformatics. 23(13):273–281. [DOI] [PubMed] [Google Scholar]
- Lazar C, Gatto L, Ferro M, Bruley C, Burger T. 2016. Accounting for the multiple natures of missing values in label‐Free quantitative proteomics data sets to compare imputation strategies. J Proteome Res. 15(4):1116–1125. [DOI] [PubMed] [Google Scholar]
- Le XC, Wang Y. 2014. Analytical methods in toxicology. Anal Chem 86(24):11929. [DOI] [PubMed] [Google Scholar]
- Leptos KC, Sarracino DA, Jaffe JD, Krastins B, Church GM. 2006. MapQuant: Open‐source software for large‐scale protein quantification. Proteomics. 6(6):1770–1782. [DOI] [PubMed] [Google Scholar]
- Li XJ, Yi EC, Kemp CJ, Zhang H, Aebersold R. 2005. A software suite for the generation and comparison of peptide arrays from sets of data collected by liquid chromatography‐mass spectrometry. Mol Cell Proteomics 4(9):1328–1340. [DOI] [PubMed] [Google Scholar]
- Lin Y, Liu H, Liu ZH, et al. 2013. Development and evaluation of an entirely solution‐based combinative sample preparation method for membrane proteomics. Anal Biochem 432(1):41–48. [DOI] [PubMed] [Google Scholar]
- Lin Y, Liu Y, Li JJ, et al. 2010. Evaluation and optimization of removal of an acid‐insoluble surfactant for shotgun analysis of membrane proteome. Electrophoresis 31(16):2705–2713. [DOI] [PubMed] [Google Scholar]
- Lin Y, Zhou J, Bi D, Chen P, Wang XC, Liang SP. 2008. Sodium‐deoxycholate‐assisted tryptic digestion and identification of proteolytically resistant proteins. Anal Biochem 377(2):259–266. [DOI] [PubMed] [Google Scholar]
- Listgarten J, Neal RM, Roweis ST, Wong P, Emili A. 2007. Difference detection in LC‐MS data for protein biomarker discovery. Bioinformatics 23(2):E198–E204. [DOI] [PubMed] [Google Scholar]
- Liu NQ, Stingl C, Look MP, et al. 2014. Comparative proteome analysis revealing an 11‐protein signature for aggressive triple‐negative breast cancer. J Natl Cancer Inst. 106(2):d376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chang J, Liu M, et al. 2017. Quantitative proteomics profiling reveals activation of mTOR pathway in trastuzumab resistance. Oncotarget. 8(28):45793–45806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Q, Page JS, Tang KQ, Smith RD. 2007. MicroSPE‐nanoLC‐ESI‐MS/MS using 10‐mu m‐i.d. Silica‐based monolithic columns for proteomics. Anal Chem 79(2):540–545. [DOI] [PubMed] [Google Scholar]
- Ma F, Liu F, Xu W, Li L. 2018. Surfactant and chaotropic agent assisted sequential extraction/on pellet digestion (SCAD) for enhanced proteomics. J Proteome Res. 17(8):2744–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JHJ, Shen SC, Wang JJ, et al. 2017. Comparative proteomic analysis of the mitochondria‐associated ER membrane (MAM) in a long‐term type 2 diabetic rodent model. Sci Rep 7:2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick P, Kuster B. 2010. Proteomics: A pragmatic perspective. Nat Biotechnol. 28(7):695–709. [DOI] [PubMed] [Google Scholar]
- Mann M, Hendrickson RC, Pandey A. 2001. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 70:437–473. [DOI] [PubMed] [Google Scholar]
- Masuda T, Tomita M, Ishihama Y. 2008. Phase transfer surfactant‐aided trypsin digestion for membrane proteome analysis. J Proteome Res 7(2):731–740. [DOI] [PubMed] [Google Scholar]
- May D, Liu Y, Law W, et al. 2008. Peptide sequence confidence in accurate mass and time analysis and its use in complex proteomics experiments. J Proteome Res 7(12):5148–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer RL, Schwarzmeier JD, Gerner MC, et al. 2018. Proteomics and metabolomics identify molecular mechanisms of aging potentially predisposing for chronic lymphocytic leukemia. Mol Cell Proteomics. 17(2):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, et al. 2014. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 86(14):7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merl J, Ueffing M, Hauck SM, von Toerne C. 2012. Direct comparison of MS‐based label‐free and SILAC quantitative proteome profiling strategies in primary retinal Muller cells. Proteomics. 12(12):1902–1911. [DOI] [PubMed] [Google Scholar]
- Meyer JG, Schilling B. 2017. Clinical applications of quantitative proteomics using targeted and untargeted data‐independent acquisition techniques. Expert Rev Proteomics 14(5):419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski A, Cox J, Mann M. 2011. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data‐Dependent LC‐MS/MS. J Proteome Res 10(4):1785–1793. [DOI] [PubMed] [Google Scholar]
- Monroe ME, Tolic N, Jaitly N, Shaw JL, Adkins JN, Smith RD. 2007. VIPER: An advanced software package to support high‐throughput LC‐MS peptide identification. Bioinformatics. 23(15):2021–2023. [DOI] [PubMed] [Google Scholar]
- Moore SM, Hess SM, Jorgenson JW. 2016. Extraction, enrichment, solubilization, and digestion techniques for membrane proteomics. J Proteome Res 15(4):1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller LN, Rinner O, Schmidt A, et al. 2007. SuperHirn—A novel tool for high resolution LC‐MS‐based peptide/protein profiling. Proteomics. 7(19):3470–3480. [DOI] [PubMed] [Google Scholar]
- Nahnsen S, Bielow C, Reinert K, Kohlbacher O. 2013. Tools for label‐free peptide quantification. Mol Cell Proteomics. 12(3):549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nel AJ, Garnett S, Blackburn JM, Soares NC. 2015. Comparative reevaluation of FASP and enhanced FASP methods by LC‐MS/MS. J Proteome Res. 14(3):1637–1642. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI. 2010. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics. 73(11):2092–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, McLachlan GJ, Hill MM. 2017. Statistical evaluation of labeled comparative profiling proteomics experiments using permutation test. Methods Mol Biol. 1549:109–117. [DOI] [PubMed] [Google Scholar]
- Noble WS. 2009. How does multiple testing correction work? Nat Biotechnol. 27(12):1135–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JL, Porter NA, Caprioli RM. 2003. Mass spectrometry of intracellular and membrane proteins using cleavable detergents. Anal Chem. 75(23):6642–6647. [DOI] [PubMed] [Google Scholar]
- Nounou MN, Bakshi BR, Goel PK, Shen X. 2002. Bayesian principal component analysis. J Chemom 16(11):576–595. [Google Scholar]
- Nouri‐Nigjeh E, Sukumaran S, Tu C, et al. 2014. Highly multiplexed and reproducible ion‐current‐based strategy for large‐scale quantitative proteomics and the application to protein expression dynamics induced by methylprednisolone in 60 rats. Anal Chem 86(16):8149–8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Old WM, Meyer‐Arendt K, Aveline‐Wolf L, et al. 2005. Comparison of label‐free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics 4(10):1487–1502. [DOI] [PubMed] [Google Scholar]
- Olszowy PP, Burns A, Ciborowski PS. 2013. Pressure‐assisted sample preparation for proteomic analysis. Anal Biochem 438(1):67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, et al. 2002. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1(5):376–386. [DOI] [PubMed] [Google Scholar]
- Onile OS, Calder B, Soares NC, Anumudu CI, Blackburn JM. 2017. Quantitative label‐free proteomic analysis of human urine to identify novel candidate protein biomarkers for schistosomiasis. PLoS Negl Trop Dis 11(11):e0006045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, Furlong MT, Wu S, et al. 2012. Pellet digestion: A simple and efficient sample preparation technique for LC‐MS/MS quantification of large therapeutic proteins in plasma. Bioanalysis 4(1):17–28. [DOI] [PubMed] [Google Scholar]
- Ow SY, Salim M, Noirel J, Evans C, Rehman I, Wright PC. 2009. ITRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly. J Proteome Res 8(11):5347–5355. [DOI] [PubMed] [Google Scholar]
- Paoletti AC, Parmely TJ, Tomomori‐Sato C, et al. 2006. Quantitative proteomic analysis of distinct mammalian mediator complexes using normalized spectral abundance factors. Proc Natl Acad Sci USA 103(50):18928–18933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Yates JR, 3rd . 2010. Census for proteome quantification. Curr Protoc Bioinformatics. Chapter 13:Unit 13.12. 11–11. [DOI] [PubMed] [Google Scholar]
- Pascovici D, Handler DC, Wu JX, Haynes PA. 2016. Multiple testing corrections in quantitative proteomics: A useful but blunt tool. Proteomics 16(18):2448–2453. [DOI] [PubMed] [Google Scholar]
- Pavelka N, Fournier ML, Swanson SK, et al. 2008. Statistical similarities between transcriptomics and quantitative shotgun proteomics data. Mol Cell Proteomics 7(4):631–644. [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. 1999. Probability‐based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20(18):3551–3567. [DOI] [PubMed] [Google Scholar]
- Pernemalm M, Lewensohn R, Lehtio J. 2009. Affinity prefractionation for MS‐based plasma proteomics. Proteomics 9(6):1420–1427. [DOI] [PubMed] [Google Scholar]
- Polpitiya AD, Qian WJ, Jaitly N, et al. 2008. DAnTE: A statistical tool for quantitative analysis of −omics data. Bioinformatics 24(13):1556–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potriquet J, Laohaviroj M, Bethony JM, Mulvenna J. 2017. A modified FASP protocol for high‐throughput preparation of protein samples for mass spectrometry. PLoS ONE 12(7):e0175967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Straubinger RM. 2005. Improved sensitivity for quantification of proteins using triply charged cleavable isotope‐coded affinity tag peptides. Rapid Commun Mass Spectrom 19(19):2857–2864. [DOI] [PubMed] [Google Scholar]
- Raynie DE. 2010. Modern extraction techniques. Anal Chem 82(12):4911–4916. [DOI] [PubMed] [Google Scholar]
- Reiter L, Claassen M, Schrimpf SP, et al. 2009. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics 8(11):2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogeberg M, Vehus T, Grutle L, Greibrokk T, Wilson SR, Lundanes E. 2013. Separation optimization of long porous‐layer open‐tubular columns for nano‐LC‐MS of limited proteomic samples. J Sep Sci 36(17):2838–2847. [DOI] [PubMed] [Google Scholar]
- Ronsein GE, Pamir N, von Haller PD, et al. 2015. Parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics. J Proteom 113:388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJ. 2012. High‐sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat Methods 9(11):1084–1086. [DOI] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN, et al. 2004. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine‐reactive isobaric tagging reagents. Mol Cell Proteomics 3(12):1154–1169. [DOI] [PubMed] [Google Scholar]
- Rost HL, Rosenberger G, Navarro P, et al. 2014. OpenSWATH enables automated, targeted analysis of data‐independent acquisition MS data. Nat Biotechnol 32(3):219–223. [DOI] [PubMed] [Google Scholar]
- Roxas BA, Li Q. 2008. Significance analysis of microarray for relative quantitation of LC/MS data in proteomics. BMC Bioinformatics 9:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadygov RG, Maroto FM, Huhmer AF. 2006. ChromAlign: A two‐step algorithmic procedure for time alignment of three‐dimensional LC‐MS chromatographic surfaces. Anal Chem 78(24):8207–8217. [DOI] [PubMed] [Google Scholar]
- Saei AA, Sabatier P, Tokat UG, Chernobrovkin A, Pirmoradian M, Zubarev RA. 2018. Comparative proteomics of dying and surviving cancer cells improves the identification of drug targets and sheds light on cell Life/Death decisions. Mol Cell Proteomics 17(6):1144–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin M, Teleman J, Malmstrom J, Levander F. 2014. Data processing methods and quality control strategies for label‐free LC‐MS protein quantification. Biochim Biophys Acta 1844(1 Pt A):29–41. [DOI] [PubMed] [Google Scholar]
- Savas JN, Stein BD, Wu CC, Yates JR, 3rd . 2011. Mass spectrometry accelerates membrane protein analysis. Trends Biochem Sci 36(7):388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Wilhelm M, Hahne H, Kuster B, Bantscheff M. 2015. A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol Cell Proteomics 14(9):2394–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling B, Rardin MJ, MacLean BX, et al. 2012. Platform‐independent and label‐free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline application to protein acetylation and phosphorylation. Mol Cell Proteomics 11(5):202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle M, Bantscheff M, Kuster B. 2012. Mass spectrometry‐based proteomics in preclinical drug discovery. Chem Biol 19(1):72–84. [DOI] [PubMed] [Google Scholar]
- Schneider T. 2001. Analysis of incomplete climate data: Estimation of mean values and covariance matrices and imputation of missing values. J Clim 14(5):853–871. [Google Scholar]
- Schwammle V, Leon IR, Jensen ON. 2013. Assessment and improvement of statistical tools for comparative proteomics analysis of sparse data sets with few experimental replicates. J Proteome Res 12(9):3874–3883. [DOI] [PubMed] [Google Scholar]
- Serang O, Cansizoglu AE, Kall L, Steen H, Steen JA. 2013. Nonparametric Bayesian evaluation of differential protein quantification. J Proteome Res 12(10):4556–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, An B, Wang X, et al. 2018a. A surfactant cocktail‐aided extraction/precipitation/on‐pellet digestion (SEPOD) strategy enables rapid, efficient and reproducible sample preparation for large‐Scale quantitative proteomics. Anal Chem. [DOI] [PubMed] [Google Scholar]
- Shen SC, Jiang XS, Li J, et al. 2016. Large‐scale, ion‐current‐based proteomic investigation of the rat striatal proteome in a model of short‐ and long‐term cocaine withdrawal. J Proteome Res 15(5):1702–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Hu Q, Li J, Wang J, Qu J. 2015. Experimental null method to guide the development of technical procedures and to control false‐positive discovery in quantitative proteomics. J Proteome Res 14(10):4147–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Shen S, Li J, et al. 2017a. An IonStar experimental strategy for MS1 ion current‐Based quantification using ultrahigh‐field orbitrap: Reproducible, In‐Depth, and accurate protein measurement in large cohorts. J Proteome Res 16(7):2445–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Shen S, Li J, et al. 2018b. IonStar enables high‐precision, low‐missing‐data proteomics quantification in large biological cohorts. Proc Natl Acad Sci USA 115(21):E4767–E4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XM, Nair B, Mahajan SD, et al. 2015b. New insights into the disease progression control mechanisms by comparing long‐Term‐Nonprogressors versus normal‐progressors among HIV‐1‐positive patients using an ion current‐Based MS1 proteomic profiling. J Proteome Res 14(12):5225–5239. [DOI] [PubMed] [Google Scholar]
- Shen Y, Tolic N, Piehowski PD, et al. 2017b. High‐resolution ultrahigh‐pressure long column reversed‐phase liquid chromatography for top‐down proteomics. J Chromatogr A. 1498:99–110. [DOI] [PubMed] [Google Scholar]
- Shen YF, Strittmatter EF, Zhang R, et al. 2005a. Making broad proteome protein measurements in 1‐5min using high‐speed RPLC separations and high‐accuracy mass measurements. Anal Chem 77(23):7763–7773. [DOI] [PubMed] [Google Scholar]
- Shen YF, Zhang R, Moore RJ, et al. 2005b. Automated 20 kpsi RPLC‐MS and MS/MS with chromatographic peak capacities of 1000–1500 and capabilities in proteomics and metabolomics. Anal Chem 77(10):3090–3100. [DOI] [PubMed] [Google Scholar]
- Shen YF, Zhao R, Berger SJ, Anderson GA, Rodriguez N, Smith RD. 2002. High‐efficiency nanoscale liquid chromatography coupled on‐line with mass spectrometry using nanoelectrospray ionization for proteomics. Anal Chem 74(16):4235–4249. [DOI] [PubMed] [Google Scholar]
- Shruthi BS, Vinodhkumar P, Selvamani. 2016. Proteomics: A new perspective for cancer. Adv Biomed Res 5:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. 2018. Cancer statistics, 2018. CA Cancer J Clin 68(1):7–30. [DOI] [PubMed] [Google Scholar]
- Sielaff M, Kuharev J, Bohn T, et al. 2017. Evaluation of FASP, SP3, and iST protocols for proteomic sample preparation in the low microgram range. J Proteome Res 16(11):4060–4072. [DOI] [PubMed] [Google Scholar]
- Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. 2006. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem 78(3):779–787. [DOI] [PubMed] [Google Scholar]
- Smith R, Ventura D, Prince JT. 2015. LC‐MS alignment in theory and practice: A comprehensive algorithmic review. Brief Bioinform 16(1):104–117. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Limma: linear models for microarray data In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, eds. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer, New York, NY: Statistics for Biology and Health; 2005:397–420. [Google Scholar]
- Sonnett M, Yeung E, Wuhr M. 2018. Accurate, sensitive, and precise multiplexed proteomics using the complement reporter ion cluster. Anal Chem 90(8):5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100(16):9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm M, Bertsch A, Gropl C, et al. 2008. OpenMS‐An open‐source software framework for mass spectrometry. Bmc Bioinformatics 9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomi T, Corthals GL, Nevalainen OS, Elo LL. 2015. Using peptide‐level proteomics data for detecting differentially expressed proteins. J Proteome Res 14(11):4564–4570. [DOI] [PubMed] [Google Scholar]
- Tabb DL, Fernando CG, Chambers MC. 2007. MyriMatch: Highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res 6(2):654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Schafer J, Kuhn K, et al. 2003. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS (vol 15, pg1895, 2003). Anal Chem 75(18):4942–4942. [DOI] [PubMed] [Google Scholar]
- Ting L, Cowley MJ, Hoon SL, Guilhaus M, Raftery MJ, Cavicchioli R. 2009. Normalization and statistical analysis of quantitative proteomics data generated by metabolic labeling. Mol Cell Proteomics 8(10):2227–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting YS, Egertson JD, Bollinger JG, et al. 2017. PECAN: Library‐free peptide detection for data‐independent acquisition tandem mass spectrometry data. Nat Methods 14(9):903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping ME, Bishop CM. 1999. Probabilistic principal component analysis. J R Stat Soc 61(3):611–622. [Google Scholar]
- Titz B, Elamin A, Martin F, et al. 2014. Proteomics for systems toxicology. Comput Struct Biotechnol J 11(18):73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou CC, Avtonomov D, Larsen B, et al. 2015. DIA‐Umpire: Comprehensive computational framework for data‐independent acquisition proteomics. Nat Methods 12(3):258–264. 257 p following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C, Li J, Sheng Q, Zhang M, Qu J. 2014a. Systematic assessment of survey scan and MS2‐based abundance strategies for label‐free quantitative proteomics using high‐resolution MS data. J Proteome Res 13(4):2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C, Mojica W, Straubinger RM, et al. 2017a. Quantitative proteomic profiling of paired cancerous and normal colon epithelial cells isolated freshly from colorectal cancer patients. Proteomics Clin Appl 11:(5–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C, Shen S, Sheng Q, Shyr Y, Qu J. 2017b. A peptide‐retrieval strategy enables significant improvement of quantitative performance without compromising confidence of identification. J Proteomics 152:276–282. [DOI] [PubMed] [Google Scholar]
- Tu C, Sheng Q, Li J, et al. 2014b. ICan: An optimized ion‐current‐based quantification procedure with enhanced quantitative accuracy and sensitivity in biomarker discovery. J Proteome Res 13(12):5888–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu CJ, Mammen MJ, Li J, et al. 2014c. Large‐Scale, ion‐current‐bsed proteomics investigation of bronchoalveolar lavage fluid in chronic obstructive pulmonary disease patients. J Proteome Res 13(2):627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98(9):5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J. 2016. The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc 11(12):2301–2319. [DOI] [PubMed] [Google Scholar]
- Valikangas T, Suomi T, Elo LL. 2018. A systematic evaluation of normalization methods in quantitative label‐free proteomics. Brief Bioinform 19(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wu WW, Zhang Z, Masilamani S, Shen R‐F. 2009. Decoy methods for assessing false positives and false discovery rates in shotgun proteomics. Anal Chem 81(1):146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Shi T, Qian WJ, et al. 2016. The clinical impact of recent advances in LC‐MS for cancer biomarker discovery and verification. Expert Rev Proteomics 13(1):99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang L, Yang P, Chen G. 2008. Infrared‐assisted tryptic proteolysis for peptide mapping. Proteomics 8(13):2579–2582. [DOI] [PubMed] [Google Scholar]
- Wang X, Niu J, Li J, et al. 2018. Temporal effects of combined birinapant and paclitaxel on pancreatic cancer cells investigated via large‐scale, ion‐current‐based quantitative proteomics (IonStar). Mol Cell Proteomics 17(4):655–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasinger VC, Zeng M, Yau Y. 2013. Current status and advances in quantitative proteomic mass spectrometry. Int J Proteomics. 2013:180605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb‐Robertson B‐JM, Matzke MM, Datta S, et al. 2014. Bayesian proteoform modeling improves protein quantification of global proteomic measurements. Mol Cell Proteomics 13(12):3639–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb‐Robertson BJ, Wiberg HK, Matzke MM, et al. 2015. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry‐based label‐free global proteomics. J Proteome Res 14(5):1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisser H, Choudhary JS. 2017. Targeted feature detection for data‐dependent shotgun proteomics. J Proteome Res 16(8):2964–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger CD, Coon JJ. 2013. A proteomics search algorithm specifically designed for high‐resolution tandem mass spectra. J Proteome Res 12(3):1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter D, Steen H. 2011. Optimization of cell lysis and protein digestion protocols for the analysis of HeLa S3 cells by LC‐MS/MS. Proteomics 11(24):4726–4730. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR. 2016. Quantitative evaluation of filter aided sample preparation (FASP) and multienzyme digestion FASP protocols. Anal Chem 88(10):5438–5443. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Dus‐Szachniewicz K, Ostasiewicz P, Ziolkowski P, Rakus D, Mann M. 2015. Absolute proteome analysis of colorectal mucosa, adenoma, and cancer reveals drastic changes in fatty acid metabolism and plasma membrane transporters. J Proteome Res 14(9):4005–4018. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Mann M. 2012. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal Chem 84(6):2631–2637. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Mann M. 2016. A proteomics approach to the protein normalization problem: selection of unvarying proteins for MS‐based proteomics and western blotting. J Proteome Res 15(7):2321–2326. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nature Methods 6(5):359‐U360. [DOI] [PubMed] [Google Scholar]
- Xie F, Liu T, Qian WJ, Petyuk VA, Smith RD. 2011. Liquid chromatography‐mass spectrometry‐based quantitative proteomics. J Biol Chem 286(29):25443–25449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B, Zhao J, Brown JS, Blackwell J, Carr PW. 2000. High‐temperature ultrafast liquid chromatography. Anal Chem 72(6):1253–1262. [DOI] [PubMed] [Google Scholar]
- Yao W, Yin X, Hu Y. 2007. A new algorithm of piecewise automated beam search for peak alignment of chromatographic fingerprints. J Chromatogr A 1160(1‐2):254–262. [DOI] [PubMed] [Google Scholar]
- Ye X, Li L. 2012. Microwave‐assisted protein solubilization for mass spectrometry‐based shotgun proteome analysis. Anal Chem 84(14):6181–6191. [DOI] [PubMed] [Google Scholar]
- Yi L, Piehowski PD, Shi TJ, Smith RD, Qian WJ. 2017. Advances in microscale separations towards nanoproteomics applications. J Chromatogr A 1523:40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Kall L, Zubarev RA. 2016a. DeMix‐Q: quantification‐Centered data processing workflow. Mol Cell Proteomics 15(4):1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Pirmoradian M, Zubarev R, Kall L. 2017. Covariation of peptide abundances accurately reflects protein concentration differences. Mol Cell Proteomics 16(5):936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, VerBerkmoes NC, Langston MA, Uberbacher E, Hettich RL, Samatova NF. 2006. Detecting differential and correlated protein expression in label‐free shotgun proteomics. J Proteome Res 5(11):2909–2918. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xin L, Shan B, et al. 2012. PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics 11(4):M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JQ, Gonzalez E, Hestilow T, Haskins W, Huang YF. 2009. Review of peak detection algorithms in liquid‐chromatography‐mass spectrometry. Current Genomics 10(6):388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu Z, Wang Y, et al. 2016b. Quantitative proteomics reveals molecular mechanism of gamabufotalin and its potential inhibition on Hsp90 in lung cancer. Oncotarget 7(47):76551–76564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, An B, Qu Y, et al. 2018. Sensitive, high‐throughput, and robust trapping‐micro‐LC‐MS strategy for the quantification of biomarkers and antibody biotherapeutics. Anal Chem 90(3):1870–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Faca V, Hanash S. 2011. Mining the plasma proteome for disease applications across seven logs of protein abundance. J Proteome Res 10(1):46–50. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wen Z, Washburn MP, Florens L. 2010. Refinements to label free proteome quantitation: How to deal with peptides shared by multiple proteins. Anal Chem 82(6):2272–2281. [DOI] [PubMed] [Google Scholar]
- Zhou JY, Dann GP, Shi T, et al. 2012a. Simple sodium dodecyl sulfate‐assisted sample preparation method for LC‐MS‐based proteomics applications. Anal Chem 84(6):2862–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou WD, Liotta LA, Petricoin EF. 2012b. The spectra count label‐free quantitation in cancer proteomics. Cancer Genomics Proteomics 9(3):135–142. [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Shen XM, Qu J, Straubinger RM, Jusko WJ. 2018a. Proteomic analysis of combined gemcitabine and birinapant in pancreatic cancer cells. Front Pharmacol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Zhao R, Piehowski PD, et al. 2018b. Subnanogram proteomics: Impact of LC column selection, MS instrumentation and data analysis strategy on proteome coverage for trace samples. Int J Mass Spectrom 427:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]