

Abstract
Melodamide A, a phenolic amide from the leaves of Melodorum fruticosum Lour., has previously shown pronounced anti‐inflammatory activity. In order to rapidly isolate larger quantities for biological testing, a fast, one‐step isolation method by centrifugal partition chromatography was developed within this study. Fractionation of the dichloromethane extract was performed with a two‐phase solvent system consisting of n‐hexane, ethyl acetate, methanol, and water (3:7:5:5, v/v), leading to the isolation of melodamide A with a purity of >90% and a yield of 6.7 w% within 32 min. The developed method can also be used in dual mode for the enrichment of further constituents like flavonoids or chalcones. In order to support the centrifugal partition chromatography method development, additionally, a high‐performance liquid chromatography method was established and validated to determine quantities of melodamide A in plant material and crude extracts. Analysis of M. fruticosum leaves and a dichloromethane extract obtained from this plant material showed a total melodamide A content of 0.19 ± 0.008 and 8.9 ± 0.249 w%, respectively.
Keywords: centrifugal partition chromatography, high performance liquid chromatography, melodamide A, Melodorum fruticosum
Article Related Abbreviations
- CPC
centrifugal partition chromatography
- DAD
diode array detector
- DCM
dichloromethane
- EtOAc
ethyl acetate
- ICH
International Conference on Harmonization
- MeOH
methanol
- n‐hex
n‐hexane
- PA
peak area
1. INTRODUCTION
Melodorum fruticosum Lour., a member of the Annonaceae family, is a slender evergreen tree native to southeast Asia, more specifically indigenous to Vietnam, Laos, Cambodia, and Thailand 1. M. fruticosum has edible fruits which can be used to make wine and beverages 2, 3 while the fragrant flowers are used to make perfume 1. Dried flowers are said to be slightly heart stimulating and described as a blood tonic in Thailand 4. The leaves are used in Vietnamese traditional medicine to aid digestion 1. The flowers and bark of M. fruticosum are known to contain essential oils, heptenes, and butenolides with antifungal, antioxidant, and cytotoxic properties 5, 6, 7, 8, 9, 10. Constituents isolated from a methanolic leaf extract, in particular a phenolic amide, melodamide A (1), showed strong inhibition of superoxide anion generation (IC50 = 5.25 µM) and elastase release (39.17% inhibition at 10 µM) in human neutrophils 11. Recently we could show that 1 is also a potent inhibitor of interleukin‐8 release (IC50 = 6.6 µM) and tumor necrosis factor alpha production (IC50 = 4.5 µM) in human neutrophils 12. Due to the promising pharmacological activities of 1, sufficient quantities of this compound are required to perform further biological assays. Two methods have previously described the isolation of compound 1 from the leaves of M. fruticosum, i.e. partitioning of the extract between immiscible liquids followed by two silica gel column chromatography steps 11 and column chromatography with silica gel followed by a second separation step with Sephadex LH‐20 12. Since both methods are time consuming and require the usage of large amounts of solvents, a method for the rapid and economically viable isolation of this phenolic amide from crude extracts is of considerable interest.
Centrifugal partition chromatography (CPC) is an efficient method for the preparative isolation of natural products whereby separation is achieved by partition of a sample between two immiscible solvents 13, 14. It boasts a number of advantages over other popular isolation techniques, i.e. (i) since there is no need for a solid support, there is no risk of irreversible adsorption to the stationary phase; (ii) the risk of sample denaturation, like in the case of silica interactions, is low; (iii) samples are recovered completely; (iv) tailing is minimal; and (v) it is an economically viable technique, requiring low solvent consumption 15, 16. In natural products research, CPC has been used to isolate a variety of natural compounds including alkaloids, carotenoids, terpenoids, flavonoids, saponins, and anthraquinones 14, 17, 18 and has been effectively employed in the isolation of unstable natural products such as carnosic acid and carnosol, thereby circumventing stability issues related to classical isolation techniques 19.
In the course of this study, an efficient one‐step CPC method for the isolation of compound 1 from a crude dichloromethane (DCM) M. fruticosum leaf extract was established, without the need for pretreatment or further fractionation. Moreover, since to date, there is no method for the quantification of this bioactive compound available in the literature, a validated HPLC‐diode array detector (DAD) method for the quantification of 1 in plant material or crude extracts was developed in order to support the CPC method establishment.
2. MATERIALS AND METHODS
2.1. Materials
2.1.1. Solvents and reagents
All solvents used for extraction, HPLC, and CPC were provided by VWR International (Darmstadt, Germany). Ultrapure water was produced onsite by a Sartorius Arium 611 UV water purification system (Sartorius, Göttingen, Germany).
2.1.2. Plant material
Leaves from M. fruticosum Lour. were collected in the Tam thành commune, Phú ninh district, Quảng nam province of Vietnam in August 2014 and identified by Dr. Võ Văn Chi from the Department of Pharmacognosy, Faculty of Pharmacy, University of Medicine and Pharmacy at Hồ Chí Minh City, Vietnam. A voucher specimen (BW‐20141210‐03) of the air‐dried plant material was deposited at the Institute of Pharmacy/Pharmacognosy of the University of Innsbruck, Austria.
2.2. Quantification of melodamide A using HPLC
2.2.1. HPLC conditions
HPLC analysis was performed on a Shimadzu UFLCxR system (Shimadzu, Tokyo, Japan) equipped with quaternary pumps, auto sampler, column oven, and DAD using an Agilent Zorbax SB‐C18 column (150 × 4.6 mm id) with a particle size of 3.5 µm. The mobile phase consisted of water (A) and acetonitrile (B). The solvent composition was set to: t = 0 min B: 15%; t = 1 min B: 15%; t = 20 min B: 45%; t = 25 min B: 70%; t = 30 min B: 70%; t = 30.1 min B: 15%; t = 40 min B: 15%. The flow rate was 1.0 mL/min, the injection volume was set to 10 µL, the column oven temperature was 35°C, and the detection wavelength was set to 230 nm.
2.2.2. HPLC method validation
The HPLC‐DAD method was validated in accordance with the International Conference on Harmonization (ICH) guidelines for Validation of Analytical Procedures Q2 (R1) 20 for linearity, repeatability, reproducibility, recovery, LOQ, and LOD for melodamide A (1). For quantitative determination, two stock solutions containing 1 mg of compound 1 were dissolved in methanol (MeOH) and diluted to 200, 150, 100, 50, 25, and 12.5 µg/mL, respectively.
2.2.3. Sample preparation
Extracts for HPLC quantification and validation were prepared by extracting 100 mg powdered air‐dried leaf material in 10 mL DCM by sonication for 10 min in an ultrasonic bath followed by filtration through cotton wool. This procedure was repeated four times with the recovered plant material, respectively. The filtrates from each extraction step were combined and evaporated to dryness. The residue was dissolved in MeOH, transferred to a 25 mL volumetric flask and filled up to 25 mL with MeOH. Two milliliters of the solution were withdrawn, the solvent was partly evaporated, and the remaining fluid was transferred quantitatively to a 1 mL volumetric flask and filled up with MeOH, leading to a final concentration equivalent to 8 mg of used plant material/mL of sample. The extracted plant material was recovered and extracted one final time, as described above, to yield a blank sample which was analyzed by HPLC to verify exhaustive extraction of the leaf material.
2.3. Extraction and one‐step isolation of melodamide A using centrifugal partition chromatography
2.3.1. Extract preparation
The air‐dried leaves of M. fruticosum (952 g) were first ground to a fine powder, followed by exhaustive extraction with DCM by sonication and evaporation of the solvent under reduced pressure to yield 63.8 g crude DCM extract.
2.3.2. Selection and preparation of the two‐phase solvent system
The partition coefficient (K value) of melodamide A (1) and corresponding values of other constituents present in the M. fruticosum DCM extract were determined by HPLC analysis as follows: various two‐phase solvent systems were prepared by adding selected solvents to a stoppered 20 mL test tube, vigorously shaking the test tube by hand, and allowing the two phases to settle. For each solvent system, 10 mg of extract was weighed in another 20 mL stoppered test tube before 5 mL from each phase of the pre‐equilibrated two‐phase solvent system were added. The test tube was vigorously shaken by hand and left to settle. The upper and lower phases of solvent systems which provided satisfactory settling speeds, i.e. a settling time of less than 30 s, were analyzed by HPLC. The K values of the compounds present in the extract were obtained by dividing their peak areas (PAs) in the upper phases by the PAs of the same compounds in the lower phases. A solvent system containing n‐hexane (n‐hex), ethyl acetate (EtOAc), MeOH, and water (3:7:5:5, v/v) was selected for the isolation of compound 1 by CPC on the basis of the calculated K value of 1 compared to the K values of other compounds present in the extract while also considering the lipophilic nature and subsequent solubility of the DCM extract.
2.3.3. Centrifugal partition chromatography apparatus
Semi‐preparative CPC separation was performed on a FCPC 200 apparatus (Kromaton, Angers, France) with a total rotor volume of 192 mL equipped with an 803C pump system (Gilson, Villiers‐la‐Bel, France) and a 20 mL sample loop.
2.3.4. Separation of the M. fruticosum dichloromethane extract in dual mode
A two‐phase solvent system (1 L) containing n‐hex, EtOAc, MeOH, and water (3:7:5:5, v/v) was prepared by thorough mixing in a separating funnel before the phases were separated and degassed in an ultrasonic bath for 10 min prior to use. Starting with descending mode, the rotor (192 mL volume) was completely filled with stationary phase (upper organic phase; 280 mL) at a flow rate of 4 mL/min. Rotation was set to 900 rpm and the mobile phase (lower aqueous phase; 300 mL) was pumped into the system at a rate of 4 mL/min until equilibrium was achieved. After that, 503.7 mg of the crude DCM extract was dissolved in 10 mL upper phase, filtered through a 0.45 µM polytetrafluoroethylene (PTFE) prefilter, and injected into the sample loop. Elution was switched from descending mode to ascending mode after 40 min and continued for 60 min, followed by extrusion with MeOH at 8 mL/min for 30 min. Fractions were collected every 2 min for both the descending and the ascending mode with a fraction collector in 10 mL tared test tubes resulting in 50 fractions, each containing 8 mL eluent, which were left to dry under chemical air before weighing. The eluent from extrusion (240 mL) was collected separately (fraction 51) and evaporated to dryness under reduced pressure.
3. RESULTS AND DISCUSSION
Melodamide A (1) from the leaves of M. fruticosum has shown promising biological activity on the pro‐inflammatory functions of human neutrophils 11, 12. Therefore, the main objectives of this study were the development and validation of a HPLC‐DAD method to quantify levels of the bioactive phenolic amide 1 in plant material and crude extracts and the development of a CPC method to allow for the rapid, solvent‐saving, and economically viable isolation of compound 1 in larger quantities. The HPLC‐DAD method allows for the assessment of new plant material or extracts to ensure the feasibility of proceeding with isolation by quantifying the levels of 1. The development of the CPC method for the isolation of compound 1 was focused on separating the desired product from other constituents of varying polarities contained in the crude DCM extract, which include phenolic amides, flavonoids, and chalcones 12, in one step without the need for pretreatment or further fractionation.
3.1. HPLC method development
It is imperative that levels of compound 1 are determined in material selected for extraction and purification, to ensure that sufficient quantities of the target compound are contained. Since there are currently no methods described in the literature for the quantitative analysis of 1, a HPLC‐DAD method was developed to allow for the quantitative determination of the compound in plant material and crude extracts.
The DCM extract from the M. fruticosum leaves, dissolved in MeOH (1 mg/mL), was used as a reference sample during method development. A gradient employing water (A) and acetonitrile (B) as mobile phase was used to separate compound 1 from other compounds present in the crude extract. The initial experiments were performed to compare different reversed phase HPLC columns. The best peak shapes and separations were achieved using an Agilent Zorbax SB‐C18 (150 × 4.6 mm id, 3.5 µm particle size) HPLC column, which was thus selected for further method development. Varying the oven temperature from 25 to 40°C in 5°C increments afforded the best separation at a temperature of 35°C. The optimized gradient selected for validation at a flow rate of 1 mL/min consisted of: 0 min, 15% B; 1 min, 15% B; 20 min, 45% B; 25 min, 70% B; 30 min, 70% B; 30.1 min, 15% B; and 40 min, 15% B. Finally, the injection volume was set to 10 µL and a wavelength of 230 nm was selected for compound detection. The chromatogram of the HPLC‐DAD analysis of the M. fruticosum DCM extract using the optimized method is shown in Figure 1. As we have reported previously, the main peaks present in the chromatogram of the DCM extract could be assigned to 14 constituents, i.e. melodamide A (1), 4′,7‐dihydroxy‐5‐methoxyflavanone (2), haplamide (3), 4′‐hydroxy‐5,7‐dimethoxyflavanone (4), (2S)‐5‐hydroxy‐4′,7‐dimethoxyflavanone (5), 7‐hydroxy‐4′,5‐dimethoxyflavanone (6), 7‐hydroxy‐5‐methoxyflavanone (7), N‐trans‐cinnamoyltyramine (8), 5‐hydroxy‐6‐(2‐hydroxybenzyl)‐4′,7‐dimethoxyflavanone (9), (2S)‐4′,5,7‐trimethoxyflavanone (10), 2′,4′‐dihydroxy‐4,6′‐dimethoxychalcone (11), 2′,4′‐dihydroxy‐4,6′‐dimethoxydihydrochalcone (12), 2′,6′‐dihydroxy‐4′‐methoxychalcone (13), and 2′,4′‐dihydroxy‐3′‐(2‐hydroxy‐benzyl)‐4,6′‐dimethoxychalcone (14) 12.
Figure 1.
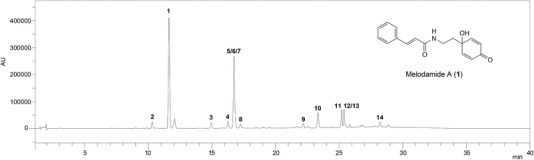
HPLC‐DAD analysis of the DCM extract of M. fruticosum leaves at a concentration of 1 mg/mL. Conditions: stationary phase: Agilent Zorbax SB‐C18 (150 × 4.6 mm, 3.5 µm particle size); mobile phase: solvent A: water; solvent B: acetonitrile; gradient: 0 min, 15% B; 1 min, 15% B; 20 min, 45% B; 25 min, 70% B; 30 min, 70% B; 30.1 min, 15% B; 40 min, 15% B; flow rate: 1 mL/min; temperature: 35°C; injection volume: 10 µL; detection: DAD: 230 nm
3.2. HPLC method validation
The developed HPLC‐DAD method was validated towards specificity, linearity and range, repeatability, precision, and accuracy for the quantification of compound 1 according to the ICH guidelines for Validation of Analytical Procedures Q2 (R1) 20.
3.2.1. Specificity
No coelution of 1 with other components of the DCM extract was observed, as determined through the peak purity option using LabSolutions software version 5.81 SP1. Therefore, the method was deemed to be specific for the quantification of compound 1.
3.2.2. Linearity and range
Calibration curves of 1 were plotted by linear regression of analyte concentration (cA) against PA at 230 nm resulting in a linear range from 12.5 to 200 µg/mL. Consequently, correlation coefficient values (R 2) were determined. The six‐point calibration curve for compound 1 revealed values of y = 30350x−1570.6 and R2 = 0.9990. Thus, the proposed method exhibits good linearity in the required range.
The LOD and the LOQ were calculated from the SD of the response and the slope of the calibration curve following the ICH guidelines Q2 (R1) 20. LOD and LOQ were found to be 2.71 and 8.21 µg/mL, respectively.
3.2.3. Repeatability
The repeatability of the HPLC‐DAD method was confirmed by repeated injection (n = 6) of 10 µL of a stock solution containing 100 µg/mL of compound 1 which showed a %RSD of 0.058 in terms of PA and 0.016 %RSD for retention time shift.
3.2.4. Precision
The intraday and interday precision of the HPLC‐DAD method was determined by analyzing three replicates of 1 at three different concentrations (25, 100, and 200 µg/mL) on three different days over a period of one week, respectively. The calculated %RSD values were less than 2% (Table 1) and thus the intraday and interday precision met the acceptance criteria according to the ICH guidelines 20. The method was therefore deemed reliable and repeatable for the quantification of compound 1.
Table 1.
Evaluation of intraday (n = 3) and interday precision (n = 9) of the HPLC method
| Intraday precision (%RSD) | ||||
|---|---|---|---|---|
| Concentration (µg/mL) | Day 1 | Day 2 | Day 3 | Interday precision (%RSD) |
| 25 | 0.23 | 0.08 | 0.17 | 0.34 |
| 100 | 0.07 | 0.07 | 0.73 | 0.75 |
| 200 | 0.10 | 0.33 | 0.08 | 0.56 |
3.2.5. Accuracy
Accuracy of the HPLC‐DAD method was evaluated by analyzing the DCM leaf extract spiked with known concentrations of 1 (10, 25, and 50 µg/mL). Analysis of three extraction replicates, measured three times, respectively, afforded recovery rates of 98.90 ± 5.22% for the extract spiked with 10 µg/mL of compound 1, 103.21 ± 1.73% for the extract spiked with 25 µg/mL of compound 1, and 102.6 ± 3.27% for the high spike sample (50 µg/mL). These results show that the method has suitable accuracy.
3.3. Quantification of melodamide A in M. fruticosum leaves and crude dichloromethane extract
The established and validated HPLC‐DAD method was used for the quantification of melodamide A (1) in dried leaves of M. fruticosum and a DCM extract obtained from this material.
For measuring the levels of 1 in the dried leaf material, the DCM extract obtained from 100 mg dried M. fruticosum leaves (in triplicate) was analyzed three times by HPLC‐DAD. The quantity of compound 1 in the investigated plant material was found to be 0.19 ± 0.008 w%. This is the first reported quantification of 1 in the leaves of M. fruticosum, the only plant currently known to contain this bioactive phenolic amide.
Furthermore, the quantity of compound 1 in the crude DCM extract, which was generated in a larger scale and intended to be used for the isolation of 1 by CPC, was also determined. Triplicate extract samples dissolved in MeOH at a concentration of 1 mg/mL were measured three times by HPLC‐DAD, respectively. The content of 1 was found to be 8.9 ± 0.249 w%, showing that it is a major component of the crude DCM extract. Thus, the extract was considered to be a good source for the isolation of compound 1.
3.4. Selection of the two‐phase solvent system for centrifugal partition chromatography
To develop a fast method for the rapid isolation of larger quantities of melodamide A (1) from the crude DCM extract of the M. fruticosum leaf material, CPC was chosen due to its advantages over other isolation techniques. The first crucial step in the method development for a successful separation by CPC was the selection of a suitable two‐phase solvent system. An important criterion for the identification of a solvent system is the partition coefficient (K value) of the compound of interest, which should ideally be between 0.5 and 1 15. Moreover, to achieve a good separation of the compound of interest and avoid coelution of other extract constituents, the K values of the other compounds should be as different as possible to the K value of the compound of interest. Therefore, initially, solvent systems of varying polarities and compositions were evaluated. A total number of 29 solvent systems including 14 systems from the n‐hex/EtOAc/MeOH/water family 21, covering a range of different polarities, showed satisfactory settling speeds after the addition of the DCM extract and vigorous shaking to distribute the extract between the two phases and were thus subjected to HPLC‐DAD analysis (Table 2). The K values of melodamide A (1) and other constituents of the DCM extract (2–14) were determined for these solvent systems with the developed HPLC‐DAD method (K values of 1, Table 2, K values of other extract constituents (2–14), Supporting Information Table S1). Seven out of the 29 systems, i.e. three systems from the n‐hex/EtOAc/MeOH/water family and four solvent systems comprised of methyl tert‐butyl ether, dimethoxyethane, and water, exhibited a K value of 1, the compound of main interest, between 0.25 and 1.25, and were thus further considered, whereas all other solvent systems were disregarded. The seven selected solvent systems displayed K values of the other extract constituents (2–14) between 0.32 and 94.41, whereas up to four compounds could not be detected in the lower phase, respectively. Moreover, the DCM extract was not fully soluble in the aqueous layers, suggesting the selection of a less polar solvent system for CPC. One of the less polar ones, i.e. a solvent system containing n‐hex/EtOAc/MeOH/water in a ratio of 3:7:5:5 v/v, gave a K value of compound 1 of 0.26 and K values of the other constituents present in the extract between 0.32 and 13.70, whereas one compound, i.e. compound 14, could not be detected in the lower phase. The difference between the K value of 1 of 0.26 and the K values of the other compounds is big enough, suggesting a suitable separation of compound 1 from the other components in the extract could be achieved, whereas at the same time, the K values of the extract constituents are not too high, suggesting that, if desired, in addition to 1, also some of the other constituents could be enriched using this solvent system. Therefore, this solvent system was selected for CPC separation.
Table 2.
Partition coefficients (K) of compound 1 in two‐phase solvent systems
| Solvent system | Ratio (v/v) | K a |
|---|---|---|
| n‐hex/EtOAc/MeOH/water | 9:1:9:1 | 0.00 |
| n‐hex/EtOAc/MeOH/water | 8:2:8:2 | 0.01 |
| n‐hex/EtOAc/MeOH/water | 7:3:7:3 | 0.00 |
| n‐hex/EtOAc/MeOH/water | 7:3:6:4 | 0.01 |
| n‐hex/EtOAc/MeOH/water | 6:4:6:4 | 0.01 |
| n‐hex/EtOAc/MeOH/water | 7:3:5:5 | 0.01 |
| n‐hex/EtOAc/MeOH/water | 5:5:5:5 | 0.05 |
| n‐hex/EtOAc/MeOH/water | 3:7:5:5 | 0.26 |
| n‐hex/EtOAc/MeOH/water | 4:6:4:6 | 0.23 |
| n‐hex/EtOAc/MeOH/water | 3:7:4:6 | 0.49 |
| n‐hex/EtOAc/MeOH/water | 4:10:4:10 | 1.21 |
| n‐hex/EtOAc/MeOH/water | 2:8:2:8 | 2.98 |
| n‐hex/EtOAc/MeOH/water | 1:9:1:9 | 7.18 |
| n‐hex/EtOAc/MeOH/water | 0:10:0:10 | 13.65 |
| EtOAc/DME/water | 2:1:1 | 5.69 |
| MTBE/DME/water | 1:2:1 | 0.92 |
| MTBE/DME/water | 2:1:2 | 1.25 |
| MTBE/DME/water | 2:2:1 | 0.77 |
| MTBE/DME/water | 2:2:2 | 1.04 |
| n‐hep/DCM/ACN | 10:3:7 | 0.01 |
| n‐hex/EtOAc/MeOH/water/DME | 7:3:6:4:2 | 0.01 |
| MTBE/DME/Ace/water | 4:1:1:4 | 1.28 |
| n‐hex/ACN | 1:1 | 0.00 |
| MTBE/EtOAc/water | 2:1:2 | 4.67 |
| MTBE/EtOH/water | 2:1:2 | 4.12 |
| MTBE/MeOH/water | 2:1:2 | 2.19 |
| MTBE/n‐prop/water | 2:1:2 | 18.99 |
| MTBE/2‐prop/water | 2:1:2 | 10.19 |
| MTBE/n‐BuOH/water | 2:1:2 | 45.42 |
Calculation based on the analysis results using the developed HPLC method at 230 nm
2‐prop, 2‐propanol; Ace, acetone; DME, dimethoxyethane; EtOH, ethanol absolute; MTBE, methyl tert‐butyl ether; n‐BuOH, n‐butanol absolute; n‐hep, n‐heptane; n‐prop, n‐propanol
3.5. One‐step isolation of melodamide A using centrifugal partition chromatography
With the main aim of isolating melodamide A (1) directly from the crude DCM extract of M. fruticosum leaves, but also to enable an enrichment of further extract constituents with a diverse range of polarities, dual elution mode followed by extrusion mode was chosen for CPC separation. The method was started in descending mode, using the upper, organic phase of the biphasic solvent system containing a ratio of 3:7:5:5 v/v of n‐hex/EtOAc/MeOH/water as stationary phase. The retention of the stationary phase was 51% at equilibrium with a pressure of 32 bar. After the injection of 503.7 mg of the crude M. fruticosum extract, separation in descending mode was continued for 40 min. The eluate was collected in 20 fractions (F1–F20), of which the first ten tubes only contained mobile phase. Although complete isolation of compound 1 from the crude extract was achieved using descending mode, further compound separation was conducted in ascending mode for 60 min, using the aqueous phase as the stationary phase, to enrich further extract constituents with different polarities. During elution in ascending mode, the pressure dropped to 16 bar. The ascending mode elute afforded 30 fractions (F21‐F50) containing mixtures of additional extract constituents allowing for further phytochemical isolation and purification if desired. Finally, extrusion with MeOH was performed for 30 min to ensure that the elution of compounds from the column was as complete as possible. The extrusion elute was also collected (F51). Based on the weights of the obtained fractions, which contained some sample after drying (F11–F51) (Figure 2), the recovery rate was calculated to be 90% of the used extract.
Figure 2.
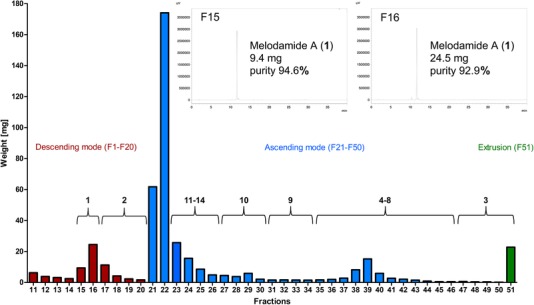
Distribution of the weights of fractions 11 to 51, obtained from the one‐step CPC separation of the M. fruticosum DCM extract, and the main extract constituents. Red, blue, and green colors indicate fractions derived from the descending mode (F11–F20), the ascending mode (F21–F50), and the extrusion mode elute (F51), respectively. The chromatogram inserts show the HPLC‐DAD analyses of fractions 15 and 16 comprising melodamide A (1) with a purity of >90%
Analysis of fractions 11–51 by HPLC‐DAD revealed that melodamide A (1) with a purity of >90% was obtained in descending mode. In particular, fractions 15 and 16 comprised 9.4 and 24.5 mg of compound 1 with a purity of 94.6 and 92.9%, respectively, giving a total of 33.9 mg of compound 1 from 503.7 mg crude extract (6.7% yield, 75.6% recovery) (Figure 2).
Furthermore, HPLC‐DAD analysis of all fractions concluded that 4′,7‐dihydroxy‐5‐methoxyflavanone (2) was enriched in fractions 17–20, haplamide (3) was comprised in fractions 47–51, compounds 4–8 were present in fractions 35–46, and the flavanones 9 and 10 were enriched in fractions 31–34 and 27–30, respectively, while the chalcones 11–14 were collected in fractions 23–26. Therefore, if desired, these fractions could be pooled for further isolation. If the mentioned compounds are not of interest, the CPC separation can also be stopped directly after the elution of 1 after 32 min without mode switch followed by an extrusion step with stationary phase in order to save time for a next sample injection.
4. CONCLUDING REMARKS
The previously shown anti‐inflammatory potential of melodamide A (1), present in the leaves of M. fruticosum, has evoked the need for a method to quantify levels of this phenolic amide in plant material and crude extracts as well as a separation method with minimal fractionation steps, allowing for its rapid isolation to provide sufficient quantities for further biological assessment.
Within this work, a HPLC‐DAD method for the quantification of compound 1 in extracts and plant material with high precision, accuracy, and selectivity was generated and validated. The calibration curves showed a promising linearity over a range of 12.5–200 µg/mL of compound 1 with a regression coefficient not lower than R 2 = 0.9990. The obtained %RSD values for the intra‐ and interday precision of the proposed method were <1%. The accuracy, determined by recovery experiments, ranged from −1.10 to +3.21%. Therefore, this HPLC method can be applied to quantify levels of 1 in M. fruticosum leaves prior to phytochemical isolation. The total melodamide A (1) content of the M. fruticosum leaves used in this study was determined to be 0.19 ± 0.008 w%. The DCM extract used for the isolation of compound 1 contained 8.9 ± 0.249 w% of 1.
Furthermore, within this study, a fast, one‐step separation method using CPC was developed and successfully applied to isolate compound 1 at a purity of >90% and a yield of 6.7 w% from the crude DCM extract of M. fruticosum, the only plant currently known to contain this bioactive phenolic amide.
This study describes the first reported quantification of compound 1 in the leaves of M. fruticosum as well as the first reported one‐step isolation of 1 from a crude plant extract. The validated HPLC‐DAD method should provide a useful tool for assessing plant material prior to extraction and compound isolation to ensure that sufficient quantities of 1 are present in the material. Moreover, the presented CPC method is a fast, solvent‐saving, and economically viable technique for the isolation of 1, which also has the potential for further scale‐up to isolate sufficient material for biological testing.
CONFLICT OF INTEREST
The authors have declared no conflict of interest.
Supporting information
Supplementary material
ACKNOWLEDGMENTS
This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sector.
Engels NS, Waltenberger B, Schwaiger S, Huynh L, Tran H, Stuppner H. Melodamide A from Melodorum fruticosum — Quantification using HPLC and one‐step‐isolation by centrifugal partition chromatography. J Sep Sci 2019;42:3165–3172. 10.1002/jssc.201900392
REFERENCES
- 1. Võ, V. C. , Dictionary of Common Plants, Science and Technique Publishing House, Hà Noi; 2003. [Google Scholar]
- 2. Nanasombat, S. , Thonglong, J. , Jitlakha, J. , Formulation and characterization of novel functional beverages with antioxidant and anti‐acetylcholinesterase activities. Funct. Foods Health Dis. 2015, 5, 1–16. [Google Scholar]
- 3. Tatdao, P. , Norraset, S. , Tiwawan, S. , Physico‐chemical and sensory properties of musts and wines from Melodorum fruticosum Lour. Int. Food Res. J. 2014, 21, 39–43. [Google Scholar]
- 4. Quattrocchi, U. , CRC World Dictionary of Medicinal and Poisonous Plants, CRC Press, Boca Raton; 2012. [Google Scholar]
- 5. Pripdeevech, P. , Chukeatirote, E. , Chemical compositions, antifungal and antioxidant activities of essential oil and various extracts of Melodorum fruticosum L. flowers. Food Chem. Toxicol. 2010, 48, 2754–2758. [DOI] [PubMed] [Google Scholar]
- 6. Jung, J. H. , Chang, C. J. , Smith, D. L. , McLaughlin, J. L. , Pummangura, S. , Chaichantipyuth, C. , Patarapanich, C. , Additional bioactive heptenes from Melodorum fruticosum . J. Nat. Prod. 1991, 54, 500–505. [DOI] [PubMed] [Google Scholar]
- 7. Jung, J. H. , Pummangura, S. , Chaichantipyuth, C. , Patarapanich, C. , Fanwick, P. E. , Chang, C.‐J. , McLaughlin, J. L. , New bioactive heptenes from Melodorum fruticosum (Annonaceae). Tetrahedron 1990, 46, 5043–5054. [DOI] [PubMed] [Google Scholar]
- 8. Jung, J. H. , Pummangura, S. , Chaichantipyuth, C. , Patarapanich, C. , McLaughlin, J. L. , Bioactive constituents of Melodorum fruticosum . Phytochemistry 1990, 29, 1667–1670. [Google Scholar]
- 9. Tuchinda, P. , Udchachon, J. , Reutrakul, V. , Santisuk, T. , Taylor, W. C. , Farnsworth, N. R. , Pezzuto, J. M. , Kinghorn, A. D. , Bioactive butenolides from Melodorum fruticosum . Phytochemistry 1991, 30, 2685–2689. [Google Scholar]
- 10. Chaichantipyuth, C. , Tiyaworanan, S. , Mekaroonreung, S. , Ngamrojnavanich, N. , Roengsumran, S. , Puthong, S. , Petsom, A. , Ishikawa, T. , Oxidized heptenes from flowers of Melodorum fruticosum . Phytochemistry 2001, 58, 1311–1315. [DOI] [PubMed] [Google Scholar]
- 11. Chan, H.‐H. , Hwang, T.‐L. , Thang, T. D. , Leu, Y.‐L. , Kuo, P.‐C. , Nguyet, B. T. M. , Dai, D. N. , Wu, T.‐S. , Isolation and synthesis of melodamide A, a new anti‐inflammatory phenolic amide from the leaves of Melodorum fruticosum . Planta Med. 2013, 79, 288–294. [DOI] [PubMed] [Google Scholar]
- 12. Engels, N. S. , Waltenberger, B. , Michalak, B. , Huynh, L. , Tran, H. , Kiss, A. K. , Stuppner, H. , Inhibition of pro‐inflammatory functions of human neutrophils by constituents of Melodorum fruticosum leaves. Chem. Biodivers. 2018, 15, e1800269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pauli, G. F. , Pro, S. M. , Friesen, J. B. , Countercurrent separation of natural products. J. Nat. Prod. 2008, 71, 1489–1508. [DOI] [PubMed] [Google Scholar]
- 14. Friesen, J. B. , McAlpine, J. B. , Chen, S.‐N. , Pauli, G. F. , Countercurrent separation of natural products: an update. J. Nat. Prod. 2015, 78, 1765–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ito, Y. , Golden rules and pitfalls in selecting optimum conditions for high‐speed counter‐current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [DOI] [PubMed] [Google Scholar]
- 16. Marston, A. , Hostettmann, K. , Developments in the application of counter‐current chromatography to plant analysis. J. Chromatogr. A 2006, 1112, 181–194. [DOI] [PubMed] [Google Scholar]
- 17. Bojczuk, M. , Żyżelewicz, D. , Hodurek, P. , Centrifugal partition chromatography — a review of recent applications and some classic references. J. Sep. Sci. 2017, 40, 1597–1609. [DOI] [PubMed] [Google Scholar]
- 18. Karkoula, E. , Angelis, A. , Koulakiotis, N.‐S. , Gikas, E. , Halabalaki, M. , Tsarbopoulos, A. , Skaltsounis, A.‐L. , Rapid isolation and characterization of crocins, picrocrocin, and crocetin from saffron using centrifugal partition chromatography and LC–MS. J. Sep. Sci. 2018, 41, 4105–4114. [DOI] [PubMed] [Google Scholar]
- 19. Grace, M. H. , Qiang, Y. , Sang, S. , Lila, M. A. , One‐step isolation of carnosic acid and carnosol from rosemary by centrifugal partition chromatography. J. Sep. Sci. 2017, 40, 1057–1062. [DOI] [PubMed] [Google Scholar]
- 20. International Conference on Harmonization , International Federation of Pharmaceutical Manufacturers and Associations (IFPMA), Geneva 1996.
- 21. Oka, F. , Oka, H. , Ito, Y. , Systematic search for suitable two‐phase solvent systems for high‐speed counter‐current chromatography. J. Chromatogr. A 1991, 538, 99–108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material