

HER2 directed therapies have improved prognosis for HER2‐positive breast cancer, although patients with advanced disease ultimately develop resistance. This article reports a phase Ib trial of durvalumab in combination with trastuzumab in resistant HER2‐positive advanced breast cancer.
Keywords: Trastuzumab, Durvalumab, HER2 metastatic breast cancer, Immunotherapy
Abstract
Background.
Immune checkpoint inhibitors are active in a broad range of cancers, including programmed death ligand 1 (PD‐L1)‐positive, triple‐negative, metastatic breast cancer (MBC). Antibody‐dependent cell‐mediated cytotoxicity is a mechanism of action of trastuzumab. We performed a phase Ib trial of durvalumab and trastuzumab in HER2‐positive MBC previously treated with chemotherapy and anti‐HER2 antibodies to assess safety, efficacy, and correlative endpoints.
Patients and Methods.
Patients with HER2‐positive MBC were enrolled on a standard 3 + 3 design. Dose level 1 was durvalumab (1,125 mg intravenously day 1) and trastuzumab (8 mg/kg intravenously loading, then 6 mg/kg day 1) on a q3 weekly cycle. An expansion cohort at the recommended phase II dose (RP2D) performed tumor biopsies at baseline and after cycle 1. The primary endpoint was to establish the RP2D.
Results.
Fifteen patients were accrued from April to December 2016, of which 14 were evaluable for response. Median age was 54 years (range 40–86); the majority had visceral disease (87%) and at least three prior (adjuvant and/or metastatic) lines of chemotherapy (73%), including trastuzumab (93%), pertuzumab (60%), and trastuzumab‐emtansine (93%) for MBC. No dose‐limiting toxicities were observed at dose level 1 (n = 6) or dose expansion (n = 9) during cycle 1. One patient developed a grade ≥3 immune‐related adverse event (grade 4 diabetes mellitus). No responses by RECIST were seen, with 4 of 14 patients (29%) demonstrating stable disease as best response at week 6 (median duration, 2.7 months). All patients had <1% PD‐L1 expression on either archival tissue (7/15) or prestudy biopsy (8/15). In the dose expansion cohort, evaluable pretreatment and on‐treatment tumor biopsies (n = 5) showed minimal CD8 cell infiltration.
Conclusion.
The RP2D of durvalumab and trastuzumab is standard full doses of both agents. No significant clinical activity was observed in patients with heavily pretreated HER2‐positive PD‐L1‐negative MBC.
Implications for Practice.
This phase Ib trial with associated correlative endpoints provides insights into the lack of activity of the combination of durvalumab and trastuzumab in heavily pretreated HER2‐positive metastatic breast cancer (MBC). No significant clinical activity was observed in patients with heavily pretreated HER2‐positive programmed death ligand 1 (PD‐L1)‐negative MBC with evidence of cytotoxic T‐cell exhaustion. Furthermore, all patients had no expression of PD‐L1 in the tumor cells. These data support the importance of PD‐L1 as an important selection biomarker and the need to assess the tumor microenvironment for immune regulatory cells. Further work is needed to understand how to activate the “cold” tumors to be able to combine current immune‐oncology agents.
摘要
背景。免疫检查点抑制剂在众多癌症中具有活性,包括程序性死亡配体 1 (PD‐L1) 阳性、三阴性、转移性乳腺癌 (MBC)。抗体依赖性细胞介导的细胞毒性是曲妥珠单抗的作用机制。我们进行了一项关于度伐单抗联合曲妥珠单抗治疗先前曾接受化疗和抗 HER2 抗体治疗的 HER2 阳性MBC患者的 Ib 期试验,旨在对安全性、疗效和相关终点进行评估。
患者和方法。HER2 阳性MBC患者以标准 3 + 3 设计入组。剂量水平 1 为度伐单抗(第 1 天静脉给药 1 125 mg)和曲妥珠单抗(第 1 天静脉给药 8 mg/kg负荷量,之后的剂量为 6 mg/kg维持量),每3周为 1 个周期。按照推荐的 II 期研究剂量 (RP2D) 给药的扩展队列在基线时以及在经过第 1 个周期后接受肿瘤活检。主要终点旨在确定 RP2D。
结果。从 2016 年 4 月至 12 月期间,本研究共纳入 15 名患者,其中,14 名患者可进行疗效评估。中位年龄为 54 岁(范围为 40–86 岁);大多数患者患有内脏疾病 (87%),既往至少采用 3 线(辅助性和/或转移性)化疗方案 (73%),包括针对MBC的曲妥珠单抗 (93%)、帕妥珠单抗 (60%) 以及曲妥珠单抗‐emtansine (93%)。在第 1 个周期内,在剂量水平 1 组 (n = 6) 或剂量扩展组 (n = 9) 均未观察到剂量限制性毒性。一名患者出现 ≥3 级的免疫相关不良反应(4 级糖尿病)。未发现实体瘤疗效评价标准 (RECIST) 中的任何缓解,在第 6 周时,14 名患者中有 4 名患者 (29%) 表现出最佳缓解,即疾病稳定(中位持续时间为 2.7 个月)。所有患者在标本组织检查 (7/15) 或前期研究活检 (8/15) 中均出现 <1% PD‐L1 表达。在剂量扩展队列中,可评估的治疗前和治疗中肿瘤活检 (n = 5) 均显示出最小 CD8 细胞浸润。
结论。度伐单抗和曲妥珠单抗的 RP2D 是这两种药物的标准全剂量。在既往曾多次接受治疗的 HER2 阳性 PD‐L1 阴性MBC患者中,未观察到显著的临床活性。
实践意义:这项具有相关终点的 Ib 期试验证明度伐单抗联合曲妥珠单抗治疗既往曾多次接受治疗的 HER2 阳性转移性乳腺癌 (MBC) 患者缺乏活性。在既往曾多次接受治疗的 HER2 阳性程序性死亡配体 1 (PD‐L1) 阴性MBC患者中,有证据表明细胞毒性 T 细胞衰竭,未观察到显著的临床活性。此外,所有患者在肿瘤细胞中均未出现 PD‐L1 表达。这些数据证实了 PD‐L1 作为重要选择标志物的重要性以及对免疫调节细胞的肿瘤微环境进行评估的必要性。了解如何激活“冷”肿瘤还需要进一步开展工作,以便能够联合使用现有的各种免疫肿瘤药物。
Introduction
The HER2 receptor is amplified and/or overexpressed in 15%–20% of breast cancers. The advent of HER2‐directed therapies, such as trastuzumab, pertuzumab, and trastuzumab‐emtansine (T‐DM1), has markedly improved prognosis for early‐stage [1], [2] and advanced‐stage [3], [4] HER2‐positive breast cancer. Patients with advanced disease, however, ultimately develop resistance to currently approved HER2‐targeted therapies, and response to further systemic therapy is typically short lived [5], [6].
The immune system is increasingly recognized as an important determinant of outcome with HER2‐targeted therapy. Higher levels of tumor‐infiltrating lymphocytes (TILs) are associated with an improved pathological complete response rate to neoadjuvant HER2‐targeted therapy and improved disease‐free survival in early‐stage disease [7], [8], [9]. Higher TILs were prognostic for longer overall survival but no differential benefit with the addition of pertuzumab to docetaxel chemotherapy and trastuzumab in the first‐line CLEOPATRA trial for patients with HER2‐positive advanced disease [10]. Increased expression of immune activation gene‐expression signatures has also been shown to be predictive of benefit from trastuzumab in the adjuvant setting [11].
The programmed death 1 (PD‐1) protein is a co‐inhibitory receptor known to be expressed on activated T cells, which, when bound to its ligand PD‐L1, limits T‐cell antitumor activity in the tumor microenvironment [12]. Dual blockade of HER2 and PD‐1/PD‐L1 can significantly improve the activity of HER2 antibody therapy in immunocompetent mouse models of HER2‐positive breast cancer [13]. Durvalumab is a monoclonal antibody against PD‐L1 that is active in a variety of tumor types, including urothelial, non‐small cell lung, melanoma, head and neck, and gastroesophageal cancers [14].
We hypothesized that the addition of durvalumab to trastuzumab might induce immune activation and resensitize resistant HER2‐positive advanced breast cancers to further trastuzumab therapy. Here, we report a phase Ib trial of durvalumab in combination with trastuzumab in resistant HER2‐positive advanced breast cancers. The primary objective was to establish the recommended phase II dose (RP2D) for the combination; secondary objectives included an evaluation of safety, response, and clinical benefit rate in an exploratory cohort and an assessment of pharmacodynamics markers of immune activation in serial tumor biopsies.
Materials and Methods
Study Population
Eligibility criteria for trial enrollment required patients to have HER2‐positive (immunohistochemistry [IHC] 3+ and/or fluorescence in situ hybridization amplified as assessed locally and by their laboratory standards) advanced breast cancer for which no curative therapy exists. Patients must have had prior exposure to a taxane, trastuzumab, and pertuzumab (although exceptions to the requirement for prior pertuzumab could be waived) and preferably also prior exposure to T‐DM1. Other prior treatment inclusion criteria included not being eligible for further trastuzumab treatment as per provincial or formulary guidelines and at least two lines of anti‐HER2 regimens in the setting of metastatic breast cancer (MBC). All patients were required to be at least 18 years of age and to have measurable disease, Eastern Cooperative Oncology Group performance status of 0 to 2, and standard baseline organ function measurements. There was no limit to the number of prior cytotoxic or other systemic therapy regimens, but patients could not have had previous treatment with PD‐1 or PD‐L1 inhibitors or other immune‐based therapy. Patients enrolled to the RP2D/expansion cohort must have had accessible disease suitable for biopsy and consent to a biopsy prior to treatment and at the end of cycle 1. Paired biopsies were also strongly recommended in the dose‐finding stage of the study. All patients consented to release of archival tissue (if patients did not have archival tissue, then a biopsy prior to treatment was acceptable). Patients could not have had active or prior documented autoimmune or inflammatory disorders. Live attenuated vaccination was not allowed within 30 days prior to registration or within 30 days of receiving durvalumab. Patients with active infections or untreated, uncontrolled, or symptomatic cardiovascular conditions were not eligible. All patients provided written informed consent approved by their institutional research ethics board before any protocol‐directed procedures were performed. The trial was registered on Clinicaltrials.gov (identifier, NCT02649686).
Study Treatment
Durvalumab was administered intravenously on day 1 once every 3 weeks, at a flat dose starting at dose level 1 (1,125 mg) with one dose level reduction planned (dose level − 1: 750 mg) if necessary. Trastuzumab was administered intravenously at 8 mg/kg (loading dose cycle 1 only) and then at 6 mg/kg for subsequent cycles to a maximum of six cycles (treatment beyond six cycles in patients deriving ongoing clinical benefit as determined by their treating physician was possible after consultation with trial committee). No dose modification of trastuzumab was allowed. A standard 3 + 3 dose escalation schedule was used in the dose‐finding portion of the study.
If no dose‐limiting toxicity (DLT) was observed in the first three patients during the first cycle of therapy, then dose level 1 would be declared as the RP2D. If one of the first three patients (but not two or more) at dose level 1 experienced a DLT, then an additional three patients at that dose level were added. The maximum administered dose (MAD) was defined as the dose level at which at least two of six or at least two of three patients experienced dose‐limiting toxicity. The next lower dose below the MAD was then declared the recommended phase II dose.
The study included a dose expansion cohort at the RP2D for safety assessment and to perform serial core biopsies in a larger series of patients for pharmacodynamic assessment of PD‐L1 tumor expression and stromal TIL status.
Study Procedures
Patients were assessed clinically on day 1 of each cycle with standard hematological and biochemical bloodwork performed weekly during cycle 1 and then on day 1 of each subsequent cycle. Radiological imaging was performed at baseline and at the end of every second cycle, and response was assessed using RECIST 1.1 [15]. An assessment of left ventricular ejection function was performed at baseline, upon completion of six cycles of study therapy, and as clinically indicated.
Adverse events were assessed by Common Terminology Criteria for Adverse Events, version 4.0. DLT was defined as any of the following occurring during cycle 1 of each dose level: grade 3 or higher diarrhea/colitis or pneumonitis, liver transaminase elevation >8 × upper limit of normal (ULN) or total bilirubin >5 × ULN, other grade 3 toxicity (excluding fatigue, infusion reaction, endocrine disorder, inflammatory reaction, electrolyte abnormalities, or myelosuppression) that did not resolve to grade 1 within 3 days with intensive management, any grade 4 immune‐related adverse event, and toxicity requiring infliximab. In order for a toxicity to be considered a DLT, the toxicity must be considered related (possibly, probably, or definitely) to durvalumab. Late toxicities that were determined to be immune related were also considered when defining the RP2D.
Immunohistochemistry for PD‐L1 and CD8
Paraffin sections at 4‐um thickness were dried in a 60°C oven overnight before staining. IHC was performed according to the manufacturer's guidelines using BenchMark XT, an automated slide strainer (Ventana Medical Systems, Oro Valley, AZ) with standard antigen retrieval (CC1, Tris/Borate/EDTA, pH 8.0, #950‐124) (760‐2018). For CD8 (clone SP57, Ventana/Roche), Ventana Ultraview Universal DAB Detection Kit (#760‐500) was used. For PD‐L1, the slides were incubated in PD‐L1 antibody (clone 263, Ventana 790‐4905) for 16 minutes. Ventana OptiView Detection Kit (#860‐099) was used. Both kits contain a cocktail of enzyme labeled secondary antibodies that locate the bound primary antibody. The complex was then visualized with hydrogen peroxide substrate and 3,3’‐diaminobenzidinetetrahydrochloride chromogen, which produced a dark brown precipitate that is readily detected by light microscope. The slides were counterstained with Gill modified hematoxylin and bluing in phosphate‐buffered saline, dehydrated in graded alcohol, cleared in xylene, and cover‐slipped in Permount.
IHC Scoring for PD‐L1 and CD8
Tumor areas of the entire specimen were examined at low power (×4 objective magnification) for PD‐L1 staining. Evaluation of viable tumor cells with partial or complete membranous staining at any intensity for PD‐L1 was determined at higher magnification (×10 to ×40 magnification). The overall percent of tumor staining, or tumor proportion score, was measured. For CD8, the overall predominant pattern of infiltration was assessed at low power. Four patterns of CD8‐positive lymphocytic infiltration were observed: minimal (low number of lymphocytic infiltration), marginal (lymphocytic aggregation at periphery of tumor), stromal (lymphocytic infiltration within tumor stroma), and tumoral (lymphocytes present within and directly contacting tumor cells). CD8‐positive lymphocyte density was quantified by averaging the number of lymphocytes counted in 10 high‐power fields (at ×40 objective magnification).
Statistics
This was a phase I study with the primary objective designed to determine the recommended phase II dose of durvalumab in combination with trastuzumab. The final sample size was dependent upon the number of dose levels required to reach the recommended phase II dose. Once the recommended phase II dose was established, there was an option to enroll 9 to 12 additional patients to further evaluate toxicity and response and to perform correlative studies of the combination.
Results
Fifteen patients with HER2‐positive metastatic breast cancer were enrolled and treated at three Canadian centers between April and December 2016 (CONSORT diagram, supplemental online Fig. 1). The baseline characteristics of these 15 patients are detailed in Table 1. Overall, the study cohort was a relatively heavily pretreated population of patients in the adjuvant and/or the metastatic setting. The median age was 54 years (range 40–86 years); the majority had visceral disease (87%); 20% had documented brain metastases at baseline; at least three prior (adjuvant and/or metastatic) lines of chemotherapy (73%), including trastuzumab (93%), pertuzumab (60%), and T‐DM1 (93%) were delivered for MBC in the study population. The majority of the cohort was estrogen receptor‐negative (73%), and two thirds had elevated lactate dehydrogenase at study entry. All patients were enrolled to dose level 1 (durvalumab 1,125 mg day 1 q3w and trastuzumab 8 mg/kg day 1 cycle 1, then 6 mg/kg day 1 q3w cycles 2–6). No DLTs were noted in the first three patients, although the first patient experienced grade 2 alanine transaminase rise in cycle 1 (considered related to end of cycle 1 fresh biopsy of liver). To further evaluate the safety of this dose level, the first cohort was expanded to enroll an additional three patients. No DLTs were observed in any of these additional patients, so dose level 1 was expanded per protocol to enroll an additional nine patients in the dose expansion phase. No DLTs were observed in these nine patients after the first cycle of study treatment.
Table 1. Patient characteristics (n = 15).
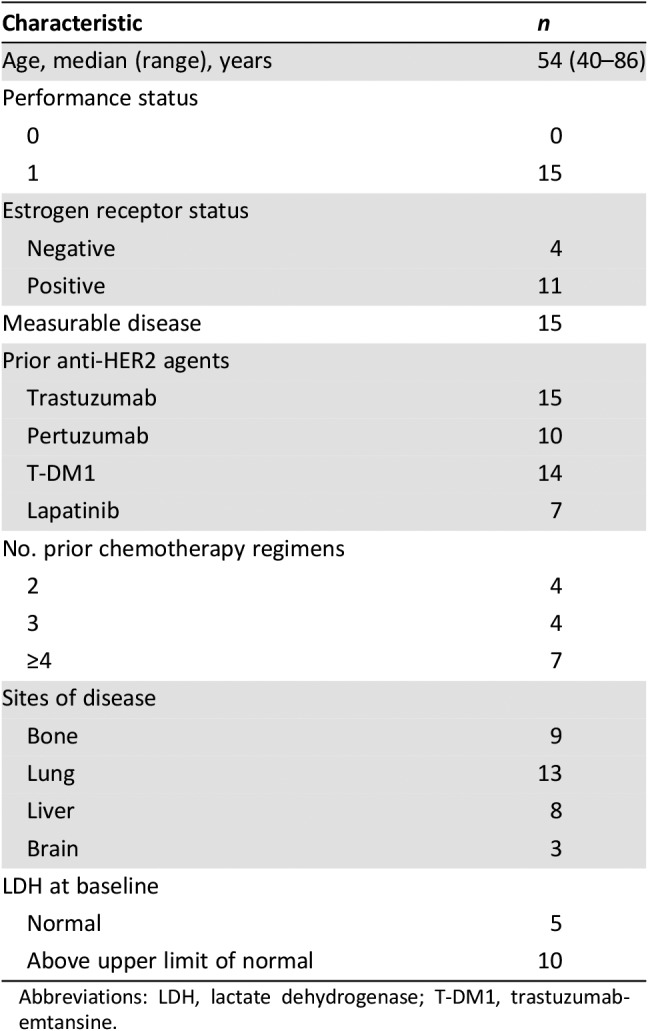
Abbreviations: LDH, lactate dehydrogenase; T‐DM1, trastuzumab‐emtansine.
All 15 patients were evaluable for nonhematologic toxicity. The most commonly reported adverse events (any causality) were fatigue, nausea, constipation, cough, headache, anorexia, back pain, peripheral sensory neuropathy, and dizziness, and were either grade 1 or 2 in severity, with the exception of one event of grade 3 nausea. Adverse events considered related to both durvalumab and trastuzumab were diarrhea, arthralgia, paresthesia, and rash (Table 2). Adverse events related to durvalumab alone were nausea, vomiting, chills, fatigue, fever, dizziness, headache, pneumonitis, pain of skin, and diabetes. All adverse events considered related to protocol treatment were classified as grade 1–2, with the exception of diabetes (grade 4, type 1 diabetes presenting with diabetic ketoacidosis). This patient received two cycles of study treatment before demonstration of progressive disease. She presented to the local emergency room approximately 1 month after last study drug administration with symptoms of confusion and weakness and a blood glucose level of 30.6 mmol/L. She was treated for diabetic ketoacidosis with fluids and insulin, discharged from hospital 1 week later, and remained dependent on exogenous insulin control on further follow‐up. This immune‐related adverse event was determined to be related to durvalumab and is a known immune‐related adverse event related to checkpoint inhibitors.
Table 2. Adverse events related to durvalumab.
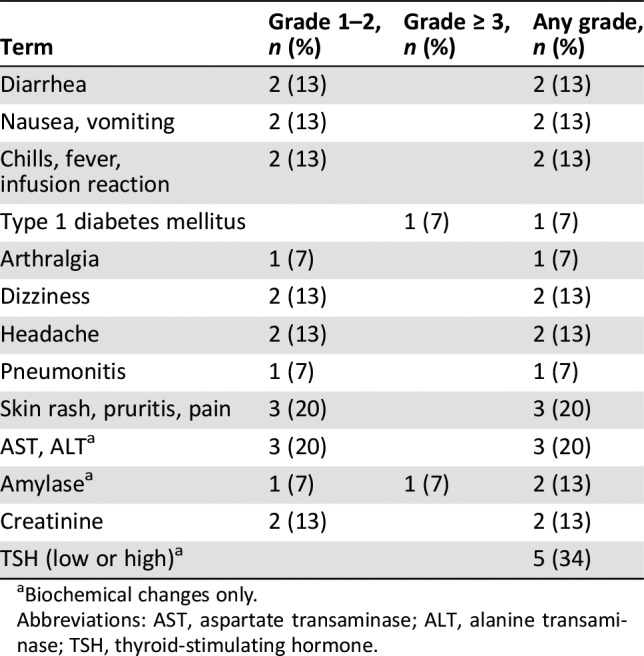
Biochemical changes only.
Abbreviations: AST, aspartate transaminase; ALT, alanine transaminase; TSH, thyroid‐stimulating hormone.
All 15 patients were also evaluable for hematologic and biochemical toxicity. Most changes were grade 1–2. Two patients experienced grade 3 lymphocytopenia, one patient experienced grade 3 amylase elevation, and two patients had grade 4 hyperglycemia. One patient had grade 3 amylase elevation and grade 4 hyperglycemia related to dexamethasone given (for the treatment of brain metastases) during the 4‐week posttreatment period. Thyroid‐stimulating hormone (TSH) reductions of <1.0 x lower limit of normal (LLN) were documented in four patients, with one patient having TSH of <0.5 x LLN. None of these patients had previous history of hypothyroidism, and none of these events was considered clinically significant, as patients recovered without thyroid replacement therapy. No cardiac toxicity was noted through study treatment. No patients required dose reductions of either durvalumab or trastuzumab. Adverse events felt to be related to durvalumab are summarized in Table 2.
No responses by RECIST were seen, with 4 of 14 patients (29%) demonstrating stable disease as best response at week 6 (median duration, 2.7 months; range, 2.6–2.9 months). Three patients received more than four cycles of therapy, with the longest duration of study treatment being six cycles of combination therapy. Best tumor shrinkage over study treatment is illustrated in the spider plot as categorized by RECIST 1.1 and by iRECIST (Fig. 1) [16]. The median progression‐free survival (PFS) was 1.35 months (95% confidence interval [CI], 1.25–1.71 months). The estimated 6‐month PFS was 0%. The estimated 6‐month overall survival (OS) was 51.6%, and 1‐year OS was 17.2%.
Figure 1.

Spider plot of response and duration of response of the study cohort.
Abbreviations: PD, progressive disease; SD, stable disease.
All patients had <1% PD‐L1 tumor expression on either archival tissue (7/15) or prestudy biopsy (8/15). In the dose expansion cohort, serial pretreatment and on‐treatment tumor biopsies with sufficient tumor for IHC assessment (n = 5) showed minimal CD8 cell infiltration. A full description of the correlative IHC results per patient is presented in Table 3. It is interesting to note that in our limited correlative assessment, there was no change in TILs after one cycle of an immune‐oncology (IO) agent and trastuzumab.
Table 3. Pharmacodynamic assessment of biopsies and/or archival tissue.

Archival tissue 10%–24% but pretreatment biopsy <1%.
Moderate on archival tissue but minimal on pretreatment biopsy.
Abbreviations: PD‐1, programmed death 1; PD‐L1, PD‐1 ligand; TIL, tumor‐infiltrating lymphocyte.
Discussion
Despite the initial substantial benefits seen with adjuvant trastuzumab, with longer follow‐up, relapses continue to occur over time. At the first analysis of the National Surgical Adjuvant Breast and Bowel Project B‐31 and North Central Cancer Treatment Group N9831 trials, the 2‐year disease‐free survival (DFS) was 87.1% [17]. With longer follow‐up at year 10, there is now an observed 73.7% DFS rate [18]. In advanced‐stage disease, despite significant improvements in progression‐free survival and overall survival with dual HER2 antibody blockade (pertuzumab and trastuzumab with docetaxel) and with an antibody drug conjugate (T‐DM1), progression of disease and death ultimately occurs in the vast majority of patients [3], [5]. Thus, additional therapeutic agents are still needed in the management of HER2‐positive breast cancer, particularly those that may overcome resistance or resensitize response to continued targeted HER2 therapy.
One proposed mechanism of action of anti‐HER2 monoclonal antibodies is through engagement of the adaptive immune response. The PD‐1 pathway is an important immune checkpoint regulatory protein. Binding of PD‐1 by either of its ligands, PD‐L1 or PD‐L2, suppresses activation of effector T cells [19]. Although this interaction functions to protect against excessive inflammation under normal conditions, it is hypothesized that upregulation of the PD‐1 pathway allows cancer cells to evade the adaptive immune response [20]. Inhibitory antibodies targeting PD‐1, such as pembrolizumab and nivolumab, demonstrate robust antitumor activity across multiple tumor types and are approved for the treatment of multiple advanced malignancies [21], [22], [23]. In a randomized phase III trial of a PD‐L1 inhibitor (atezolizumab) in combination with nab‐paclitaxel versus placebo with nab‐paclitaxel, a significant improvement in PFS (hazard ratio, 0.62; p < .001) was demonstrated in the PD‐L1‐positive metastatic triple‐negative breast cancer population [24]. Within the trial in which PD‐L1 status was stratified, the hazard ratio for PFS in the PD‐L1‐negative cohort was 0.95 (95% CI, 0.79–1.15), supporting PD‐L1 as an important biomarker selection criterion. Durvalumab is a selective, high‐affinity, human immunoglobulin G1K monoclonal antibody that blocks PD‐L1 binding to PD‐1 and CD80 (B7.1), allowing T cells to then recognize and induce tumor cell kill. Durvalumab in a phase I/II trial of 61 patients (regardless of PD‐L1 status) with advanced‐stage urothelial cancers demonstrated an overall response rate of 31.0% (95% CI, 17.6–47.1), with all the responses seen only in the PD‐L1‐positive cohort (46.4% response rate in this subgroup), with a median duration of response not yet reached at time of publication [25]. Preclinical studies have suggested that checkpoint inhibition in combination with trastuzumab may overcome immune‐mediated mechanisms of trastuzumab resistance [13].
In our phase Ib study we were able to establish that the recommended phase II dose of the combination of trastuzumab and durvalumab is standard full dosing of both monoclonal antibodies delivered on an every‐3‐week schedule. Overall, the rates of grade 3–4 toxicity were low, with the spectrum of adverse events as expected with either agent. We did not observe any significant cardiotoxicity, and aside from asymptomatic changes in TSH, the only clinically significant treatment‐related adverse event (AE) was one grade 4 immune‐related AE (diabetes mellitus). We did not observe any significant clinical activity in this small dose‐determination study, as we saw no documented responses and less than one third of patients achieved stable disease as best response at week 6. Furthermore, the duration of stable disease (as defined in the protocol) was of short duration. In this relatively heavily pretreated population of patients with HER2‐positive MBC exposed to several lines of anti‐HER2 therapies, immune checkpoint blockade with trastuzumab does not appear to be the best therapeutic strategy to overcome acquired resistance. As our correlative studies were performed after enrollment was complete, we retrospectively identified that our study population included only patients with nonimmunogenic tumors, characterized by no PD‐L1 tumor expression and minimal stromal cytotoxic T‐cell infiltration prior to treatment. Approximately half of our patient population had demonstrated liver metastases, which have been demonstrated to have low/no PDL‐1 expression and minimal TILs relative to lung and lymph node metastases. All these factors likely contributed to a less than optimal environment for effective activity of durvalumab in combination with trastuzumab.
One other study investigating the safety and efficacy of a PD‐1 inhibitor (pembrolizumab) in combination with trastuzumab in HER2‐positive MBC has also recently reported [26]. The PANACEA study, a phase Ib–II trial, enrolled both a PD‐L1‐positive (defined as ≥1% tumor or stromal staining) and a PD‐L1‐negative cohort. Similar to our trial, all patients had documented progressive disease on trastuzumab or T‐DM1. With a median duration of follow‐up of 13.6 months, no responses were documented in the PD‐L1‐negative cohort (n = 12), whereas in the PD‐L1‐positive phase II component of the study (n = 40), a 15% response rate was documented (90% CI, 7%–29%). Median duration of response was 3.5 months, with five patients continuing on treatment with no evidence of progression at time of reporting. In a post hoc analysis of the combined PD‐L1‐positive cohort, the median PFS was 2.7 months (90% CI, 2.6–4.0). Baseline stromal TILs were also low in the PD‐L1‐negative patients relative to the PD‐L1‐positive patient population. The authors were also able to demonstrate that the baseline presence of stromal TILs of ≥5% may be a potential biomarker for enhanced clinical activity in the PD‐L1‐positive cohort.
Lastly, the majority of the clinical trials investigating IO agents in breast cancer have been focused on the triple‐negative MBC subtype. The KEYNOTE‐012 trial enrolled 32 women with PD‐L1‐positive (≥1% stromal or tumor staining) triple‐negative MBC on a phase Ib trial of single‐agent pembrolizumab [27]. The overall response rate was 18.5% (95% CI, 6.3%–38.1 %) with no apparent correlation with number of prior lines of therapy. The investigators were able to demonstrate that increasing PD‐L1 staining in stroma or tumor was statistically associated with an increased probability of an objective response. In contrast, in the KEYNOTE‐086 trial in which 170 patients with triple‐negative MBC previously exposed to at least one line of chemotherapy were enrolled, the overall response rate was 5% (95% CI, 2%–9%), and median PFS was 2.0 months (95% CI, 1.9–2.0 months) regardless of PD‐L1 status [28]. In a single‐agent study of atezolizumab (a PD‐L1 inhibitor), a clear trend for greater efficacy was seen when the IO agent was given in the first‐line setting relative to second‐line setting or later [29]. Lastly, in the GeparNuevo randomized phase II neoadjuvant trial assessing the activity of durvalumab in combination with a taxane‐anthracycline‐based regimen in triple‐negative breast cancer, the addition of durvalumab did numerically increase the pathologic complete response rate (53% vs. 44%; adjusted p = .182) and overall was well tolerated relative to placebo [30].
Conclusion
The recommended phase II dose of durvalumab and trastuzumab is standard full doses of both agents with no new safety signals emerging. No significant clinical activity was observed in our small cohort of patients with heavily pretreated HER2‐positive MBC that lacked PD‐L1 tumor expression and had evidence of cytotoxic T‐cell exhaustion prior to treatment. Based on our study and others in advanced‐stage breast cancer, it appears that a better therapeutic strategy would be to study checkpoint inhibitors in an earlier‐line setting and to enrich for patients with more immunogenic tumors, characterized by some degree of PD‐L1 expression and the presence of stromal T‐cell infiltration prior to treatment exposure.
Acknowledgments
This trial was supported by a grant to the Canadian Cancer Trials Group from the Canadian Cancer Society Research (grant 704970) and from AstraZeneca (which also provided durvalumab).
Author Contributions
Conception/design: Stephen Chia, Phillipe L. Bedard, Lesley Seymour
Provision of study material or patients: Stephen Chia, Phillipe L. Bedard, John Hilton, Eitan Amir, Karen Gelmon, Rachel Goodwin, Diego Villa, Ming Tsao, Lesley Seymour
Collection and/or assembly of data: Stephen Chia, Phillipe L. Bedard, John Hilton, Eitan Amir, Karen Gelmon, Rachel Goodwin, Diego Villa, Dongsheng Tu, Ming Tsao, Lesley Seymour
Data analysis and interpretation: Stephen Chia, Phillipe L. Bedard, Michael Cabanero, Dongsheng Tu, Ming Tsao, Lesley Seymour
Manuscript writing: Stephen Chia, Phillipe L. Bedard, John Hilton, Eitan Amir, Karen Gelmon, Rachel Goodwin, Diego Villa, M. Cabanero, Dongsheng Tu, Ming Tsao, Lesley Seymour
Final approval of manuscript: Stephen Chia, Phillipe L. Bedard, John Hilton, Eitan Amir, Karen Gelmon, Rachel Goodwin, Diego Villa, Michael Cabanero, Dongsheng Tu, Ming Tsao, Lesley Seymour
Disclosures
Stephen Chia: AstraZeneca (RF), Hoffmann LaRoche (C/A); Phillipe L. Bedard: Bristol‐Myers Squibb, Genentech/Roche, Pfizer, Sanofi (C/A), AstraZeneca, Bristol‐Myers Squibb, Genentech/Roche, GlaxoSmithKline, Immunomedics, Merck, Mersana, Nektar, Novartis, Oncothyreon, Pfizer, PTC Therapeutics, Sanofi, Seattle Genetics, Servier, SignalChem Life Sciences (RF); John Hilton: AstraZeneca, Bristol‐Myers Squibb, Novartis, Puma, Pfizer (C/A); Eitan Amir: Genentech/Roche (ET), AstraZeneca, Apobiologiz, Agendia, Myriad Genetics (C/A); Karen Gelmon: Pfizer, AstraZeneca, Eli Lilly & Co., Novartis, Genomic Health, Nanostring, Merck, Mylan, Bristol‐Myers Squibb (C/A), AstraZeneca, Roche, Bristol‐Myers Squibb (RF), Genetech (ET); Rachel Goodwin: Ipsen, Novertis, Servier, Taiho, Amgen, Celgene (C/A), Pfizer, Novartis, Ipsen (RF); Diego Villa: Roche, Celgene, Abbvie, Seattle Genetics, Lundbeck, Gilead, AstraZeneca, Jannsen, Nanostring (H, SAB); Ming Tsao: Roche, Celgene, Abbvie, Seattle Genetics, Lundbeck, Gilead, AstraZeneca, Jannsen, Nanostring (H, SAB); Lesley Seymour: AstraZeneca (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Dawood S, Broglio K, Buzdar AU et al. Prognosis of women with metastatic breast cancer by HER2 status and trastuzumab treatment: An institutional‐based review. J Clin Oncol 2009;28:92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.von Minckwitz G, Procter M, de Azambuja E et al. Adjuvant pertuzumab and trastuzumab in early HER2‐positive breast cancer. N Engl J Med 2017;377:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swain SM, Baselga J, Kim SB et al. Pertuzumab, trastuzumab, and docetaxel in HER2‐positive metastatic breast cancer. N Engl J Med 2015;372:724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diéras V, Miles D, Verma S et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2‐positive advanced breast cancer (EMILIA): A descriptive analysis of final overall survival results from a randomised, open‐label, phase 3 trial. Lancet Oncol 2017;18:732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krop IE, Kim SB, Martin AG et al. Trastuzumab emtansine versus treatment of physician's choice in patients with previously treated HER2‐positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomised open‐label phase 3 trial. Lancet Oncol 2017;18:743–754. [DOI] [PubMed] [Google Scholar]
- 6.Vici P, Pizzuti L, Michelotti A et al. A retrospective multicentric observational study of trastuzumab emtansine in HER2 positive metastatic breast cancer: A real‐world experience. Oncotarget 2017;8:56921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salgado R, Denkert C, Campbell C et al. Tumor‐infiltrating lymphocytes and associations with pathological complete response and event‐free survival in HER2‐positive early‐stage breast cancer treated with lapatinib and trastuzumab: A secondary analysis of the NeoALTTO trial. JAMA Oncol 2015;1:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denkert C, Von Minckwitz G, Brase JC et al. Tumor‐infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2–positive and triple‐negative primary breast cancers. J Clin Oncol 2014;33:983–991. [DOI] [PubMed] [Google Scholar]
- 9.Loi S, Michiels S, Salgado R et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann Oncol 2014;25:1544–1550. [DOI] [PubMed] [Google Scholar]
- 10.Luen SJ, Salgado R, Fox S et al. Tumour‐infiltrating lymphocytes in advanced HER2‐positive breast cancer treated with pertuzumab or placebo in addition to trastuzumab and docetaxel: A retrospective analysis of the CLEOPATRA study. Lancet Oncol 2017;18:52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varadan V, Gilmore H, Miskimen K et al. Immune signatures following single dose trastuzumab predict pathologic response to preoperative trastuzumab and chemotherapy in HER2‐positive early breast cancer. Clin Cancer Res 2016;22:3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fife BT, Pauken KE, Eagar TN et al. Interactions between PD‐1 and PD‐L1 promote tolerance by blocking the TCR–induced stop signal. Nature Immunology 2009;10:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stagg J, Loi S, Divisekera U et al. Anti–ErbB‐2 mAb therapy requires type I and II interferons and synergizes with anti–PD‐1 or anti‐CD137 mAb therapy. Proc Natl Acad Sci USA 2011;108:7142–7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segal NH, Antonia SJ, Brahmer JR et al. Preliminary data from a multi‐arm expansion study of MEDI4736, an anti‐PD‐L1 antibody. J Clin Oncol 2014;32(suppl 15):3001A. [Google Scholar]
- 15.Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 16.Seymour L, Bogaerts J, Perrone A et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 2017;18:e143–e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romond EH, Perez EA, Bryant J et al. Trastuzumab plus adjuvant chemotherapy for operable HER2‐positive breast cancer. N Engl J Med 2005;353:1673–1684. [DOI] [PubMed] [Google Scholar]
- 18.Perez EA, Romond EH, Suman VJ et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2‐positive breast cancer: Planned joint analysis of overall survival from NSABP B‐31 and NCCTG N9831. J Clin Oncol 2014;32:3744–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ribas A. Tumor immunotherapy directed at PD‐1. N Engl J Med 2012;366:2517–2519. [DOI] [PubMed] [Google Scholar]
- 20.Iwai Y, Ishida M, Tanaka Y et al, Involvement of PD‐L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD‐L1 blockade. Proc Natl Acad Sci USA 2002;99:12293–12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolchok JD, Chiarion‐Sileni V, Gonzalez R et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Motzer RJ, Escudier B, DF McDermott et al. Nivolumab versus everolimus in advanced renal‐cell carcinoma. N Engl J Med 2015;373:1803–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reck M, Rodríguez‐Abreu D, Robinson AG et al. Pembrolizumab versus chemotherapy for PD‐L1–positive non–small‐cell lung cancer. N Engl J Med 2016;375:1823–1833. [DOI] [PubMed] [Google Scholar]
- 24.Schmid P, Adams S, Rugo HS et al. Atezolizumab and nab‐paclitaxel in advanced triple negative breast cancer. N Engl J Med 2018;379:2108–2121. [DOI] [PubMed] [Google Scholar]
- 25.Massard C, Sosman JA, Sznol M et al. Atezolizumab, an anti‐programmed death‐ligand 1 antibody, in metastatic renal cell carcinoma: Long‐term safety, clinical activity, and immune correlates from a phase Ia study. J Clin Oncol 2016;10:34:833–842. [DOI] [PubMed] [Google Scholar]
- 26.Loi S, Giobbe‐Hurder A, Gombos A et al. Pembrolizumab plus trastuzumab in trastuzumab‐resistant advanced HER2‐positive breast cancer (PANACEA): A single arm, multi‐centre, phase Ib‐2 trial. Lancet Oncol 2019;20:371–382. [DOI] [PubMed] [Google Scholar]
- 27.Nanda R, Chow LQ, Dees EC et al. Pembrolizumab in patients with advanced triple‐negative breast cancer: Phase Ib KEYNOTE‐012 study. J Clin Oncol 2016;20:34:2460–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams S, Schmid P, Rugo HS et al. ASCO 2017. Phase 2 study of pembrolizumab (pembro) monotherapy for previously treated metastatic triple‐negative breast cancer (mTNBC): KEYNOTE‐086 cohort A. J Clin Oncol 2017;35(suppl 15):1008A. [Google Scholar]
- 29.Schmid P, Cruz C, Braiteh F et al. Atezolizumab in metastatic TNBC (mTNBC): Long‐term clinical outcomes and biomarker analyses. Cancer Res 2017;77(suppl 13):2986A. [Google Scholar]
- 30.Loibl S, Untch M, Burchardi N et al. Randomized phase II neoadjuvant study (GeparNuevo) to investigate the addition of durvalumab to a taxane‐anthracycline containing chemotherapy in triple negative breast cancer. J Clin Oncol 2018;36(suppl 15):104A. [Google Scholar]