

Abstract
Retinol dehydrogenases catalyze the rate-limiting step in the biosynthesis of retinoic acid, a bioactive lipid molecule that regulates the expression of hundreds of genes by binding to nuclear transcription factors, the retinoic acid receptors. Several enzymes exhibit retinol dehydrogenase activities in vitro; however, their physiological relevance for retinoic acid biosynthesis in vivo remains unclear. Here, we present evidence that two murine epidermal retinol dehydrogenases, short-chain dehydrogenase/reductase family 16C member 5 (SDR16C5) and SDR16C6, contribute to retinoic acid biosynthesis in living cells and are also essential for the oxidation of retinol to retinaldehyde in vivo. Mice with targeted knockout of the more catalytically active SDR16C6 enzyme have no obvious phenotype, possibly due to functional redundancy, because Sdr16c5 and Sdr16c6 exhibit an overlapping expression pattern during later developmental stages and in adulthood. Mice that lack both enzymes are viable and fertile but display accelerated hair growth after shaving and also enlarged meibomian glands, consistent with a nearly 80% reduction in the retinol dehydrogenase activities of skin membrane fractions from the Sdr16c5/Sdr16c6 double-knockout mice. The up-regulation of hair-follicle stem cell genes is consistent with reduced retinoic acid signaling in the skin of the double-knockout mice. These results indicate that the retinol dehydrogenase activities of murine SDR16C5 and SDR16C6 enzymes are not critical for survival but are responsible for most of the retinol dehydrogenase activity in skin, essential for the regulation of the hair-follicle cycle, and required for the maintenance of both sebaceous and meibomian glands.
Keywords: vitamin A, retinol, carotenoid, dehydrogenase, retinoic acid, retinaldehyde, retinol dehydrogenase epidermal (RDHE), short-chain dehydrogenase/reductase family 16C member 5 (SDR16C5), hair follicle, epidermis, meibomian gland
Introduction
All-trans-retinoic acid (RA)4 is the major bioactive form of vitamin A that influences a broad spectrum of physiological processes including embryogenesis and epithelial homeostasis (1–4). In the nucleus, RA regulates gene expression primarily through binding to nuclear transcription factors, retinoic acid receptors (RARs α, β, and γ), which act as heterodimers with retinoid X receptors (5, 6). In the cytoplasm, RA regulates the activity of extracellular signal-regulated kinase (7) and exhibits numerous other extranuclear activities (8). RA is synthesized from the alcohol form of vitamin A (all-trans-retinol) via a two-step process. In the first step, retinol dehydrogenases oxidize all-trans-retinol to all-trans-retinaldehyde, which is oxidized further by retinaldehyde dehydrogenases (RALDHs) to RA (reviewed in Ref. 9).
The oxidation of retinol to retinaldehyde is the rate-limiting step in RA biosynthesis (10). Several members of the short-chain dehydrogenase/reductase (SDR) superfamily of proteins catalyze this reaction in vitro (9), but only one of the retinoid-active SDRs characterized to date, murine retinol dehydrogenase 10 (RDH10), is known to be indispensable for RA biosynthesis, because embryos lacking functional RDH10 do not survive past E12.5 (11). RDH10-null embryos display numerous abnormalities including forelimb, craniofacial, neural, and heart defects (12, 13). The severity of the phenotype indicates that RDH10 functions as the major murine retinol dehydrogenase during mid-embryogenesis. However, whereas the ablation of RDH10 eliminates most of the retinol dehydrogenase activity during the early stages of development, RA synthesis persists in the neural tube of RDH10-null embryos at E9.5 and E10.5 (14, 15). Importantly, Rdh10−/− embryos can be rescued by supplementation of maternal diets with retinaldehyde between embryonic stages E7.5 and E9.5. Thus, RDH10 appears to be dispensable during later stages of development and transition to adulthood (11). These data point toward the existence of RDH10-independent sources of RA. However, the identities of additional retinol dehydrogenases accounting for this residual retinaldehyde synthesis remain elusive.
RDH10 belongs to the 16C family of the SDR superfamily of proteins (16, 17). Notably, two other genes encoding members of the SDR16C family are located adjacent to the gene encoding RDH10 in the human genome on chromosome 8: retinol dehydrogenase epidermal 2 (RDHE2, SDR16C5) and retinol dehydrogenase epidermal 2-similar (RDHE2S, SDR16C6) (18, 19). The deduced RDHE2 and RDHE2S proteins share the highest sequence homology (∼43%) with RDH10 (SDR16C4). As we reported previously, the single ortholog of human genes encoding RDHE2 and RDHE2S in Xenopus laevis functions as a retinol dehydrogenase in vivo and is essential for embryonic development in frogs (20). These findings imply that mammalian RDHE2 and RDHE2S complement RDH10 in generating retinaldehyde for RA biosynthesis. However, it remains to be established whether the in vivo function of amphibian rdhe2 is conserved by its mammalian orthologs. This study was undertaken to assess the catalytic properties of mammalian RDHE2 and RDHE2S as compared with amphibian rdhe2, to determine the expression patterns of RDHE2 and RDHE2S in mice, and to establish whether these enzymes are essential for RA biosynthesis in mammals.
Results
Kinetic characterization of epidermal retinol dehydrogenases
To compare the catalytic properties of murine RDHE2 (SDR16C5) and RDHE2S (SDR16C6) as retinol dehydrogenases with those of frog rdhe2, we expressed each enzyme as a fusion protein with the C-terminal His6 tag in insect Sf9 cells using the baculovirus expression system. Western blot analysis showed that the recombinant SDRs were expressed at comparable protein levels in the microsomal fractions of Sf9 cells (Fig. 1A). Therefore, the microsomal fractions containing the RDHE proteins were used for characterization of their retinoid activities. Activity assays revealed that murine RDHE2S and Xenopus rdhe2 both catalyzed the oxidation of all-trans-retinol to all-trans-retinaldehyde in the presence of NAD+. The activity rates with NADP+ were at the limit of detection (data not shown); hence, NAD+ was the preferred cofactor for both enzymes. RDHE2S exhibited a higher apparent Km value and a lower apparent Vmax value for the oxidation of all-trans-retinol than Xenopus rdhe2 (Table 1). Thus, Xenopus rdhe2 exhibited a higher catalytic efficiency (Vmax/Km) than murine RDHE2S. Both enzymes also catalyzed the reduction of all-trans-retinaldehyde to all-trans-retinol in the presence of NADH, but their catalytic efficiencies in the reductive direction were lower than in the oxidative direction (Table 1). In addition to all-trans-retinol, both RDHE2S and Xenopus rdhe2 recognized 11-cis-retinol as substrate (Table 1).
Figure 1.

Expression and characterization of frog and murine RDHE2 and RDHE2S. A, Western blot analysis of microsomal fractions (5 μg) isolated from control Sf9 cells (mock) or cells expressing murine RDHE2, murine RDHE2S, or Xenopus rdhe2. His tag antibodies were used at a 1:1,000 dilution. B, Western blot analysis of HEK293 cell lysates (20 μg) containing murine RDHE2 or RDHE2S co-expressed with human RALDH1. RDHE2 and RDHE2S were detected using FLAG tag antibodies at a 1:3,000 dilution. RALDH1 was detected using HA antibodies at a 1:50 dilution. C, HPLC analysis of all-trans-retinaldehyde (RAL) production from all-trans-retinol (10 μm). Cells were incubated for 11 h. D, HPLC analysis of RA production from all-trans-retinol (10 μm). Samples are as indicated. Mock, cells transfected with empty vector. *, p < 0.01; **, p < 0.001; ***, p < 0.0001; mean ± S.D. (error bars), n = 3.
Table 1.
Kinetic constants of SDR16C enzymes
Kinetic constants for each enzyme were determined using the same preparation of microsomes containing the corresponding enzyme; therefore, the differences in the catalytic rates of each enzyme towards various substrates and cofactors reflect the properties of the enzymes rather than differences in protein expression levels. UD, undetectable under the conditions of the assay; ND, not determined. The activity of murine RDHE2 toward 10 μm all-trans-retinol in the presence of 1 mm NAD+ was not detectable with up to 10 μg of RDHE2 microsomal preparation.
| SDR | Substrate/Cofactor | Apparent Km | Apparent Vmax | Vmax/Km |
|---|---|---|---|---|
| μm | nmol·min−1·mg−1 | |||
| Xenopus rdhe2 | All-trans-retinol | 0.6 ± 0.1 | 19.5 ± 0.6 | 32.5 |
| NAD+ | 108 ± 27 | 21 ± 1 | ||
| All-trans-retinaldehyde | 0.6 ± 0.1 | 3.6 ± 0.1 | 6 | |
| NADH | 8.4 ± 1.7 | 4.1 ± 0.2 | ||
| 11-cis-Retinol | 3.3 ± 0.4 | 3.9 ± 0.2 | 1.8 | |
| RDHE2 (SDR16C5) | All-trans-retinol | UD | UD | |
| All-trans-retinaldehyde | N.D. | N.D. | ||
| RDHE2S (SDR16C6) | All-trans-retinol | 0.87 ± 0.21 | 8.7 ± 0.6 | 10 |
| NAD+ | 460 ± 30 | 5.8 ± 0.2 | ||
| All-trans-retinaldehyde | 3.9 ± 0.2 | 5.4 ± 0.1 | 1.4 | |
| NADH | 11 ± 3 | 2.6 ± 0.1 | ||
| 11-cis-Retinol | 0.86 ± 0.14 | 1.34 ± 0.07 | 1.6 | |
| 9-cis-Retinol | UD | UD |
Unexpectedly, no activity toward all-trans-retinol was detected using the microsomal preparation of recombinant murine RDHE2 protein (Table 1). Some SDRs show dual subcellular distribution between microsomal and mitochondrial fractions (21); therefore, we tested whether the mitochondria isolated from Sf9 cells expressing RDHE2 or RDHE2S exhibited a retinol dehydrogenase activity. No activity was detected using RDHE2-containing mitochondria, whereas 10 μg of mitochondria from RDHE2S-expressing Sf9 cells exhibited a much lower activity (0.03 nmol·min−1·mg−1) than RDHE2S microsomes under the same assay conditions (10 μm retinol and 1 mm NAD+) (Table 1), suggesting that RDHE2S is localized primarily in the microsomal membranes.
Retinol dehydrogenase activities of murine RDHE2 and RDHE2S in intact cells
The finding that RDHE2 was inactive when assayed using either the microsomal or mitochondrial fractions of Sf9 cells prompted us to examine whether RDHE2 was catalytically active in the context of intact living cells. HEK293 cells were transiently transfected with pCS105 expression vectors encoding RDHE2 with a C-terminal FLAG tag. RDHE2S, which was previously shown to oxidize retinol in living cells (18), was also cloned into pCS105 expression vector in frame with FLAG tag and used as a positive control. The two enzymes were co-expressed with RALDH1 to enhance the relatively low endogenous retinaldehyde dehydrogenase activity of HEK293 cells (Fig. 1B), which caused accumulation of unprocessed retinaldehyde in the cells under the conditions of this experiment. Remarkably, when expressed in intact cells, RDHE2 was found to exhibit a retinol dehydrogenase activity, increasing the production of retinaldehyde from retinol (Fig. 1C). This activity of RDHE2 resulted in a 3-fold increase in RA biosynthesis relative to mock-transfected cells (Fig. 1D). In comparison, expression of RDHE2S at similar protein levels, as judged based on comparable staining intensity for FLAG tag (Fig. 1B), resulted in a 15-fold increase in RA production (Fig. 1D). These experiments confirmed that RDHE2S is a more active enzyme than RDHE2, but most importantly, they revealed that, although inactive toward retinol in isolated microsomes, RDHE2 was able to function as a retinol dehydrogenase in intact cells, confirming that RDHE2 is a catalytically active enzyme.
Expression patterns of murine RDHE2 and RDHE2S
To identify the tissues where RDHE2 and RDHE2S contribution could be essential for RA biosynthesis, we carried out analysis of their gene expression patterns. Tissue distribution of Rdhe2 and Rdhe2s transcripts in adult mice was analyzed by qPCR. The two transcripts exhibited a largely overlapping expression pattern with the highest expression levels in skin, followed by tongue, intestine, and esophagus (Fig. 2A). In addition, we examined the distribution of the PCR-amplified full-length mRNA corresponding to Rdhe2 and Rdhe2s genes. This analysis largely confirmed the pattern determined by qPCR, with the highest expression levels observed in skin, esophagus, stomach, and tongue and trace amounts of transcripts detected in adipose tissue, intestine, colon, and possibly testis (Fig. 2A, inset).
Figure 2.

Expression profile of Rdhe2 and Rdhe2s transcripts in mouse tissues. A, qPCR analysis of Rdhe2 and Rdhe2s expression in adult tissues of two animals. Inset, PCR-amplified full-length Rdhe2 and Rdhe2s transcripts. Lanes 1 and 2, tongue; lanes 3 and 4, esophagus; lanes 5 and 6, stomach; lanes 7 and 8, skin; lane 9, adipose tissue; lanes 10 and 11, intestine; lanes 12 and 13, brain; lanes 14 and 15, testis; lanes 16 and 17, heart; lanes 18 and 19, lung; lanes 20 and 21, liver; lanes 22 and 23, kidney; lanes 24 and 25, eyes; lanes 26 and 27, colon. B, semiquantitative PCR analysis of temporal expression patterns of Rdhe2 and Rdhe2s transcript fragments in embryonic mouse tissues. C57BL/J6 embryos were collected at E9.5–E14.5 stages of development as indicated. Five μg of mRNA was used for reverse transcription with random hexamer primers, and one-twentieth of the resulting cDNA was used per reaction. Gapdh was used as a loading control.
To determine whether Rdhe2 and Rdhe2s are expressed during embryogenesis, we employed semiquantitative PCR. RNA was isolated from WT C57BL/6J embryos between stages E9.5 and E14.5 in 1-day increments and utilized for the synthesis of cDNA, which then served as a template for PCR amplification using gene-specific primers (Fig. 2B). PCR products encoding Rdhe2 were observed starting at E12.5, increasing in abundance through E14.5. Products encoding Rdhe2s were detected earlier in development at E10.5 and persisted through E14.5. Thus, both Rdhe2 and Rdhe2s were expressed from middle to late gestation.
Generation of RDHE2 and RDHE2S gene knockout mouse models
The ultimate proof that RDHE2 and RDHE2S are essential for RA biosynthesis would have to come from gene knockout studies. Because RDHE2S exhibited a severalfold higher catalytic activity as a retinol dehydrogenase than RDHE2 in in vitro assays, we decided to begin the in vivo functional analysis of these enzymes by focusing on RDHE2S. A RDHE2S-null mouse line was generated that carries a lacZ reporter element under the control of the native Rdhe2s promoter (Fig. S1A). Upon Cre and Flp excision, exon 4 in the Rdhe2s was deleted, and a frameshift mutation was introduced, which created a stop codon prior to catalytic residues (Fig. S1B). Mice were genotyped using allele-specific primers, with expected product size of 570 bp for WT mice and a 248-bp product for Rdhe2s−/− mice (Fig. S1C). Importantly, Rdhe2s−/− mice were obtained at Mendelian frequencies (28 of 100 genotyped), were fertile, and displayed no distinguishable phenotype, at least when maintained on a regular laboratory chow diet that contains 24.2 IU of vitamin A per gram. qPCR analysis of gene expression in skin of Rdhe2s−/− mice versus WT littermates did not reveal any significant differences (Fig. S2).
Because Rdhe2 and Rdhe2s transcripts showed an overlapping expression pattern during development and adulthood, we reasoned that the lack of obvious phenotype in RDHE2S-null mice could be due to functional redundancy with RDHE2. To test this hypothesis, we generated two independent strains of Rdhe2−/−;Rdhe2s−/− double-knockout mice (DKO) lacking both RDHE2 and RDHE2S using CRISPR-mediated gene editing (Fig. 3). The first strain, DKO1, lacks exons 5–7 of Rdhe2 and exons 1–5 of Rdhe2s. The second strain, DKO2, lacks exon 5 in Rdhe2 and has an 8-bp deletion in exon 2 of Rdhe2s, causing a frameshift (Fig. 3A). The exact sequences resulting from these excisions are shown in Fig. S3. The absence of RDHE2 and RDHE2S proteins was confirmed by immunoblotting of microsomal fractions from skin of DKO mice with commercially available antibodies against murine RDHE2S (Fig. 3B, top) and antibodies against Xenopus rdhe2 (Fig. 3B, bottom), which recognize both murine RDHE2 and RDHE2S proteins.
Figure 3.

Generation of mice deficient in RDHE2 (SDR16C5) and RDHE2S (SDR16C6). A, CRISPR-Cas9–mediated targeting and screening strategy. Positions of guide RNA targets are indicated by black arrows. Screening PCR primers are shown as gray arrows. PCR products are shown as gray lines connecting the primers. Primer pairs 1–5 do not produce a PCR product from the WT allele, because the distance between the primer annealing sites is too large. Pair 4 amplifies a chimeric sequence from DKO1 allele, where rearrangement resulted in a shortening of the distance between the annealing sites. Long-range PCR with pair 6 primers results in a 4,912-bp WT product and a shorter 1,837-bp product from DKO2 allele. The exact positions of CRISPR-induced deletions and the sequence of the insertion in DKO1 allele are shown in Fig. S3. B, Western blot analysis of RDHE2 and RDHE2S in skin microsomes (50 μg) of female DKO1 (n = 3, #105, #107, and #108) and WT (n = 3, #104, #106, and #110) mice using RDHE2S antibodies (top) and Xenopus rdhe2 antibodies (bottom), both at a 1:3,000 dilution. Note the difference in mobility of murine RDHE2 versus murine RDHE2S; *, a nonspecific band. C, eye phenotype of DKO mice. Note the puffy eyelids and partially closed eyes in the DKO animal. This phenotype was observed in all mice of both sexes on chow diet as well as on vitamin A–deficient diet (n > 100 animals). Shown are littermates DKO1#20 and WT#18.
The DKO mice have been backcrossed for six generations to WT C57BL/6 mice to resolve any potential off-target mutations. Both strains of mice are fertile and produce homozygous and heterozygous offspring at the expected Mendelian ratio. Interestingly, DKO mice can be readily distinguished from WT littermates by the appearance of their eyes, which have puffy eyelids and an almond-like shape, compared with the round eyes in a similarly aged control animal (Fig. 3C).
Skin phenotype of DKO mouse models
The highest expression level of Rdhe2 and Rdhe2s was observed in skin. Therefore, we began the investigation of the functional role of RDHE2 and RDHE2S by analyzing the retinol dehydrogenase activities of membrane fractions isolated from skin of DKO mice. Compared with WT mice, the retinol dehydrogenase activity of the microsomal fractions from DKO skin was reduced by ∼10-fold (p = 0.001), whereas the mitochondrial activity was reduced by ∼6-fold (p = 0.02) (Fig. 4A).
Figure 4.

Skin phenotype of DKO mice. A, analysis of retinol dehydrogenase activities. Microsomal and mitochondrial fractions (50 μg) from DKO mice and WT littermates on chow diet were incubated with 3 μm retinol and 1 mm NAD for 15 min. Note the significant decrease in activity in fractions from DKO mice. B, qPCR analysis of RA-regulated or retinoid metabolic genes in dorsal skin of male and female mice from two different Rdhe2−/−;Rdhe2s−/− strains (DKO1 (n = 5) and DKO2 (n = 4)). Levels of transcripts were determined by normalizing to Gapdh; *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, immunohistochemical detection of RDHE2 and RDHE2S in skin sections from male P60 WT and DKO2 littermates utilizing Xenopus rdhe2 antibodies at a 1:800 dilution. Arrow, sebaceous gland. Images are representative of three biological replicates, with six skin sections stained from each mouse. There were four representative images with a total of 15 hair follicles captured for each mouse. Scale bar, 100 μm, 200-fold magnification. D, detection of Rdhe2s expression domains in E14.5 Rdhe2s+/− (HET) embryos using β-gal staining. Note the strong staining in whisker pads and nasal pits. E, detection of Rdhe2s expression domains in dorsal skin of adult Rdhe2s+/− (HET) male mice. Strong staining is observed in sebaceous glands (arrow) and epidermis (arrowhead). Scale bar, 50 μm, 400-fold magnification. F, Rdhe2 detection by in situ hybridization in tail skin from WT C57BL/J6 male mouse. Like Rdhe2s, Rdhe2 is expressed in sebaceous gland (arrow). G, hematoxylin and eosin (H&E) staining of dorsal skin from WT and DKO2 male littermates at 10 weeks old. Note the enlarged hair follicles and thicker skin in DKO animals. Top, 60-fold magnification; bottom, 100-fold magnification; scale bar, 100 μm. H, hematoxylin and eosin staining of dorsal skin from WT and DKO2 male littermates at P50. The dermal papilla (arrowhead) appears to be larger in DKO skin with a thicker keratinocyte strand above, suggesting an entry into anagen phase. The images are representative of seven biological replicates with six skin sections stained from each mouse. There are three representative images with a total of 10 hair follicles captured for each mouse. Scale bar, 100 μm, 200-fold magnification. Error bars, S.D.
Such a dramatic reduction in skin retinol dehydrogenase activity would be expected to have a major impact on RA biosynthesis and expression of RA target genes in DKO skin, possibly resulting in abnormal skin appearance. However, upon visual inspection, there were no discernable differences in the appearance of skin or hair between DKO and WT littermates. We reasoned that the differences in RA signaling between WT and DKO mice might be exacerbated under the conditions of vitamin A deficiency. To produce vitamin A–deficient mice, a cohort of pregnant dams was placed on a vitamin A–deficient diet at mid-gestation, and the pups were maintained on vitamin A–deficient diet until sacrificed. The vitamin A–deficient status was confirmed by analyzing the hepatic levels of retinyl esters (0.45 ± 0.28 nmol/mg) and retinol (0.026 ± 0.022 nmol/mg). Dorsal skin samples collected from these mice were examined for expression levels of RA-regulated and retinoid metabolic genes by qPCR. Surprisingly, the expression patterns of these genes in skin of DKO mice appeared to be only partially consistent with reduced RA signaling (Fig. 4B). For example, the RA-inducible Cyp26b1 was down-regulated, but the expression levels of several other genes known to be regulated by RA (Dhrs3 and Rarβ) did not change or were significantly increased (Lrat, Stra6, Rbp1, Raldh2, and Dhrs9) (22–25).
Skin is a highly heterogeneous tissue composed of keratinocytes, hair follicles, sebaceous glands, fibroblasts, etc. The differentiation and growth of many of these cell types are affected by RA levels (4). To determine the exact localization of RDHE2 and RDHE2S in skin, we carried out immunohistological analysis of skin sections using custom-made antibodies against Xenopus rdhe2 that recognize both murine proteins (18). This analysis showed that RDHE2 and RDHE2S proteins are abundant in sebaceous gland and in epidermis (Fig. 4C). This pattern was confirmed at the level of gene expression based on the activity of the Rdhe2s gene promoter, which was detected by β-gal staining assays in developing vibrissae and nasal pits of E14.5 Rdhe2s+/− embryos (Fig. 4D) and also in sebaceous glands of adult Rdhe2s+/− mice (Fig. 4E). Likewise, Rdhe2 transcripts were detected in sebaceous glands and epidermis of adult WT C57BL/J6 mice by in situ hybridization (Fig. 4F).
To determine whether the absence of RDHE2 and RDHE2S affected the structure of skin, we sectioned DKO and WT dorsal skin samples from mice on a vitamin A–deficient (VAD) diet and analyzed skin histology. The most obvious difference between DKO and WT skin from littermates was the larger size of sebaceous glands and a longer hair shaft in DKO skin (Fig. 4G). This observation suggested that the hair follicles in the skin of DKO mice were in growth phase (anagen), whereas in WT skin they were in resting (telogen) phase. Furthermore, whereas the WT mice had characteristic telogen features at P50, such as a condensed dermal papilla and lack of an inner root sheath, the DKO littermate dorsal skin was in anagen, demonstrated by the enlarged dermal papilla and thickening of the keratinocyte strand between the dermal papilla and club hair (Fig. 4H). To obtain independent and quantitative evidence that hair follicles were in anagen phase in skin of DKO mice, we carried out qPCR analysis of genes expressed in the pilosebaceous unit (26). Of 18 genes tested, three were down-regulated (Cd34, Krt15, and Lgr6), whereas most of the others were up-regulated (Fig. 5).
Figure 5.

qPCR analysis of genes expressed in hair follicles and sebaceous glands. qPCR was performed using skin from animals (∼10 weeks old) of both sexes from two independent DKO strains fed vitamin A–deficient diet as described under “Experimental procedures.” WT and DKO1 females, n = 5; WT and DKO2 males, n = 4; *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.D.
The elevated expression of hair-follicle growth and differentiation marker genes in DKO skin relative to littermate WT skin suggested that the hair follicles of DKO mice enter anagen sooner than in WT skin. We tested this hypothesis by shaving the backs of male and female mice and monitoring their hair regrowth. As shown in Fig. 6, both sexes displayed an accelerated hair regrowth. However, in males, this was noticeable only during the second synchronized hair cycle at the age of 60–62 days, whereas in females, the difference was obvious already in the first cycle at P29. Importantly, the difference in the hair regrowth was observed not only on VAD diet (Fig. 6A) but also on chow diet (Fig. 6B). The daily progression of hair growth in the two strains versus matched WT littermates on VAD diet is shown in Fig. S4.
Figure 6.
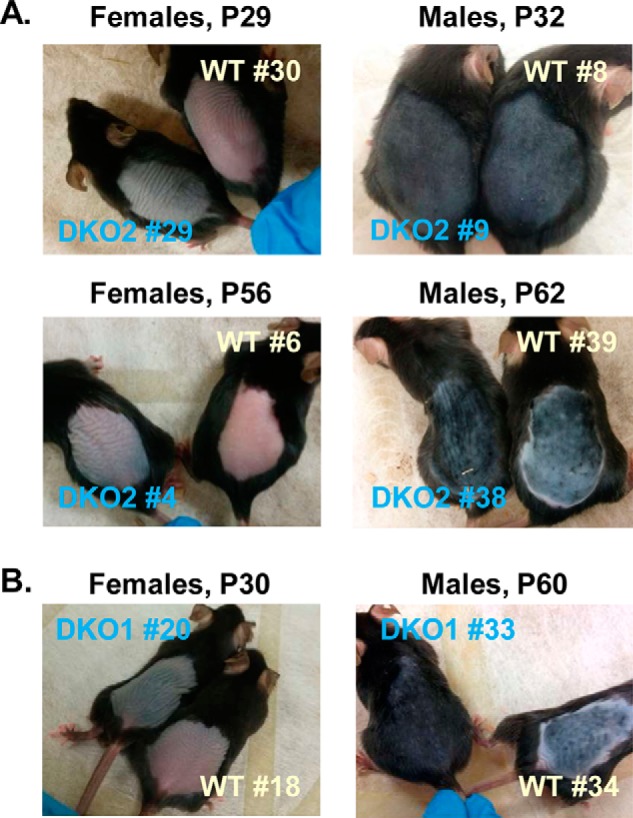
Hair regrowth in DKO and WT mice on vitamin A–deficient diet (A) and on chow diet (B). A, DKO2 and their littermate female mice were shaved at P25 (#29) and P50 (#4) after birth and photographed at P29 (4 days of hair growth) and P56 (6 days of hair growth), respectively. Shown are representative images of two sets of younger females and two sets of older females on VAD diet. DKO2 and their littermate male mice were shaved at P25 (#9) and P49 (#38) and photographed at P32 (7 days of hair growth) and P62 (13 days of hair growth), respectively. Shown are representative images of four sets of younger males and three sets of older males on VAD diet. B, female DKO1 and its WT female littermate were shaved at P25 and photographed at P30 (5 days of hair growth). Shown are representative images of six sets of females on chow diet. Male DKO1 and its WT male littermate were shaved at P49 and photographed at P60 (11 days of hair growth). Shown are representative images of four sets of males on chow diet. Images are available for additional shaved single animals from DKO1 and DKO2 strains that did not have WT littermates. The two independent strains showed a very similar external phenotype on vitamin A–deficient diet and on chow diet.
Some members of the SDR superfamily exhibit broad substrate specificity (27). Specifically, SDR9C family members, which include mouse RDH1 and human RoDH4, exhibit higher activity toward hydroxysteroids than retinol (28, 29). Previously, we examined the activity of human RDHE2 toward a panel of hydroxysteroids but found no activity (20). To determine whether murine orthologs exhibit activity toward steroid substrates, we compared the activity of microsomal fractions from RDHE2/E2S DKO mice and WT mice toward various functional groups in androgens, progestins, corticosteroid, and estradiol: androsterone (3α-OH oxidation with NAD+; 17-keto reduction with NADH), dihydrotestosterone/NAD+ (17β-OH oxidation), 3α-hydroxy-5α-pregnan-20-one (3α-OH oxidation with NAD+; 20-keto reduction with NADH); Δ4-pregnene-3,20-dione (3 and 20-one reduction with NADH), androst-5-en-3β-ol-17-one (3β-OH oxidation with NAD)+; estradiol (3β-OH and 17β-OH oxidation with NAD+; and corticosterone (11β-OH and 21-OH oxidation with NAD+; 3-keto reduction with NADH) (90). In contrast to retinol as substrate, where we saw an ∼80% drop in activity of DKO microsomes, no decrease was observed in the activity of the same microsomes toward any of the substrates tested relative to WT microsomes (Fig. S5). In fact, there was a slight increase in the activity of DKO microsomes toward some of the steroid substrates, which could be related to the differences in hair-follicle growth phase in DKO mice versus WT mice. The lack of a decrease in steroid dehydrogenase activity further strengthens our conclusion that RDHE/E2S function as retinol dehydrogenases.
Eye phenotype of DKO mouse models
To determine the underlying cause of the almond-shaped appearance of eyes in DKO mice, their eyes were dissected and compared with littermate controls. No difference in either size or weight of eyeballs was observed. However, histological analysis revealed that both the upper and lower eyelids in DKO mice were significantly thicker and longer compared with WT animals (Fig. 7A), which explains why the eyes appeared partially closed. The expansion of eyelids appeared to be due to the larger size of meibomian glands in DKO mice. To determine the localization of RDHE2 and RDHE2S, sections of eyelids from WT mice were immunostained with custom-made antibodies against murine RDHE2S (Fig. 7B) and commercially available antibodies against RDHE2 (Fig. 7C). Both antibodies detected the corresponding proteins in WT meibomian glands, whereas DKO eyelids showed little or no staining. Thus, the lack of expression of RDHE2 and RDHE2S resulted in enlargement of meibomian glands of DKO animals.
Figure 7.
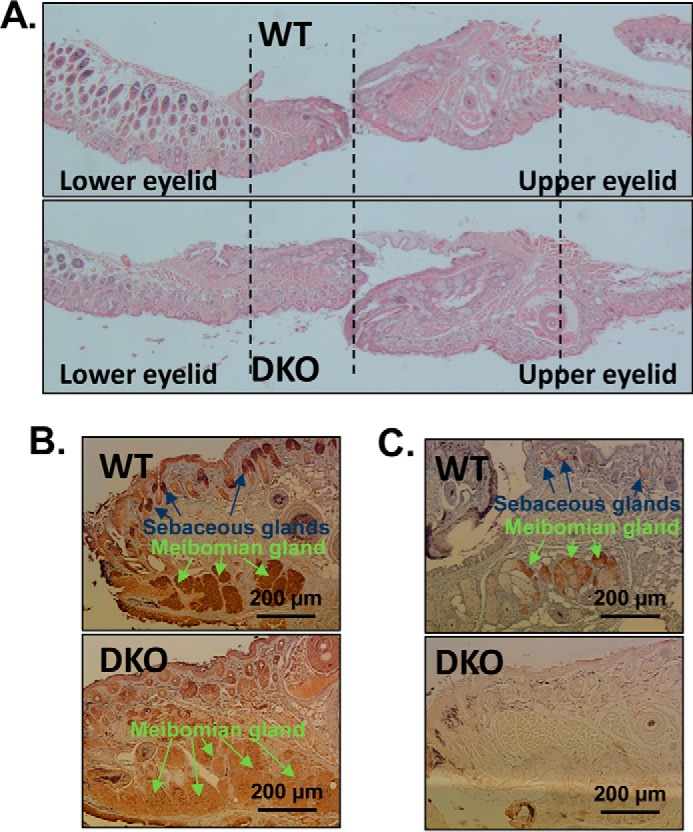
Eye phenotype of DKO mice. A, histology of whole eye lids. Magnification, ×60. Note the longer and thicker appearance of lower and upper eyelids from DKO mice. Four biological replicas were analyzed from littermates WT#38/DKO1#37, WT#39/DKO2#40, WT#41/DKO2#38 and WT#18/DKO1#20. B, immunohistology of eyelid sections using custom-made RDHE2S antibodies at a 1:400 dilution. C, immunohistology of eyelid sections using commercial SDR16C5 (RDHE2) antibodies (Sigma αSDR16C5 #HPA 025224, lot R11757) at a 1:200 dilution. Both RDHE2S and RDHE2 are localized in meibomian glands and sebaceous glands in eyelids. As determined by measuring a representative image of eyelid sections using the image-processing program ImageJ (33), the DKO meibomian glands are nearly twice the size (198%) of the WT meibomian glands. Overall, the eyelids of DKO mice are 122.5 ± 1.3% the size of WT eyelids (n = 3 pairs of littermates). Scale bar, 200 μm, 100-fold magnification.
Discussion
Several members of the SDR superfamily of proteins can oxidize retinol to retinaldehyde in vitro; however, with the exception of RDH10, their physiological relevance for RA biosynthesis remains unclear. This study provides convincing evidence that mammalian RDHE2 and RDHE2S function as physiologically relevant retinol dehydrogenases. The results of this study are important because they highlight the existence of tissue-specific retinol dehydrogenases that are essential for RA biosynthesis in specific cell types while not being critical for survival, as in the case with RDH10 during midgestation (11–15). In fact, because RDH10-null embryos can be rescued by supplementation with retinaldehyde (11), the precursor for RA, our findings suggest that other retinol dehydrogenases (e.g., RDHE2 and RDHE2S) may be as important for postnatal development and maintenance of specific adult tissues as RDH10.
Analysis of substrate and cofactor specificity carried out in this study shows that, in agreement with their functions as oxidative enzymes, epidermal retinol dehydrogenases prefer NAD(H) as cofactors and are more catalytically efficient in the oxidative direction with all-trans-retinol than in the reductive direction with all-trans-retinaldehyde as substrate. Interestingly, like human RDH10 (30), RDHE2S recognizes 11-cis-retinol as substrate in addition to all-trans-retinol. A recent study provided evidence that RDH10 contributes to the oxidation of 11-cis-retinol in mouse retina, but mice deficient in both RDH10 and RDH5 still convert 11-cis-retinol to 11-cis-retinaldehyde in the visual cycle (31). The identities of the remaining 11-cis-retinol dehydrogenases are currently unknown. If RDHE2S is expressed in mouse retina, this enzyme could complement RDH10 and RDH5 in oxidizing 11-cis-retinol.
A surprising finding of this study is that the retinol dehydrogenase activity of murine RDHE2 is undetectable in the isolated microsomal or mitochondrial fractions of RDHE2-expressing Sf9 cells. However, RDHE2 is fully functional as a retinol dehydrogenase when expressed in mammalian living cells. This observation suggests that murine RDHE2 requires additional cellular factors to exhibit its full enzymatic potential. As shown in our previous study, a member of the SDR16C family, human DHRS3 (SDR16C1), requires the presence of human RDH10 (SDR16C4) to display a measurable retinaldehyde reductase activity and, in turn, activates RDH10 (32). However, co-expression of murine RDHE2 or RDHE2S with human DHRS3, which is 97.7% identical to murine DHRS3 in HEK293 cells, had no activating effect on DHRS3 (19), indicating that RDHE2 and RDHE2S do not interact with DHRS3. Like murine RDHE2, human RDHE2 (SDR16C5) also displays a rather low activity (34). Future affinity purification assays may be warranted to identify possible partners of RDHE2.
It is also interesting that whereas RDHE2S exhibits a higher retinol dehydrogenase activity than RDHE2, a single Rdhe2s gene knockout does not display the same phenotype as the DKO mice. Rdhe2s−/− mice have normal eyelids and regular hair growth (data not shown). This observation indicates that first, despite its lower enzymatic activity, RDHE2 is essential for RA biosynthesis in mice and, second, the functions of these two enzymes may be redundant because of the overlapping expression pattern. The reason for such an overlap is unclear at this time. It should be noted that in humans, only RDHE2 represents a functional gene encoding a stable protein with enzymatic activity, whereas human RDHE2S (SDR16C6) appears to have lost its function and is classified as pseudogene (34). It is possible that in humans, RDHE2, which exhibits a wider tissue distribution pattern (https://www.ncbi.nlm.nih.gov/gene/195814#gene-expression) than either murine Rdhe2 (https://www.ncbi.nlm.nih.gov/gene/242285) or Rdhe2s (https://www.ncbi.nlm.nih.gov/gene/?term=Mus+musculus+Sdr16c6) fulfills the mission of both enzymes.
This study shows that in mice, RDHE2 and RDHE2S together account for up to 80% of the total membrane-associated retinol dehydrogenase activity in skin. Thus, although present in skin, RDH10 is not the major retinol dehydrogenase in this tissue. The absence of RDHE2 and RDHE2S results in significant changes in the hair cycle of mice. Hair cycle occurs in three phases: anagen (growth), catagen (regression), and telogen (resting) (35–47). Typically, development of dorsal hair follicles continues until postnatal day 16, when the first catagen occurs on days 17–20. Telogen spans days 21–25, and anagen lasts from day 28 to day 42. This is followed by the second cycle, during which hair follicles continue to cycle in a nearly synchronized manner. Anagen phase is characterized by thickening of the dermal and epidermal layers of the skin, increased size of hair follicles, extension of follicles deep into the dermal adipose tissue, and initiation of melanin synthesis. All of these features were observed in the dermis and epidermis of DKO skin but not in skin of WT littermates. Consistent with histological assessment, DKO skin showed increased expression of several markers of anagen phase (26). For example, a strong indication of the anagen phase is the induction of the hedgehog signaling pathway. In DKO skin, both Gli1 and Gli2 transcriptional regulators in the hedgehog pathway were up-regulated. Hair cycles are fueled by HF stem cells that reside in the “bulge” niche located at the base of the telogen phase HF (48). In addition to Gli1, which is expressed in the hair bulge and germ (49, 50), several other markers of the bulge stem cell compartment were up-regulated in DKO skin. Among these, the most notable are SRY (sex-determining region Y)-box 9 (SOX-9), which is indispensable for hair homeostasis (51, 52); T-box 1 (Tbx1), which is highly enriched in the bulge of developing and cycling HFs (53); and leucine-rich repeat-containing G protein–coupled receptor 5 (Lgr5), a marker of hair-follicle stem cells (54).
The enlargement of sebaceous glands was confirmed by severalfold higher expression of melanocortin-5 receptor (Mc5r), a marker of sebocyte differentiation (55); Prdm1 gene, which encodes BLIMP1, believed to be a marker of terminally differentiated sebocytes (56) and/or a sebocyte progenitor marker (57); peroxisome proliferator–activated receptor γ (Pparγ), which plays an important role in sebaceous gland development; and stearyl CoA-desaturase 1 (Scd1) and perilipin 2 (Plin2), which are necessary for sebocyte differentiation and sebum production in sebocytes (reviewed in Ref. 58).
One of the most up-regulated genes in both males (DKO2 strain) and females (DKO1 strain) was the Lef1 gene in the Wnt signaling pathway. It is well-known that the regulation of the hair-follicle cycle involves cross-talk of the Wnt/β-catenin, Hedgehog, and Notch signaling pathways (59–64). However, nonprotein factors, and in particular vitamin A (see Ref. 65; reviewed in Ref. 66), are also critical for maintenance of skin health. In this respect, there is evidence for mutual antagonism between Wnt/β-catenin signaling and RA signaling (67). β-Catenin positively regulates CYP26A1, which degrades RA, and conversely, β-catenin undergoes retinoid-dependent interaction with RAR, resulting in competitive inhibition of TCF-binding sites (67, 68). Inhibition of Wnt/β-catenin signaling leads to up-regulation of RA-inducible cellular RA-binding protein type 2 (CRABP2), suggesting increased RA signaling (69). It is noteworthy, however, that whereas RA signaling is clearly essential for epidermal differentiation, the mechanisms by which it acts are largely unexplored. This study and the generation of RDHE2 and RDHE2S DKO mouse models should facilitate the investigation of the role RA plays in the regulation of hair-follicle growth and differentiation and in general maintenance of epidermis.
Interestingly, 13-cis-retinoic acid (isotretinoin) is clinically used for acne treatment and believed to inhibit sebocyte differentiation and lipid synthesis (70, 71) by isomerizing to all-trans-retinoic acid (58). The enlarged sebaceous glands observed in DKO mice are consistent with the decreased levels of RA biosynthesis in skin. We have also observed enlarged meibomian glands in DKO eyelids. Meibomian glands are modified sebaceous glands that secrete meibum, a lipid-rich fluid that forms a superficial oily layer on the tear film to prevent the film from evaporating. Of note, severe bilateral lower eyelid retraction and dry eye symptoms were observed in patients with the long-term use of topical retinoids for cosmetic purposes (72–74). Thus, reduced RA biosynthesis in eyelids of DKO mice appears to be consistent with the observed enlargement of their meibomian glands. Previous studies have also noted that systemic use of retinoids for acne treatment or skin rejuvenation was associated with histopathological changes in the eyelids and degenerative changes in the meibomian gland acini (73, 74).
It is worth pointing out that, whereas the in vivo phenotype is clearly consistent with the reduced RA signaling in DKO skin as a result of the 80% reduction in retinol dehydrogenase activity, qPCR analysis of endogenous RA-regulated genes produced a very mixed picture, with some RA-inducible genes being down-regulated, whereas others were unchanged or even up-regulated. Histological analysis of skin sections revealed the cause of this seemingly puzzling outcome. Because the reduced RA signaling led to enlargement of sebaceous glands, which express several RA-sensitive genes (Dhrs9, Lrat, and Stra6), the transcript levels of these genes were likely increased as a result of an expanded number of sebocytes. Thus, measurement of total levels of RA in skin could be misleading, because it would not reflect the localized differences in cell type–specific RA levels in tissue as heterogeneous as skin.
Overall, the results of this study establish RDHE2 and RDHE2S as physiologically relevant retinol dehydrogenases in mammals, which, despite their importance for RA biosynthesis, are not critical for survival during embryogenesis. In view of these findings, the physiological impact of mutations in these genes or changes in their expression can be better appreciated considering their role in RA biosynthesis. As discussed previously (34), genome-wide association studies have linked the chromosomal region harboring RDHE2 (SDR16C5) and seven other genes to stature and growth in cattle, humans, and pigs (35–82) and beak deformity in chickens (83). RDHE2 was also identified as the important candidate gene in the pig growth trait by an integrative genomic approach (84). Interestingly, expression of SDR16C5 was reported to be 4-fold higher than RDH10 in human lung (pooled RNA from five individuals) (85). Altered SDR16C5 expression is frequently noted in various pathophysiological conditions. For example, SDR16C5 was decreased in triple-negative breast cancer patient samples (86). Evidence provided by this study that mammalian RDHE2/E2S function as physiologically relevant retinol dehydrogenases implicates the decrease in RA biosynthesis as at least one of the causes leading to these pathophysiological outcomes.
Experimental procedures
Expression constructs
All primer sequences with corresponding restriction sites used for generation of constructs are listed in Table S1. Constructs encoding FLAG-tagged murine RDHE2S and X. laevis rdhe2 in pCMV-Tag 4A vector were described previously (18, 20). Murine Rdhe2 cDNA was obtained by RT-PCR of mouse skin and liver mRNA and cloned into SalI and XbaI sites of pBluescript II SK (−) vector.
To generate FLAG-tagged RDHE2 expression construct, the corresponding cDNA was cloned into pCMV-Tag 4A vector in frame with the C-terminal FLAG tag. Subsequently, the FLAG-tagged construct was amplified using primers specified in Table S1 and cloned into pCS105 and pVL1393 vectors. In addition, a His-tagged RDHE2 construct was generated using a modified pVL1393 vector containing an in-frame C-terminal His6 tag (87).
Human HA-tagged RALDH1 expression construct in pcDNA3.1-neo (CMV promoter) was a generous gift of Dr. Sylvie Mader (Department of Biochemistry, University of Montreal, Canada). All expression constructs and plasmids were verified by sequencing.
Cell culture models
For activity assays in intact cells, SDR constructs were expressed in human HEK293 cells following the protocols published previously (30, 32). The transfected cells were incubated with all-trans-retinol or all-trans-retinaldehyde as indicated. Retinoids were extracted and analyzed by normal phase HPLC as described previously (30). For kinetic analysis, RDHE2, RDHE2S, and frog rdhe2 were expressed in Sf9 insect cells using pVL1393 transfer vector and the BaculoGold Baculovirus Expression System (BD Biosciences). The subcellular fractions were isolated by differential centrifugation as described (87). The membrane pellets were resuspended in 90 mm KH2PO4, 40 mm KCl (pH 7.4), and 20% glycerol (w/v).
In vitro activity assays and immunoblotting
Activity assays using subcellular fractions of Sf9 cells were performed as described previously (30). The Km and Vmax values for all-trans-retinol, 9-cis-retinol, 11-cis-retinol, or all-trans-retinaldehyde were obtained using seven concentrations of each substrate (0.0625–8 μm) in the presence of 1 mm NAD(H). The Km and Vmax values for NAD+ and NADH were determined at fixed concentration of all-trans-retinol or all-trans-retinaldehyde (5 μm) and seven concentrations of NAD+ or NADH (1–500 μm).
Western blot analysis was performed using rabbit polyclonal antibodies against FLAG epitope (Sigma-Aldrich) and β-actin (Abcam, Cambridge, UK) and mouse monoclonal antibodies against His epitope (Clontech, BD Biosciences) and HA epitope (received as a gift from Dr. Hengbin Wang, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham School of Medicine).
Protein samples were separated in 12% polyacrylamide gels in the presence of SDS and transferred to Amersham Biosciences Hybond P polyvinylidene difluoride membranes (GE Healthcare). Following transfer, membranes were blocked with 4% BSA in TBS with Tween 20 (TBST) and incubated with rabbit polyclonal or mouse monoclonal antibodies diluted with 4% BSA in TBST overnight at 4 °C. Dilutions of primary antibodies are indicated in the figure legends. Membranes were rinsed with TBST and incubated for 1 h at room temperature in goat anti-rabbit antibody or goat anti-mouse antibody conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), both of which were diluted 1:10,000 with 4% BSA in TBST. Protein visualization was achieved with Pierce ECL Western blotting substrate (Thermo Fisher Scientific).
Generation of Rdhe2s−/− mouse model
Rdhe2s+/− mice were generated using the Sdr16c6tm1a(KOMP)Wtsi “knockout-first” allele obtained from the KOMP repository [https://www.komp.org/pdf.php?projectID=41879,5 SangerID design 43965]. The knockout-first allele contains an IRES:lacZ trapping cassette and a floxed promoter-driven neo cassette inserted into the intron of a gene, disrupting gene function. The targeting vector (KOMP design 43965) was digested with AsiSI and SalI enzymes, which resulted in a 1,431-bp truncation of the 5′ homology arm. Electroporation of the linearized vector in mouse embryonic stem cells (ESCs), selection of ESC-derived clones, and isolation of genomic DNA were performed by the University of Alabama at Birmingham Transgenic and Genetically Engineered Models facility. ESC-derived clones carrying a targeted insertion of the knockout-first cassette were identified by long-range PCR spanning 5′ and 3′ homology arms using the SequalPrep long PCR kit (Invitrogen, Life Technologies, Inc.) with primers listed in Table S1. Chimeras were generated by the University of Alabama at Birmingham Transgenic and Genetically Engineered Models facility. Male chimeras were crossed to WT C57Bl56/J mice, and pups were genotyped to identify heterozygote founders carrying a knockout-first allele. Crossing of these mice to FLPeR mice carrying a gene encoding FLP recombinase resulted in conversion of the knockout-first allele to a conditional allele, restoring gene activity. Subsequent crossing of FLP-excised mice to EIIa-cre mice, which express Cre recombinase prior to implantations, resulted in a mouse with a deletion of exon 4 and a frameshift mutation (Fig. S1). Genotyping of mice crossed to FLPeR mice was accomplished using DNA isolated from tails and the primers listed in Table S1.
Generation of Rdhe2−/−;Rdhe2s−/− mouse models
Murine Sdr16c6 is located immediately upstream of murine Sdr16c5 gene. For both genes, CRISPR guide RNAs were designed to target exons 2 and 5, which encode the conserved cofactor and substrate-binding site motifs, respectively (Table S2, Fig. 3A, and Fig S3). Guide RNAs were designed as described previously (88). Six F0 pups were born following CRISPR-Cas9 microinjections. For the initial screening, the targeted exons 2 and 5 were PCR-amplified from genomic DNA isolated from tail snips and separated by electrophoresis in polyacrylamide gel. In mouse #21855, the electrophoretic mobility shift assay revealed heterogeneous bands in PCR products corresponding to exon 2 of Sdr16c5 and exon 5 of Sdr16c6, suggesting the formation of heteroduplexes. In mouse #21853, PCR product for exon 5 of Sdr16c5 was completely absent, indicating that CRISPR-Cas9 injections generated a larger deletion(s) in this animal and that both copies of the Sdr16c6-Sdr16c5 locus are mutated.
Subcloning and resequencing of the targeted exons in mouse #21855 identified a 3-bp deletion in exon 2 of Sdr16c5, as well as 1- and 3-bp deletions in exon 5 of Sdr16c6. The loss of a single amino acid resulting from the 3-bp deletion may not lead to a complete loss of enzymatic activity of SDR16C5 (RDHE2), and the frameshift-causing 1-bp deletion in Sdr16c6 was expected to result in a knockout of the Sdr16c6 gene only. As we already had an established knockout strain of Sdr16c6, mouse #21855 was not used as a founder.
To precisely define the alleles generated in mouse #21853, we performed a series of PCR amplifications with primer pairs as indicated in Fig. 3A and in Table S1. We assumed that the products would only be generated if the primers' annealing sites were brought close enough as a result of deletion, because the distance between the annealing sites in the WT type allele is too large for efficient amplification. The primer pair 4 yielded a 585-bp product in mouse #21853, but not in other F0 animals. The product contained a chimeric sequence composed of incomplete exon 5 of Sdr16c6, incomplete exon 5 of Sdr16c5 gene, and a 298-bp insertion corresponding to an inverted partial sequence of exon 2 of Sdr16c5 (Fig. 3A and Fig. S3). This result indicated that mouse #21853 contained a large 56,057-bp deletion covering the region between CRISPR targets in exon 5 of Sdr16c6 and exon 5 of Sdr16c5. The ORF of the chimeric transcript encodes a truncated polypeptide (Fig. S3). We have designated this double Sdr16c6−/−;Sdr16c5−/− knock-out allele as DKO1.
The existence of a full-length PCR product for exon 5 of Sdr16c6, which was obtained during initial F0 screening was inconsistent with the DKO1 allele, which retains only part of this exon. This result suggested that mouse #21853 carries a second mutant allele. PCR amplification of individual exons 4 and 6 of Sdr16c5 yielded fragments of the expected size, indicating that a shorter-than-DKO1 deletion occurred in the second allele of the same animal. This shorter deletion resulted in a loss of a single exon 5 in Sdr16c5. Long-range PCR amplification with primers spanning exons 4–6 yielded a 4,912-bp product in WT mouse but a shorter product in mouse #21853. Sequencing of this product confirmed the deletion of 1,837 bp, which covers exon 5 of Sdr16c5 and a portion of flanking intronic sequences. This allele was designated DKO2 (Fig. 3A and Fig. S3).
Thus, mouse #21853 carried two different CRISPR-Cas9–generated alleles, DKO1 and DKO2. This animal was used as the founder, and crossed to WT females to isolate DKO1 and DKO2 strains. Resequencing of individual exons in the DKO2 strain revealed a short 8-bp deletion in exon 2 of Sdr16c6, in addition to deleted exon 5 in Sdr16c5. Thus, the ORFs of both Sdr16c6 and Sdr16c5 in DKO2 allele are predicted to encode truncated proteins (Fig. 3A and Fig. S3). DKO2 represents a second double-knockout strain.
Mice were maintained on either on LabDiet NIH-31 (PMI Nutrition International) containing 22 IU vitamin A/g or on vitamin A–deficient diet Teklad TD.86143 (Envigo) in a facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care. Mice were euthanized by CO2 inhalation followed by cervical dislocation, in accordance with institutional animal care and use committee guidelines at the University of Alabama (Birmingham, AL).
β-Galactosidase staining and in situ hybridizations
Adult mice were arranged in mating pairs, and females were checked for vaginal plugs at noon of the following day. The presence of a plug was considered to represent a developmental stage of E0.5. Genotypes were determined using DNA isolated from yolk sacs. Full-length mouse Rdhe2 cDNA in pBluescript II SK (−) vector was used for generation of antisense probes for in situ hybridization. Probes were synthesized using linearized template, T3 RNA polymerase (Promega), and digoxigenin RNA labeling mix (Roche Applied Science). Skin was isolated from the backs of 4-month-old male C57BL/J6 mice, fixed overnight in 4% paraformaldehyde at 4 °C, and embedded in paraffin. The paraffin-embedded skin was cut into 10-μm sections, which were placed on Superfrost Plus microscope slides (Fisher). In situ hybridization was carried out following standard procedures. β-Galactosidase staining was performed on E14.5 Rdhe2s+/− embryos and skin isolated from 4-month-old Rdhe2s+/− male mouse tails according to the protocols published previously (89).
qPCR analysis
To determine the expression pattern of Rdhe2 and Rdhe2s genes, two male WT mice fed a VAD diet for 10 weeks were sacrificed with CO2; tissues were collected and stored at −80 °C until RNA extraction. TRIzol reagent (Ambion, catalog no. 15596018) was used for extraction of RNA from all tissues except skin, for which the AurumTM total RNA Fatty and Fibrous Tissue Pack (Bio-Rad, catalog no. 732-6870) was employed. Three μg of RNA per tissue was used for reverse transcription; cDNA was purified with QIAquick Spin columns (Qiagen, catalog no. 1018215). qPCR was performed with 25 ng of cDNA per reaction. Rdhe2 and Rdhe2s expression was normalized to Gapdh and presented as a relative expression of fold-difference from the expression level in the stomach for each gene, which was set to 1.
For analysis of gene expression in skin, ∼75 mg of skin tissue was homogenized, and RNA was extracted with the AurumTM total RNA Fatty and Fibrous Tissue Pack. The concentration of extracted RNA was determined using a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). First-strand cDNA was synthesized from 3.0 μg of total RNA with the Superscript III first-strand synthesis kit (Invitrogen) according to the manufacturer's protocol. For real-time RT-PCRs, the cDNA was diluted 15-fold. Sequences of the primers are available by request. Real-time PCR analysis was conducted on a Roche LightCycler®480 detection system (Roche Applied Science) with SYBR Green as probe (LightCycler®480 CYBR Green I Master, Roche Applied Science). Relative gene expression levels were calculated using the comparative Ct method by normalization to reference genes. Unpaired t test was used to test for statistical significance.
Hair regrowth
The diet for DKO2 heterozygote breeder dams was switched to a VAD diet at mid-gestation (12–14 days post-conception), and the pups were kept on a VAD diet until they were sacrificed at 10 weeks of age. At 8 weeks after birth, WT and DKO2 littermates' dorsal hair was clipped and depilated with Nair® lotion. Littermates were checked daily for hair regrowth until they were sacrificed. Animals that had abnormal hair due to excessive grooming by female breeders were not used for the analysis of hair regrowth.
Statistical analysis
Statistical significance was determined using a two-tailed unpaired t test.
Author contributions
L. W., O. V. B., M. K. A., S.-A. L., R. A. K., K. M. P., and N. Y. K. conceptualization; L. W., O. V. B., M. K. A., A. K., S.-A. L., R. A. K., K. M. P., and N. Y. K. data curation; L. W., O. V. B., M. K. A., A. K., S.-A. L., R. A. K., K. M. P., and N. Y. K. formal analysis; L. W., O. V. B., M. K. A., S.-A. L., K. R. G., R. A. K., and N. Y. K. validation; L. W., O. V. B., M. K. A., A. K., S.-A. L., K. R. G., and N. Y. K. investigation; L. W., O. V. B., M. K. A., S.-A. L., K. R. G., K. M. P., and N. Y. K. visualization; L. W., O. V. B., M. K. A., A. K., S.-A. L., K. R. G., and N. Y. K. methodology; L. W., O. V. B., M. K. A., S.-A. L., K. R. G., R. A. K., and N. Y. K. writing-original draft; L. W., O. V. B., M. K. A., A. K., K. R. G., R. A. K., K. M. P., and N. Y. K. writing-review and editing; R. A. K. and N. Y. K. supervision; R. A. K. and N. Y. K. project administration; N. Y. K. resources; N. Y. K. funding acquisition.
Supplementary Material
Acknowledgments
Human HA-tagged RALDH1 expression construct in pcDNA3.1-neo (CMV promoter) was a generous gift of Dr. Sylvie Mader (Department of Biochemistry, University of Montreal, Canada). HA epitope antibodies were received as a gift from Dr. Hengbin Wang (Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham School of Medicine). We are grateful to Dr. Anil K. Challa (University of Alabama, Birmingham, AL) for help with performing CRISPR-mediated gene mutations. Services provided in this publication through the University of Alabama at Birmingham Transgenic and Genetically Engineered Models facility (R. A. K.) are supported by National Institutes of Health Grants P30 CA13148, P30 AR048311, P30 DK074038, P30 DK05336, and P60 DK079626.
This work was supported in part by NIAAA, National Institutes of Health, Grant AA012153 (to N. Y. K.).The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- RA
- all-trans-retinoic acid
- SDR
- short-chain dehydrogenase/reductase
- RDH
- retinol dehydrogenase
- RALDH
- retinaldehyde dehydrogenase
- E
- embryonic day
- P
- postnatal day
- RAR
- retinoic acid receptor
- qPCR
- quantitative PCR
- DKO
- double-knockout
- VAD
- vitamin A–deficient
- ESC
- embryonic stem cell
- CMV
- cytomegalovirus.
References
- 1. Al Tanoury Z., Piskunov A., and Rochette-Egly C. (2013) Vitamin A and retinoid signaling: genomic and nongenomic effects. J. Lipid Res. 54, 1761–1775 10.1194/jlr.R030833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shannon S. R., Moise A. R., and Trainor P. A. (2017) New insights and changing paradigms in the regulation of vitamin A metabolism in development. Wiley Interdiscip. Rev. Dev. Biol. 6 10.1002/wdev.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Metzler M. A., and Sandell L. L. (2016) Enzymatic metabolism of vitamin A in developing vertebrate embryos. Nutrients 8, E812 10.3390/nu8120812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Everts H. B. (2012) Endogenous retinoids in the hair follicle and sebaceous gland. Biochim. Biophys. Acta 1821, 222–229 10.1016/j.bbalip.2011.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balmer J. E., and Blomhoff R. (2002) Gene expression regulation by retinoic acid. J. Lipid Res. 43, 1773–1808 10.1194/jlr.R100015-JLR200 [DOI] [PubMed] [Google Scholar]
- 6. Benbrook D. M., Chambon P., Rochette-Egly C., and Asson-Batres M. A. (2014) History of retinoic acid receptors. Subcell. Biochem. 70, 1–20 10.1007/978-94-017-9050-5_1 [DOI] [PubMed] [Google Scholar]
- 7. Wei L. N. (2016) Cellular retinoic acid binding proteins: genomic and non-genomic functions and their regulation. Subcell. Biochem. 81, 163–178 10.1007/978-94-024-0945-1_6 [DOI] [PubMed] [Google Scholar]
- 8. Iskakova M., Karbyshev M., Piskunov A., and Rochette-Egly C. (2015) Nuclear and extranuclear effects of vitamin A. Can. J. Physiol. Pharmacol. 93, 1065–1075 10.1139/cjpp-2014-0522 [DOI] [PubMed] [Google Scholar]
- 9. Kedishvili N. Y. (2013) Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 54, 1744–1760 10.1194/jlr.R037028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Napoli J. L. (1986) Retinol metabolism in UC-PKI cells: characterization of retinoic acid synthesis by an established mammalian cell line. J. Biol. Chem. 261, 13592–13597 [PubMed] [Google Scholar]
- 11. Rhinn M., Schuhbaur B., Niederreither K., and Dollé P. (2011) Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment. Proc. Natl. Acad. Sci. U.S.A. 108, 16687–16692 10.1073/pnas.1103877108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sandell L. L., Sanderson B. W., Moiseyev G., Johnson T., Mushegian A., Young K., Rey J.-P., Ma J.-X., Staehling-Hampton K., and Trainor P. A. (2007) RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 21, 1113–1124 10.1101/gad.1533407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ashique A. M., May S. R., Kane M. A., Folias A. E., Phamluong K., Choe Y., Napoli J. L., and Peterson A. S. (2012) Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling. Genesis 50, 415–423 10.1002/dvg.22002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cunningham T. J., Chatzi C., Sandell L. L., Trainor P. A., and Duester G. (2011) Rdh10 mutants deficient in limb field retinoic acid signaling exhibit normal limb patterning but display interdigital webbing. Dev. Dyn. 240, 1142–1150 10.1002/dvdy.22583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sandell L. L., Lynn M. L., Inman K. E., McDowell W., and Trainor P. A. (2012) RDH10 oxidation of vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PLoS One 7, e30698 10.1371/journal.pone.0030698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu B. X., Chen Y., Chen Y., Fan J., Rohrer B., Crouch R. K., and Ma J.-X. (2002) Cloning and characterization of a novel all-trans-retinol short-chain dehydrogenase/reductase from the RPE. Invest. Ophthalmol. Vis. Sci. 43, 3365–3372 [PubMed] [Google Scholar]
- 17. Persson B., Bray J. E., Bruford E., Dellaporta S. L., Favia A. D., Gonzalez Duarte R. G., Jörnvall H., Kallberg Y., Kavanagh K. L., Kedishvili N., Kisiela M., Maser E., Mindnich R., Orchard S., Penning T. M., et al. (2009) The SDR (Short-Chain Dehydrogenase/Reductase and Related Enzymes) Nomenclature Initiative. Chem. Biol. Interact. 178, 94–98 10.1016/j.cbi.2008.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Belyaeva O. V., Lee S.-A., Adams M. K., Chang C., and Kedishvili N. Y. (2012) Short chain dehydrogenase/reductase Rdhe2 is a novel retinol dehydrogenase essential for frog embryonic development. J. Biol. Chem. 287, 9061–9071 10.1074/jbc.M111.336727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Belyaeva O. V., Chang C., Berlett M. C., and Kedishvili N. Y. (2015) Evolutionary origins of retinoid active short-chain dehydrogenases/reductases of SDR16C family. Chem. Biol. Interact. 234, 135–143 10.1016/j.cbi.2014.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee S.-A., Belyaeva O. V., and Kedishvili N. Y. (2009) Biochemical characterization of human epidermal retinol dehydrogenase 2. Chem. Biol. Interact. 178, 182–187 10.1016/j.cbi.2008.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Labrie F., Simard J., Luu-The V., Pelletier G., Bélanger A., Lachance Y., Zhao H. F., Labrie C., Breton N., and de Launoit Y. (1992) Structure and tissue-specific expression of 3β-hydroxysteroid dehydrogenase/5-ene-4-ene isomerase genes in human and rat classical and peripheral steroidogenic tissues. J. Steroid Biochem. Mol. Biol. 41, 421–435 10.1016/0960-0760(92)90368-S [DOI] [PubMed] [Google Scholar]
- 22. Ross A. C., and Zolfaghari R. (2011) Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 31, 65–87 10.1146/annurev-nutr-072610-145127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Byrne S. M., and Blaner W. S. (2013) Retinol and retinyl esters: biochemistry and physiology. J. Lipid Res. 54, 1731–1743 10.1194/jlr.R037648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee L. M., Leung C. Y., Tang W. W., Choi H. L., Leung Y. C., McCaffery P. J., Wang C. C., Woolf A. S., and Shum A. S. (2012) A paradoxical teratogenic mechanism for retinoic acid. Proc. Natl. Acad. Sci. U.S.A. 109, 13668–13673 10.1073/pnas.1200872109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee S. A., Belyaeva O. V., Wu L., and Kedishvili N. Y. (2011) Retinol dehydrogenase 10 but not retinol/sterol dehydrogenase(s) regulates the expression of retinoic acid-responsive genes in human transgenic skin raft culture. J. Biol. Chem. 286, 13550–13560 10.1074/jbc.M110.181065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gonzales K. A. U., and Fuchs E. (2017) Skin and its regenerative powers: an alliance between stem cells and their niche. Dev. Cell 43, 387–401 10.1016/j.devcel.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Napoli J. L. (2001) 17β-Hydroxysteroid dehydrogenase type 9 and other short-chain dehydrogenases/reductases that catalyze retinoid, 17β- and 3α-hydroxysteroid metabolism. Mol. Cell Endocrinol. 171, 103–109 10.1016/S0303-7207(00)00392-0 [DOI] [PubMed] [Google Scholar]
- 28. Zhang M., Chen W., Smith S. M., and Napoli J. L. (2001) Molecular characterization of a mouse short chain dehydrogenase/reductase active with all-trans-retinol in intact cells, mRDH1. J. Biol. Chem. 276, 44083–44090 10.1074/jbc.M105748200 [DOI] [PubMed] [Google Scholar]
- 29. Gough W. H., VanOoteghem S., Sint T., and Kedishvili N. Y. (1998) cDNA cloning and characterization of a new human microsomal NAD+-dependent dehydrogenase that oxidizes all-trans-retinol and 3α-hydroxysteroids. J. Biol. Chem. 273, 19778–19785 10.1074/jbc.273.31.19778 [DOI] [PubMed] [Google Scholar]
- 30. Belyaeva O. V., Johnson M. P., and Kedishvili N. Y. (2008) Kinetic analysis of human enzyme RDH10 defines the characteristics of a physiologically relevant retinol dehydrogenase. J. Biol. Chem. 283, 20299–20308 10.1074/jbc.M800019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sahu B., Sun W., Perusek L., Parmar V., Le Y. Z., Griswold M. D., Palczewski K., and Maeda A. (2015) Conditional ablation of retinol dehydrogenase 10 in the retinal pigmented epithelium causes delayed dark adaption in mice. J. Biol. Chem. 290, 27239–27247 10.1074/jbc.M115.682096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adams M. K., Belyaeva O. V., Wu L., and Kedishvili N. Y. (2014) The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J. Biol. Chem. 289, 14868–14880 10.1074/jbc.M114.552257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams M. K., Lee S. A., Belyaeva O. V., Wu L., and Kedishvili N. Y. (2017) Characterization of human short chain dehydrogenase/reductase SDR16C family members related to retinol dehydrogenase 10. Chem. Biol. Interact. 276, 88–94 10.1016/j.cbi.2016.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Myung P., and Ito M. (2012) Dissecting the bulge in hair regeneration. J. Clin. Invest. 122, 448–454 10.1172/JCI57414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chase H. B., Rauch R., and Smith V. W. (1951) Critical stages of hair development and pigmentation in the mouse. Physiol. Zool. 24, 1–8 10.1086/physzool.24.1.30152098 [DOI] [PubMed] [Google Scholar]
- 37. Plikus M. V., Mayer J. A., de la Cruz D., Baker R. E., Maini P. K., Maxson R., and Chuong C. M. (2008) Cyclic dermal BMP signalling regulates stem cell activation during hair regeneration. Nature 451, 340–344 10.1038/nature06457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Müller-Röver S., Handjiski B., van der Veen C., Eichmüller S., Foitzik K., McKay I. A., Stenn K. S., and Paus R. (2001) A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J. Invest. Dermatol. 117, 3–15 10.1046/j.0022-202x.2001.01377.x [DOI] [PubMed] [Google Scholar]
- 39. Chen C. C., Plikus M. V., Tang P. C., Widelitz R. B., and Chuong C. M. (2016) The modulatable stem cell niche: tissue interactions during hair and feather follicle regeneration. J. Mol. Biol. 428, 1423–1440 10.1016/j.jmb.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsu Y. C., Li L., and Fuchs E. (2014) Emerging interactions between skin stem cells and their niches. Nat. Med. 20, 847–856 10.1038/nm.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lay K., Kume T., and Fuchs E. (2016) FOXC1 maintains the hair follicle stem cell niche and governs stem cell quiescence to preserve long-term tissue-regenerating potential. Proc. Natl. Acad. Sci. U.S.A. 113, E1506–E1515 10.1073/pnas.1601569113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schneider M. R., Schmidt-Ullrich R., and Paus R. (2009) The hair follicle as a dynamic miniorgan. Curr. Biol. 19, R132–R142 10.1016/j.cub.2008.12.005 [DOI] [PubMed] [Google Scholar]
- 43. Barker N., Tan S., and Clevers H. (2013) Lgr proteins in epithelial stem cell biology. Development 140, 2484–2494 10.1242/dev.083113 [DOI] [PubMed] [Google Scholar]
- 44. Goldstein J., and Horsley V. (2012) Home sweet home: skin stem cell niches. Cell Mol. Life Sci. 69, 2573–2582 10.1007/s00018-012-0943-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jensen K. B., Collins C. A., Nascimento E., Tan D. W., Frye M., Itami S., and Watt F. M. (2009) Lrig1 expression defines a distinct multipotent stem cell population in mammalian epidermis. Cell Stem Cell 4, 427–439 10.1016/j.stem.2009.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Genander M., Cook P. J., Ramsköld D., Keyes B. E., Mertz A. F., Sandberg R., and Fuchs E. (2014) BMP signaling and its pSMAD1/5 target genes differentially regulate hair follicle stem cell lineages. Cell Stem Cell 15, 619–633 10.1016/j.stem.2014.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ahmed N. S., Ghatak S., El Masry M. S., Gnyawali S. C., Roy S., Amer M., Everts H., Sen C. K., and Khanna S. (2017) Epidermal E-cadherin dependent β-catenin pathway is phytochemical inducible and accelerates anagen hair cycling. Mol. Ther. 25, 2502–2512 10.1016/j.ymthe.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cotsarelis G., Sun T. T., and Lavker R. M. (1990) Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 61, 1329–1337 10.1016/0092-8674(90)90696-C [DOI] [PubMed] [Google Scholar]
- 49. Levy V., Lindon C., Harfe B. D., and Morgan B. A. (2005) Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev. Cell 9, 855–861 10.1016/j.devcel.2005.11.003 [DOI] [PubMed] [Google Scholar]
- 50. Brownell I., Guevara E., Bai C. B., Loomis C. A., and Joyner A. L. (2011) Nerve-derived sonic hedgehog defines a niche for hair follicle stem cells capable of becoming epidermal stem cells. Cell Stem Cell 8, 552–565 10.1016/j.stem.2011.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nowak J. A., Polak L., Pasolli H. A., and Fuchs E. (2008) Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell 3, 33–43 10.1016/j.stem.2008.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vidal V. P., Chaboissier M. C., Lützkendorf S., Cotsarelis G., Mill P., Hui C. C., Ortonne N., Ortonne J. P., and Schedl A. (2005) Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr. Biol. 15, 1340–1351 10.1016/j.cub.2005.06.064 [DOI] [PubMed] [Google Scholar]
- 53. Chen T., Heller E., Beronja S., Oshimori N., Stokes N., and Fuchs E. (2012) An RNA interference screen uncovers a new molecule in stem cell self-renewal and long-term regeneration. Nature 485, 104–108 10.1038/nature10940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jaks V., Barker N., Kasper M., van Es J. H., Snippert H. J., Clevers H., and Toftgård R. (2008) Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet. 40, 1291–1299 10.1038/ng.239 [DOI] [PubMed] [Google Scholar]
- 55. Zhang L., Li W. H., Anthonavage M., and Eisinger M. (2006) Melanocortin-5 receptor: a marker of human sebocyte differentiation. Peptides 27, 413–420 10.1016/j.peptides.2005.05.030 [DOI] [PubMed] [Google Scholar]
- 56. Sellheyer K., and Krahl D. (2010) Blimp-1: a marker of terminal differentiation but not of sebocytic progenitor cells. J. Cutan. Pathol. 37, 362–370 10.1111/j.1600-0560.2009.01434.x [DOI] [PubMed] [Google Scholar]
- 57. Horsley V., O'Carroll D., Tooze R., Ohinata Y., Saitou M., Obukhanych T., Nussenzweig M., Tarakhovsky A., and Fuchs E. (2006) Blimp1 defines a progenitor population that governs cellular input to the sebaceous gland. Cell 126, 597–609 10.1016/j.cell.2006.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zouboulis C. C., Picardo M., Ju Q., Kurokaw I., Törőcsik D., Bíró T., and Schneider M. R. (2016) Beyond acne: current aspects of sebaceous gland biology and function. Rev. Endocr. Metab. Disord. 17, 319–334 10.1007/s11154-016-9389-5 [DOI] [PubMed] [Google Scholar]
- 59. Gat U., DasGupta R., Degenstein L., and Fuchs E. (1998) De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated β-catenin in skin. Cell 95, 605–614 10.1016/S0092-8674(00)81631-1 [DOI] [PubMed] [Google Scholar]
- 60. Oro A. E., and Higgins K. (2003) Hair cycle regulation of Hedgehog signal reception. Dev. Biol. 255, 238–248 10.1016/S0012-1606(02)00042-8 [DOI] [PubMed] [Google Scholar]
- 61. Silva-Vargas V., Lo Celso C., Giangreco A., Ofstad T., Prowse D. M., Braun K. M., and Watt F. M. (2005) β-Catenin and Hedgehog signal strength can specify number and location of hair follicles in adult epidermis without recruitment of bulge stem cells. Dev. Cell 9, 121–131 10.1016/j.devcel.2005.04.013 [DOI] [PubMed] [Google Scholar]
- 62. Vauclair S., Nicolas M., Barrandon Y., and Radtke F. (2005) Notch1 is essential for postnatal hair follicle development and homeostasis. Dev. Biol. 284, 184–193 10.1016/j.ydbio.2005.05.018 [DOI] [PubMed] [Google Scholar]
- 63. Estrach S., Cordes R., Hozumi K., Gossler A., and Watt F. M. (2008) Role of the Notch ligand Delta1 in embryonic and adult mouse epidermis. J. Invest. Dermatol. 128, 825–832 10.1038/sj.jid.5701113 [DOI] [PubMed] [Google Scholar]
- 64. Blanpain C., Lowry W. E., Pasolli H. A., and Fuchs E. (2006) Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 20, 3022–3035 10.1101/gad.1477606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wolbach S. B., and Howe P. R. (1978) Nutrition Classics. The Journal of Experimental Medicine 42: 753–77, 1925. Tissue changes following deprivation of fat-soluble A vitamin. S. Burt Wolbach and Percy R. Howe. Nutr. Rev. 36, 16–19 10.1111/j.1753-4887.1978.tb03675.x [DOI] [PubMed] [Google Scholar]
- 66. Chambon P. (1996) A decade of molecular biology of retinoic acid receptors. FASEB J. 10, 940–954 10.1096/fasebj.10.9.8801176 [DOI] [PubMed] [Google Scholar]
- 67. Mongan N. P., and Gudas L. J. (2007) Diverse actions of retinoid receptors in cancer prevention and treatment. Differentiation 75, 853–870 10.1111/j.1432-0436.2007.00206.x [DOI] [PubMed] [Google Scholar]
- 68. Easwaran V., Pishvaian M., Salimuddin, and Byers S. (1999) Cross-regulation of β-catenin-LEF/TCF and retinoid signalling pathways. Curr. Biol. 9, 1415–1418 10.1016/S0960-9822(00)80088-3 [DOI] [PubMed] [Google Scholar]
- 69. Collins C. A., and Watt F. M. (2008) Dynamic regulation of retinoic acid-binding proteins in developing, adult and neoplastic skin reveals roles for β-catenin and Notch signalling. Dev. Biol. 324, 55–67 10.1016/j.ydbio.2008.08.034 [DOI] [PubMed] [Google Scholar]
- 70. Clarke S. B., Nelson A. M., George R. E., and Thiboutot D. M. (2007) Pharmacologic modulation of sebaceous gland activity: mechanisms and clinical applications. Dermatol. Clin. 25, 137–146 10.1016/j.det.2007.01.004 [DOI] [PubMed] [Google Scholar]
- 71. Pan J., Wang Q., and Tu P. (2017) A topical medication of all-trans-retinoic acid reduces sebum excretion rate in patients with forehead acne. Am. J. Ther. 24, e207–e212 10.1097/MJT.0000000000000390 [DOI] [PubMed] [Google Scholar]
- 72. Winkler K. P., Black E. H., and Servat J. (2017) Lower eyelid retraction associated with topical retinol use. Ophthal. Plast. Reconstr. Surg. 33, 483 10.1097/IOP.0000000000000996 [DOI] [PubMed] [Google Scholar]
- 73. Kremer I., Gaton D. D., David M., Gaton E., and Shapiro A. (1994) Toxic effects of systemic retinoids on meibomian glands. Ophthalmic Res. 26, 124–128 10.1159/000267402 [DOI] [PubMed] [Google Scholar]
- 74. Ding J., Kam W. R., Dieckow J., and Sullivan D. A. (2013) The influence of 13-cis-retinoic acid on human meibomian gland epithelial cells. Invest. Ophthalmol. Vis. Sci. 54, 4341–4350 10.1167/iovs.13-11863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pryce J. E., Hayes B. J., Bolormaa S., and Goddard M. E. (2011) Polymorphic regions affecting human height also control stature in cattle. Genetics 187, 981–984 10.1534/genetics.110.123943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nishimura S., Watanabe T., Mizoshita K., Tatsuda K., Fujita T., Watanabe N., Sugimoto Y., and Takasuga A. (2012) Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 13, 40 10.1186/1471-2156-13-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gudbjartsson D. F., Walters G. B., Thorleifsson G., Stefansson H., Halldorsson B. V., Zusmanovich P., Sulem P., Thorlacius S., Gylfason A., Steinberg S., Helgadottir A., Ingason A., Steinthorsdottir V., Olafsdottir E. J., Olafsdottir G. H., et al. (2008) Many sequence variants affecting diversity of adult human height. Nat. Genet. 40, 609–615 10.1038/ng.122 [DOI] [PubMed] [Google Scholar]
- 78. Weedon M. N., Lango H., Lindgren C. M., Wallace C., Evans D. M., Mangino M., Freathy R. M., Perry J. R. B., Stevens S., Hall A. S., Samani N. J., Shields B., Prokopenko I., Farrall M., Dominiczak A., et al. (2008) Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 40, 575–583 10.1038/ng.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lettre G., Jackson A. U., Gieger C., Schumacher F. R., Berndt S. I., Sanna S., Eyheramendy S., Voight B. F., Butler J. L., Guiducci C., Illig T., Hackett R., Heid I. M., Jacobs K. B., Lyssenko V., et al. (2008) Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 40, 584–591 10.1038/ng.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Karim L., Takeda H., Lin L., Druet T., Arias J. A. C., Baurain D., Cambisano N., Davis S. R., Farnir F., Grisart B., Harris B. L., Keehan M. D., Littlejohn M. D., Spelman R. J., Georges M., and Coppieters W. (2011) Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat. Genet. 43, 405–413 10.1038/ng.814 [DOI] [PubMed] [Google Scholar]
- 81. Littlejohn M., Grala T., Sanders K., Walker C., Waghorn G., Macdonald K., Coppieters W., Georges M., Spelman R., Hillerton E., Davis S., and Snell R. (2012) Genetic variation in PLAG1 associates with early life body weight and peripubertal weight and growth in Bos Taurus. Anim. Genet. 43, 591–594 10.1111/j.1365-2052.2011.02293.x [DOI] [PubMed] [Google Scholar]
- 82. Jiao S., Maltecca C., Gray K. A., and Cassady J. P. (2014) Feed intake, average daily gain, feed efficiency, and real-time ultrasound traits in Duroc pigs: II. Genomewide association. J. Anim. Sci. 92, 2846–2860 10.2527/jas.2014-7337 [DOI] [PubMed] [Google Scholar]
- 83. Bai H., Zhu J., Sun Y., Liu R., Liu N., Li D., Wen J., and Chen J. (2014) Identification of genes related to beak deformity of chickens using digital gene expression profiling. PLoS One 9, e107050 10.1371/journal.pone.0107050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Xiong X., Yang H., Yang B., Chen C., and Huang L. (2015) Identification of quantitative trait transcripts for growth traits in the large scales of liver and muscle samples. Physiol. Genomics 47, 274–280 10.1152/physiolgenomics.00005.2015 [DOI] [PubMed] [Google Scholar]
- 85. Ashmore J. H., Luo S., Watson C. J. W., and Lazarus P. (2018) Carbonyl reduction of NNK by recombinant human lung enzymes: identification of HSD17β12 as the reductase important in (R)-NNAL formation in human lung. Carcinogenesis 39, 1079–1088 10.1093/carcin/bgy065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Qi F., Qin W.-X., and Zang Y.-S. (2019) Molecular mechanism of triple-negative breast cancer-associated BRCA1 and the identification of signaling pathways. Oncol. Lett. 17, 2905–2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Belyaeva O. V., Stetsenko A. V., Nelson P., and Kedishvili N. Y. (2003) Properties of short-chain dehydrogenase/reductase RalR1: characterization of purified enzyme, its orientation in the microsomal membrane, and distribution in human tissues and cell lines. Biochemistry 42, 14838–14845 10.1021/bi035288u [DOI] [PubMed] [Google Scholar]
- 88. Challa A. K., Boitet E. R., Turner A. N., Johnson L. W., Kennedy D., Downs E. R., Hymel K. M., Gross A. K., and Kesterson R. A. (2016) Novel hypomorphic alleles of the mouse tyrosinase gene induced by CRISPR-Cas9 nucleases cause non-albino pigmentation phenotypes. PLoS One 11, e0155812 10.1371/journal.pone.0155812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nagy A., Gertsenstein M., Vintersten K., and Behringer R. (2003) Manipulating the Mouse Embryo: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 90. Chetyrkin S. V., Hu J., Gough W. H., Dumaual N., and Kedishvili N. Y. (2001) Further characterization of human microsomal 3α-hydroxysteroid dehydrogenase. Arch. Biochem. Biophys. 386, 1–10 10.1006/abbi.2000.2203 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.