

Summary
Amplicon sequencing of the 16S rRNA gene is the predominant method to quantify microbial compositions and to discover novel lineages. However, traditional short amplicons often do not contain enough information to confidently resolve their phylogeny. Here we present a cost‐effective protocol that amplifies a large part of the rRNA operon and sequences the amplicons with PacBio technology. We tested our method on a mock community and developed a read‐curation pipeline that reduces the overall read error rate to 0.18%. Applying our method on four environmental samples, we captured near full‐length rRNA operon amplicons from a large diversity of prokaryotes. The method operated at moderately high‐throughput (22286–37,850 raw ccs reads) and generated a large amount of putative novel archaeal 23S rRNA gene sequences compared to the archaeal SILVA database. These long amplicons allowed for higher resolution during taxonomic classification by means of long (∼1000 bp) 16S rRNA gene fragments and for substantially more confident phylogenies by means of combined near full‐length 16S and 23S rRNA gene sequences, compared to shorter traditional amplicons (250 bp of the 16S rRNA gene). We recommend our method to those who wish to cost‐effectively and confidently estimate the phylogenetic diversity of prokaryotes in environmental samples at high throughput.
Introduction
The 16S rRNA gene has been used for decades to phylogenetically classify bacteria and archaea (Woese and Fox, 1977). The gene excels in this respect because of its universal occurrence, resistance to horizontal gene transfer and high degree of conservation (Woese, 1987; Green and Noller, 1997). Highly conserved regions are interspersed with highly variable regions, allowing for phylogenetic classification at species and higher taxonomic levels. In addition, the gene has proven to be an excellent target for studies aiming to quantify the taxonomic composition of microbial communities via high‐throughput PCR amplicon sequencing (Doolittle, 1999). Primers are usually designed such that they anneal to stretches of conserved sites that flank a variable region, in effect of capturing the informative variable region of a large fraction of the microbial community. 16S rRNA gene amplicon surveys are now a standard method in microbial ecology and have led to important insights into the taxonomic makeup of many different environments. Examples include oceanic waters (Sogin et al., 2006), deep sea sediments (Jorgensen et al., 2012), hot springs (Hou et al., 2013) and the human gut (Turnbaugh et al., 2007).
Despite its many advantages, the 16S rRNA gene is limited in its number of phylogenetically informative sites. 16S rRNA gene‐based phylogenetic analyses are therefore sensitive to stochastic error and exhibit limited resolution (Brown et al., 2001; Delsuc et al., 2005). Studies aiming to resolve deeper evolutionary relationships between taxa often favour large data sets of conserved protein coding genes over the 16S rRNA gene to overcome such error and increase resolution (Brown et al., 2001; Wolf et al., 2001; Brochier et al., 2002; Matte‐Tailliez et al., 2002). Another frequently used method is to concatenate the 16S rRNA gene with the larger 23S rRNA gene (Williams et al., 2012; Ferla et al., 2013; Zaremba‐Niedzwiedzka et al., 2017). However, both methods typically require that genome sequences are available for the taxa in question. While it is now possible to obtain genomic data via metagenomic binning (Albertsen et al., 2013; Alneberg et al., 2014), relatively expensive deep sequencing and computationally demanding metagenome assembly is required. In addition, draft genomes acquired via metagenomic binning approaches often lack rRNA genes (Hugenholtz et al., 2016; Nelson and Mobberley, 2017), which obstructs linking the obtained metagenomic bins to environmental lineages observed in 16S rRNA amplicon surveys. One possible solution is to obtain 16S and 23S rRNA gene sequences simultaneously via PCR amplicon sequencing. This approach exploits that both rRNA genes are often neighbouring (67% in known bacterial genomes, 74% in known archaeal genomes—Supporting Information Table S1). However, sequences generated by currently available high‐throughput sequencing methods are too short to capture such long amplicons.
With the introduction of Pacific Bioscience's single‐molecule real‐time (SMRT) sequencing technology, sequencing long amplicons at moderately high throughput became realistic. Its relatively high sequencing error rates (15%) are now substantially reduced via circular consensus sequencing (ccs). A number of pioneering studies have already used the technology to successfully obtain near full‐length 16S rRNA gene sequences with low error rates (Schloss et al., 2016; Singer et al., 2016; Wagner et al., 2016). Here we go one step further and obtain near full‐length 16S and 23S rRNA genes by sequencing a large part of the rRNA operon. We develop a read curation pipeline that deals with PacBio‐specific issues and evaluate error rates with a phylogenetically diverse mock community. We apply our method to four diverse environmental samples and compare our method with classic partial 16S rRNA amplicon sequencing with respect to taxonomic classification and resolving deeper phylogenetic relationships of novel taxa.
Results and discussion
Reducing the mean error rates
Here we present a method for generating and sequencing amplicons of approximately 4000 bp containing near full‐length 16S and 23S rRNA genes from environmental taxa. Because the method uses PacBio sequencing technology, which exhibits higher error rates compared to Illumina, we developed a read curation pipeline (Fig. 1 and Supporting Information Fig. S1) that attempts to reduce the mean error rate. To evaluate the error rate, we applied the method to a synthetic ‘mock community’ composed of the genomic DNA of 38 phylogenetically diverse archaeal and bacterial lineages for which complete genomes are available that encode at least one 16S‐ITS‐23S cluster. A single SMRT cell RSII run generated 22,286 ccs reads. We observed a mean error rate of 2.45%. The large majority of errors were deletions (77.7%), followed by insertions (15.8%) and substitutions (6.5%) (Fig. 1). Of these, 868 ccs reads (3.9%) had no errors.
Figure 1.
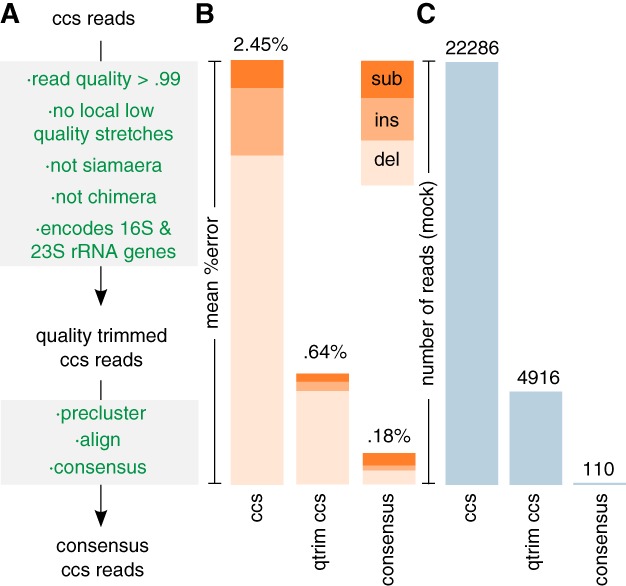
A. Read curation pipeline. ‘ccs’ = circular consensus sequence, ‘qtrim’ = quality trimmed. For a more detailed overview of the pipeline see Supporting Information Fig. S1.
B. Observed mean error rates. ‘del’ = deletions, ‘ins’ = insertions, ‘sub’ = substitutions.
C. Number of (remaining) ccs reads of the mock community per stage of the read curation pipeline.
We sought to reduce the error rate by removing reads that are highly erroneous because of several reasons. First, as was observed by (Schloss et al., 2016), error rates were strongly correlated with the ‘read quality’ values that are calculated by the SMRT analysis software (Supporting Information Fig. S2). We removed 11,664 reads (52.3%) with an associated read quality value of lower than 0.99. Second, we removed 106 high quality reads (1.0%) containing local stretches (≥ 30 bp) of consecutive low‐quality base‐calls (Phred ≤18) with an enriched number of errors (Supporting Information Fig. S3). Third, we observed 1201 high quality ccs reads (11.3%) that have been previously referred to as ‘siamaeras’ (Hackl et al., 2014). The first half of these reads consists of the expected amplicon, while the second half consists of the reverse complement of the first half, but missing a primer at the breakpoint (Supporting Information Fig. S4). Siamaeras most likely stem from damaged amplicons that have a long overhang. The overhang forms a hairpin, which anneals to the complementary strand. As a result, the SMRTbell adapter is blocked from ligating there during the library preparation, and the read processing software will interpret the concatenation of both strands as a single insert (Supporting Information Fig. S4). Since siamaeras are about twice the expected read length, they are easily detected and removed by setting a length cutoff (here 5 kbp). However, this does not remove all siamaeras. Some stem from partial rather than full amplicons and may as a result be shorter than the length cutoff. Partial amplicons (which start or end with a primer and cover ≤3500 bp of the entire locus) account for a substantial fraction (3727 ccs reads, 35.1%) of the high‐quality ccs reads. The cause for these type of ccs reads is unclear. To detect partial siamaeras, we use another property of siamaeras: that they start and end with the same primer. Curiously, the large majority (1185 ccs reads, 98.7%) of identified siamaeras start and end with the reverse primer, implying that choice of primers affects siamaera formation. Fourth, since our method is PCR based, we need to detect and remove chimeras. Chimeras are formed when incomplete extensions from one locus or amplicons from that locus anneal to another locus or amplicons from that locus, either within the same genome or on another genome. A frequently used method to detect chimeras uses a de novo approach (Edgar et al., 2011). A query read is split into four equally sized ‘chunks’ and compared against more abundant reads of the same dataset to find two candidate parents. If an artificial sequence constructed of fragments from the two parents is more similar to the query than each original parent, and at least one of the parent reads is at least two times more abundant than the query, the read is deemed chimeric. The method thus assumes that chimeric amplicons are less abundant than non‐chimeric amplicons. This assumption works well for Illumina data, but needs to be adjusted for PacBio data. The lower throughput, longer reads and higher error rate mean that virtually all reads are unique (and thus have an abundance of ‘one’). As a result, a chimera will have the same abundance as a parent and will not be detected. In addition, because by default only four chunks are used, chimeras with breakpoints in the first or last ∼1000 bp may be missed. Thus, to account for the nature of the PacBio data, we used an abundance ratio threshold of one and increased the number of chunks to 16. Among the remaining 5030 reads, 91 (1.8%) were identified as chimeric. Finally, since some reads may stem from loci other than 16S‐ITS‐23S, we removed 23 reads (0.5%) that did not contain both genes. After these operations, the error rate was reduced to 0.64% (Fig. 1). Deletions were still the most prominent type of error (84.4%), followed by insertions (8.3%) and substitutions (7.2%). Among these ‘qtrim ccs’ reads, only 1 was without error. The other 867 raw ccs reads without error were short (≤ 3 kbp, median 1001 bp) and removed by the pipeline. Though a substantially improved error rate, we recommend that users do not submit rRNA genes predicted from qtrim ccs reads to reference databases, as they may still contain a relatively large number of errors (∼8 per 16S rRNA gene, ∼15–18 per 23S rRNA gene).
We sought to reduce the error rate further. We argued that if the remaining read errors are still randomly distributed over the reads, then reads originating from the same locus or the same genome will have errors at different positions. As a result, true base‐calls would outnumber erroneous base‐calls per site and their consensus would have a reduced error rate. We grouped reads putatively originating from the same genome by clustering the reads at a 99% identity threshold. This is similar to the preclustering method used by Schloss and co‐workers (Schloss et al., 2016) that constructs a final set of high quality reads by selecting the most abundant (and hence most accurate) read per 99% precluster. Since there is virtually no abundance information of longer PacBio reads (there are nearly no identical reads), we preferred a consensus‐based method. When considering consensus‐ccs reads generated from 110 preclusters with at least three reads, the error rate dropped to 0.18%. Deletions were no longer as prominent (44.9%), now more comparable to insertions (17.5%) and substitutions (37.5%) (Fig. 1). Remaining deletions were often found after long homopolymers (≥ 3 bp) (unpublished observation), implying that deletion errors are not randomly distributed (also observed by Tedersoo et al., (2017)). Of these, 30 consensus‐ccs reads were without error (27.2%). It should be noted that our consensus method to reduce read error rate is appropriate for our mock community because all taxa are phylogenetically distinct. For environmental samples that likely contain a degree of strain microdiversity, 99% preclusters will most likely contain reads originating from various strains in addition to from various loci of the same strain. The consensus‐ccs reads will thus represent not the 16S‐ITS‐23S sequence of one strain or locus, but of multiple closely related strains and/or loci. Thus, we recommend that users only generate consensus‐ccs reads from samples with no microdiversity and do not submit rRNA genes derived from consensus‐ccs reads to reference databases.
Here, we made use of the PacBio RSII sequencer. During the execution of this study, PacBio has released the Sequel sequencer, which has a substantially higher throughput. We estimated that the Sequel is about 4× more cost effective than the RSII with respect to the protocol presented here. Since the principal sequencing chemistry remains unchanged, our protocol is perfectly suitable for the Sequel, and we expect similarly accurate reads but at a substantially higher throughput.
Taxonomic classification of environmental sample reads
We applied our method to four environmental samples: ‘TNS08’, a sediment sample of a shallow hydrothermal vent field (5083 qtrim ccs reads), ‘SALA’, a black biofilm from an old silver mine, (4069 qtrim ccs reads), ‘PM3’, a marine sediment sample (6950 qtrim ccs reads) and ‘P19’, a hot spring sediment sample (8634 qtrim ccs reads). Taxonomic classification was done by comparing the qtrim ccs reads to the SILVA SSU database. Note that percentages stated next do not necessarily reflect biological relative abundances; our method only captures taxa with 16S‐ITS‐23S loci and is subject to primer biases (see also Supporting Information). All samples are characterized by a relatively high fraction of archaeal sequences (Fig. 2 and Supporting Information Fig. S5). In particular, TNS08 appears to be dominated by archaeal sequences: 33.6% THSCG (Aigarchaeota), 11.8% ANME‐1 (Euryarchaeota), 11.6% Bathyarchaeota, 7.1% Thermoprotei (Crenarchaeota), 4.3% Group C3 (Thaumarchaeota) and 3.6% Candidate division YNPFFA. In addition, 23.6% of the reads stemmed from unclassified archaeal lineages. SALA on the other hand has a larger bacterial representation, including Nitrospirae (6.1%) and various lineages of Actinobacteria (8.7%) and Proteobacteria (18.0%; of which 32.6% Desulfurellaceae). Archaeal sequences are solely represented by Thaumarchaeota, including Marine Group I (50.5% of which 40.0% Candidatus Nitrosoarchaeum) and SAGMCG‐1 (3.1%). PM3 sequences were rich in archaeal lineages MBG‐D/Izemarchaea (32.3%; Euryarchaeota), Group C3 (19.1%; Thaumarchaeota) and Bathyarchaeota (3.5%). Atribacteria (5.5%), OPB41 (20.6%; sole representative of the Actinobacteria) and Anaerolineaceae (2.5%) constituted the most abundant bacterial sequences. Finally, archaea in P19 were mostly represented by Thaumarchaeota (unclassified Thaumarchaeota: 34.4%, AK59: 5.8%, OPPD003: 3.0%), Aigarchaeota (4.0%, solely represented by Candidatus Caldiarchaeum) and unclassified Archaea (5.0%), while bacterial sequences are mostly represented by 8.0% Venenivibrio (Aquificae), 7.6% Ignavibacteria, 5.1% Thermodesulfovibrio (Nitrospirae), 7.0% Dictyoglomus (Dictyoglomi) and 6.9% unclassified Bacteria.
Figure 2.
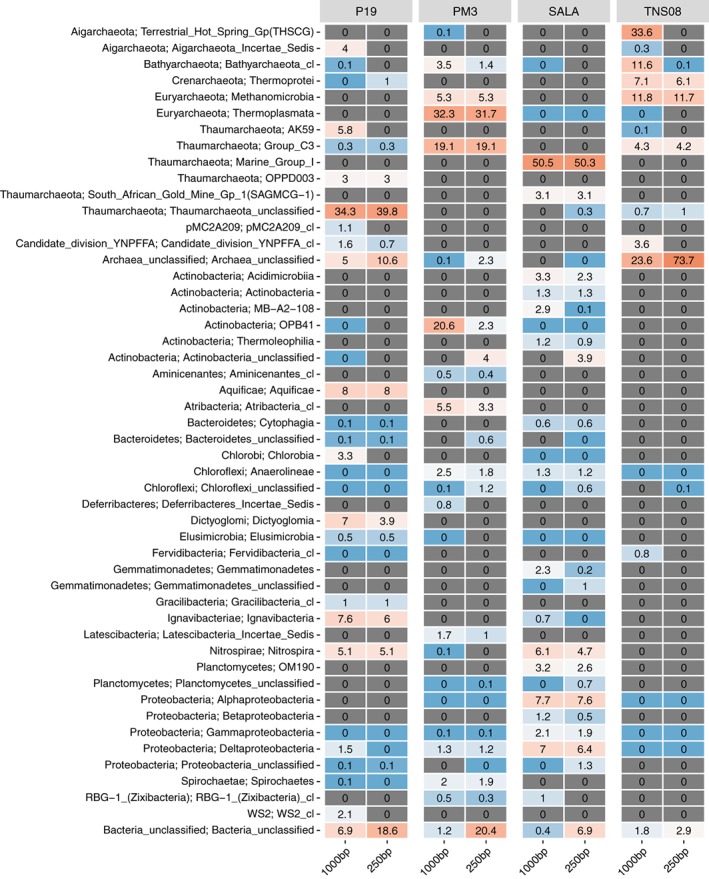
Relative abundance (%) estimates of environmental samples based on qtrim ccs reads. Top 50 most abundant lineages (phylum; class) across the four samples are shown. Colours in a blue‐to‐red gradient reflect low‐to‐high relative abundances. Unobserved lineages are indicated with grey.
Long‐read information increases taxonomic resolution
We compared the taxonomic resolution obtained from near full‐length 16S rRNA genes encoded on the ccs reads (∼1000 bp) with that of shorter 16S rRNA gene fragments (250 bp) that simulate more common contemporary Illumina‐like read lengths. The estimated taxonomic compositions using 250 bp fragments (Fig. 2 and Supporting Information Fig. S5) generally resemble that of ∼1000 bp fragments. However, large differences are found at assignments to the unclassified Archaea and unclassified bacteria phyla: P19 (10.6% vs 5.0%) and TNS08 (73.7% vs 23.6%) see a large decrease in unclassified Archaea assignments when using ∼1000 bp fragments, while P19 (18.6% vs 6.9%), PM3 (20.4% vs 1.2%) and SALA (6.9% vs 0.4%) see a large decrease in unclassified bacteria assignments. Taxonomic assignments seem to transfer particularly to AK59 Thaumarchaeota (0 vs 5.8%) and Chlorobia (0 vs 3.3%) in P19, OPB41 (2.3 vs 20.6%) in PM3 and THSCG (0 vs 33.6%) and Bathyarchaeota (0.1 vs 11.6%) in TNS08 when using ∼1000 bp fragments. The extra sequence information can thus aid with the identification of more specified lineages in particular samples that would otherwise not be classified. This result is in‐line with (Schloss et al., 2016), which showed that the fraction of reads classified at genus or species levels increased with increasing read coverage of the 16S rRNA gene.
Phylogenetic diversity of captured clades in the environmental samples
When investigating environmental samples with a large degree of novel diversity, taxonomic classification can yield a good general overview of present higher order taxonomic levels. However, their degree of phylogenetic diversity and their respective placements in the tree of life often remain unresolved. Here, we clustered the qtrim ccs reads into 97% OTUs per sample and incorporated rRNA genes predicted from the centroids into separate bacterial and archaeal, concatenated ‘16S + 23S’ phylogenetics data sets, which further comprised representative reference bacterial and archaeal 16S and 23S rRNA gene sequences, and inferred maximum likelihood phylogenies (Fig. 3 and Supporting Information Figs. S6, S7). TNS08 reads are characterized by a rich archaeal diversity of Crenarchaeota (mainly Thermoprotei and Candidate division YNPFFA), Aigarchaeota (mainly THSCG), some diversity of Thaumarchaeota (mainly Group C3 and unclassified Thaumarchaeota), Bathyarchaeota and unclassified Archaea. SALA reads, despite being dominated by Marine Group I archaea (Fig. 2), feature a rich bacterial diversity of Proteobacteria, FCB group (Bacteroidetes, Gemmatimonadetes), Actinobacteria, PVC group (Planctomycetes, Omnitrophica), Chloroflexi, Nitrospirae and Acidobacteria. PM3 reads are home to both diverse archaea and bacteria. Among archaea, we observed Bathyarchaeota and Asgard archaea, as well as a diverse clade sister to Bathyarchaeota and a rich microdiversity of Izemarchaea (Marine Benthic Group D; part of ‘Thermoplasmata’ in Fig. 2). Among bacteria, we observed several lineages with extensive (micro)diversity closely related to, respectively, deltaproteobacterium Desulfatiglans anilini, CPR bacterium ‘Aminicenantes bacterium SCGC AAA252‐G21’, ‘Latescibacteria bacterium SCGC AAA252‐D10’, Candidate division JS1, Bacteroidetes, Spirochaetes, Actinobacteria and Chloroflexi. P19 reads display an overall lesser degree of diversity and most notably include various diverse lineages related to Thaumarchaeota and Aigarchaeota, a lineage branching deep relative to MSBL1 archaea and moderately diverse bacterial lineages related to ‘Nitrospirae bacterium JdFR‐88’ and Dictyoglomus thermophilum.
Figure 3.
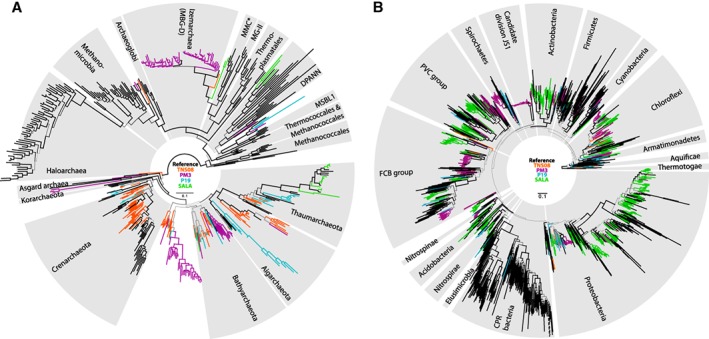
Captured archaeal (A) and bacterial (B) phylogenetic diversity in the environmental samples. Maximum likelihood phylogenetic trees (IQTREE, under GTR + R10) are based on the concatenation of 16S and 23S rRNA gene alignments. Branches represent reference rRNA gene sequences (black) and 97% OTU centroids of environmental rRNA gene sequences (orange: TNS08, purple: PM3, blue: P19, green: SALA). Grey branches to lead to clades present in multiple environmental samples. Branch thickness is proportional to non‐parametric bootstrap support. Branches that have been shortened for aesthetic purposes are dashed. *MMC: Methanomassiliicoccales. MG‐II: Marine Group II. MBG‐D: Marine Benthic Group D, MSBL1: Mediterranean Sea Brine Lakes 1.
Phylogenetic signal of 16S‐ITS‐23S reads
Combining the 16S and the 23S rRNA gene sequences in phylogenetic analyses should in theory increase the phylogenetic signal compared to using only the sequence of the 16S rRNA gene because the number of informative sites increases (Williams et al., 2012; Ferla et al., 2013; Zaremba‐Niedzwiedzka et al., 2017). To assess the degree of increase in phylogenetic signal of ‘16S‐ITS‐23S’ qtrim ccs reads compared to that of common ‘partial 16S’ reads with regards to phylogenetic placement, we incorporated rRNA genes predicted from the centroids in two additional phylogenetic data sets: ‘16S’, containing full‐length reference 16S rRNA genes, supplemented with near full‐length read predicted 16S rRNA genes, and ‘16S_250bp’, containing full‐length reference 16S rRNA genes, supplemented with read predicted 16S rRNA genes shortened to 250 bp (spanning the V4 region) to simulate Illumina‐like reads. Separate bacterial and archaeal data sets were constructed. We then inferred maximum likelihood phylogenies for all data sets and compared the overall topology and statistical support of the obtained trees between 16S + 23S, 16S and 16S_250bp data sets. For both archaea and bacteria, the 16S + 23S phylogeny featured, when compared to 16S and 16S_250bp phylogenies, (i) a general substantial increase in statistical support at both deep and shallow levels of the tree (Fig. 3, 4, Supporting Information Figs. S6, S7), (ii) a higher degree of monophyly of established clades and (iii) a clearer identification of clades not affiliated with any reference taxa (Supporting Information Figs. S6, S7). In addition, our analysis illustrates that the ‘16S + 23S’ phylogeny allows for the recognition of environmental lineages affiliated with clades of interests that in ‘16S_250bp’ and ‘16S’ phylogenies remain unresolved. For example, the position of OTUs 165, 189 and 287 of the PM3 sample were unresolved in the ‘16S_250bp’ (placed next to Euryarchaeota and DPANN archaea with weak support) and ‘16S’ phylogenies (placed within Asgard with negligible support) but were firmly identified as a member of the Asgard archaea in the ‘16S + 23S’ phylogeny (Supporting Information Fig. S6). Similarly, the position of OTUs 26, 92, 331, 457 and 484 of the SALA sample were unresolved in the ‘16S_250bp’ (placed among Delta‐ and Epsilonproteobacteria without support) and ‘16S’ phylogenies (placed within Alphaproteobacteria and Deltaproteobacteria with negligible support) but were firmly identified as the members of the Alphaproteobacteria in the ‘16S + 23S’ phylogeny (Supporting Information Fig. S7). This suggests that these lineages, which are potentially important for understanding the origins of eukaryotes and mitochondria, respectively, would not have been identified with standard 16S rRNA gene amplicon assays.
Figure 4.
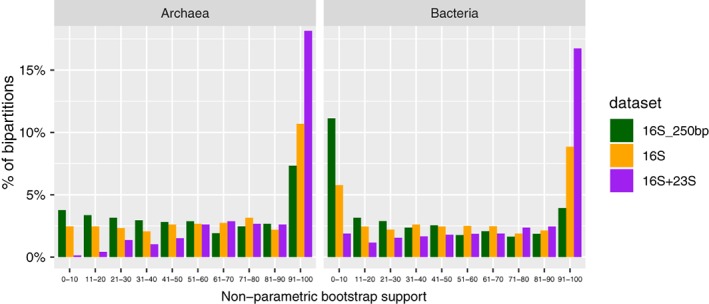
Non‐parametric bootstrap support distributions in maximum likelihood phylogenies of ‘16S_250bp’, ‘16S’ and ‘16S + 23S’ data sets (see Methods ‐ Phylogenetic analyses). Separate comparisons for Archaea and bacteria data sets are shown.
High throughput capture of 23S rRNA gene sequences
The 16S rRNA gene has long been a key tool in phylogenetic and ecological analyses (Lane et al., 1985; Weisburg et al., 1991). Previous studies have used PCR primers targeting the 16S rRNA gene to assess microbial diversity from various environments (Turnbaugh et al., 2007; Hou et al., 2013), and a large amount of (full or partial) 16S rRNA gene sequences in public databases has been collected. Although amplicon studies targeting the 23S rRNA gene have been performed in the past (Zimmermann et al., 2005; Hunt et al., 2006), the retrieval of 23S rRNA gene sequences relies mostly on genome sequencing projects. This is especially visible for the archaeal domain, for which only 1271 reference 23S rRNA sequences (distributed over 412 97% OTUs, calculated with the same clustering algorithm used for environmental reads) are available in the SILVA database [LSURef release 128; (Pruesse et al., 2012)]. This study generated archaeal 23S rRNA gene sequences from 16,069 qtrim ccs reads (generally between 2400 and 2800 bp long, and expected error rates of ∼0.6% ‐ Fig. 1), distributed over 263 97% OTUs. Bacterial LSURef is more extensive, with 130,923 entries distributed over 8707 97% OTUs. This work generated bacterial 23S rRNA gene sequences from 8635 qtrim ccs reads, distributed over 883 97% OTUs. Based on these comparisons, we conclude that our method has the potential to capture a large amount of novel 23S rRNA gene diversity, especially for archaea. We envision that the increased availability of near full‐length 23S rRNA gene sequences, in combination with linked near full‐length 16S rRNA gene sequences, could aid in resolving the phylogenies of particular bacterial and archaeal lineages of interest.
Internal transcribed spacer
In addition to the 16S and 23S rRNA genes, our method also captures the internal transcribed spacer (ITS). The ITS region occasionally harbours additional genes, often tRNA genes (Acinas et al., 2004). In case longer genes situated here are of interest, the length cutoff in the pipeline (here: 5000 bp) would have to be appropriately adjusted. In addition, the ITS region is typically subject to higher evolutionary rates and it is tempting to use the ITS to differentiate 16S and 23S rRNA genes stemming from different loci from the same strain or from different strains. However, there is a large variation in degree of ITS sequence divergence: on the one hand, different ITS copies between closely related genomes may be a 100% identical (Supporting Information Fig. S8). On the other hand, different ITS copies within the same genome may be <50% identical (Supporting Information Fig. S8). It is therefore not possible to distinguish with certainty whether ccs reads with identical or highly different ITS sequences stem from the same locus, same strain, or from different strains. Identical ccs reads may thus stem from different strains, while dissimilar ccs reads could stem from the same strain. We therefore caution to make any firm conclusions about levels of strain diversity based on this assay alone.
Conclusions
The method presented here allows users to generate good quality, near full‐length 16S and 23S rRNA gene sequences from environmental taxa and to use these to more confidently determine their taxonomic classification and phylogenetic context compared to standard, Illumina‐based 16S rRNA gene sequencing assays. Quality trimmed ccs reads are relatively low in sequencing error and could serve as reference sequences in future phylogenetic analyses. We envision that our method represents a cost‐effective approach for generation of novel near full‐length rRNA gene sequences and for a relatively straightforward, PCR‐based exploration of phylogenetic diversity of environmental samples. Combining 16S and 23S rRNA sequence information increases the phylogenetic resolution for both deeper branches (allowing one to more confidently identify novel deeply diverging lineages) and shallower branches (allowing one to study the phylogenetic relationships of closely related rRNA gene sequences) of the tree. Moreover, our method also captures the ITS region, which may contain additional genes of interest.
In addition to the benefits outlined above, our method can be used in complement with present‐day metagenomics approaches aiming to investigate the microbial diversity of environmental samples using high throughput sequencing techniques. While these methods have proven their use in the recent past, e.g. in their ability to reconstruct high‐quality draft genomes from metagenomic datasets using various binning strategies, they typically perform poorly with respect to assembling full length rRNA gene sequences. The underlying reason for this resides in the repetitive nature of rRNA gene sequences along with alternate sequence composition and coverage that confuse assembly and binning algorithms (Ghurye et al., 2016; Hugenholtz et al., 2016; Nelson and Mobberley, 2017). Near full‐length 16S and 23S rRNA sequence data generated by our method can be used to complement genome bins that are lacking these genes completely or partially.
Our method is restricted to taxa with neighbouring 16S and 23S rRNA genes. However, this limitation can be overcome by using primer sets that specifically target the 16S or the 23S rRNA gene. Such amplicons are expected to capture more diversity and exhibit lower mean error rates, but at the expense of 16S‐23S rRNA gene linkage information. In particular assays specifically targeting archaeal 23S rRNA genes could be valuable as current archaeal 23S rRNA gene databases are severely underrepresented. One can alternatively specifically sequence full‐length 16S rRNA genes with the recently developed method of (Karst et al., 2018). Their method is based on unique molecular sequence tagging, is high throughput, free of primer bias and operates at a low error rate (∼0.17%). However, their current protocol is more labor intensive and is based on Illumina synthetic long read sequencing which is limited to ∼2000 bp. It is thus unable to capture full‐length 23S rRNA genes or entire 16S‐ITS‐23S loci.
In the current study we have used PacBio long‐read sequencing technology to sequence long (∼4 kb) amplicons. However, other long‐read sequencing technologies such as Oxford Nanopore could be appropriate as well. Indeed, one study (Kerkhof et al., 2017) has already attempted to sequence 16S‐ITS‐23S loci with the MinION. However, their raw sequence data suffered from high error rates (81 ± 5% average similarity to references) and though a consensus method was proposed to reduce error rates, it was based on the assumption that reads with the same best BLAST hit stem from the same strain/locus. Its usefulness to obtain high quality 16S‐ITS‐23S sequences from novel lineages is thus questionable. In contrast, protocols such as the recently developed NanoAmpli‐Seq (Calus et al., 2018) present a promising way to obtain long and yet good quality sequences. NanoAmpli‐Seq entails a similar consensus sequence generating step as presented here which can reduce read error rates from ∼2.0% to ∼0.5%. However, as discussed above, when applied to complex natural communities there is a risk of generating multispecies or multiloci consensus sequences. Reads stemming from the exact same 16S‐ITS‐23S locus could potentially be recognized through a unique molecular sequence tag method in combination with Oxford Nanopore sequencing, as was briefly investigated by (Karst et al., 2018). However, current throughput with the MinION appears to be too low to retrieve sufficient number of reads per molecular tag, and error rates did not drop below ∼1%.
Experimental procedures
Fraction of reference genomes with neighbouring 16S and 23S rRNA genes
We estimated the fraction of the natural diversity of bacteria and archaea with neighbouring 16S and 23S rRNA genes by using all reference genomes available in NCBI RefSeq (May 2017; 12,596 bacteria and 364 archaea, one representative per species). We required that the 16S rRNA gene was situated upstream of the 23S rRNA gene, and that the total length of the ‘16S‐ITS‐23S’ part of the rRNA operon was less than 6500 bp.
Primer design
For the forward primer, we used the A519F (5’‐CAGCMGCCGCGGTAA‐3′, derived from https://academic.oup.com/nar/article/41/1/e1/1164457) primer. It covers a large fraction of the known bacterial and archaeal diversity and has shown robust performance in previous 16S rRNA gene amplicon studies (Spang et al., 2015; Baker et al., 2016).
For our reverse primer, we designed one in such a manner that it would anneal to the 3′ end of the 23S rRNA gene and would cover as large a diversity of bacteria and archaea as possible. First, we took the full, 150,000 nt long SILVA LSURef (release 119) alignment (SILVA_119_LSURef_tax_silva_full_align_trunc.fasta, available at the SILVA archive) and removed all sequences that corresponded to eukaryotes or were shorter than 2200 bp (not counting gaps). We removed all sites with more than 10% gaps (trimAl v1.4 ‐gt 0.90 (Capella‐Gutiérrez et al., 2009)). The archaea are heavily underrepresented in the LSURef 119 database (43,822 bacterial entries vs 629 archaeal entries), and any primer based on the current alignment may have a strong bias towards bacteria. To prevent this and further prevent a bias towards species that have entries for a disproportionate number of strains, we clustered all bacterial entries into 90% OTUs with UCLUST (Edgar, 2010) and rebuilt the alignment with one representative sequence (the centroid) per OTU and supplemented it with all archaeal entries.
We fed this alignment to WEBLOGOv3 (http://weblogo.threeplusone.com/) and ran it in both ‘bit’ and ‘probability’ modes with otherwise default settings. The bit mode visualizes highly conserved sites, while the probability mode visualizes the relative occurrence of each base per site. We then used both logos to design candidate primers. They were required to (i) anneal to a highly conserved region on the 3′ region of the 23S rRNA gene, (ii) lack degenerate bases in the 3′ end and generally contain as few degeneracies as possible, (iii) have a predicted melting temperature (TM) within 5°C of the predicted TM of the forward primer, (iv) have a 3′ terminal G or C to facilitate primer extension and (v) have a low probability of forming homodimers or cross‐dimers with the forward primer under PCR conditions. Expected TM and probability of primer‐dimers were evaluated with Thermo Fisher Scientific's online MULTI PRIMER ANALYSER.
Taxonomic coverage of all candidate primers was then evaluated with SILVA's TestProbe. In the end, primer ‘U2428R’ (5’‐CCRAMCTGTCTCACGACG‐3′) was selected. It covers 98.9% of all bacterial, and 89.5% of all archaeal 23S rRNA gene entries in the SILVA 128 LSURef release.
Mock community
We constructed a synthetic ‘mock community’ sample composed of genomic DNA from 38 phylogenetically distinct and diverse bacteria and archaea. To be included, taxa were required to have a complete genome available and have at least one 16S‐ITS‐23S locus (a neighbouring 16S and 23S rRNA gene with an ITS <1 kbp). For a complete list, see Table S2. Genomic DNA for these taxa was ordered from DSMZ (Brunswick, Germany) and quantified with the Quant‐IT PicoGreen dsDNA Assay kit (ThermoFisher) using the FLUOstar Omega microplate reader (BMG Labtech). The genomic DNAs of all selected taxa were pooled in such a manner that the gDNA of each taxon contributed an estimated equal number of 16S‐ITS‐23S loci to the final mock community. For example, the mock would have ten times more Desulfovibrio gDNA than Bacillus gDNA, because Desulfovibrio encodes a single 16S‐ITS‐23S locus, whereas Bacillus encodes 10 such loci. To calculate for each species the volume fraction in the mock, we first converted the measured concentrations in ng/μl to concentrations in ‘operonmol’/μl (Table S2). An operonmol here is defined as the number of 16S‐ITS‐23S loci present in a genome multiplied with the molecular weight of the genome. The volume fraction is then calculated by dividing the inverse, μl/operonmol with the sum of μl/operonmol of all taxa.
Environmental samples
Four environmental samples were used in this study: ‘P19’, ‘PM3’, ‘TNS08’ and ‘SALA’. P19 is a sediment sample obtained from hot spring Radiata Pool, Ngatamariki, New Zealand. PM3 is a sediment sample taken from 1.25 m below the sea floor using a gravity core at Aarhus Bay, Denmark. TNS08 is a sediment sample taken from a shallow submarine hydrothermal vent field near Taketomi Island, Japan. SALA is a sample of a black biofilm that was taken at 60 m depth in an old silver mine near Sala, Sweden. Detailed descriptions of DNA extractions and further DNA sample cleaning of the samples P19 (or ‘P1.0019’), PM3 and TNS08 (or ‘617–1‐3’) can be found in (Zaremba‐Niedzwiedzka et al., 2017). DNA extraction of SALA was done with the FastDNA 50 ml spin kit for soil (MP Biomedicals).
PCR
We used primers A519F and U2428R to amplify the rRNA operon between position ∼520 of the 16S rRNA gene and position ∼2430 of the 23S rRNA gene in the mock community and the extracted DNA from the four environmental samples. All PCR reactions were set up with the Q5 High‐Fidelity DNA Polymerase kit (New England Biolabs) according to manufacturer's recommended reaction mix except for a final Q5 concentration of 0.04 U/μl instead of 0.02 U/μl. Each reaction included the Q5 High GC Enhancer and was done with 2 ng of template DNA, unless otherwise specified. Cycling conditions were as follows: denaturation at 98°C for 30 s, followed by 30 cycles of amplification (denaturation at 98°C for 10 s, annealing at 64°C for 30 s, extension at 72°C for 3 min and 30 s) and a final extension at 72°C for 10 min. By default, each sample was amplified in three parallel 50 μl reactions. Deviations from the default reaction: the mock community amplifications were done in 25 μl reactions with 1 ng of template DNA, the TNS08 sample was amplified in two, 34 cycle parallel reactions with 0.2 ng of template, the PM3 sample was amplified in five parallel reactions, and the P19 sample was amplified in nine parallel reactions, of which three were done with 34 cycles. Note that TNS08 and P19 read error rates will be higher compared to the mock, as they were done with 4 additional PCR cycles. PCR products were cleaned with AMPure XP beads (Beckman Coulter) according to Illumina's Nextera DNA Library Prep, Clean Up Library protocol (page 15–16). We used a 2:1 PCR product:beads volume ratio, and eluted in ddH2O. In addition to removing PCR reagents, bead purification also removes short DNA fragments such as primers, primer‐dimers and potential small unspecific PCR products. All purified PCR products were then pooled by sample and quantified with the Qubit dsDNA HS (High‐Sensitivity) Assay kit (ThermoFisher Scientific).
PacBio sequencing
Libraries were prepared by ligating SMRTbell adapters onto the PCR products as described in the ‘Procedures & Checklist ‐ 5 kb Template Preparation and Sequencing’ protocol (without the fragmentation step). Each library was loaded onto the SMRT cells with MagBead loading (one library per SMRT cell) and sequenced on a Pacific Bioscience (PacBio) RSII SMRT DNA Sequencing System with the P6‐C4 chemistry and a movie length of 240 min. Circular consensus (ccs) reads were generated from the movies with ‘ReadsOfInsert’ protocol, implemented in the SMRT Analysis v2.3.0 (Patch 5).
Read curation pipeline
For a graphical overview of the curation pipeline that includes explanations on each curation operation, see Fig. 1 and Supporting Information Fig. S1). We started by discarding ccs reads that had an associated predicted read quality (the ‘rq’ tag in the ccs.bam file) of less than 0.99. We further discarded any high quality read that had an internal window of at least 30 bp with an average phred score of lower than 18 (stretch of low quality, see (Fichot and Norman, 2013)) with an in‐house script (see Data availability statement). All non‐ambiguous base calls among the remaining ccs reads with phred score of 0 were changed to ‘N’ with the mothur v.1.37.4 (Schloss et al., 2009) http://fastq.info function (pacbio = T). The mothur trim.seqs() function was then used to discard all reads with 10 or more consecutive identical base calls (maxhomop = 10), reads shorter than 3000 bp (minlength = 3000) or longer than 5000 bp (maxlength = 5000), reads with more than two mismatches to primers (pdiffs = 2) and simultaneously trim recognized primer sequences from the starts and ends of reads (keepforward = F). We further used the demultiplexing capacity of trim.seqs() to recognize and consecutively remove reads (siamaeras ‐ see http://github.com/BioInf-Wuerzburg/proovread (Hackl et al., 2014)) that started and ended with the same primer. Up to this point each ccs read still represents the positive or negative strand, depending on which strand the polymerase initiated the sequencing. In the next step we ‘polarized’ the reads, meaning that after polarization all reads are in the same direction and represent the same strand. To recognize reads derived from opposite strands, we used the ‐‐adjustdirection function of MAFFT v7.050b (Katoh and Standley, 2013). We detected chimeras de novo and subsequently removed them with mothur's chimera.uchime (reference = self, chunks = 16, abskew = 1) and remove.seqs(), respectively. On all reads that passed we predicted the partial 16S and 23S rRNA genes and their associated ITS with RNAmmer v1.2 (Lagesen et al., 2007). RNAmmer was run in bacterial and archaeal modes. Per read, we chose the gene predictions (bacterial or archaeal) with the highest score. All reads that have both 16S and 23S rRNA genes predicted are referred to as ‘quality trimmed ccs reads’.
Consensus calling
We preclustered all quality trimmed ccs reads with VSEARCH v2.4.3 at 99% identity level (Rognes et al., 2016) (−‐cluster_fast, −‐id 0.99, −‐sizeout). Next, we generated majority rule consensus reads for each precluster of size 3 or larger. Such preclusters were aligned with MAFFT Q‐INS‐i (−‐kimura 1) and each alignment was consequently used as input for mothur's consensus.seqs (cutoff = 51). Gaps were removed in the resulting consensus, yielding final precluster consensus sequences.
Error rate evaluation
The degree of erroneous base calls was evaluated by comparing the ccs reads derived from the mock community with the reference loci that comprises the 16S rRNA gene, the 23S rRNA gene and ITS (henceforth referred to as ‘16S‐ITS‐23S’) sequences at different points in the read curation pipeline (Fig. 1). We extracted reference 16S‐ITS‐23S sequences from the mock community taxa genomes and only included instances smaller than 6000 bp. The reference was further supplemented with 16S‐ITS‐23S sequences from taxa that were found to contaminate the mock community (Veillonella parvula DSM 2008, Moellerella wisconsensis ATCC 35017, Staphylococcus epidermidis ATCC 12228 and Streptococcus pneumoniae R6). For Moellerella, the 16S and 23S rRNA genes were encoded on different contigs (derived from a metagenome assembly of the mock community, data not shown) thus do not necessarily form a 16S‐ITS‐23S locus. However, since we identified ccs reads with Moellerella 16S and 23S rRNA genes, we conclude that they do. We patched the reference 16S and 23S rRNA gene sequences with the ITS sequence from the highest quality Moellerella ccs read and included it in our reference. Ccs reads were compared with the reference by mapping them onto the reference with BLASR v3.1 (−minMatch 15, −maxMatch 20, −bestn 1) (Chaisson and Tesler, 2012). Number of substitutions, insertions and deletions were extracted from the CIGAR string and the NM tag in the output SAM file and used to calculate the overall error rate (sum of substitutions, insertions and deletions divided by the alignment length).
OTU clustering
Operational taxonomic unit (OTU) clustering of quality‐trimmed ccs reads was done per environmental sample with VSEARCH (−‐cluster_size, −‐strand both, −‐id 0.97, −‐sizeout). Read names were appended with ‘;size = <read_quality>‘prior to clustering. This ensured that the ‐‐cluster_size algorithm ranked reads from highest to lowest read quality prior to clustering and that the highest quality read became the centroid of the OTU.
Phylogenetic analyses
Two separate reference phylogenetics datasets were constructed, one for Bacteria and one for Archaea: All 16S (≥ 900 bp) and 23S (≥ 1500 bp) rRNA gene sequences available in SILVA (release 128) https://academic.oup.com/nar/article/35/21/7188/2376260, JGI and NCBI (April 2017) were included. The genetic redundancy of the datasets was reduced by clustering the 16S rRNA gene sequences with VSEARCH at 85% identity for Bacteria, and 95% identity for Archaea (−‐cluster_fast) and keeping only the centroid sequences and their corresponding 23S rRNA gene sequences. A lower identity threshold was used for Bacteria to keep the phylogenetics dataset computationally tractable. The 16S and 23S rRNA gene sequences were aligned separately using MAFFT L‐INS‐i 7.309 (−‐maxiterate 0, −‐adjustdirection), trimmed using trimAl (−gt 0.5), and a maximum likelihood phylogenetic tree was inferred from the concatenated 16S and 23S rRNA gene alignment using IQ‐TREE 1.6.beta4 (Nguyen et al., 2015) under the GTR + R10 model (Bacteria) or the GTR + R8 model (Archaea) as chosen by the IQTREE's ModelFinder (Kalyaanamoorthy et al., 2017). Misclassified taxa (Bacteria classified as Archaea and vice versa) were removed from the dataset. The final reference datasets contained 588 bacterial and 227 archaeal taxa.
To assess general phylogenetic diversity of the samples and the increased phylogenetic signal of ‘16S‐ITS‐23S’ quality trimmed ccs reads relative to ‘partial 16S’ Illumina reads, we built two ‘16S + 23S’, two ‘16S’ and two ‘16S_250bp’ phylogenetic datasets (one for Bacteria and one for Archaea per dataset): ‘16S + 23S’ consisted of the reference 16S and 23S rRNA gene sets plus all 16S and 23S rRNA gene sequences predicted from the 97% OTU centroid quality trimmed ccs reads of all 4 samples. The ‘16S’ dataset is the 16S rRNA gene only equivalent of ‘16S + 23S’. The ‘16S_250bp’ dataset is equal to ‘16S’, except that all predicted 16S rRNA gene sequences were shortened to the first 250 bp (spanning the V4 region) with the fastx_trimmer algorithm (FastX Toolkit; after removing all ambiguous characters). All reference sequences of archaeal and bacterial ‘16S + 23S’, ‘16S’ and ‘16S_250bp’ datasets were aligned with MAFFT L‐INS‐i (−‐maxiterate 1000, −‐adjustdirection). Sample rRNA gene sequences were then aligned against these reference alignments with MAFFT L‐INS‐i (−‐addfragments, −‐maxiterate 1000). All alignments were trimmed with trimAl (gap threshold of 30% for ‘16S + 23S’ and ‘16S’, and of 20% for ‘16S_250bp’). After checking that the trimmed 16S and 23S rRNA gene alignments of the ‘16S + 23S’ dataset represented the same strand, they were concatenated into a supermatrix alignment. Maximum likelihood phylogenies were then inferred for all alignments, using IQTREE v1.5.3 with ModelFinder (−mset GTR ‐m TESTNEW) and a 100 non‐parametric bootstraps (−b 100).
Estimations of taxonomic compositions
Relative abundances of bacterial and archaeal phyla in environmental samples and the mock community captured by the quality‐trimmed ccs reads were estimated with mothur's classify.seqs(), using the SILVA database as a reference (see “rRNA gene prediction”). The process was repeated for quality‐trimmed ccs reads shortened to the first 250 bp (as described in ‘Phylogenetic analyses’). Taxonomic composition bar charts were constructed for phyla with ≥0.5% estimated abundance with ggplot2 (Wickham, 2016) and taxonomic heatmap was constructed for the top 50 most abundant lineages using AmpVis2 (tax_show = 50, tax_aggregrate = ‘Class’, tax_add = ‘Phylum’) (Andersen et al., 2018).
ITS sequence divergence
All ITS sequences were extracted from all mock community reference genomes and their closely related strains for which complete genome data were available. Pairwise identities were calculated with VSEARCH (−‐allpairs_global, −‐acceptall, −‐blast6out), separated in between‐strain comparisons and within‐strain comparisons, and plotted with ggplot2 (Wickham, 2016).
Data availability
The raw ccs sequences generated and analysed in the current study are available in the Sequence Read Archive repository under BioProject PRJNA498591. Detailed software commands and custom scripts used in the read curation pipeline are available on GitHub (https://github.com/novigit/broCode/tree/master/pbamp).
Author contributions
TJGE conceived the study. JM and IS designed the primers. JM, IS and LJ designed and constructed the mock community, and developed the PCR protocol. LJ extracted DNA from the environmental samples. IB and IVP performed PacBio sequencing. JM and MES developed the read curation pipeline. AEL and JM performed the phylogenetic analyses. JM, AEL and TJGE interpreted the results and wrote the manuscript. All authors edited and approved the manuscript.
Conflicts of interests
The authors declare that they have no conflicts of interests.
Abbreviations
- ITS
Internal Transcribed Spacer
- OTU
Operational Taxonomic Unit
- SMRT sequencing
Single Molecule Real Time sequencing
Supporting information
Fig. S1. Read curation pipeline and mothur function settings. See ‘Read curation pipeline’ in Methods for a detailed description of the pipeline. See http://mothur.org/wiki/Sequence_processing for more detailed descriptions and explanations of the mothur functions.
Fig. S2. Relationship between ccs read quality and observed error rate
Fig. S3. Example of a ccs read with a local stretch of low quality base calls that are enriched in sequencing errors. Bars represent Phred scores, red base calls correspond to errors. Red bars indicate local stretch of low quality base calls
Fig. S4. Hypothetical mechanism leading to a siamaeric read. In the bottom two examples of siamaeric reads, recognized by BLASTN dot plots
Fig. S5. Bar charts reflecting estimated relative abundances of bacterial and archaeal phyla with ≥0.5% abundance. *_1000bp: relative abundances estimated from ∼1000 bp 16S rRNA gene sequence. *_250bp: relative abundances estimated from 250 bp 16S rRNA gene sequence fragments spanning the V4 region.
Fig. S6. Unrooted maximum likelihood phylogenies inferred from archaeal ‘16s_250bp’, ‘16S’ and ‘16S + 23S’ datasets. ‘size=’ indicates the number of qtrim ccs reads in the respective 97% OTUs. Taxa of major taxonomic groups are coloured. Branch values indicate non‐parametric bootstrap support. Indicates taxa that are discussed in the main text.
Fig. S7. Unrooted maximum likelihood phylogenies inferred from bacterial ‘16s_250bp’, ‘16S’ and ‘16S + 23S’ datasets. ‘size=’ indicates the number of qtrim ccs reads in the respective 97% OTUs. Taxa of major taxonomic groups are coloured. Branch values indicate non‐parametric bootstrap support. Indicates taxa discussed in the main text
Fig. S8. Sequence similarities of internal transcribed spacer (ITS) copies either between different strains of the same species (left) or between different ITS copies within the same strain (right). Bacterial and archaeal species used are those that are present in the mock community and their close relatives with complete genomes available.
Fig. S9. Bar chart reflecting the estimated relative abundances of genomes that are part of the mock community.
Fig. S10. Relationships between the %GC (left) or length (right) of the 16S‐ITS‐23S loci of the mock community genomes and their quality‐trimmed ccs read counts.
Table S1. 16S‐ITS‐23S loci in Archaea and Bacteria
Table S2. Composition of the mock community.
Acknowledgements
We thank F. Homa, K. Katoh, M. Hammond, J. Vosseberg, C. Bergin, A.M. Divne, C. Stairs and the technical support of New England Biolabs and Pacific Biosciences for useful advice and insightful discussions. We thank the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) at Uppsala University and the Swedish National Infrastructure for Computing (SNIC) at the PDC Center for High‐Performance Computing for providing computational resources. This work was supported by grants of the European Research Council (ERC Starting grant 310039‐PUZZLE_CELL), the Swedish Foundation for Strategic Research (SSF‐FFL5) and the Swedish Research Council (VR grant 2015‐04959) awarded to TJGE.
References
- Acinas, S.G. , Marcelino, L.A. , Klepac‐Ceraj, V. , and Polz, M.F. (2004) Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol 186: 2629–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertsen, M. , Hugenholtz, P. , Skarshewski, A. , Nielsen, K.L. , Tyson, G.W. , and Nielsen, P.H. (2013) Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538. [DOI] [PubMed] [Google Scholar]
- Alneberg, J. , Bjarnason, B.S. , de Bruijn, I. , Schirmer, M. , Quick, J. , Ijaz, U.Z. , et al (2014) Binning metagenomic contigs by coverage and composition. Nat Methods 11: 1144–1146. [DOI] [PubMed] [Google Scholar]
- Andersen, K.S.S. , Kirkegaard, R.H. , Karst, S.M. , and Albertsen, M. (2018) ampvis2: an R package to analyse and visualise 16S rRNA amplicon data. bioRxiv : 299537. [Google Scholar]
- Baker, B.J. , Saw, J.H. , Lind, A.E. , Lazar, C.S. , Hinrichs, K.‐U. , Teske, A.P. , and Ettema, T.J.G. (2016) Genomic inference of the metabolism of cosmopolitan subsurface archaea, Hadesarchaea. Nat Microbiol 1: 16002. [DOI] [PubMed] [Google Scholar]
- Brochier, C. , Bapteste, E. , Moreira, D. , and Philippe, H. (2002) Eubacterial phylogeny based on translational apparatus proteins. Trends Genet 18: 1–5. [DOI] [PubMed] [Google Scholar]
- Brown, J.R. , Douady, C.J. , Italia, M.J. , Marshall, W.E. , and Stanhope, M.J. (2001) Universal trees based on large combined protein sequence data sets. Nat Genet 28: 281–285. [DOI] [PubMed] [Google Scholar]
- Calus, S.T. , Ijaz, U.Z. , and Pinto, A.J. (2018) NanoAmpli‐Seq: a workflow for amplicon sequencing for mixed microbial communities on the nanopore sequencing platform. Gigascience 7 10.1093/gigascience/giy140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutiérrez, S. , Silla‐Martínez, J.M. , and Gabaldón, T. (2009) trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics 25: 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson, M.J. , and Tesler, G. (2012) Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsuc, F. , Brinkmann, H. , and Philippe, H. (2005) Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet 6: 361–375. [DOI] [PubMed] [Google Scholar]
- Doolittle, W.F. (1999) Phylogenetic classification and the universal tree. Science 284: 2124–2128. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. , Haas, B.J. , Clemente, J.C. , Quince, C. , and Knight, R. (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferla, M.P. , Thrash, J.C. , Giovannoni, S.J. , and Patrick, W.M. (2013) New rRNA gene‐based phylogenies of the Alphaproteobacteria provide perspective on major groups, mitochondrial ancestry and phylogenetic instability. PLoS One 8: e83383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichot, E.B. , and Norman, R.S. (2013) Microbial phylogenetic profiling with the Pacific biosciences sequencing platform. Microbiome 1: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye, J.S. , Cepeda‐Espinoza, V. , and Pop, M. (2016) Metagenomic assembly: overview, challenges and applications. Yale J Biol Med 89: 353–362. [PMC free article] [PubMed] [Google Scholar]
- Green, R. , and Noller, H.F. (1997) Ribosomes and translation. Annu Rev Biochem 66: 679–716. [DOI] [PubMed] [Google Scholar]
- Hackl, T. , Hedrich, R. , Schultz, J. , and Förster, F. (2014) Proovread : large‐scale high‐accuracy PacBio correction through iterative short read consensus. Bioinformatics 30: 3004–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, W. , Wang, S. , Dong, H. , Jiang, H. , Briggs, B.R. , Peacock, J.P. , et al (2013) A comprehensive census of microbial diversity in hot springs of Tengchong, Yunnan Province China using 16S rRNA gene pyrosequencing. PLoS One 8: e53350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugenholtz, P. , Skarshewski, A. , and Parks, D.H. (2016) Genome‐based microbial taxonomy coming of age. Cold Spring Harb Perspect Biol 8: a018085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt, D.E. , Klepac‐Ceraj, V. , Acinas, S.G. , Gautier, C. , Bertilsson, S. , and Polz, M.F. (2006) Evaluation of 23S rRNA PCR primers for use in phylogenetic studies of bacterial diversity. Appl Environ Microbiol 72: 2221–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, S.L. , Hannisdal, B. , Lanzén, A. , Baumberger, T. , Flesland, K. , Fonseca, R. , et al (2012) Correlating microbial community profiles with geochemical data in highly stratified sediments from the Arctic Mid‐Ocean ridge. Proc Natl Acad Sci U S A 109: E2846–E2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy, S. , Minh, B.Q. , Wong, T.K.F. , von Haeseler, A. , and Jermiin, L.S. (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14: 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst, S.M. , Dueholm, M.S. , McIlroy, S.J. , Kirkegaard, R.H. , Nielsen, P.H. , and Albertsen, M. (2018) Retrieval of a million high‐quality, full‐length microbial 16S and 18S rRNA gene sequences without primer bias. Nat Biotechnol 36: 190–195. [DOI] [PubMed] [Google Scholar]
- Katoh, K. , and Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkhof, L.J. , Dillon, K.P. , Häggblom, M.M. , and McGuinness, L.R. (2017) Profiling bacterial communities by MinION sequencing of ribosomal operons. Microbiome 5: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen, K. , Hallin, P. , Rødland, E.A. , Stærfeldt, H.‐H. , Rognes, T. , and Ussery, D.W. (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35: 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, D.J. , Pace, B. , Olsen, G.J. , Stahl, D.A. , Sogin, M.L. , and Pace, N.R. (1985) Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A 82: 6955–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matte‐Tailliez, O. , Brochier, C. , Forterre, P. , and Philippe, H. (2002) Archaeal phylogeny based on ribosomal proteins. Mol Biol Evol 19: 631–639. [DOI] [PubMed] [Google Scholar]
- Nelson, W.C. , and Mobberley, J.M. (2017) Biases in genome reconstruction from metagenomic data. PeerJ Prepr. https://peerj.com/preprints/2953/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L.‐T. , Schmidt, H.A. , von Haeseler, A. , and Minh, B.Q. (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol Biol Evol 32: 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse, E. , Peplies, J. , and Glöckner, F.O. (2012) SINA: accurate high‐throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28: 1823–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes, T. , Flouri, T. , Nichols, B. , Quince, C. , and Mahé, F. (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ Prepr. https://peerj.com/articles/2584/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss, P.D. , Jenior, M.L. , Koumpouras, C.C. , Westcott, S.L. , and Highlander, S.K. (2016) Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 4: e1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss, P.D. , Westcott, S.L. , Ryabin, T. , Hall, J.R. , Hartmann, M. , Hollister, E.B. , et al (2009) Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, E. , Bushnell, B. , Coleman‐Derr, D. , Bowman, B. , Bowers, R.M. , Levy, A. , et al (2016) High‐resolution phylogenetic microbial community profiling. ISME J 10: 2020–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin, M.L. , Morrison, H.G. , Huber, J.A. , Welch, D.M. , Huse, S.M. , Neal, P.R. , et al (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere.”. Proc Natl Acad Sci U S A 103: 12115–12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang, A. , Saw, J.H. , Jørgensen, S.L. , Zaremba‐Niedzwiedzka, K. , Martijn, J. , Lind, A.E. , et al (2015) Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521: 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedersoo, L. , Tooming‐Klunderud, A. , and Anslan, S. (2017) PacBio metabarcoding of fungi and other eukaryotes: errors, biases and perspectives. New Phytol 217: 1370–1385. [DOI] [PubMed] [Google Scholar]
- Turnbaugh, P.J. , Ley, R.E. , Hamady, M. , Fraser‐Liggett, C. , Knight, R. , and Gordon, J.I. (2007) The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature 449: 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, J. , Coupland, P. , Browne, H.P. , Lawley, T.D. , Francis, S.C. , and Parkhill, J. (2016) Evaluation of PacBio sequencing for full‐length bacterial 16S rRNA gene classification. BMC Microbiol 16: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisburg, W.G. , Barns, S.M. , Pelletier, D.A. , and Lane, D.J. (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173: 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2016) Ggplot2: Elegant Graphics for Data Analysis: Springer. [Google Scholar]
- Williams, T.A. , Foster, P.G. , Nye, T.M.W. , Cox, C.J. , and Embley, T.M. (2012) A congruent phylogenomic signal places eukaryotes within the Archaea. Proc Biol Sci 279: 4870–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese, C.R. (1987) Bacterial evolution. Microbiol Rev 51: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese, C.R. , and Fox, G.E. (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A 74: 5088–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, Y.I. , Rogozin, I.B. , Grishin, N.V. , Tatusov, R.L. , and Koonin, E.V. (2001) Genome trees constructed using five different approaches suggest new major bacterial clades. BMC Evol Biol 1: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba‐Niedzwiedzka, K. , Caceres, E.F. , Saw, J.H. , Bäckström, D. , Juzokaite, L. , Vancaester, E. , et al (2017) Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541: 353–358. [DOI] [PubMed] [Google Scholar]
- Zimmermann, J. , Gonzalez, J.M. , Saiz‐Jimenez, C. , and Ludwig, W. (2005) Detection and phylogenetic relationships of highly diverse uncultured acidobacterial communities in Altamira cave using 23S rRNA sequence analyses. Geomicrobiol J 22: 379–388. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Read curation pipeline and mothur function settings. See ‘Read curation pipeline’ in Methods for a detailed description of the pipeline. See http://mothur.org/wiki/Sequence_processing for more detailed descriptions and explanations of the mothur functions.
Fig. S2. Relationship between ccs read quality and observed error rate
Fig. S3. Example of a ccs read with a local stretch of low quality base calls that are enriched in sequencing errors. Bars represent Phred scores, red base calls correspond to errors. Red bars indicate local stretch of low quality base calls
Fig. S4. Hypothetical mechanism leading to a siamaeric read. In the bottom two examples of siamaeric reads, recognized by BLASTN dot plots
Fig. S5. Bar charts reflecting estimated relative abundances of bacterial and archaeal phyla with ≥0.5% abundance. *_1000bp: relative abundances estimated from ∼1000 bp 16S rRNA gene sequence. *_250bp: relative abundances estimated from 250 bp 16S rRNA gene sequence fragments spanning the V4 region.
Fig. S6. Unrooted maximum likelihood phylogenies inferred from archaeal ‘16s_250bp’, ‘16S’ and ‘16S + 23S’ datasets. ‘size=’ indicates the number of qtrim ccs reads in the respective 97% OTUs. Taxa of major taxonomic groups are coloured. Branch values indicate non‐parametric bootstrap support. Indicates taxa that are discussed in the main text.
Fig. S7. Unrooted maximum likelihood phylogenies inferred from bacterial ‘16s_250bp’, ‘16S’ and ‘16S + 23S’ datasets. ‘size=’ indicates the number of qtrim ccs reads in the respective 97% OTUs. Taxa of major taxonomic groups are coloured. Branch values indicate non‐parametric bootstrap support. Indicates taxa discussed in the main text
Fig. S8. Sequence similarities of internal transcribed spacer (ITS) copies either between different strains of the same species (left) or between different ITS copies within the same strain (right). Bacterial and archaeal species used are those that are present in the mock community and their close relatives with complete genomes available.
Fig. S9. Bar chart reflecting the estimated relative abundances of genomes that are part of the mock community.
Fig. S10. Relationships between the %GC (left) or length (right) of the 16S‐ITS‐23S loci of the mock community genomes and their quality‐trimmed ccs read counts.
Table S1. 16S‐ITS‐23S loci in Archaea and Bacteria
Table S2. Composition of the mock community.
Data Availability Statement
The raw ccs sequences generated and analysed in the current study are available in the Sequence Read Archive repository under BioProject PRJNA498591. Detailed software commands and custom scripts used in the read curation pipeline are available on GitHub (https://github.com/novigit/broCode/tree/master/pbamp).