

Abstract
Biocatalytic oxyfunctionalisation reactions are traditionally conducted in aqueous media limiting their production yield. Here we report the application of a peroxygenase in neat reaction conditions reaching product concentrations of up to 360 mM.
Keywords: Biocatalysis, Epoxidation, Peroxygenases, Neat reaction conditions, Cascade reactions
Very neat! Peroxygenases catalyse epoxidation of C=C‐double bonds under non‐aqueous conditions.
Epoxides are important building blocks in organic synthesis. The ring opening of epoxides leads to useful ɑ‐ or β‐substituted alcohols.1 As a result, a broad range of catalytic methods for the epoxidation of C=C‐double bonds have been established.2 Compared to this variety, only few biocatalytic methods are known. The chemoenzymatic epoxidation of alkenes using lipase‐borne peracids for example is receiving tremendous interest but yields racemic products.3 Amongst the stereospecific epoxidation methods the use of flavin‐dependent styrene monooxygenases4 and P450 monooxygenases5 are most prominent.
The latter approaches rely on reductive activation of molecular oxygen using reduced nicotinamide cofactors (NAD(P)H) as source of reducing equivalents (Scheme 1). This not only implies complicated and vulnerable electron transport chains but also, due to the exclusive water‐solubility of the cofactors, largely limits these processes to aqueous reaction media.
Scheme 1.
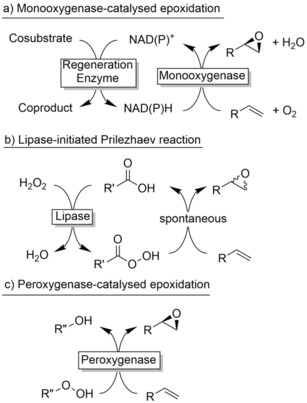
Comparison of biocatalytic epoxidation reactions.
The majority of the alkenes of interest are however hydrophobic, limiting the final product titres to the lower millimolar range. This is inacceptable from an economic and an environmental point‐of‐view. Current solutions focus around two‐liquid‐phase‐system approaches (2LPS).6
Ideally, (bio) catalytic epoxidation reactions should occur in organic media (even neat) to enable high product concentrations. In this respect, peroxygenases represent a promising solution.7 Peroxygenases are heme‐thiolate enzymes enabling P450 monooxygenase‐like oxyfunctionalisation reactions. In contrast to monooxygenases, peroxygenases do not rely on (water‐soluble) redox partners but on (organic) peroxides, enabling their potential application in non‐aqueous media. Pioneering works by Pu, Wang and Zhang8 and Hofrichter9 have established peroxygenase‐catalysed epoxidation reactions using hydrogen peroxide or organic hydroperoxides as oxidants, albeit in aqueous reaction media thereby limiting the reagent concentration to the lower millimolar range.
Klibanov and co‐workers reported peroxidase‐reactions under non‐aqueous conditions.10 Unfortunately, these contributions have not yet found widespread attention in the biocatalysis community.
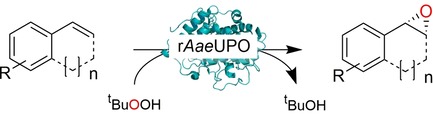
We therefore set out to establish peroxygenase‐catalysed, selective oxyfunctionalisation reactions in neat reaction media. As model peroxygenase we chose an evolved recombinant peroxygenase from Agrocybe aegerita (rAaeUPO)11 to catalyse the epoxidation of styrene and its derivatives.9
As oxidant, we chose tert‐butyl hydroperoxide (tBuOOH) due to its high solubility in hydrophobic media.
To employ rAaeUPO in neat reaction media, we first immobilised it covalently on an epoxide‐modified polyacrylic matrix (Immobead IB‐COV‐1). Under non‐optimised conditions, 72.8 % of the enzyme was immobilised (for further details see SI section 4). The remaining catalytic activity, however was only 3 % (Figure S1). Further development will have to focus on optimised immobilisation procedures yielding higher activity.
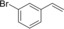
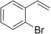
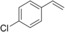
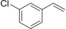
Nevertheless, having the immobilised enzyme in hand, we first explored its substrate scope (Table 1). Pleasingly, all of the styrene derivatives tested were converted with satisfactory to excellent turnover numbers for the biocatalyst. In accordance with previous reports9, 12 wild‐type rAaeUPO converted the majority of styrenes non‐stereoselectively giving (near racemic) epoxides, one notable exception being cis‐ß‐methylstyrene, which was converted highly stereoselectively into (1R,2S)‐cis‐β‐methylstyrene oxide. It is also interesting to note that in some cases, the desired epoxide was not stable and spontaneously rearranged into the corresponding carbonyl compound (for further details see SI section 5.3).
Table 1.
Substrate scope of the epoxidation of styrene derivatives with immobilised rAaeUPO. Data are an average of duplicates and corrected from potential substrate evaporation.[a]
|
| ||||||
|---|---|---|---|---|---|---|
|
Substrate |
Epoxy product [mM] |
ee [%] |
Carbonyl product[b] [mM(%)] |
Time [h] |
TON[c] |
|
|
|
1 a |
16 |
12 |
2 (12) |
42 |
3203 |
|
|
2 a |
80 |
30 |
47 (37) |
86 |
22598 |
|
|
3 a |
9 |
35 |
1 (11) |
86 |
1779 |
|
|
4 a |
36 |
>99 |
2 (4) |
21 |
6762 |
|
|
5 a |
59 |
9 |
21 (26) |
60 |
14235 |
|
|
6 a |
24 |
50 |
102 (81) |
62 |
22420 |
|
|
7 a |
10 |
39 |
90 (90) |
49 |
17794 |
|
|
8 a |
8 |
12 |
22 (73) |
62 |
5338 |
|
|
9 a |
16 |
17 |
12 (43) |
42 |
4982 |
|
|
10 a |
15 |
15 |
22 (59) |
22 |
6584 |
|
|
11 a |
11 |
15 |
25 (69) |
42 |
6406 |
|
|
12 a |
3 |
42 |
1 (23) |
42 |
712 |
|
|
13 a |
14 |
–[d] |
60 (81) |
62 |
13167 |
|
|
14 a |
36 |
28 |
59 (62) |
97 |
16904 |
|
|
15 a |
4 |
6 |
25 (86) |
49 |
5160 |
|
|
16 a |
none |
– |
– |
– |
0 |
|
|
17 a |
136 |
42 |
147 (52) |
108 |
50356 |
|
|
18 a |
none |
– |
33 (100) |
69 |
5872 |
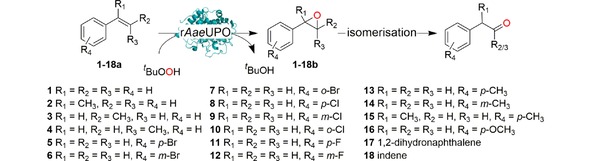
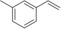
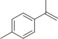
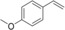
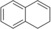
[a] Reaction conditions: [rAaeUPO]=5.62 μM, tBuOOH dosing rate=5 mM/h, room temperature, 20 rpm in overhead rotator. [b] The concentrations of carbonyl product were calculated using the calibration curves of the epoxides. Carbonyl products are aldehyde or ketone in β position from the ring opening of the epoxides, [c] TON=[product]/[enzyme], [d] n.d.=not determined.
Nevertheless, very significant product concentrations of up to 100 mM were achieved. The catalytic performance of rAaeUPO in terms of turnover numbers (TON=amount of product divided by the amount of enzyme, [mol×mol−1]) was excellent.
To identify parameters influencing the productivity of the reaction, we systematically varied the biocatalyst loading and the tBuOOH feeding rate in the epoxidation of cis‐β‐methylstyrene (Figure 1). The initial rate of the epoxidation reaction correlated directly with the dosing rate of tBuOOH. This, however did not necessarily translate in higher product titres. Most probably, increasing feed rates of the oxidant also increased the undesired oxidative inactivation of the enzyme's active site.13 This is supported by the observation that the robustness of the reactions (i. e. the duration of product accumulation) inversely correlated with the tBuOOH feed rate (Table S2).
Figure 1.
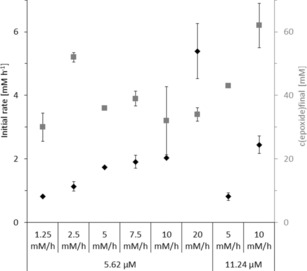
Characterisation of tBuOOH feeding rate and enzyme concentration comparing the initial reaction rates (black diamonds) and final product concentrations (grey squares). General conditions: room temperature, 20 rpm in overhead rotator. Data presented are an average of duplicates and corrected from potential substrate evaporation (see the Supporting Information, Section 5.1)
As mentioned above, epoxides are versatile building blocks for the synthesis of a broad range of products. Amino alcohols, for example, are common structural motifs in many pharmaceutically active ingredients.14 We therefore envisioned a chemoenzymatic cascade reaction comprising the rAaeUPO‐catalysed, stereoselective epoxidation of cis‐β‐methylstyrene followed by the chemical oxirane‐opening with methyl amine yielding (pseudo)ephedrine (Scheme 2).
Scheme 2.
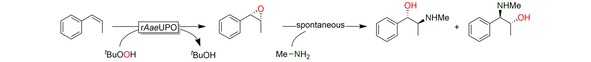
Envisioned chemoenzymatic cascade to obtain (pseudo)ephedrine from cis‐ß‐methylstyrene.
The epoxidation reaction was performed on a 10 mL scale with gradual tBuOOH feed (Figure 2). Although a conservative tBuOOH feed rate of 5 mM h−1 was applied, inactivation of the biocatalyst represented a major challenge for the reaction, necessitating further provision of the reaction with fresh enzyme (indicated by arrows in Figure 2). It is also interesting to note that in contrast to previous experiments using rAaeUPO in aqueous reaction media using H2O2,15 the peroxide utilisation efficiency was only approximately 50 %. It will be interesting to further investigate this increased catalase activity of rAaeUPO.
Figure 2.
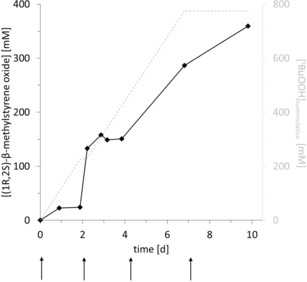
Time‐course of the rAaeUPO‐catalysed epoxidation of cis‐β‐methylstyrene. Conditions: 10 mL cis‐β‐methylstyrene, [rAaeUPO@IB‐COV‐1]total=41.8 μM (added in equal portions at the times indicated by the arrows), room temperature. tBuOOH was added continuously according to the feed profile shown as a dashed grey line.
From this experiment, 360 mM of (1R,2S)‐β‐methylstyrene oxide were obtained. The turnover number of the enzyme was more than 8500. Next to the desired product, the reaction mixture also contained some benzaldehyde, originating from rAaeUPO‐catalysed C=C‐bond cleavage.16 To avoid negative influences of this by‐product, the desired product was purified chromatographically and subjected to chemical ring‐opening with methylamine resulting in pseudoephedrine (58.2 %), ephedrine (7 %) and isoephedrine (34.8 %) (Scheme S2).
Overall, with this contribution we have demonstrated that peroxygenase‐catalysed epoxidations can be performed under neat reaction conditions. This opens up new possibilities for the preparative scale‐application of this promising class of enzymes. Product concentrations of up to 360 mM have been achieved representing one of the highest product titres obtained with oxidoreductase catalysis17 and certainly the highest product concentration with isolated enzymes.6a, 6b, 18
Nevertheless, some issues remain to be solved en route to a truly preparatively useful system. First and foremost, more active immobilisates of rAaeUPO need to be found. The activity recovery of the peroxygenase needs to be improved to obtain more active catalysts.19 We are confident that from the wealth of immobilisation methods available today,20 a suitable method will be found in our ongoing research. Also, more enantioselective rAaeUPO versions are highly desirable to broaden the scope of the reaction.
Experimental Section
Enzyme preparation. Recombinant expression and purification of the evolved unspecific peroxygenase mutant from A. aegerita in P. pastoris was performed following a previously described procedure.9
Immobilisation protocol. Immobeads (IB‐COV‐1) from ChiralVision was used to immobilise rAaeUPO. The beads were washed before usage and then mixed with rAaeUPO . Immobilisation was carried out for 5 hours using overhead rotator. After 5 hours, the immobilisation mixture was stored at 6 °C for 12 hours without stirring or shaking. After overnight incubation, the supernatant was removed, and the beads 3 times washed. The washing fractions were pooled. The peroxygenase concentration was determined via CO difference spectra in the supernatant and the washing fraction to calculate the amount of immobilised peroxygenase. A detailed description of the immobilisation of the enzymes is available in the Supporting Information.
Enzymatic reaction conditions. Reactions were performed in GC vial of 1.5 mL at room temperature. Immobilised rAaeUPO was first weighed in the vial according to the concentration of enzyme wanted, then pure substrate was added to the vial. Before each samples were taken, the vial was weighed in order to estimate the loss of substrate by evaporation. tBuOOH was added in the vial via a tube connected to a syringe pump. An overhead rotator from neoLab was mixing the reactions at 20 rpm. At intervals, aliquots were withdrawn, extracted with ethyl acetate, dried over MgSO4 and analysed by chiral gas chromatography. Details of gas chromatography and temperature profiles are shown in Supporting Information.
Chemical ring opening. 10 mg of pure epoxide were diluted in 200 μL of MeNH2 (40 % in water). The reaction was mixed during 20 hours at 60 °C. The reaction was then extracted with dichloromethane and analysed on NMR.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the European Research Council (ERC Consolidator Grant No. 648026) is gratefully acknowledged.
M. C. R. Rauch, F. Tieves, C. E. Paul, I. W. C. E. Arends, M. Alcalde, F. Hollmann, ChemCatChem 2019, 11, 4519.
References
- 1. Pineschi M., Eur. J. Org. Chem. 2006, 2006, 4979–4988. [Google Scholar]
- 2.
- 2a. McGarrigle E. M., Gilheany D. G., Chem. Rev. 2005, 105, 1563–1602; [DOI] [PubMed] [Google Scholar]
- 2b. Davis R. L., Stiller J., Naicker T., Jiang H., Jørgensen K. A., Angew. Chem. 2014, 53, 7406–7426, [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 7406–7426. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Burek B. O. O., Bormann S., Hollmann F., Bloh J., Holtmann D., Green Chem. 2019, 21, 3232–3249; [Google Scholar]
- 3b. Dong J., Fernández-Fueyo E., Hollmann F., Paul C., Pesic M., Schmidt S., Wang Y., Younes S., Zhang W., Angew. Chem., 2018, 130, 9380–9404, [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2018, 57, 9238–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Romero E., Castellanos J. R. Gómez, Gadda G., Fraaije M. W., Mattevi A., Chem. Rev. 2018, 118, 1742–1769; [DOI] [PubMed] [Google Scholar]
- 4b. Heine T., van Berkel W., Gassner G., van Pée K.-H., Tischler D., Biology 2018, 7, 42; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Gygli G., van Berkel W. J. H., Curr. Biotechnol. 2015, 4, 100–110; [Google Scholar]
- 4d. Huijbers M. M. E., Montersino S., Westphal A. H., Tischler D., van Berkel W. J. H., Arch. Biochem. Biophys. 2014, 544, 2–17. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Urlacher V. B., Girhard M., Trends Biotechnol. 2019; [DOI] [PubMed] [Google Scholar]
- 5b. Wang J. B., Reetz M. T., Nat. Chem. 2015, 7, 948–949. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Toda H., Imae R., Itoh N., Adv. Synth. Catal. 2014, 356, 3443–3450; [Google Scholar]
- 6b. Toda H., Imae R., Itoh N., Tetrahedron: Asymmetry 2012, 23, 1542–1549; [Google Scholar]
- 6c. Park J. B., Buehler B., Habicher T., Hauer B., Panke S., Witholt B., Schmid A., Biotechnol. Bioeng. 2006, 95, 501–512. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Hofrichter M., Ullrich R., Curr. Opin. Chem. Biol. 2014, 19, 116–125; [DOI] [PubMed] [Google Scholar]
- 7b. Wang Y., Lan D., Durrani R., Hollmann F., Curr. Opin. Chem. Biol. 2017, 37, 1–9. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Wang L., Wei S., Pan X., Liu P., Du X., Zhang C., Pu L., Wang Q., Chem. Eur. J. 2018, 24, 2741–2749; [DOI] [PubMed] [Google Scholar]
- 8b. Zhang C., Liu P.-X., Huang L.-Y., Wei S.-P., Wang L., Yang S.-Y., Yu X.-Q., Pu L., Wang Q., Chem. Eur. J. 2016, 22, 10969–10975. [DOI] [PubMed] [Google Scholar]
- 9. Kluge M., Ullrich R., Scheibner K., Hofrichter M., Green Chem. 2012, 14, 440–446. [Google Scholar]
- 10.
- 10a. Dordick J. S., Marletta M. A., Klibanov A. M., Proc. Natl. Acad. Sci. USA 1986, 83, 6255–6257; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Zaks A., Klibanov A. M., Science 1984, 224, 1249–1251. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Molina-Espeja P., Ma S., Mate D. M., Ludwig R., Alcalde M., Enz. Microb. Technol. 2015, 73–74, 29–33; [DOI] [PubMed] [Google Scholar]
- 11b. Molina-Espeja P., Garcia-Ruiz E., Gonzalez-Perez D., Ullrich R., Hofrichter M., Alcalde M., Appl. Environ. Microbiol. 2014, 80, 3496–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Churakova E., Kluge M., Ullrich R., Arends I., Hofrichter M., Hollmann F., Angew. Chem. Int. Ed. 2011, 50, 10716–10719; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10904–10907. [Google Scholar]
- 13. Valderrama B., Ayala M., Vazquez-Duhalt R., Chem. Biol. 2002, 9, 555–565. [DOI] [PubMed] [Google Scholar]
- 14. Grogan G., Curr. Opin. Chem. Biol. 2018, 43, 15–22. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Tieves F., Willot S. J.-P., van Schie M. M. C. H., Rauch M. C. R., Younes S. H. H., Zhang W., de Santos P. G., Robbins J. M., Bommarius B., Alcalde M., Bommarius A., Hollmann F., Angew. Chem. 2019, 131, 7955–7959, [Google Scholar]; Angew. Chem. Int. Ed. 2019, 58, 7873–7877; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Ni Y., Fernández-Fueyo E., Baraibar A. G., Ullrich R., Hofrichter M., Yanase H., Alcalde M., van Berkel W. J. H., Hollmann F., Angew. Chem., 2016, 128, 809–812, [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2016, 55, 798–801. [DOI] [PubMed] [Google Scholar]
- 16. Mutti F. G., Lara M., Kroutil M., Kroutil W., Chem. Eur. J. 2010, 16, 14142–14148. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Bühler B., Bollhalder I., Hauer B., Witholt B., Schmid A., Biotechnol. Bioeng. 2003, 82, 833–842; [DOI] [PubMed] [Google Scholar]
- 17b. Bühler B., Bollhalder I., Hauer B., Witholt B., Schmid A., Biotechnol. Bioeng. 2003, 81, 683–694; [DOI] [PubMed] [Google Scholar]
- 17c. Schmid A., Hofstetter K., Feiten H.-J., Hollmann F., Witholt B., Adv. Synth. Catal. 2001, 343, 732–737. [Google Scholar]
- 18.
- 18a. Hofstetter K., Lutz J., Lang I., Witholt B., Schmid A., Angew. Chem. Int. Ed. 2004, 43, 2163–2166; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2215–2218; [Google Scholar]
- 18b. Fernández-Fueyo E., Ni Y., Gomez Baraibar A., Alcalde M., van Langen L. M., Hollmann F., J. Mol. Catal. B. Enzym. 2016, 134. [Google Scholar]
- 19. Molina-Espeja P., Santos-Moriano P., García-Ruiz E., Ballesteros A., Plou F. J., Alcalde M., Int. J. Mol. Sci. 2019, 20, 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hanefeld U., Gardossi L., Magner E., Chem. Soc. Rev. 2009, 38, 453–468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary