

Abstract
Zirconacyclopentadienes are versatile precursors for a large number of heteroles, which are accessible by Zr‐element exchange reactions. The vast majority of reports describe their preparation by the use of Negishi′s reagent, which is a species that is formed in situ. The zirconacyclopentadiene is then formed by the addition of one equivalent of a diyne or two equivalents of a monoyne moiety to this Negishi species. Another route involves Rosenthal′s reagent (Cp2Zr(py)Me3SiC≡CSiMe3), which then reacts with a diyne or monoyne moiety. In this work, the efficiency of both routes was compared in terms of reaction time, stability of the product in the reaction mixture, and yield. The synthetic implications of using both routes are evaluated. Novel zirconacyclopentadienes were synthesized, characterized directly from the reaction mixture, and crystal structures could be obtained in most cases.
Keywords: metallacycles, Negishi′s reagent, Rosenthal′s reagent, substituent effects, zirconium
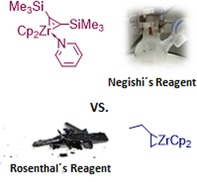
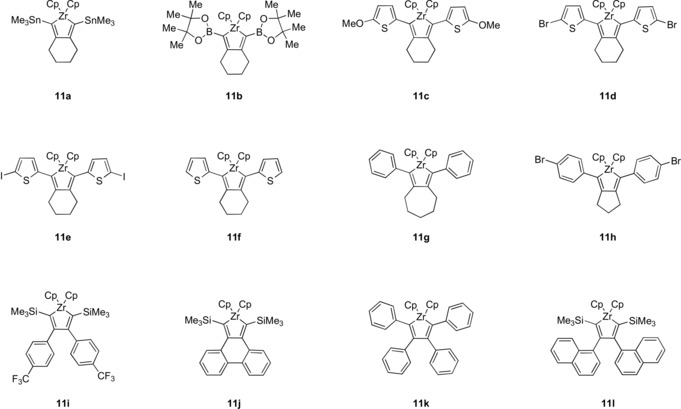
Negishi vs. Rosenthal: Two “Cp2Zr” sources, the Negishi and the Rosenthal reagents, were compared with respect to yield, reaction, time, and product stability. In total, twelve compounds with different substituents were used for the reductive coupling by these zirconium sources.
Introduction
Heteroles are five‐membered cycles derived from cyclopentadiene or its derivatives. In these molecules, the sp3 carbon is replaced by a heteroatom. They have become very important during the past few decades in the areas of biology,1 materials science,2 and medicinal chemistry.1
The most common of these heterocycles are aromatic (e.g., thiophene, pyrrole, or furan),3 although others are formally anti‐aromatic (e.g., boroles).4 Some are non‐aromatic with other forms of conjugations such as σ*–π* conjugation, which is exclusive to heavier element containing heterocycles (for example, group 14 metalloles).5
There are many established routes for the synthesis of these classical aromatic heteroles,6 but the breakthrough to access the non‐classical non‐ or anti‐aromatic congeners was achieved by the use of zirconacyclopentadienes or other metallacyclopentadienes7 as precursor molecules.8 In zirconacyclopentadienes, the sp3 atom of a cyclopentadiene is replaced by a Zr atom with further ligands saturating the coordination sphere of the zirconium. These ligands are Cp rings in most cases. Starting with a zirconacyclopentadiene (e.g., 1 a), many metalloles can be synthesized, in which group 14 elements (E=Si, Ge, Sn, Pb) or other main group elements are introduced into the cycle by transmetalation reactions.5a, 9 Moreover, zirconacyclopentadienes show other synthetic uses (Scheme 1). For example, if the zirconacyclopentadiene core (e.g., 1 b) is part of a large polyaromatic system, protonation with HCl gives dienes such as compound 3, which are otherwise difficult to access.10 Furthermore, if zirconacyclopentadienes are included in polymeric systems (e.g., 1 c), they can be converted into stable aromatic building blocks as in polymer 4.11
Scheme 1.
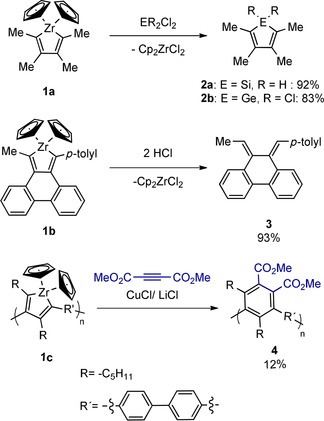
Transformation of zirconacyclopentadienes into metalloles and extended diene systems.9b–9d Use of polyzirconacyclopentadienes for the synthesis of stable polymers.10, 11
Zirconacyclopentadienes 1 are typically formed by a reductive coupling of alkynes 5 by the zirconocene species “Cp2Zr” 8 (Scheme 2).12 Such zirconacyclopentadienes can be also synthesized by the reaction of two monoynes with the active zirconocene species 8.
Scheme 2.
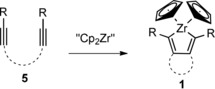
General synthesis of zirconacyclopentadienes of type 1.
In general, there are different methods to form this “Cp2Zr” species from reagents that are mixtures of Cp2ZrCl2/Na, Cp2ZrCl2/Mg, or Cp2ZrCl2/Ln.9c Further reagents are Takahashi′s (“Cp2ZrEt2”),13 Negishi′s (“Cp2ZrBu2”),14 and Rosenthal′s reagent (Cp2Zr(py)Me3SiC≡CSiMe3).15 However, the vast majority of reports describe the formation of the active “Cp2Zr” species by using either of the latter two methods.
The synthesis of zirconacyclopentadienes via Negishi′s reagent includes an in situ formed Zr reagent, coordinated by THF, which is not stable for long times at room temperature and which can be not isolated in pure form.12, 14, 16 The reaction between Cp2ZrCl2 (6) and two equivalents of nBuLi yields the intermediate 7 and insoluble LiCl (Scheme 3). The zirconocene 7 then decomposes into n‐butane and a number of active species collectively labelled as “Cp2Zr” (8),17 which is Negishi′s reagent. The advantages of using this reagent include the use of relatively cheap precursors, time‐saving in situ handling of the precursor reagents, and the high reactivity of the Negishi species. However, the latter may turn into a disadvantage when the dialkynes bear functional groups such as aromatic rings with halogen atoms. In a side reaction, these can undergo halogen–lithium exchange reactions if the concentration of nBuLi is not exactly known and an excess is used. Also, the tolerance towards other functional groups and heterocycles as substituents is often not given.10b Finally, the reaction conditions limit the choice of solvent, because Cp2ZrCl2 (6) is insoluble in non‐polar solvents such as n‐pentane or n‐hexane.10b
Scheme 3.
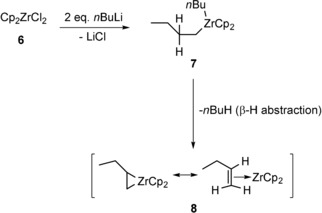
Formation of the “Cp2Zr” Negishi's reagent 8.12
Another possibility for the formation of zirconacyclopentadienes is the use of Rosenthal′s reagent 9 (Figure 1).15, 18 The synthesis of Rosenthal's zirconocene is based on the substitution of THF by pyridine of the complex Cp2Zr(THF)Me3SiC≡CSiMe3. Rosenthal and co‐workers exchanged THF with pyridine because the zirconocene complex of THF is not as stable as with pyridine in solution and in pure form. There are two ways to prepare such a complex: one is based on the reduction of Cp2ZrCl2 with magnesium in the presence of bis(trimethylsilyl)acetylene in THF at room temperature (in a yield of 66 %).15b An improved method is based on the formation of the complex via Negishi's conditions (Scheme 3), elaborated by Tilley and co‐workers (in a yield of 85 %).
Figure 1.
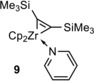
Structure of the Rosenthal's reagent 9.
The “Cp2Zr” species, stabilized by a pyridine and a bis(trimethylsilyl)acetylene ligand, is a thermally stable compound that can be easily crystallized and stored under inert conditions.10b, 19
This means that for further syntheses with the Rosenthal reagent, much milder reaction conditions will be given to form further metalloles in general; some examples for the synthesis of functionalized metalloles already exist.20 The great solubility in different solvents including non‐polar ones renders this reagent particularly useful for applications in macrocyclic and polymer chemistry, but mostly non‐coordinating solvents are used in the literature. Furthermore, only volatile byproducts (pyridine and bis(trimethylsilyl)acetylene) are formed during the reaction and can be therefore easily removed compared with LiCl.10b, 18a, 21
However, a direct, quantitative, and practicability comparison between the two reagents has not been made so far. The aim of this work was to investigate the exact differences between the use of two sources of “Cp2Zr” in the preparation of zirconacyclopentadienes 11 a–11 h starting with eight dialkynes 10 a–10 h (Scheme 4) and zirconacyclopentadienes 11 i–11 l from four alkynes 10 i–10 l (Scheme 5).22 A particular focus was the compatibility of the reagents with the alkynes that are functionalized.
Scheme 4.
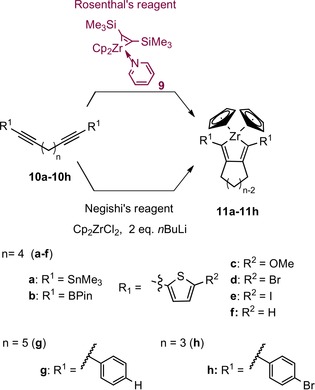
Synthetic path to form the zirconacyclopentadienes 11 a–11 h by using two different sources of “Cp2Zr” and eight types of dialkynes 10 a–10 h.
Scheme 5.
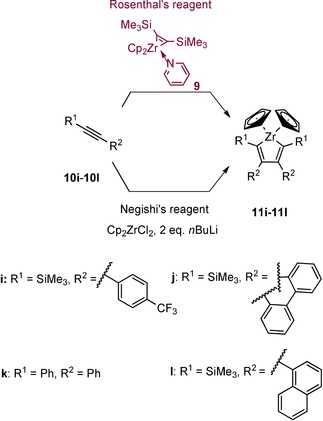
Synthetic path to form the zirconacyclopentadienes 11 i–11 l by using two different sources of “Cp2Zr” and four alkynes 10 i–10 l.
In the case of the dialkynes 10 a and 10 b, the use of Negishi′s reagent for the formation of the zirconacyclopentadienes has already been documented; the isolated yields are 55 % for zirconacyclopentadiene 11 a 23 and 48 % for compound 11 b.9d They are included in this study as benchmarks.
Results and Discussion
The “Results and Discussion” part of the synthesis of the alkynes can be found as a part of the Supporting Information.
Synthesis of zirconacyclopentadienes: Monitoring of reaction progress by 1H NMR spectroscopy
Reaction of the diynes and monoynes 10 a–10 l, respectively, with the “Cp2Zr” species, derived either from Negishi's or Rosenthal′s reagent, should lead to the formation of the zirconacyclopentadienes 11 a–11 l (Scheme 4 and Scheme 5). The progress of the reactions under both conditions was followed by 1H NMR spectroscopy by using naphthalene as an internal standard for quantification.
In the case of Negishi′s reagent, the reactive “Cp2Zr” species was formed by adding nBuLi to a solution of zirconocene dichloride in THF at −78 °C. The time started with the addition of the solution of the respective alkynes 10 a–10 l in THF. The cooling bath was then removed, allowing the reaction to reach room temperature (ca. 22 °C in our laboratories, see the Supporting Information for details). A defined sample was taken out after 10 min, 30 min, 1 h, 3 h, and 22 h, and the solvent was removed immediately under reduced pressure at 22 °C. Then, naphthalene in C6D6 as an internal standard was added in equimolar proportions for the 1H NMR measurements.
The formation of the zirconacyclopentadiene 11 a via this route was confirmed as reported in the literature;23 after 30 min of reaction, the product was formed with a yield of 85 %. The 1H NMR spectrum showed a resonance at 6.00 ppm, which was indicative for the cyclopentadienyl groups (Cp) of zirconacyclopentadiene 11 a (Figure 2). Such a downfield shift of the Cp signal in the 1H NMR spectrum in comparison to the chemical shift of these protons of the starting material (Cp2ZrCl2 (C6D6): δ=5.88 ppm) suggests zirconacyclopentadiene ring formation, because the shift is affected by the C−C double bonds inside the zirconacyclopentadiene.
Figure 2.
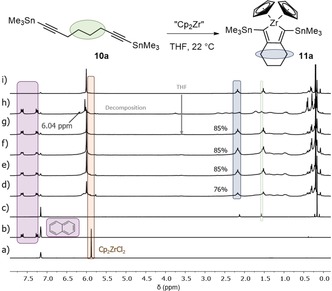
1H NMR spectra (recorded at 300 K, 200 MHz in C6D6) of the reaction monitoring for the synthesis of zirconacyclopentadiene 11 a (Negishi's reagent) with naphthalene as standard (1 equiv). a) Starting material Cp2ZrCl2, b) naphthalene, c) starting material 10 a at t=0 min. Reaction monitoring after d) t=10 min, e) t=30 min, f) t=1 h, g) t=3 h, and h) t=22 h. i) Zirconacyclopentadiene 11 a previously isolated.23
However, over 22 h, the Cp signal shifted from 6.00 ppm to 6.04 ppm and the methylene groups were not as well defined as before, which may indicate that the product was not stable in the reaction mixture.
In the case of zirconacyclopentadiene 11 b with BPin moieties attached, 20 % yield was observed after 10 min of reaction; after 3 h the yield increased to 58 %. However, after 22 h, the zirconacyclopentadiene decomposed (for the corresponding 1H NMR spectra see Figure S61 in the Supporting Information).
For the reaction of Negishi's reagent and the dialkynes 10 c–10 f with thiophene moieties, quite a different outcome was observed. In the case of the reaction of diyne 10 c, the zirconacyclopentadiene 11 c was formed after 10 min with a yield of 15 %, but after 30 min the yield decreased to 9 % and after 1 h to 2 % (Figure 3). After 3 h of reaction, the product appeared to be entirely decomposed to the starting material 10 c, indicating that the zirconacyclopentadiene 11 c was unstable under the conditions of this synthetic route. This finding was confirmed by integration of the peak corresponding to the methoxy group compared with the standard, which indicated that 98 % of the starting material was present in the reaction mixture at 3 h.
Figure 3.
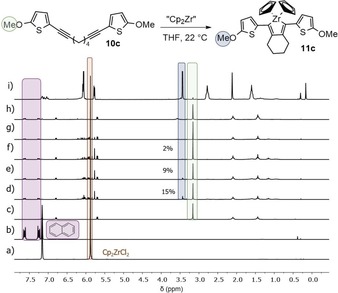
1H NMR spectra (recorded at 300 K, 200 MHz in C6D6) of the reaction monitoring for the synthesis of zirconacyclopentadiene 11 c (Negishi's reagent) with naphthalene as standard (1 equiv). a) Starting material Cp2ZrCl2, b) naphthalene, c) starting material 10 c. Reaction monitoring after d) t=10 min, e) t=30 min, f) t=1 h, g) t=3 h, and h) t=22 h. i) Zirconacyclopentadiene 11 c that was previously isolated.
For the formation of the zirconacyclopentadienes 11 d and 11 f by the reaction of the Negishi′s reagent and diynes 10 d or 10 f, respectively, something similar could be observed. After 10 min reaction time, 26 % was generated for compound 11 d and 82 % for 11 f. After 30 min, the yield increased to 28 % for 11 d whereas for 11 f, it decreased to 80 %. The yield decreased after 1 h to 21 % for 11 d and after 3 h to 65 % for 11 f. After 22 h, the signals present were attributed to the starting materials 10 d and 10 f and decomposition of the zirconium part into different structures16 was observed; a spectrum of different Cp signals appeared in the 1H NMR spectra (see Figures S63 and S66 in the Supporting Information). The zirconacyclopentadiene 11 e was not formed at any time of the reaction, just starting material 10 e and decomposition of the zirconium reagent was observed throughout the reaction monitoring by 1H NMR spectroscopy (see Figure S64 in the Supporting Information).
However, if bis(trimethylsilyl)acetylene (BTMSA) is first added to the Negishi′s reagent followed by the diyne 10 e, it could be observed that the zirconacyclopentadiene 11 e was formed in a yield of 41 % after 60 min. Still, after 22 h, the zirconacyclopentadiene 11 e was decomposed again (Figure 4). The difference in both reactions of diyne 10 e with Negishi′s reagent was the BTMSA, which seemed to be the crucial factor determining whether the product 11 e can be formed or not.
Figure 4.
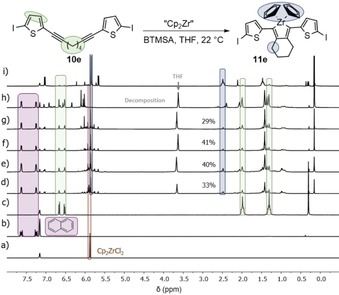
1H NMR spectra (recorded at 300 K, 600 MHz in C6D6) of the reaction monitoring of the formation of zirconacyclopentadiene 11 e at 22 °C with naphthalene as an internal standard. a) Starting material Cp2ZrCl2, b) naphthalene, c) starting material 10 e at t=0 min. Reaction monitoring after d) t=10 min, e) t=30 min, f) t=1 h, g) t=3 h, and h) t=22 h. i) Zirconacyclopentadiene 11 e previously isolated.
In the case of the reaction of Negishi's reagent with the starting materials 10 h and 10 k, their respective zirconacyclopentadienes 11 h and 11 k were formed with high yields of 99 % and 97 % after 10 min, respectively (see Figures S67 and S70 in the Supporting Information). Contrary to previous results, both were still stable within the reaction mixture after 22 h with yields of 99 % and 70 %. For the conversion of the alkynes 10 i, 10 j, and 10 l into the zirconacyclopentadienes 11 i, 11 j, and 11 l, the yield rose to 94 %, 88 %, and 80 % after 10 min, 10 min, and 30 min, respectively, but decreased after 22 h to 67 %, 65 %, and 26 % (see Figures S68, S69, and S71 in the Supporting Information). This means that these particular zirconacyclopentadienes were insufficiently stable within the reaction mixture over time: care would need to be taken to terminate the reaction with the appropriate Zr exchange at an optimal reaction time.24
The yield of zirconacyclopentadiene 11 g could not be determined by 1H NMR measurements (monitoring of reaction progress by 1H NMR spectroscopy), because this compound could not be completely redissolved after isolation. The compound instead formed gel‐like needles with the NMR solvents. Therefore, the yield of the zirconacyclopentadiene 11 g had to be determined after complete isolation after 3 h from the reaction mixture. It could be successfully synthesized with a yield of 88 % (purity 90 %). A long 1H NMR measurement and HR‐MS confirmed the identity of the product.
Thus, Negishi′s reagent proved to be incompatible with all compounds that bear thiophenyl motifs. In general, the synthesis was successful with yields between 28 % and 98 % for all other zirconacyclopentadienes.
The reactions with Rosenthal's reagent were placed inside a glovebox under a nitrogen atmosphere. To a solution of Rosenthal's zirconocene 9 in toluene, the alkynes 10 a–l, respectively, were added as a solution in toluene. After removing the solvent of each sample for the reaction monitoring, naphthalene in C6D6 as an internal standard was added in equimolar proportions and the reactions were monitored immediately by 1H NMR spectroscopy. Rosenthal′s reagent is mostly used in non‐coordinating solvents (n‐pentane, n‐hexane, toluene, or benzene) in the literature, a reason for which could be that the original zirconium complex undergoes ligand‐exchange reactions and might be less stable again. Also, for further transmetalation reactions, which can be done by one‐pot reaction, it is often desired to have non‐coordinating solvents. Therefore, toluene was used as the solvent, but two further experiments in THF were also conducted.
Figure 5 shows the progress of the formation of the zirconacyclopentadiene 11 a. After 10 min, a signal at 6.00 ppm appears, which can be assigned to the Cp protons of the desired zirconacyclopentadiene, whereas the Cp signals at 5.44 ppm of the Rosenthal's reagent disappear. Contrary to the synthesis with the Negishi reagent, the product was still stable after 22 h.
Figure 5.
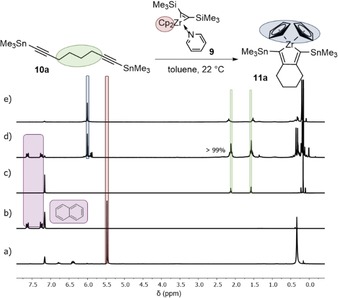
1H NMR spectra (recorded at 300 K, 200 MHz in C6D6) of the reaction monitoring of the formation of zirconacyclopentadiene 11 a at 22 °C with naphthalene as an internal standard. a) Rosenthal's reagent 9, b) naphthalene, c) starting material 10 a, d) monitoring after 10 min. e) Zirconacyclopentadiene 11 a.23
The transformation of the alkynes 10 b–l in toluene to their respective zirconacyclopentadienes 11 b–l proceeded in the same manner and were completed in all cases after 10 min; the products were stable for more than 22 h with yields ranging from 83 % to 99 % (For the 1H NMR spectra of the reaction monitoring of the zirconacyclopentadienes 11 b–l, see the Supporting Information). In the cases of the zirconacyclopentadienes 11 i and 11 l, the yield dropped from 97 % and 95 % to 83 % and 90 %, respectively, after 22 h (see Figures S82 and S85 in the Supporting Information). It appears that these two compounds might undergo a reversion of the cyclization as was observed in most of the synthesis examples under the Negishi conditions. Zirconacyclopentadiene 11 g could be obtained with a yield of 91 % (purity 92 %) after 3 h of reaction time after complete isolation from the reaction mixture. Because of the solubility problems, the identity was determined again with a long 1H NMR measurement and a HR‐MS measurement. Zirconacyclopentadienes 11 d and 11 h were also synthesized by using Rosenthal′s reagent in THF. The yield for both reactions was >99 % after 10 min, but after 22 h dropped to 63 % and 83 %, respectively. After 22 h, both 1H NMR spectra showed again signals of the alkyne starting materials 10 d and 10 h, thus, the zirconium complex is not quite as stable as in toluene. This might arise because the coordinating THF in excess can induce the reverse reaction.
To investigate the stability of the zirconacyclopentadienes in solution in the presence of lithium chloride (which is a byproduct of the Negishi reagent), isolated zirconacyclopentadiene 11 e was stirred in a mixture of LiCl in toluene for 22 h and defined samples were taken after 10 min, 30 min, 60 min, 180 min, and 22 h. It was observed that a precipitate formed and, after 22 h, the zirconacyclopentadiene 11 e was completely decomposed (Figure 6).
Figure 6.
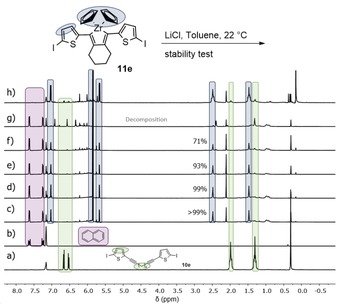
1H NMR spectra (recorded at 300 K, 600 MHz in C6D6) of the reaction monitoring of the stability test of zirconacyclopentadiene 11 e with naphthalene as standard (1 equiv). a) Naphthalene, b) starting material 10 e. Reaction monitoring after c) 10 min, d) 30 min, e) 1 h, f) 3 h, g) 22 h, and h) isolated zirconacyclopentadiene 11 e.
Table 1 summarizes the 1H NMR signals of the Cp groups and the analytical yields of all zirconacyclopentadienes obtained with both routes.
Table 1.
Summary of 1H NMR signals of the Cp rings in C6D6 and the highest conversion [%] for the zirconacyclopentadienes 11 a–l synthesized by using the Rosenthal or Negishi reagents.
|
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
Entry |
Product |
1H δ Cp |
Rosenthal |
Negishi |
||||
|
|
|
[ppm] |
Max. conversion [%][a] in toluene |
t [min] |
Remaining yield after 22 h [%] |
Max. conversion [%][a] |
t [min] |
Remaining yield after 22 h [%] |
|
1 |
11 a |
6.00 |
>99 |
10 |
>99 |
85 |
10 |
0 |
|
2 |
11 b |
6.37 |
97 |
10 |
97 |
56 |
180 |
0 |
|
3 |
11 c |
6.05 |
98 |
10 |
96 |
15 |
30 |
0 |
|
4 |
11 d |
5.84 |
91 (>99 % in THF) |
10 |
88 (63 % in THF) |
28 |
30 |
0 |
|
5 |
11 e |
5.85 |
>99 |
10 |
>99 |
0 (41 % with BTMSA) |
–(60 min) |
0 |
|
6 |
11 f |
6.00 |
93 |
10 |
93 |
83 |
10 |
0 |
|
7 |
11 g |
5.93 |
91 |
180[b] |
– |
88 |
180[c] |
– |
|
8 |
11 h |
5.70 |
>99 (>99 % in THF) |
10 |
>99 (83 % in THF) |
98 |
10 |
98 |
|
9 |
11 i |
6.09 |
97 |
10 |
90 |
94 |
10 |
67 |
|
10 |
11 j |
5.94 |
96 |
10 |
96 |
88 |
10 |
65 |
|
11 |
11 k |
6.01 |
>99 |
10 |
>99 |
97 |
10 |
70 |
|
12 |
11 l |
6.35 |
95 |
10 |
83 |
80 |
30 |
26 |
[a] The conversion was calculated based on the consumption of the starting material. [b] Yield obtained after complete isolation of the product after 3 h. [c] Yield obtained after complete isolation of the product after 3 h.
In general, it seems that for longer reaction times, the Rosenthal reagent is more stable in solution than the Negishi reagent. This is most likely due to the stabilizing ligands (bis(trimethylsilyl)acetylene and pyridine). This might be also explained by the slower and lower conversion for five (11 a–11 e) of the examples, because a certain amount of the active “Cp2Zr” species might decompose before it can undergo the cyclization reaction. The extra experiment of Negishi′s reagent with diyne 10 e, where BTMSA was added beforehand, shows also that stabilizing ligands are important for the prevention of decomposition of the active zirconium species and the formation of the zirconacyclopentadienes. For the other seven (11 f–11 l) examples, the conversion with the Negishi reagent was nearly as fast and high‐yielding as with Rosenthal′s reagent (Table 1).
An important fact to recognize is also that the zirconacyclopentadienes (11 a–11 f) were more stable over time (monitoring until 22 h) under the mild Rosenthal conditions than under the harsher Negishi conditions. In the six examples 11 a–11 f, the product was completely decomposed under Negishi's conditions. However, in the case of the compounds 11 h–11 l, with less reactive motifs such as phenyl groups or trimethylsilyl groups, the stability under the Negishi conditions was almost equal; the remaining yields after 22 h were higher than 26 %. The maximum conversion for all reactions with Rosenthal′s reagent were nearly quantitative after only 10 min and the remaining yields were higher than 83 % (toluene) and 63 % (THF).
For each of the two reagents, it could be observed that the conversion for all alkynes was different. One reason for this could be the stability of the alkyne complex of the Zr species in general. The formation of the zirconacyclopentadiene is an equilibrium reaction, which means for some of the alkynes the equilibrium might be on the side of the alkyne complex, but for some it might be on the side of the starting material. This can be seen by the fact that the maximal conversion for all the alkynes is different for the reaction with Rosenthal′s reagent and the same is also for the route with Negishi′s reagent. This observation may be explained by the different binding behavior of the alkynes to the zirconium, which means the yield of the zirconacyclopentadiene should be higher for a good‐binding alkyne. For the reaction with Negishi′s reagent, this can be seen more extremely in terms of yields (Table 1, maximum conversion and remaining yield after 22 h) than for the reaction with Rosenthal′s reagent owing to the fact that there are stabilizing ligands such as pyridine and/or bis(trimethylsilyl)acetylene from Rosenthal′s reagent remaining in the solution. Therefore, in the equilibrium, there are stable starting materials and products. In the case of the Negishi reagent, the starting material is unstable; thus, if the reverse reaction occurs, decomposition of the alkyne complex cannot be prevented. Because of the decomposition of the active “Cp2Zr” species over time, the yield decreases and new zirconacyclopentadiene cannot be formed.
The visible formation of a solid after 22 h in all of the reactions with Negishi's reagent might result from LiCl slowly precipitating; however, it could also be a result of the decomposition of the “Cp2Zr” species and the reversibility of the cyclization reaction to the starting materials.19 Another point in terms of decomposition of the zirconacyclopentadienes under Negishi′s conditions could be the LiCl in the solution. Zirconacyclopentadiene 11 e, which performed worst with Negishi's reagent, was tested for its stability. In a mixture of LiCl and pure zirconacyclopentadiene 11 e in toluene, we observed indeed that LiCl can induce decomposition. It appears again that the stability of the alkyne complex in the reaction mixture is crucial, because in the study with Negishi′s reagent, the zirconacyclopentadiene did not decompose in all cases. The choice of solvent also clearly has an effect on the overall stability, which can be seen from the reactions with Rosenthal′s reagent in THF.
In addition, comparing both methods, the handling of the Rosenthal's reagent was much easier compared with Negishi's reagent; especially when exact stoichiometry is needed. On the one hand, it was sufficient to prepare merely one stock solution for several reactions without the need for lower temperatures for the formation of such zirconacyclopentadienes. The in situ formation of Negishi′s reagent, on the other hand, requires lower temperatures and therefore temperature controls to prevent the reagent from decomposing.16
Additionally, air stability tests were conducted for all products and can be found as a part of the Supporting Information.
Structures in the solid state
The molecular structures of 11 d, 11 f, and 11 h–11 l are shown in Figure 7 (and are also part of the Supporting Information) and confirm the identity of the compounds. Single crystals were obtained from a saturated toluene solution. Selected bond lengths and angles of all structures can be found as a part of the Supporting Information (Tables S6–8). In all compounds, the Zr atom is incorporated into almost planar five‐membered central rings (e.g., Zr(1)‐C(1)‐C(2)‐C(7) is 8.3(2)° for 11 d). In all cases where the zirconacyclopentadiene ring bears aryl groups in the 2‐ and 5‐positions, these groups are twisted to the plane (e.g., Zr(1)‐C(1)‐C(10)‐C(11) is −23.0(3)° for 11 d). In the compounds 11 i–11 l, the aryl groups in the 3‐ and 4‐positions of the zirconacyclopentadiene ring are almost completely twisted with respect to the plane (e.g., C(1)‐C(2)‐C(20)‐C(21) is 83.36(2)° for 11 l), whereas the annulated six‐ or five‐membered rings in that position show puckered confirmations.
Figure 7.
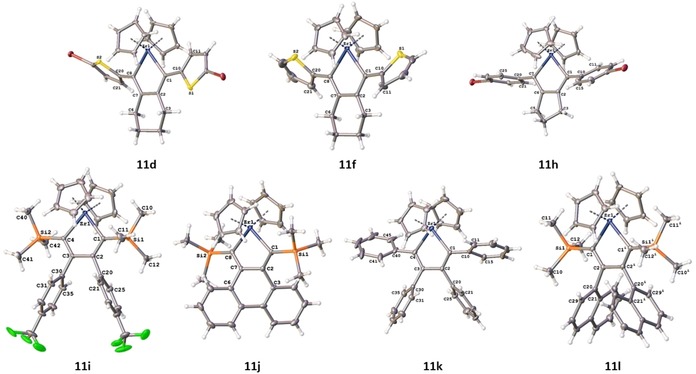
Molecular structures of 11 d, 11 f, 11 h–11 l showing 50 % probability ellipsoids and the crystallographic numbering scheme. Only the major parts of the disordered molecule of 11 f are shown for clarity. For 11 h, only one independent molecule is shown. Only the major parts of the disordered molecule of 11 i are shown for clarity.
Conclusions
In summary, it could be observed that the use of Rosenthal′s reagent was much more efficient for the synthesis of zirconacyclopentadienes in most of the cases with respect to yield, stability of the zirconacyclopentadiene, and reaction time when compared with Negishi′s reagent. More particularly, our study has shown the high importance of stabilizing ligands in the reaction mixture. In addition to reliably producing high yields, Rosenthal's reagent is very functional group tolerant; but in general, it was shown that halides also do not react under Negishi′s conditions, when nBuLi is titrated and used in exact amounts. However, the advantages of using Negishi′s reagent include using relatively cheap precursors, time‐saving in situ handling of the precursor reagents, and a highly reactive species. But, overall, Rosenthal′s zirconocene is a thermally stable reagent that can be prepared with great convenience.
Experimental Section
General methods and materials
All reactions were carried out by using standard Schlenk techniques under a dry, inert nitrogen or argon atmosphere unless noted otherwise. Some reactions were performed inside a nitrogen‐filled glovebox from Inert, Innovative Technology, Inc. (<0.1 ppm O2 and <0.1 ppm H2O).
All dry solvents were taken from the solvent purification system (SPS), degassed by freeze–pump–thaw cycles and stored under a nitrogen atmosphere unless noted otherwise. All chemicals were commercially available and were used without further purification unless noted otherwise. nBuLi was titrated by the Lin and Paquette method25 for exact concentrations.
Analytical instruments
1H NMR, 13C{1H} NMR, 11B{1H} NMR, 119Sn{1H} NMR, 19F NMR, and 29Si{1H} NMR spectra were recorded with a Bruker Avance Neo 500, Bruker Avance Neo 600, or Bruker DPX‐200 spectrometer at 300 K. All 1H NMR and 13C{1H} NMR were referenced against the solvent residual proton signals (1H), or the solvent itself (13C). The reference for the 119Sn{1H} NMR spectra was calculated based on the 1H NMR spectrum of TMS. 11B{1H} NMR and 19F NMR spectra were referenced against BF3 ⋅Et2O in CDCl3. 29Si{1H} NMR spectra were referenced against TMS in CDCl3. All chemical shifts (δ) are given in parts per million (ppm) and all coupling constants (J) in Hz. Electron impact (EI) ionization mass spectra were obtained with the double focusing mass spectrometer MAT 95+ or MAT 8200 from FINNIGAN mat. Samples were measured by direct inlet or indirect inlet methods with a source temperature of 200 °C. The ionization energy of the electron impact ionization was 70 eV. All signals were reported with the quotient from mass to charge (m/z).
Crystallography
Intensity data of 11 d, 11 f, 11 h–11 l were collected with a Bruker Venture D8 diffractometer at 100 K with MoKα (0.7107 Å) radiation. All structures were solved by intrinsic phasing and refined based on F 2 by use of the SHELX26 program package as implemented in OLex21.2.27 All non‐hydrogen atoms were refined by using anisotropic displacement parameters. Hydrogen atoms attached to carbon atoms were included in geometrically calculated positions by using a riding model. Crystal and refinement data are collected in Tables S3–5 in the Supporting Information.
A rotational disorder was resolved for the thiophene groups of compound 11 f. The refinement led to a split atom model for each group with refined occupancies of 76:24 and 53:47, respectively. Compound 11 h comprises two crystallographically independent conformers. The fluorine atoms of the two CF3 groups of compound 11 i were disordered and were refined with split occupancies of 71:29 and 57:43, respectively. The C−F distances were restrained to be equal. Compound 11 k crystallized with one molecule of toluene per asymmetric unit. The toluene solvate molecule was disordered over two positions with split occupancies of 65:35. Compound 11 l crystallized with a half molecule in the asymmetric unit. The Zr atom is located on the Wyckoff position 4e of space group C2/c.
CCDC https://summary.ccdc.cam.ac.uk/structure-summary?doi=10.1002/chem.201902255 (11 d, 11 h, 11 i, 11 j, 11 k, 11 l, respectively) contain the supplementary crystallographic data for this paper. These data are provided free of charge by http://www.ccdc.cam.ac.uk/.
General procedure for the synthesis of the zirconacyclopentadienes (11 a–l)
An equimolar solution of Rosenthal's reagent (9) and alkyne (10 a–l) in toluene was stirred at 22 °C for 10 min. The solvent was removed under inert conditions. After filtration over Celite, the final product was afforded.
Bis(h 5 ‐cyclopentadienyl)‐1,3‐bis(trimethylstannyl)‐4,5,6,7‐tetrahydro‐2H‐benzo[c]zirconacyclopentadiene 11 a: 10 a (100 mg, 230 μmol), Rosenthal′s reagent (109 mg, 230 μmol), toluene (5 mL), 22 °C, yield: 141 mg, 94 %. 1H NMR (500 MHz, C6D6): δ=6.00 (s, 10 H, Cp), 2.22–2.15 (m, 4 H, c), 1.57–1.50 (m, 4 H, d), 0.20 ppm (d, 2 J Sn‐H=24 Hz, 18 H, e); 13C{1H} NMR (126 MHz, C6D6): δ=206.1 (a), 150.7 (b), 111.4 (Cp), 38.7 (c), 23.1(d), −6.7 ppm (e; 1 J Sn‐C=146 Hz); 119Sn{1H} NMR (187 MHz, CDCl3): δ=−81.3 ppm; HR‐MS (EI, C24H36Sn2Zr): m/z calcd: 653.99153; found: 653.99188 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=654 (5) [M]+., 155 [M−C12H12]+., (100).
Bis(h 5 ‐cyclopentadienyl)‐1,3‐bis(4,4,5,5‐tetrametyhl‐1,3,2‐dioxaborolan‐2‐yl)‐4,5,6,7‐tetrahydro‐2H‐benzo[c]zirconacyclopentadiene 11 b: 10 b (100 mg, 280 μmol), Rosenthal′s reagent (132 mg, 280 μmol), toluene (5 mL), 22 °C, yield: 151 mg, 93 %. 1H NMR (500 MHz, C6D6): δ=6.37 (s, 10 H, Cp), 2.59–2.54 (m, 4 H, c), 1.65–1.58 (m, 4 H, d), 1.10 ppm (s, 24 H, f); 13C{1H} NMR (126 MHz, C6D6): δ 147.0 (b), 111.7 (Cp), 81.5(e), 35.4 (c), 24.9 (f), 23.5 (d) ppm;28 11B NMR (160 MHz, C6D6): δ=30.9 ppm; HR‐MS (EI, C30H42‐ 10/11B2O4 90Zr): m/z calcd: 577.23471; found: 577.23484 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=578 (13) [M]+., 83 (100).
Bis(h 5 ‐cyclopentadienyl)‐1,3‐bis(5‐methoxythiophen‐2‐yl)‐4,5,6,7‐tetrahydro‐2H‐benzo[c]zirconacyclopentadiene 11 c: 10 c (150 mg, 454 μmol), Rosenthal′s reagent (213 mg, 454 μmol), toluene (6 mL), 22 °C, yield: 233 mg, 93 %. 1H NMR (500 MHz, C6D6): δ=6.07 (d, 3 J=3.8 Hz, 2 H, g), 6.05 (s, 10 H, Cp), 5.77 (d, 3 J=3.7 Hz, 2 H, f), 3.44 (s, 6 H, i), 2.80–2.74 (m, 4 H, c), 1.63–1.57 ppm (m, 2 H, d); 13C{1H} NMR (126 MHz, C6D6): δ=177.9 (a), 165.7 (h), 141.3 (b), 136.8 (e), 119.9 (f), 112.1 (Cp), 104.1 (g), 59.7 (i), 30.7 (c), 24.4 ppm (d); HR‐MS (EI, C28H28O2S2 90Zr): m/z calcd: 550.05723; found: 550.05796 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=550 (74) [M]+., 220 (100) [Cp2Zr]+..
Bis(h 5 ‐cyclopentadienyl)‐1,3‐bis(5‐bromothiophen‐2‐yl)‐4,5,6,7‐tetrahydro‐2H‐benzo[c]zirconacyclopentadiene 11 d: 10 d (150 mg, 352 μmol), Rosenthal′s reagent (166 mg, 352 μmol), toluene (6 mL), 22 °C, yield: 210 mg, 93 %. 1H NMR (600 MHz, C6D6): δ=6.82 (d, 3 J=3.8 Hz, 2 H, g), 5.84 (s, 10 H, Cp), 5.69 (d, 3 J=3.8 Hz, 1 H, f), 2.49 (m, 4 H, c), 1.50–1.46 ppm (m, 4 H, d); 13C{1H} NMR (126 MHz, C6D6): δ=177.0 (a), 151.8 (e), 143.0 (b), 130.2 (g), 121.8 (f), 112.3 (Cp), 109.4 (h), 30.1 (c), 23.8 ppm (d); HR‐MS (EI, C26H22 79Br2S2 90Zr): m/z calcd: 645.85712; found: 645.85714 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=646 (38) [M]+., 301 (100).
Bis(h 5 ‐cyclopentadienyl)‐1,3‐bis(5‐iodothiophen‐2‐yl)‐4,5,6,7‐tetrahydro‐2H‐benzo[c]zirconacyclopentadiene 11 e: 10 e (150 mg, 287 μmol), Rosenthal′s reagent (137 mg, 287 μmol), toluene (6 mL), 22 °C, yield: 194 mg, 91 %. 1H NMR (500 MHz, C6D6): δ=7.03 (d, 3 J=3.7 Hz, 2 H, g), 5.85 (s, 10 H, Cp), 5.66 (d, 3 J=3.7 Hz, 2 H, f), 2.52–2.45 (m, 4 H, c), 1.50–1.45 ppm (m, 4 H, d); 13C{1H} NMR (126 MHz, C6D6): δ=176.9 (a), 156.1 (e), 142.8 (b), 137.1 (g), 129.1 (f), 112.1 (Cp), 70.0 (h), 29.9 (c), 23.6 ppm (d); HR‐MS (EI, C26H22I2S2 90Zr): m/z calcd: 741.82939; found: 741.82934 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=742 (44) [M]+., 347 (100).
Bis(h 5 ‐cyclopentadienyl)‐1,3‐di(thiophen‐2‐yl)‐4,5,6,7‐tetrahydro‐2H‐benzo[c]zirconacyclopentadiene 11 f: 10 f (150 mg, 555 μmol), Rosenthal′s reagent (261 mg, 555 μmol), toluene (6 mL), 22 °C, yield: 259 mg, 95 %. 1H NMR (500.1 MHz, C6D6): δ=6.96 (dd, 3 J=5.2 Hz, 4 J=1.0 Hz, 2 H, h), 6.90 (dd, 3 J=5.2, 3.5 Hz, 2 H, g), 6.21 (dd, 3 J=3.5 Hz, 4 J=1.0 Hz, 2 H, f), 6.00 (s, 10 H, Cp), 2.70–2.64 (m, 2 H, c), 1.57–1.54 ppm (m, 2 H, d); 13C{1H} NMR (126 MHz, C6D6): δ=177.9 (a), 150.3 (e), 142.4 (b), 127.3 (g), 123.0 (h), 121.4 (f), 112.2 (Cp), 30.1 (c), 24.0 ppm (d); HR‐MS (EI, C26H24S2 90Zr): m/z calcd: 490.03610; found: 490.03669 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=490 (19) [M]+., 220 (100) [Cp2Zr]+..
Bis(h 5 ‐cyclopentadienyl)‐1,3‐(diphenyl)‐2,4,5,6,7,8‐hexahydrocyclohepta[c]zirconacyclopentadiene 11 g: 10 g (150 mg, 551 μmol), Rosenthal′s reagent (259 mg, 552 μmol), toluene (6 mL), 22 °C, yield: 248 mg, 91 %, 92 % purity. 1H NMR (500 MHz, C6D6): δ=7.42–7.23 (m, 4 H, g, g′), 7.09–6.95 (m, 6 H, h, h“, i), 5.93 (s, 10 H, Cp), 2.38–2.31 (m, 4 H, c), 1.61–1.50 ppm (m, 6 H, d, e); 13C{1H} NMR (126 MHz, C6D6): δ=189.3 (a), 149.6 (b), 137.9 (f), 128.6 (g, g′), 126.3, 123.3 (h, h, I′), 112.2 (Cp), 31.6 (c), 31.1 ppm (d, e); HR‐MS (EI, C31H30 90Zr): m/z calcd: 492.13891; found: 492.13982 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=492 (8) [M]+., 220 (100) [Cp2Zr]+..
Bis(h 5 ‐cyclopentadienyl)‐1,3‐bis(4‐bromophenyl)‐2,4,5,6‐tetrahydrocyclopenta[c]zirconacyclopentadiene 11 h: 10 h (150 mg, 373 μmol), Rosenthal′s reagent (176 mg, 373 μmol), toluene (6 mL), 22 °C, yield: 223 mg, 96 %. 1H NMR (600 MHz, C6D6): δ=7.49–7.44 (m, 4 H, g, g′), 6.80–6.74 (m, 4 H, f, f′), 5.70 (s, 10 H, Cp), 2.33 (t, 3 J=7.1 Hz, 4 H, c), 1.26 ppm (p, 3 J=7.1 Hz, 2 H, d); 13C{1H} NMR (151 MHz, C6D6): δ=182.2 (a), 149.0 (b), 131.7 (g, g′), 128.0 (f, f′), 126.2 (e), 117.8 (h), 110.4 (Cp), 35.6 (c), 22.5 ppm (d); HR‐MS (EI, C29H24 79/80Br2 90Zr): m/z calcd: 621.92728; found: 621.92652 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=620 (69) [M]+., 220 (100) [Cp2Zr]+..
Bis(h 5 ‐cyclopentadienyl)‐3,4‐bis(4‐(trifluoromethyl)phenyl)‐2,5‐bis(trimethylsilyl)zirconacyclopentadiene 11 i: 10 i (150 mg, 650 μmol), Rosenthal′s reagent (146 mg, 310 μmol), toluene (6 mL), 22 °C, yield: 206 mg, 94 %. 1H NMR (600 MHz, C6D6): δ=7.10 (d, 3 J=7.8 Hz, 4 H, e, e′), 6.48 (d, 3 J=7.98 Hz, 4 H, d, d′), 6.08 (s, 10 H, Cp), −0.28 ppm (s, 18 H, SiMe3); 13C{1H} NMR (151 MHz, C6D6): δ=206.3 (a), 149.4 (c) 148.2 (b), 130.2 (d, d′), 127.5 (q, 2 J C‐F=32.2 Hz, f),29 125.0 (q, 1 J C‐F=271.7 Hz, g), 124.0 (q, 3 J C‐F=3.8 Hz, e, e′), 111.7 (Cp), 2.70 ppm (SiMe3); 19F NMR (471 MHz, C6D6): δ=−62.1 ppm; 29Si{1H} NMR (99 MHz, C6D6): δ=−14.9 ppm; MS (EI, 70 eV, direct inlet, 200 °C): compound shows no molecule ion.
Bis(h 5 ‐cyclopentadienyl)‐1,3‐bis(trimethylsilyl)‐2H‐phenanthro[9,10‐c]zirconacyclopentadiene 11 j: 10 j (122 mg, 352 μmol), Rosenthal′s reagent (166 mg, 352 μmol), toluene (3 mL), 22 °C, yield: 192 mg, 96 %. 1H NMR (600 MHz, C6D6): δ=7.60 (dd, 3 J=7.6 Hz, 4 J=1.3 Hz, 2 H, e), 7.51 (dd, 3 J=7.6 Hz, 4 J=1.3 Hz, h), 7.14 (td, 3 J=7.6 Hz, 4 J=1.3 Hz, 2 H, f), 7.07 (td, 3 J=7.6 Hz, 4 J=1.3 Hz, 2 H, g), 5.94 (s, 10 H, Cp), 0.21 ppm (s, 18 H, SiMe3); 13C{1H} NMR (151 MHz, C6D6): δ=208.9 (a), 137.5 (b), 136.2 (c or d), 133.7 (d or c), 129.7 (h), 128.8 (f), 126.9 (g), 123.7 (e), 110.2 (Cp), 3.8 ppm (SiMe3); HR‐MS (EI, C32H36 28Si2 90Zr): m/z calcd: 566.13971; found: 566.13966 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=566 (5) [M]+., 220 (100) [Cp2Zr]+.
Bis(h 5 ‐cyclopentadienyl)‐2,3,4,5‐tetraphenylzirconacyclopentadiene 11 k: 10 k (150 mg, 842 μmol), Rosenthal′s reagent (198 mg, 421 μmol), toluene (6 mL), 22 °C, yield: 231 mg, 95 %. 1H NMR (500 MHz, C6D6): δ=7.06–7.01 (m, 4 H, i, i′), 7.00–6.93 (m, 4 H, d, d′), 6.86–6.82 (m, 4 H, e, e′), 6.82–6.79 (m, 2 H, j), 6.76–6.71 (m, 2 H, f), 6.71–6.66 (m, 4 H, h, h′), 6.01 ppm (s, 10 H, Cp); 13C{1H} NMR (126 MHz, C6D6): δ=194.8 (a), 148.6 (g), 142.8 (b), 141.7 (c), 131.3 (i, i′), 128.3 (h, h′), 127.7 (d, d′), 127.2 (e, e′), 125.1 (f), 123.4 (j), 112.3 ppm (Cp); HR‐MS (EI, C38H30 90Zr); m/z calcd: 576.13891; found: 576.13987 (R=10 000); MS (EI, 70 eV, direct inlet, 200 °C): m/z (% relative intensity)=576 (18) [M]+., 220 (100) [Cp2Zr]+..
Bis(h 5 ‐cyclopentadienyl)‐3,4‐di(naphthalen‐1‐yl)‐2,5‐bis(trimethylsilyl)zirconacyclopentadiene 11 l: 10 l (150 mg, 669 μmol), Rosenthal′s reagent (158 mg, 335 μmol), toluene (6 mL), 22 °C, yield: 202 mg, 90 %. 1H NMR (600 MHz, C6D6): δ=8.32 (dq, 3 J=8.4 Hz, 4 J=0.9 Hz, 2 H, k), 7.46 (ddd, 3 J=8.4 Hz, 3 J=6.8 Hz, 4 J=1.3 Hz, 2 H, j), 7.38 (ddt, 3 J=8.1 Hz, 4 J=1.2 Hz, 0.6 Hz, 2 H, h), 7.21 (ddd, 3 J=8.1 Hz, 3 J=6.8 Hz, 4 J=1.3 Hz, 2 H, i), 7.02 (d, 3 J=8.1 Hz, 2 H, f), 6.84 (dd, 3 J=7.0 Hz, 4 J=1.3 Hz, 2 H, d), 6.59 (dd, 3 J=8.2 Hz, 3 J=7.0 Hz, 2 H, e), 6.34 (s, 10 H, Cp), −0.39 ppm (m, 18 H, SiMe3); 13C{1H} NMR (151 MHz, C6D6): δ=207.7 (a), 150.0 (b), 142.6 (c), 133.4 (l), 132.8 (g), 128.0 (h), 126.8 (k), 126.0 (d), 125.8 (f), 124.8 (j), 124.6 (i), 124.0 (e), 111.1 (Cp), 2.1 ppm (SiMe3); 29Si{1H} NMR (99 MHz, C6D6): δ=−14.9 ppm; MS (EI, 70 eV, direct inlet, 200 °C): compound shows no molecule ion.
Monitoring of reaction progress of the zirconacyclopentadienes (11 a–l) by 1H NMR spectroscopy
Procedure for Negishi's conditions: example 11 a
To a solution of Cp2ZrCl2 (67.7 mg, 231 μmol) in THF (2.0 mL) at −78 °C, n‐butyllithium (180 μL, 463 μmol; 2.59 m in hexanes) was added dropwise over the course of 1 min. The reaction mixture was stirred at −78 °C for 1 h and a solution of 10 a (100 mg, 231 μmol) in THF (1.0 mL) was added. The cooling bath was removed, and the reaction's time started to run. A sample (0.30 mL, 14 μmol) was taken after 10 min, 30 min, 1 h, 3 h, and 22 h and the solvent was removed immediately under inert conditions. Naphthalene in C6D6 was added (0.2 mL, 0.06 m, 14 μmol) to the sample and used as an internal standard. The reaction progress was analyzed by 1H NMR spectroscopic measurements of each sample.
Procedure for Rosenthal's condition: example 11 a
In a GB, a solution of 9 (109 mg, 230 μmol) and 10 a (100 mg, 230 μmol) in toluene (5 mL) was stirred at 22 °C. A sample of the reaction (0.3 mL, 14 μmol) was taken after 10 min, 30 min, 1 h, 3 h, and 22 h and the solvent was removed immediately under inert conditions. Naphthalene in C6D6 was added (0.2 mL, 0.06 m, 14 μmol) to the sample and used as an internal standard. The reaction progress was analyzed by 1H NMR spectroscopic measurements of each sample.
More detailed reaction monitoring conditions of both routes for the zirconacyclopentadienes 11 b–l can be found in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
S.U.‐R. thanks the German Academic Exchange Service (DAAD) and the Colfuturo Foundation for a PhD scholarship, grant number A/13/72356. This research has been supported by the Institutional Strategy of the University of Bremen, funded by the German Excellence Initiative.
S. Urrego-Riveros, I.-M. Ramirez y Medina, D. Duvinage, E. Lork, F. D. Sönnichsen, A. Staubitz, Chem. Eur. J. 2019, 25, 13318.
References
- 1. Trautwein A. W., Süßmuth R. D., Jung G., Bioorg. Med. Chem. Lett. 1998, 8, 2381–2384. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Wang C., Dong H., Hu W., Liu Y., Zhu D., Chem. Rev. 2012, 112, 2208–2267; [DOI] [PubMed] [Google Scholar]
- 2b. Facchetti A., Chem. Mater. 2011, 23, 733–758; [Google Scholar]
- 2c. Matano Y., Ohkubo H., Miyata T., Watanabe Y., Hayashi Y., Umeyama T., Imahori H., Eur. J. Inorg. Chem. 2014, 1620–1624. [Google Scholar]
- 3. Horner K. E., Karadakov P. B., J. Org. Chem. 2013, 78, 8037–8043. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Jimenez-Halla J. O. C., Matito E., Solà M., Braunschweig H., Hörl C., Krummenacher I., Wahler J., Dalton Trans. 2015, 44, 6740–6747; [DOI] [PubMed] [Google Scholar]
- 4b. Iida A., Yamaguchi S., J. Am. Chem. Soc. 2011, 133, 6952–6955. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Yamaguchi S., Itami Y., Tamao K., Organometallics 1998, 17, 4910–4916; [Google Scholar]
- 5b. Goldfuss B., Schleyer P. v. R., Organometallics 1997, 16, 1543–1552; [Google Scholar]
- 5c. Saito M., Sakaguchi M., Tajima T., Ishimura K., Nagase S., Phosphorus Sulfur Silicon Relat. Elem. 2010, 185, 1068–1076. [Google Scholar]
- 6.
- 6a. Khaghaninejad S., Heravi M. M., in Advanced Heterocyclic Chemistry Vol. 111 (Ed.: A. R. Katritzky), Academic Press, Waltham, 2014, pp. 95–146; [Google Scholar]
- 6b. Ashe A. J., Lohr L. L., Al-Taweel S. M., Organometallics 1991, 10, 2424–2431; [Google Scholar]
- 6c. Mross G., Holtz E., Langer P., J. Org. Chem. 2006, 71, 8045–8049. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Zhang Y., Liu L., Chen T., Huang Z., Zhang W.-X., Xi Z., Organometallics 2019, 38, 2174–2178; [Google Scholar]
- 7b. Wei J., Liu L., Zhan M., Xu L., Zhang W.-X., Xi Z., Angew. Chem. Int. Ed. 2014, 53, 5634–5638; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5740–5744. [Google Scholar]
- 8.
- 8a. Dubac J., Laporterie A., Manuel G., Chem. Rev. 1990, 90, 215–263; [Google Scholar]
- 8b. Ma W., Yu C., Chen T., Xu L., Zhang W.-X., Xi Z., Chem. Soc. Rev. 2017, 46, 1160–1192; [DOI] [PubMed] [Google Scholar]
- 8c. Liu L., Geng W., Yang Q., Zhang W.-X., Xi Z., Organometallics 2015, 34, 4198–4201. [Google Scholar]
- 9.
- 9a. Xi Z., Hara R., Takahashi T., J. Org. Chem. 1995, 60, 4444–4448; [Google Scholar]
- 9b. Fagan P. J., Nugent W. A., J. Am. Chem. Soc. 1988, 110, 2310–2312; [Google Scholar]
- 9c. Yan X., Xi C., Acc. Chem. Res. 2015, 48, 935–946; [DOI] [PubMed] [Google Scholar]
- 9d. He G., Kang L., Torres Delgado W., Shynkaruk O., Ferguson M. J., McDonald R., Rivard E., J. Am. Chem. Soc. 2013, 135, 5360–5363; [DOI] [PubMed] [Google Scholar]
- 9e. Ramirez y Medina I.-M., Rohdenburg M., Mostaghimi F., Grabowsky S., Swiderek P., Beckmann J., Hoffmann J., Dorcet V., Hissler M., Staubitz A., Inorg. Chem. 2018, 57, 12562–12575; [DOI] [PubMed] [Google Scholar]
- 9f. Dong Z., Reinhold C. R. W., Schmidtmann M., Müller T., Organometallics 2018, 37, 4736–4743; [Google Scholar]
- 9g. Tholen P., Dong Z., Schmidtmann M., Albers L., Müller T., Angew. Chem. Int. Ed. 2018, 57, 13319–13324; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13503–13508. [Google Scholar]
- 10.
- 10a. Kiel G. R., Ziegler M. S., Tilley T., Angew. Chem. Int. Ed. 2017, 56, 4839–4844; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4917–4922; [Google Scholar]
- 10b. Nitschke J. R., Zürcher S., Tilley T. D., J. Am. Chem. Soc. 2000, 122, 10345–10352. [Google Scholar]
- 11. Mao S. S. H., Tilley T. D., Macromolecules 1997, 30, 5566–5569. [Google Scholar]
- 12. Negishi E., Holmes S. J., Tour J. M., Miller J. A., Cederbaum F. E., Swanson D. R., Takahashi T., J. Am. Chem. Soc. 1989, 111, 3336–3346. [Google Scholar]
- 13. Takahashi T., Kageyama M., Denisov V., Hara R., Negishi E., Tetrahedron Lett. 1993, 34, 687–690. [Google Scholar]
- 14. Negishi E., Cederbaum F. E., Takahashi T., Tetrahedron Lett. 1986, 27, 2829–2832. [Google Scholar]
- 15.
- 15a. Rosenthal U., Ohff A., Michalik M., Görls H., Burlakov V. V., Shur V. B., Angew. Chem. Int. Ed. Engl. 1993, 32, 1193–1195; [Google Scholar]; Angew. Chem. 1993, 105, 1228–1230; [Google Scholar]
- 15b. Rosenthal U., Ohff A., Baumann W., Tillack A., Görls H., Burlakov V. V., Shur V. B., Zeitschr. Anorg. Allg. Chem. 1995, 621, 77–83. [Google Scholar]
- 16. Dioumaev V. K., Harrod J. F., Organometallics 1997, 16, 1452–1464. [Google Scholar]
- 17. Dioumaev V. K., Harrod J. F., Organometallics 1997, 16, 2798–2807. [Google Scholar]
- 18.
- 18a. Linshoeft J., Synlett 2014, 25, 2671–2672; [Google Scholar]
- 18b. Rosenthal U., Angew. Chem. Int. Ed. 2018, 57, 14718–14735; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14932–14950. [Google Scholar]
- 19. Miller A. D., McBee J. L., Tilley T. D., J. Am. Chem. Soc. 2008, 130, 4992–4999. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Linshoeft J., Baum E. J., Hussain A., Gates P. J., Näther C., Staubitz A., Angew. Chem. Int. Ed. 2014, 53, 12916–12920; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13130–13134; [Google Scholar]
- 20b. Urrego-Riveros S., Ramirez y Medina I. M., Hoffmann J., Heitmann A., Staubitz A., Chem. Eur. J. 2018, 24, 5680–5696. [DOI] [PubMed] [Google Scholar]
- 21. Rosenthal U., Eur. J. Inorg. Chem. 2019, 895–919. [Google Scholar]
- 22. Ura Y., Yanzhong L., Xi Z., Takahashi T., Tetrahedron Lett. 1998, 39, 2787–2790. [Google Scholar]
- 23. Oouchi K., Mitani M., Hayakawa M., Yamada T., Mukaiyama T., J. Organomet. Chem. 1996, 516, 111–114. [Google Scholar]
- 24.
- 24a. Jiang B., Tilley T. D., J. Am. Chem. Soc. 1999, 121, 9744–9745; [Google Scholar]
- 24b. Lucht B. L., Buretea M. A., Tilley T. D., Organometallics 2000, 19, 3469–3475. [Google Scholar]
- 25. Lin H.-S., Paquette L. A., Synth. Commun. 1994, 24, 2503–2506. [Google Scholar]
- 26.
- 26a. Sheldrick G., Acta Crystallogr. Sect. A 2008, 64, 112–122; [DOI] [PubMed] [Google Scholar]
- 26b. Farrugia L., J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar]
- 27. Dolomanov O. V., Bourhis L. J., Gildea R. J., Howard J. A. K., Puschmann H., J. Appl. Crystallogr. 2009, 42, 339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The signal from the carbon atom bond to boron was not visible owing to the high quadrupole moment of the boron nucleus.
- 29.The signal is overlapping with the solvent signal.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary