

Drug nanoparticles hitchhike on erythrocytes to target chemotherapy to the lungs for treating metastasis.
Abstract
Despite being the mainstay of cancer treatment, chemotherapy has shown limited efficacy for the treatment of lung metastasis due to ineffective targeting and poor tumor accumulation. Here, we report a highly effective erythrocyte leveraged chemotherapy (ELeCt) platform, consisting of biodegradable drug nanoparticles assembled onto the surface of erythrocytes, to enable chemotherapy for lung metastasis treatment. The ELeCt platform significantly extended the circulation time of the drug nanoparticles and delivered 10-fold higher drug content to the lung compared with the free nanoparticles. In both the early- and late-stage melanoma lung metastasis models, the ELeCt platform enabled substantial inhibition of tumor growth that resulted in significant improvement of survival. Further, the ELeCt platform can be used to deliver numerous approved chemotherapeutic drugs. Together, the findings suggest that the ELeCt platform offers a versatile strategy to enable chemotherapy for effective lung metastasis treatment.
INTRODUCTION
Cancer has been one of the leading causes of mortality over the last few decades (1). While early detection of tumor cells in specific tissues or the blood has improved the survival of patients with cancer, current standard-of-care interventions, including surgery, radiation therapy, or chemotherapy, have limited efficacy if cancer is not detected early (1–4). Early detection, however, is not often feasible, and in most patients, tumors have metastasized to secondary locations by the time of diagnosis (2, 4).
According to the National Cancer Institute, the most common site of metastasis for a variety of primary cancers is the lung, owing to its high vascular density. Lung metastasis is highly fatal if not treated, and currently, there is no specific treatment for it (5, 6). Systemic chemotherapy is one of the standard treatment options for lung metastasis (7, 8). However, its efficacy has been far from desirable due to ineffective targeting and poor accumulation in the lungs. Nanotechnology has played a pivotal role in enhancing the treatment of advanced metastatic cancers (9–11) and therefore can be applied in the case of lung metastasis as well. However, traditional nanoparticle (NP) delivery often fails to accumulate at the desired site of action due to the existence of biological barriers that impede the intravascularly injected NPs (12–17). Active targeting using tissue-specific ligands has often been explored as a strategy to improve tissue accumulation but has only resulted in modest improvement of therapeutic efficacy and decreased translational capability due to increased cost of production (18–26).
To achieve efficient drug delivery to enable chemotherapy for effective lung metastasis treatment, we used the unique physiology of the target site and developed a two-pronged strategy [erythrocyte leveraged chemotherapy (ELeCt)]—biodegradable drug NPs assembled on the surface of erythrocyte (Fig. 1A). Erythrocytes act as a primary drug delivery system, capable of responsively dislodging the particles in the lung endothelium and tumor nodules in response to the high shear stress experienced by erythrocytes in narrow lung capillaries (27, 28). The biodegradable NPs themselves are capable of encapsulating large amounts of chemotherapeutics and having a characteristic controlled-release mechanism (29, 30). They act as a secondary drug delivery system enabling sustained delivery of the cargo. In this study, superior accumulation and therapeutic efficacy of this lung physiology-assisted NP strategy were demonstrated using a model chemotherapeutic doxorubicin (DOX). This concept was successfully used to combat lung metastasis and improve survival in early- and late-stage melanoma lung metastasis models. The ability to incorporate a plethora of current clinical chemotherapy drugs and drug combinations in the biodegradable NPs and subsequently assemble onto the erythrocytes was demonstrated. The particles also readily assembled to human erythrocytes and dislodged in a shear-dependent manner. Together, ELeCt offers a versatile, potent, and translatable platform to combat lung metastasis.
Fig. 1. Schematic illustration of the ELeCt platform and characterization of drug (DOX)–loaded biodegradable PLGA NPs.
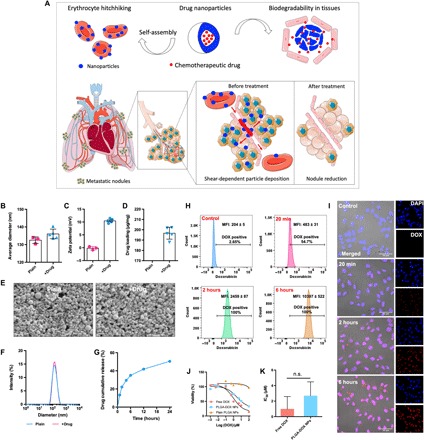
(A) Schematic illustration of the composition and mechanism of the biodegradable drug NP assembling on the erythrocyte platform (ELeCt) for lung metastasis treatment. (B to D) Average size (B), zeta potential (C), and drug loading contents (D) of plain and drug-loaded NPs. (E) SEM images showing the morphological features of the NPs. Scale bars, 200 nm. (F) Size distribution of plain and drug-loaded NPs. (G) Drug release kinetics from the biodegradable NPs in a complete medium (n = 4). (H and I) Flow cytometry histogram plots (H) and CLSM images (I) showing the interaction of drug-loaded NPs with B16F10-Luc melanoma cells. In (I), cell nuclei were stained using 4′,6-diamidino-2-phenylindole (DAPI). (J and K) Dose-response curve (J) and median inhibitory concentration (IC50) values (K) of B16F10-Luc cells after being treated with different formulations for 24 hours (n = 6). n.s., not significantly different (Student’s t test).
RESULTS
Drug-loaded biodegradable NPs could efficiently interact with target cancer cells
We used DOX as a model drug and prepared drug-loaded biodegradable polymeric [poly(lactic-co-glycolic acid) (PLGA)] NPs using the nanoprecipitation method. The drug-loaded PLGA NPs had a diameter of 136.0 ± 2.7 nm, which was slightly larger than the plain NPs (Fig. 1B). The encapsulation of DOX made the surface of the drug-loaded NPs slightly positive (10.45 ± 0.84 mV) (Fig. 1C), and this can be attributed to the presence of DOX on the NP surface. The drug-loaded PLGA NPs exhibited a high drug loading capacity (196.7 ± 5.8 mg/g) (Fig. 1D). We characterized the morphology of the NPs using scanning electron microscopy (SEM). SEM images shown in Fig. 1E revealed that both the plain and the drug-loaded PLGA NPs were spherical and relatively monodispersed. The dynamic light scattering data (Fig. 1F) confirmed the uniform size distribution of the prepared NPs. To test whether the drug could be released from the PLGA NPs, we assayed their release profile in a complete medium. A burst followed by a sustained-release profile was observed, and most of the drug was released within the first 6 hours (Fig. 1G). Efficient interaction of drug NPs with the target cancer cells is critical for successful drug delivery and efficacy. In this study, we used B16F10-Luc melanoma cells as a model to evaluate the interaction between the drug-loaded biodegradable PLGA NPs and the target cancer cells. As shown in Fig. 1H, the drug-loaded PLGA NPs appeared to be internalized by B16F10-Luc cells quickly and efficiently. Within 20 min of the incubation, a substantial portion of the cells had drug-loaded NPs in them. The confocal laser scanning microscopy (CLSM) images shown in Fig. 1I confirmed the efficient interactions between the NPs and the B16F10-Luc cells. Noticeably, the increase in DOX fluorescence within the cell nucleus suggested an effective intracellular delivery and sufficient release of the loaded drug. We further evaluated the in vitro antitumor efficacy of the drug-loaded PLGA NPs in a two-dimensional culture of the same cell line. As indicated by the dose-response curve (Fig. 1J) and IC50 (median inhibitory concentration) values (Fig. 1K), the drug-loaded PLGA NPs exhibited a slightly weaker cell killing efficacy compared with the free drug. However, the difference between them was not significant.
Drug-loaded biodegradable NPs efficiently assembled onto erythrocytes
We first evaluated whether the drug-loaded PLGA NPs could efficiently assemble onto the mouse erythrocytes. To do this, we incubated mouse erythrocytes with the NPs at a range of NP-to-erythrocyte ratios (50:1 to 800:1) and detected the binding of NPs using flow cytometry. As shown in Fig. 2 (A and B), the drug-loaded PLGA NPs indeed assembled onto the mouse erythrocytes efficiently. Particularly, 81.6% of erythrocytes were found to carry NPs when being incubated with NPs at a ratio of 200:1, and this number increased to >96% on further increasing the incubation ratio. The binding efficiency of the NPs to the erythrocytes was also quantified. Unexpectedly, a substantial portion (39.3 to 54.5%) of the incubated NPs assembled onto the mouse erythrocytes, depending on the feed ratio of the NPs to the erythrocytes (Fig. 2C). Because of this high binding efficiency and the high drug loading capacity of the NPs, the mouse erythrocytes were able to carry a high drug dose (as high as 294.1 μg per 3 × 108 erythrocytes) (Fig. 2D). In addition, the drug dose on the mouse erythrocytes could be easily tuned by manipulating the feed ratio of the NPs to the erythrocytes. Next, we visualized the assembly of drug-loaded PLGA NPs onto the mouse erythrocytes using CLSM and SEM. As shown in Fig. 2 (E and F), both the CLSM and SEM data confirmed the efficient assembly of the NPs onto the mouse erythrocytes. Meanwhile, the mouse erythrocytes maintained their biconcave shapes after being hitchhiked by the drug-loaded PLGA NPs (Fig. 2 (E and F)), indicating the assembly of the NPs had caused minimal damage to the carrier erythrocytes. To test the translational potential of the erythrocyte hitchhiking platform, we evaluated the assembly of the drug-loaded PLGA NPs onto the human erythrocytes. Both the CLSM and SEM images shown in Fig. 2 (G and H) suggested that the drug NPs could efficiently assemble onto the human erythrocytes as well. In addition, we also evaluated the assembly of drug-loaded PLGA NPs to human erythrocytes at different NP-to-erythrocyte feed ratios (200:1 to 1600:1). Similar to the murine counterparts, the drug-loaded PLGA NPs assembled onto the human erythrocytes with high efficiency (38.7 to 45.7%) at various NP-to-erythrocyte feed ratios (Fig. 2 (I and J)). Moreover, the drug dose on human erythrocytes could be tuned by changing the incubation ratio, and a very high drug dose (209.1 μg per 1.5 × 108 erythrocytes) could be hitchhiked to human erythrocytes when being incubated at a 1600:1 NP-to-erythrocyte ratio (Fig. 2K).
Fig. 2. Doxorubicin-loaded biodegradable PLGA NPs efficiently assemble onto mouse and human erythrocytes.
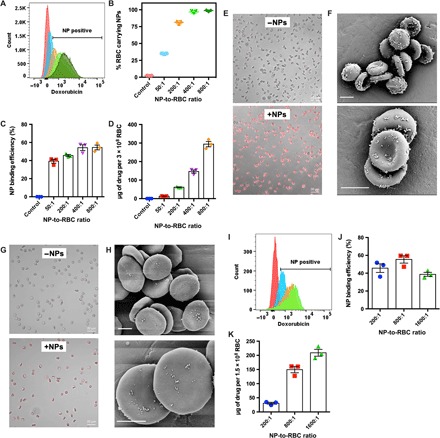
(A) Flow cytometry analysis of assembly of DOX-loaded PLGA NPs to mouse erythrocytes at different NP-to-erythrocyte ratios (left to right: 0:1, 50:1, 200:1, 400:1, and 800:1). (B) Percentage of mouse erythrocytes carrying at least one NP. (C) Nanoparticle binding efficiency and (D) drug dose on mouse erythrocytes at different NP–to–mouse erythrocyte ratios. (E) CLSM and (F) SEM images of mouse erythrocytes with drug-loaded NPs assembled on them. Scale bars in (F), 2 μm. (G) CLSM and (H) SEM images of human erythrocytes with drug-loaded NPs assembled on them. Scale bars in (H), 2 μm. (I) Flow cytometry assay of the assembly of drug-loaded NPs to human erythrocytes at different NP-to-erythrocyte ratios (left to right: 0:1, 200:1, 800:1, and 1600:1). (J) Nanoparticle binding efficiency and (K) drug dose on human erythrocytes at different NP-to-erythrocyte ratios.
ELeCt enabled enhanced and targeted delivery of the NP drugs to the lungs bearing metastasis
We first conducted a pharmacokinetic study to examine the blood circulation time of different drug formulations. As shown in Fig. 3A, by assembling drug NPs to erythrocytes, a higher drug concentration in the blood was achieved at all the tested time points, indicating an extended circulation time of the hitchhiked formulation. Mouse lung capillaries have an average diameter of 5 μm, narrowing down up to sizes as small as 1 μm, three to four times smaller than the mouse erythrocyte diameter (27). Upon intravenous administration, the drug-loaded NPs assembled onto erythrocytes are expected to detach from the carrier erythrocytes because of the high shear stress and be deposited in the narrow lung capillaries. To test this hypothesis, we first performed an in vitro shear study in which the erythrocytes carrying the drug-loaded NPs were sheared for 20 min at a low (~1 Pa) or high (6 Pa) shear stress. As shown in Fig. 3B, detachment of the drug NPs from the mouse erythrocytes was evidently shear dependent, providing a basis for specific delivery of drug NPs to the diseased lungs. Particularly, 76% of the hitchhiked drug NPs were sheared off at the lung-corresponding shear stress (6 Pa), using a rheometer. Moreover, this shear-dependent detachment of drug NPs was also observed with the human erythrocytes, bolstering the translational potential of this ELeCt platform. To test whether the drug NPs could be sheared off and deposited in the lungs that bear metastasis in vivo, we conducted a biodistribution study in mice bearing B16F10-Luc melanoma lung metastasis and quantified the amount of drug, in this case DOX. As shown in Fig. 3 (C and D), by assembly onto erythrocytes, the drug-loaded NPs delivered 16.6-fold higher drug content to the diseased lungs as compared with their free NP counterparts, 20 min after administration. Even at a longer time point (6 hours), erythrocyte hitchhiking deposited 8.7-fold higher drug content in the lungs as compared with their unhitchhiked counterparts. In addition, erythrocyte hitchhiking delivered a 6.9-fold higher drug content to the lungs with melanoma metastasis as compared with the free drug injection, 20 min after administration. Next, we investigated the distribution of the drug NPs sheared off from the carrier erythrocytes within the lungs bearing metastasis. As shown in Fig. 3E, consistent with the biodistribution data, more drug NPs were found in the lung section being treated with erythrocytes with NPs assembled on them compared with that being treated with the NPs alone. Evidently, a substantial portion, although not all, of the deposited NPs went deep into the tumor metastasis nodules, suggesting the biodegradable drug NP assembling on erythrocyte was able to precisely deliver the payload chemotherapeutic agents to their desired site of action.
Fig. 3. The ELeCt platform enables enhanced and targeted delivery of NP drugs to the lungs bearing metastasis.
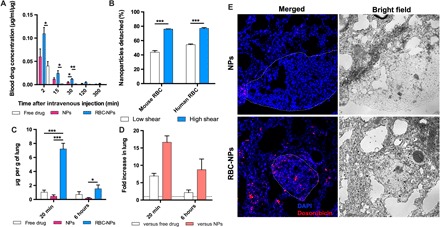
(A) Pharmacokinetics of intravenously administered DOX formulations. Extended blood circulation time of DOX was achieved by erythrocyte hitchhiking compared with using free drug or NPs alone (n = 3). Significantly different [one-way analysis of variance (ANOVA)]: *P < 0.05 and **P < 0.01. (B) Hitchhiked drug-loaded NPs could specifically detach from mouse and human erythrocytes under the lung-corresponding shear stress. Samples were sheared for 20 min (n = 3). Low shear indicates rotary shear (~1 Pa), while high shear was at 6 Pa. Significantly different (Student’s t test): ***P < 0.001. (C) Drug accumulation in the lungs of mice bearing B16F10-Luc lung metastasis at 20 min and 6 hours after intravenous administration of different DOX formulations (n = 3). Significantly different (one-way ANOVA): *P < 0.05 and ***P < 0.001. (D) Comparison of the drug concentration in the lungs of erythrocyte hitchhiking group to that of the free drug and NP-alone groups (n = 3). (E) Drug distribution in the diseased lungs 20 min after intravenous administration of DOX formulations. Dashed lines indicate the edge of metastasis nodules.
The ELeCt platform inhibited lung metastasis progression and improved survival
To evaluate the efficacy of the biodegradable drug NP assembly on the erythrocyte platform, we established a B16F10-Luc melanoma lung metastasis model and tested the antimetastatic efficacies in both the early and the late stages of the same model. We first tested the efficacy of the developed platform in controlling early-stage lung metastasis. As shown in Fig. 4A, the lung metastasis model was established by intravenously injecting B16F10-Luc cells via the tail vein. Four doses of treatments were given every other day with the first dose being administered 1 day after the tumor cell injection. The lung metastasis burden was measured by the bioluminescence intensity in the lung. As indicated by the bioluminescence images (Fig. 4B) and lung metastasis burden growth curve of individual mouse (Fig. 4C), a significantly better inhibition of the lung metastasis progression was achieved by the ELeCt as compared with using the free drug or NPs alone. Two mice remained completely free of lung metastasis after being treated with the drug NPs assembled on erythrocytes for up to day 31 after tumor inoculation. We also calculated the overall lung metastasis burden based on the bioluminescence intensity in the lungs. As shown in Fig. 4D, in the first 23 days after tumor inoculation, lung metastasis was almost completely inhibited in all mice being treated with the drug NPs assembled on erythrocytes. Particularly, as shown in Fig. 4E, on day 16, free drug and drug NPs alone resulted in a 17.2- and 1.8-fold lower average bioluminescence intensity compared with the control, respectively. In a sharp contrast, ELeCt achieved a 204.8-fold lower average bioluminescence intensity compared with the control. Similar finding was also observed on day 23. As shown in Fig. 4F, compared with using the drug NPs alone, the treatment using drug NPs assembled on erythrocytes led to a 302-fold lower average bioluminescence intensity. The Kaplan-Meier survival analysis (Fig. 4H) further confirmed the significantly improved survival benefit of the ELeCt approach over using the NPs alone. The use of the free drug or NPs alone only improved survival slightly, increasing the median survival time from 29 to 32 days. In a sharp comparison, by the treatment with drug NPs assembled on erythrocytes, the animal median survival time was extended from 29 to 61 days. Moreover, one of seven mice continued to survive for at least 70 days. We also monitored the body weight change of mice during the entire treatment period. No significant body weight loss was detected for any of the treatments, compared with a sharp decline in the body weight during the free drug treatment (Fig. 4G), indicating that only the free drug administration caused obvious toxicity at the current drug dose.
Fig. 4. The ELeCt platform inhibits lung metastasis progression and improves survival in the early-stage B16F10-Luc metastasis model.
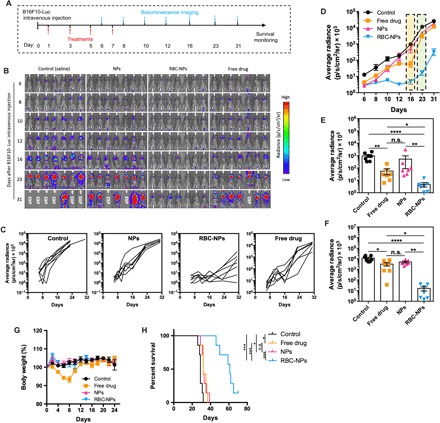
(A) Schematic chart of the treatment schedule. (B) Bioluminescence images of lung metastasis at different time points. EXP indicates “Expired.” (C) Lung metastasis progression curve as depicted from in vivo bioluminescence signal intensity. (D) Quantification of lung metastasis burden at different time points (n = 7). (E) Scatter plot comparing the lung metastasis burden in different treatment groups as depicted from bioluminescence signal intensity on day 16 (n = 7). Significantly different (Kruskal-Wallis test): *P < 0.05, **P < 0.01, and ****P < 0.0001. (F) Scatter plot comparison of the lung metastasis burden on day 23 (n = 7). Significantly different (Kruskal-Wallis test): *P < 0.05, **P < 0.01, and ****P < 0.0001. (G) Body weight change of mice during the treatment period (n = 7). (H) Survival of mice under different treatments as displayed by Kaplan-Meier curves (n = 7). Significantly different (log-rank test): *P < 0.05 and ***P < 0.001.
Next, we investigated the antimetastatic activity of the developed therapies in late-stage lung metastasis. As shown in Fig. 5A, after intravenous tumor cell injection, mice received four doses of therapies every other day with the first dose being administered a week after inoculation (day 7). According to the bioluminescence images (Fig. 5B) and lung metastasis growth curve (Fig. 5C) of individual mice, using the drug NPs alone did not lead to significant inhibition of lung metastasis progression. However, the drug NPs assembled on erythrocytes (ELeCt) were able to slow down the lung metastasis progression, although not as notably as in the early-stage metastasis model. The overall lung metastasis burden data shown in Fig. 5D confirmed the better efficacy of the hitchhiked drug NPs over using the free NPs alone. In particular, on day 16 after tumor inoculation, the hitchhiked drug NPs exhibited a 2.4-fold better efficacy in terms of inhibiting metastasis growth. On day 16, the lungs were excised, and the surface metastatic nodules on the lungs were counted. The surface nodules data shown in Fig. 5E were consistent with the bioluminescence metastasis burden data evaluated with bioluminescence. A 2.3-fold better efficacy in reducing surface nodules was achieved by assembling the drug NPs to the erythrocytes. The hematoxylin and eosin (H&E) analysis of the lungs of mice confirmed this result (fig. S1). In addition, the body weight change data shown in Fig. 2F and the H &E analysis data shown in fig. S2 suggested that no significant toxicity was associated with any of the treatments. We then conducted a separate study to evaluate the efficacy of the therapies in terms of extending the animal survival time. As shown in Fig. 5G, unlike in the early-stage metastasis model, the use of drug NPs alone did not provide any survival benefit. However, the treatment using drug NPs assembled on erythrocytes (ELeCt) significantly improved the animal survival, extending the median survival time from 28.5 to 37 days. In particular, one of eight mice that received the hitchhiked drug NPs continued to survive for at least 48 days.
Fig. 5. The ELeCt platform inhibits lung metastasis progression and extends survival in the late-stage B16F10-Luc metastasis model.
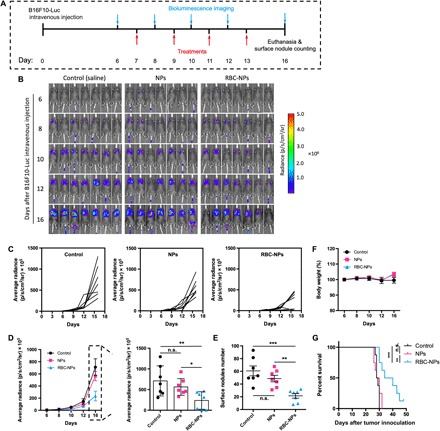
(A) Schematic illustration of the treatment schedule. (B) Bioluminescence images of lung metastasis progression at different time points. (C) Lung metastasis growth curve in mice treated with different DOX formulations. (D) Quantitative analysis of lung metastasis burden as depicted from bioluminescence signal intensity (n = 7). Significantly different (one-way ANOVA): *P < 0.05 and **P < 0.01. (E) Quantification of metastasis nodule numbers on excised lungs from mice in different treatment groups on day 16 (n = 7). Significantly different (one-way ANOVA): **P < 0.01 and ***P < 0.001. (F) Body weight change of mice during the treatment period (n = 7). (G) Kaplan-Meier survival curves of mice in different treatment groups. Significantly different (log-rank test): **P < 0.01 and ***P < 0.001.
Several chemotherapeutic agents could be loaded into biodegradable NPs and efficiently assembled onto erythrocytes
To test the feasibility of using the ELeCt platform for the delivery of other chemotherapeutic agents, we selected six other common chemotherapeutic agents or their combinations, including camptothecin, paclitaxel, docetaxel, 5-fluorouracil, gemcitabine, methotrexate, and the combination of 5-fluorouracil and methotrexate, and loaded them into the biodegradable PLGA NPs. Despite having diverse physicochemical properties (shown in fig. S3 and table S1), the different chemotherapeutic agent–loaded NPs were able to assemble onto erythrocytes (Fig. 6). These data supported that the biodegradable drug NP assembling onto erythrocytes approach (ELeCt) can potentially be a versatile platform to deliver selected chemotherapies to lung metastasis that originated from different primary tumors.
Fig. 6. Other chemotherapeutic agent–loaded biodegradable NPs can efficiently bind to erythrocytes.
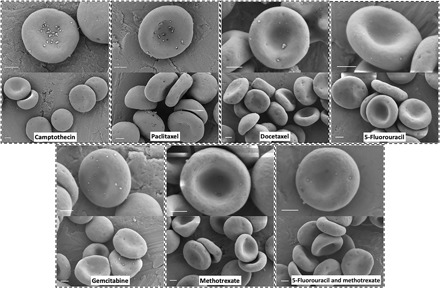
The tested chemotherapeutic agents include camptothecin, paclitaxel, docetaxel, 5-fluorouracil, gemcitabine, methotrexate, and the combination of 5-fluorouracil and methotrexate. Scale bars, 1 μm.
DISCUSSION
Because of its unique physiological features like high blood throughput and high density of narrow capillaries, lung is one of the major organs into which the evaded tumor cells from primary tumor sites can spread (31). Patients with advanced cancer (30 to 55%) have lung metastasis (32). Treating lung metastasis is more challenging than treating the primary tumors because it typically progresses more aggressively (33). Systemic chemotherapy is one standard treatment option for lung metastasis. However, its efficacy is usually far from desirable, attributed to its ineffective targeting and poor accumulation in the lungs. Conventional NP-mediated drug delivery also fails to achieve good localization with the desired site of action (34). Here, we report an erythrocyte hitchhiking platform, ELeCt, consisting of drug-loaded biodegradable NPs assembled on erythrocytes for promoting chemotherapy for effective lung metastasis treatment. Excellent studies have shown NPs hitchhiking on erythrocytes to accumulate in lungs, including recently in metastatic lungs (35); however, the ability of such a mechanism to yield survival benefits has not been known. To that end, we successfully demonstrate the ability of ELeCt to slow down the progression and improve the survival in early- and late-stage experimental melanoma metastasis models, resembling early detection and mid-to-late detection clinical scenarios, respectively.
Conventional nanomedicines use the attachment of active targeting ligands to enhance the targeted delivery of chemotherapeutic payloads (10, 11, 36–40). The ELeCt platform developed in this work exploits a completely new paradigm, taking advantage of the unique physiology of the target sites (high shear stress) and responsive dislodging of the chemotherapeutic payloads. Our in vitro drug-release data showed that the biodegradable NPs were able to have burst followed by relatively sustained drug release. Our pharmacokinetic and biodistribution data suggested that the ELeCt platform has two important features compared with the free drug and NPs alone—extended blood circulation time and improved accumulation to lung metastasis. Actually, both features are favorable for lung metastasis treatment. The extended circulation time is consistent with previous reports (27, 41). By hitchhiking to erythrocytes, NPs experience less immune recognition by the reticuloendothelial system organs, enabling them to stay in circulation for a longer time (27, 28, 41). The higher concentration of payload drug in the blood endowed by the ELeCt would allow more drug to interact with and kill the circulating tumor cells. Our in vitro shear study data evidently proved that the detachment of drug NPs from erythrocytes is shear dependent, and this is the basis for using the platform to precisely deliver payload chemotherapeutics to the target lung metastasis sites. It should be noticed that a substantial portion of the drug NPs were also detached at the low shear stress. This factor emphasized the need for investigating the surface modification of the drug NPs to modulate the binding strength of drug NPs to erythrocytes for future explorations with this technology. Our biodistribution data suggested that the biodegradable NP assembly on erythrocyte (ELeCt) platform was able to deliver a high concentration of payload chemotherapeutics to the lung metastatic sites in a short period of time. Impressively, the ELeCt platform delivered 16.6-fold more drug to the lungs bearing metastasis in 20 min compared with using the drug NPs alone. In comparison, the conventional targeted nanomedicine approach using targeting ligands can rarely achieve such high delivery enhancement (17, 42). Moreover, it usually shows a maximum tumor accumulation at a significantly longer time point (12 to 24 hours), depending on the properties of the nanomedicine (43). The quick and targeted delivery of drug NPs by the ELeCt platform would bring benefits for inhibiting tumor growth. For instance, typical nanomedicines, independent of their material origins, usually have an initial burst drug release and thus cause premature drug leakage (44), potentially attenuating the therapeutic efficacy and often leading to toxicity. The quick and targeted delivery achieved by the ELeCt platform has the potential to circumvent this issue. In addition, not unexpectedly, the lung section imaging suggested that the deposited NPs were distributed throughout the lung sections, both the inside and the outside of the lung metastatic nodules. The NPs deposited outside of the metastasis nodules have the potential to serve as a drug reservoir to release drug that can relocate to the metastatic nodules within close proximity.
Our in vivo efficacy data suggested that the enhanced and targeted delivery of chemotherapeutics by the ELeCt platform could bring benefits for inhibiting both the early-stage and the late-stage lung metastasis growth. In the early-stage lung metastasis model, the treatments using free drug or drug NPs alone exhibited some slowdown of the progression of lung metastasis. However, their antimetastatic efficacy was not potent enough to significantly extend the animal survival. In comparison, the ELeCt platform was able to provide a 100- to 300-fold better antimetastatic efficacy compared with using the free drug or drug NPs alone. Its improved antimetastatic efficacy led to a significantly extended animal survival, extending the median survival time of mice bearing lung metastasis by 32 days, compared with the control group. The data suggested that the ELeCt platform has the potential to enable chemotherapy for effective treatment of early-stage lung metastasis. In the late-stage metastasis model, the administration of drug NPs alone failed to significantly inhibit the lung metastasis growth and to improve the survival time. The ELeCt platform was able to significantly slow down the lung metastasis progression and modestly improved animal survival. Evidently, the antimetastatic efficacy of the therapies is closely related to the start time of the therapies. The efficacy of the developed therapies to treat in an even later-stage lung metastasis has not been shown yet. In addition, future studies may also need to be done to unveil the effect of drug dose and schedule of the therapies on their antimetastatic efficacy.
The exact mechanism of the drug-loaded biodegradable NP assembling on erythrocytes is not clear. Previous studies from our laboratory and others have attributed the assembly of NPs to erythrocytes to the noncovalent interactions such as electrostatic interactions, hydrophobic interactions, and H-bonds between the polymeric NPs and domains on the red blood cell (RBC) membrane (27, 28, 35). The assembly is most likely a result of balance between surface tension forces caused by the NP-induced membrane stretching and the noncovalent interactions between the cell membrane and NPs. The balance of the two factors drives stable assembly of the particles onto erythrocytes (27). However, details of this mechanism need future investigation. Our drug NP binding data suggested that the model drug–loaded NPs, in this case, DOX, could assemble onto the mouse erythrocytes at a very high binding efficiency. This feature is critical for making the ELeCt platform work. The number of erythrocytes that can be administered has an upper limit, and only having a high drug dose on individual erythrocytes can achieve the therapeutic concentration of chemotherapeutics. In addition, our data also suggested that the drug dose on erythrocytes could be tuned by changing the feed incubation ratios of drug NPs to erythrocytes, thus providing the possibility of changing drug dosage according to specific lung metastasis conditions. Other than DOX, we were able to load different commonly used chemotherapeutic agents or their combinations to the biodegradable NPs. Moreover, these drug-loaded NPs could assemble onto the mouse erythrocytes as well. This opens a new window to use the ELeCt platform to treat lung metastasis originating from different primary sites. Lung metastasis can have different primary tumor origins like breast cancer, bladder cancer, melanoma, and many others. The metastasis derived from different origins is preferably treated by specific chemotherapeutic agents (45, 46). The ELeCt platform has the potential to be a versatile platform to treat different lung metastasis by loading optimal chemotherapeutic agents according to their primary tumor origins. The impact of the chemotherapeutics’ properties on the performance of the ELeCt platform should be further investigated in future studies. Our data also suggested that the drug-loaded biodegradable NPs efficiently assembled onto human erythrocytes and were detached from them under lung-corresponding shear stress. In addition, the material used to prepare the biodegradable NPs (PLGA) is part of several FDA-approved products (47). Therefore, this platform technology has a translational potential. However, this needs to be explored further in the future.
In summary, the ELeCt platform, drug-loaded biodegradable NP assembling on erythrocyte, was developed, which enables lung physiology–assisted shear-responsive targeted delivery of chemotherapeutic agents to treat lung metastasis. The drug NPs assembled on erythrocytes could be precisely dislodged in the lungs bearing metastasis in response to the intrinsic mechanical high shear stress. Various commonly used chemotherapeutic agents could be loaded into the biodegradable NPs and further made to successfully assemble onto the erythrocytes. This platform successfully delivered one-order-of-magnitude-higher content of the model drug (DOX) to the diseased lungs as compared with using the NPs alone. This platform enabled chemotherapy to effectively inhibit lung metastasis growth and significantly improve the survival. All in all, the ELeCt platform can be a versatile strategy to treat lung metastasis originating from different primary tumors, with a strong translational potential.
MATERIALS AND METHODS
Nanoparticle preparation and characterization
PLGA NPs encapsulating DOX were prepared using a nanoprecipitation method. Briefly, 5 mg of DOX was dissolved in 500 μl of methanol and 5 μl of triethylamine. This was added to 1 ml of acetone containing 20 mg of PLGA. The mixture was then injected into 10 ml of 1% polyvinyl alcohol solution under constant stirring using a syringe pump at 1 ml/min. The particles were kept under constant stirring overnight before removing the organic solvents using rotary evaporation. The formed particles were centrifuged at 12,000g for 15 min, and the supernatant was analyzed to quantify drug loading. The particles were then resuspended in deionized water and assessed for their size, zeta potential, and polydispersity index using dynamic light scattering (Malvern Zen3600) and SEM (Zeiss FESEM Supra 55VP, Zeiss FESEM Ultra 55). The NPs were washed for a total of two washes with deionized water before their final resuspension in phosphate-buffered saline (PBS). Nanoparticles containing other chemotherapeutic drugs were prepared using the similar nanoprecipitation technique described above with minor modifications (details are shown in the Supplementary Materials).
Blood collection and processing
Murine whole blood was collected via cardiac puncture using a heparin precoated syringe and stored in BD Microtainer blood collection tubes prior to use. Whole blood was centrifuged at 1000g for 10 min at 4°C to remove the serum and the buffy coat layers from the erythrocyte compartment. The isolated erythrocytes were further washed three times with cold PBS and centrifuged at 650g for 15 min at 4°C before their final resuspension at a concentration of 10% hematocrit in PBS (erythrocyte stock solution). Human whole blood obtained from BioIVT (NY, USA) was processed and stored using the same procedure as murine blood. Freshly processed erythrocytes were used for every experiment in this study.
Assembly of drug NPs to erythrocytes and characterization
Equal volumes of erythrocyte stock solution and drug NP suspension were mixed in Axygen 1.5-ml Self-Standing Screw Cap Tubes and further thoroughly mixed by inversion and pipetting. The tubes were then allowed to rotate on a tube revolver (Thermo Fisher Scientific) for 40 min. The hitchhiked erythrocytes were then pelleted by centrifugation at 100g for 5 min at 4°C, unabsorbed particles were carefully removed, and the pellet was washed again with 1 ml of 1× PBS to remove loosely bound particles. The hitchhiked erythrocytes were finally resuspended at 10% (v/v) in 1× PBS and used for further characterization or in vivo studies.
Hitchhiking efficiency and the drug loading on erythrocytes were determined using fluorescence measurements. For quantification using fluorescence, 25 μl of erythrocytes was lysed using deionized water, and the drug content was quantified using DOX fluorescence [excitation (Ex)/emission (Em), 470/590 nm] on a plate reader (Tecan Safire 2, NC, USA). The percentage of erythrocytes carrying NPs for different NP-to-erythrocyte ratios was determined using flow cytometry (BD LSR Analyzer II, CA, USA) using DOX fluorescence (Em/Ex, 470/590 nm) and confirmed by confocal microscopy (Upright Zeiss LSM 710 NLO ready, Germany). Nanoparticle assembly to erythrocytes was confirmed using SEM (Zeiss FESEM Supra 55VP, Zeiss FESEM Ultra 55). Briefly, the hitchhiked erythrocytes were fixed using 2.5% glutaraldehyde solution and washed in an increasing ethanol gradient before being chemically dried using hexamethyldisilazane. Last, the samples were sputter coated (EMT 150T ES metal sputter coater, PA, USA) prior to imaging.
In vitro serum stability and shear studies
For serum stability studies, hitchhiked murine and human erythrocytes were incubated in 1 ml of fetal bovine serum (FBS) or human serum (from BioIVT) on a tube revolver at 12 rpm at 37°C. These conditions simulate low shear physiological environment. After incubation for 20 min, the cells were pelleted by centrifugation at 250g for 5 min and resuspended to 10% (v/v) in 1× PBS. Twenty-five microliters of erythrocytes was then lysed using deionized water, and the remaining drug content was quantified using DOX fluorescence (Ex/Em, 470/590 nm) on a plate reader (Tecan Safire 2).
For shear studies, hitchhiked murine and human erythrocytes were incubated in 10 ml of FBS or human serum. A rotatory shear (6 Pa) was applied to erythrocytes in serum using a cylindrical coquette viscometer (1 mm gap, AR-G2 rheometer, TA instruments, DE, USA) for 20 min. The samples were maintained at 37°C during the application of shear using a water jacket. These conditions simulate lung-corresponding high shear physiological environment. After 20 min, the cells were pelleted by centrifugation at 250g for 10 min and resuspended to 10% (v/v) in 1× PBS. Twenty-five microliters of erythrocytes was then lysed using deionized water, and the remaining drug content was quantified using DOX fluorescence (Ex/Em, 470/590 nm) on a plate reader (Tecan Safire 2).
Animals
Female C57BL/6 mice (7 to 9 weeks of age) were purchased from Charles River Laboratories (MA, USA). All experiments were performed according to the approved protocols by the Institutional Animal Care and Use Committee of the Faculty of Arts and Sciences, Harvard University, Cambridge.
In vivo pharmacokinetics and biodistribution studies
For the pharmacokinetics study, healthy female C57BL/6 mice were used. Free DOX, DOX-loaded NPs, and drug NPs assembled on erythrocytes (RBC-NPs) (n = 3 for all groups) were injected intravenously into the tail vein at a dose of 5.2 mg/kg. Blood samples were collected from the mice by submandibular bleed at 2 min, 15 min, 30 min, 2 hours, and 5 hours after the injection. The plasma was separated from the cellular component by centrifuging at 5000 rpm for 10 min. DOX was extracted from both the compartments (30 μl) using 150 μl of acetonitrile. The drug content was quantified using reversed-phase liquid chromatography–mass spectroscopy (LC-MS; Agilent 1290/6140 UHPLC, CA, USA) ran through an Agilent C-18 column (Poroshell 120, EC-C18, 3.0 mm by 100 mm, 2.7 μm) using a gradient mobile solvent.
For the biodistribution studies, 1 × 105 B16F10-Luc cells were injected intravenously into the tail vein of female C57BL/6 mice. Fourteen days after inoculation, mice were intravenously injected with free DOX, DOX-loaded NPs, and drug NPs assembled on erythrocytes (RBC-NPs) (n = 3 for all groups) into the tail vein at a dose of 5.2 mg/kg. Mice were euthanized at 20 min and 6 hours after the injection, and organs were harvested for further processing. Organs were rinsed using cold PBS three times to remove the residual blood. One milliliter of cold deionized water was added to each organ, and the organs were homogenized using a high shear homogenizer (IKA T 10 Basic ULTRA-TURRAX, NC, USA). DOX was extracted from the homogenates using acetonitrile (1:4 homogenate:acetonitrile), and the drug content was quantified using DOX fluorescence (Em/Ex, 470/590 nm) on a plate reader (Tecan Safire 2). The data are expressed as drug content (micrograms) normalized to the organ weight.
For NP distribution within the diseased lungs, 1 × 105 B16F10-Luc cells were injected intravenously into the tail vein of female C57BL/6 mice. Twenty-eight days after inoculation, mice were injected with DOX-loaded NPs and drug NPs assembled on erythrocytes (RBC-NPs). Twenty minutes after the injection, the mice were euthanized, and the intact lungs were collected. Lungs were washed twice with cold 1× PBS before being fixed in a 4% paraformaldehyde solution overnight. The fixed lungs were then frozen in Tissue-Tek OCT compound (Sakura Finetek) and sectioned using a cryostat (Leica CM1950, IL, USA). The sectioned tissue was mounted using Fluroshield to stain for DAPI (4′,6-diamidino-2-phenylindole) (Ex/Em, 340/488 nm) and was analyzed using a confocal microscope (Upright Zeiss LSM 710 NLO ready).
Efficacy studies on in vivo experimental lung metastasis model
An experimental lung metastasis model was established by intravenous injection of 1 × 105 B16F10-Luc cells into the tail vein of female C57BL/6 mice. Efficacy for the treatment groups was evaluated in early-stage and late-stage metastatic models. Mice were randomized on the basis of the bioluminescence intensity in the lungs 1 day before the first injection of therapies. A control (saline) group and three treatment groups (DOX-NPs, RBC-NPs, and free DOX) at a dose of 5.2 mg/kg were evaluated for their efficacy (n = 7 for all groups, unless otherwise specified).
For the early-stage metastatic model, treatments were given starting the day after the inoculation. Four injections were given over 6 days, i.e., days 1, 3, 5, and 7 after inoculation. On days 6, 8, 10, 12,18, 23, and 31 after inoculation, the mice were imaged using in vivo imaging (PerkinElmer IVIS Spectrum, MA, USA). Briefly, mice were injected intraperitoneally with 150 μl of XenoLight-d-luciferin (30 mg/ml) in saline. Fifteen minutes after the injection, mice were imaged using in vivo imaging. The average radiance (bioluminescence intensity) was evaluated using the software Living system. The animals were further monitored for their survival.
For the late-stage metastatic model, treatments were given 1 week after the inoculation. Four injections were given over 6 days, i.e., days 7, 9, 11, and 13 after the inoculation. The mice were imaged on days 6, 8, 10, 12, and 16 using in vivo imaging as described above. The average radiance was evaluated using the software Living system. On day 16, the mice were euthanized, and the lungs were excised and fixed using 10% formalin. The fixed lungs were used for counting of the surface nodules and H&E analysis. Survival in the late-stage model was evaluated by having the injection schedule as described above (n = 8 for the control and treatment groups).
Statistical analysis
All data are presented as means ± SEM. Comparison between two groups was conducted using unpaired two-tailed Student’s t test. Comparisons among multiple groups were conducted using one-way analysis of variance (ANOVA) or Kruskal-Wallis test. Kruskal-Wallis tests were performed for data that were determined to be nonparametric by the normality test. All statistical analyses were carried out using GraphPad Prism 8 software. For the analysis of Kaplan-Meier survival curves, log-rank (Mantel-Cox) analysis was used. P values represent different levels of significance: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. All the flow cytometry analyses were carried out using the FlowJo software.
Supplementary Material
Acknowledgments
Funding: This work was financially supported by Wyss Institute at Harvard University. We acknowledge funding from NIH (1R01HL143806-01). Author contributions: Z.Z., A.U., and S.M. conceived the project. Z.Z. and A.U. performed the experiments. Y.G. and J.K. helped with the LC-MS and histology analysis. Z.Z. and A.U. analyzed the data. Z.Z. prepared the graphs. Z.Z., A.U., and S.M. wrote the manuscript. All authors read and approved the manuscript. Competing interests: S.M., A.U., and Z.Z. are inventors on a patent application related to this work filed by Harvard University (no. 62/858,478, filed in June 2019). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax9250/DC1
Supplementary Materials and Methods
Fig. S1. Representative H&E staining images of lungs of mice.
Fig. S2. Representative H&E staining images of organs of mice treated with different drug formulations.
Fig. S3. Size distribution of different chemotherapeutic agent–loaded biodegradable PLGA NPs.
Table S1. Physicochemical properties of different chemotherapeutic agent–loaded biodegradable PLGA NPs.
REFERENCES AND NOTES
- 1.Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Emery J. D., Shaw K., Williams B., Mazza D., Fallon-Ferguson J., Varlow M., Trevena L. J., The role of primary care in early detection and follow-up of cancer. Nat. Rev. Clin. Oncol. 11, 38–48 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Eccles S. A., Welch D. R., Metastasis: Recent discoveries and novel treatment strategies. Lancet 369, 1742–1757 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.A. C. Society, (American Cancer Society, Atlanta, 2019).
- 5.Cardoso F., Senkus-Konefka E., Fallowfield L., Costa A., Castiglione M.; ESMO Guidelines Working Group , Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 21 ( Suppl. 5), v15–v19 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Reck M., Popat S., Reinmuth N., De Ruysscher D., Kerr K. M., Peters S.; ESMO Guidelines Working Group , Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 25 ( Suppl. 3), iii27–iii39 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Morgan G., Ward R., Barton M., The contribution of cytotoxic chemotherapy to 5-year survival in adult malignancies. Clin. Oncol. (R. Coll. Radiol.) 16, 549–560 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Chabner B. A., Roberts T. G. Jr., Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 5, 65–72 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Schroeder A., Heller D. A., Winslow M. M., Dahlman J. E., Pratt G. W., Langer R., Jacks T., Anderson D. G., Treating metastatic cancer with nanotechnology. Nat. Rev. Cancer 12, 39–50 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Peer D., Karp J. M., Hong S., Farokhzad O. C., Margalit R., Langer R., Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2, 751–760 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Shi J., Kantoff P. W., Wooster R., Farokhzad O. C., Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Z., Ukidve A., Krishnan V., Mitragotri S., Effect of physicochemical and surface properties on in vivo fate of drug nanocarriers. Adv. Drug Deliv. Rev. 143, 3–21 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Blanco E., Shen H., Ferrari M., Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 33, 941–951 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitragotri S., Burke P. A., Langer R., Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 13, 655–672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barua S., Mitragotri S., Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today 9, 223–243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nichols J. W., Bae Y. H., Odyssey of a cancer nanoparticle: From injection site to site of action. Nano Today 7, 606–618 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilhelm S., Tavares A. J., Dai Q., Ohta S., Audet J., Dvorak H. F., Chan W. C. W., Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 1, 16014 (2016). [Google Scholar]
- 18.Zhong Y., Meng F., Deng C., Zhong Z., Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules 15, 1955–1969 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Peng X. H., Wang Y., Huang D., Wang Y., Shin H. J., Chen Z., Spewak M. B., Mao H., Wang X., Wang Y., Chen Z., Nie S., Shin D. M., Targeted delivery of cisplatin to lung cancer using ScFvEGFR-heparin-cisplatin nanoparticles. ACS Nano 5, 9480–9493 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pastorino F., Brignole C., Di Paolo D., Nico B., Pezzolo A., Marimpietri D., Pagnan G., Piccardi F., Cilli M., Longhi R., Ribatti D., Corti A., Allen T. M., Ponzoni M., Targeting liposomal chemotherapy via both tumor cell–specific and tumor vasculature–specific ligands potentiates therapeutic efficacy. Cancer Res. 66, 10073–10082 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Byrne J. D., Betancourt T., Brannon-Peppas L., Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 60, 1615–1626 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Kanasty R., Dorkin J. R., Vegas A., Anderson D., Delivery materials for siRNA therapeutics. Nat. Mater. 12, 967–977 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Lammers T., Hennink W. E., Storm G., Tumour-targeted nanomedicines: Principles and practice. Br. J. Cancer 99, 392–397 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertrand N., Wu J., Xu X., Kamaly N., Farokhzad O. C., Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 66, 2–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou Y., Liu Y., Yang Z., Zhang D., Lu Y., Zheng M., Xue X., Geng J., Chung R., Shi B., Effective and targeted human orthotopic glioblastoma xenograft therapy via a multifunctional biomimetic nanomedicine. Adv. Mater. 30, 1803717 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Wang S., Yu G., Wang Z., Jacobson O., Tian R., Lin L.-S., Zhang F., Wang J., Chen X., Hierarchical tumor microenvironment-responsive nanomedicine for programmed delivery of chemotherapeutics. Adv. Mater. 30, 1803926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anselmo A. C., Gupta V., Zern B. J., Pan D., Zakrewsky M., Muzykantov V., Mitragotri S., Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS Nano 7, 11129–11137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brenner J. S., Pan D. C., Myerson J. W., Marcos-Contreras O. A., Villa C. H., Patel P., Hekierski H., Chatterjee S., Tao J.-Q., Parhiz H., Bhamidipati K., Uhler T. G., Hood E. D., Kiseleva R. Y., Shuvaev V. S., Shuvaeva T., Khoshnejad M., Johnston I., Gregory J. V., Lahann J., Wang T., Cantu E., Armstead W. M., Mitragotri S., Muzykantov V., Red blood cell-hitchhiking boosts delivery of nanocarriers to chosen organs by orders of magnitude. Nat. Commun. 9, 2684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Z., Lou S., Hu Y., Zhu J., Zhang C., A nano-in-nano polymer–dendrimer nanoparticle-based nanosystem for controlled multidrug delivery. Mol. Pharm. 14, 2697–2710 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Rezvantalab S., Drude N. I., Moraveji M. K., Güvener N., Koons E. K., Shi Y., Lammers T., Kiessling F., PLGA-Based nanoparticles in cancer treatment. Front. Pharmacol. 9, 1260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Zijl F., Krupitza G., Mikulits W., Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 728, 23–34 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.S. DS, in Thoracic Surgery, G. J, Ed. (Medscape, 2019), vol. 2019. [Google Scholar]
- 33.Altorki N. K., Markowitz G. J., Gao D., Port J. L., Saxena A., Stiles B., McGraw T., Mittal V., The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 19, 9–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramalingam S., Belani C., Systemic chemotherapy for advanced non-small cell lung cancer: Recent advances and future directions. Oncologist 13, 5–13 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Zelepukin I. V., Yaremenko A. V., Shipunova V. O., Babenyshev A. V., Balalaeva I. V., Nikitin P. I., Deyev S. M., Nikitin M. P., Nanoparticle-based drug delivery via RBC-hitchhiking for the inhibition of lung metastases growth. Nanoscale 11, 1636–1646 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Chauhan V. P., Jain R. K., Strategies for advancing cancer nanomedicine. Nat. Mater. 12, 958–962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costa S. A., Mozhdehi D., Dzuricky M. J., Isaacs F. J., Brustad E. M., Chilkoti A., Active targeting of cancer cells by nanobody decorated polypeptide micelle with bio-orthogonally conjugated drug. Nano Lett. 19, 247–254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato T., Lee D., Huang H., Cruz W., Ujiie H., Fujino K., Wada H., Patel P., Hu H.-p., Hirohashi K., Nakajima T., Sato M., Kaji M., Kaga K., Matsui Y., Chen J., Zheng G., Yasufuku K., Personalized siRNA-nanoparticle systemic therapy using metastatic lymph node specimens obtained with EBUS-TBNA in lung cancer. Mol. Cancer Res. 16, 47–57 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Guo P., Yang J., Liu D., Huang L., Fell G., Huang J., Moses M. A., Auguste D. T., Dual complementary liposomes inhibit triple-negative breast tumor progression and metastasis. Sci. Adv. 5, eaav5010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kosmides A. K., Sidhom J.-W., Fraser A., Bessell C. A., Schneck J. P., Dual Targeting Nanoparticle Stimulates the Immune System To Inhibit Tumor Growth. ACS Nano 11, 5417–5429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chambers E., Mitragotri S., Long circulating nanoparticles via adhesion on red blood cells: Mechanism and extended circulation. Exp. Biol. Med. 232, 958–966 (2007). [PubMed] [Google Scholar]
- 42.Dai Q., Wilhelm S., Ding D., Syed A. M., Sindhwani S., Zhang Y., Chen Y. Y., MacMillan P., Chan W. C. W., Quantifying the ligand-coated nanoparticle delivery to cancer cells in solid tumors. ACS Nano 12, 8423–8435 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Rosenblum D., Joshi N., Tao W., Karp J. M., Peer D., Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 9, 1410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamaly N., Yameen B., Wu J., Farokhzad O. C., Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 116, 2602–2663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steeg P. S., Targeting metastasis. Nat. Rev. Cancer 16, 201–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao Y., Xie J., Chen H., Gu S., Zhao R., Shao J., Jia L., Nanotechnology-based intelligent drug design for cancer metastasis treatment. Biotechnol. Adv. 32, 761–777 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Makadia H. K., Siegel S. J., Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 3, 1377–1397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax9250/DC1
Supplementary Materials and Methods
Fig. S1. Representative H&E staining images of lungs of mice.
Fig. S2. Representative H&E staining images of organs of mice treated with different drug formulations.
Fig. S3. Size distribution of different chemotherapeutic agent–loaded biodegradable PLGA NPs.
Table S1. Physicochemical properties of different chemotherapeutic agent–loaded biodegradable PLGA NPs.