

Abstract
Park2 is an E3 ubiquitin ligase known for its role in mitochondrial quality control via the mitophagy pathway. Park2 KO mice are protected from diet‐induced obesity and hepatic insulin sensitivity is improved in high‐fat diet (HFD)‐fed Park2 KO mice even under body weight‐matched conditions. In order to better understand the cellular mechanism by which Park2 KO mice are protected from diet‐induced hepatic insulin resistance, we determined changes in multiple pathways commonly associated with the pathogenesis of insulin resistance, namely levels of bioactive lipid species, activation of the endoplasmic reticulum (ER) stress response and changes in cytokine levels and signaling. We report for the first time that whole‐body insulin sensitivity is unchanged in regular chow (RC)‐fed Park2 KO mice, and that liver diacylglycerol levels are reduced and very‐long‐chain ceramides are increased in Park2 KO mice fed HFD for 1 week. Hepatic transcriptional markers of the ER stress response were reduced and plasma tumor necrosis factor‐α (TNFα), interleukin‐6 and −10 (IL6, IL10) were significantly increased in HFD‐fed Park2 KO mice; however, there were no detectable differences in hepatic inflammatory signaling pathways between groups. Interestingly, hepatic adenylate charge was reduced in HFD‐fed Park2 KO liver and was associated increased activation of AMPK. These data suggest that negative energy balance that contributed to protection from obesity during chronic HFD manifested at the level of the hepatocyte during short‐term HFD feeding and contributed to the improved hepatic insulin sensitivity.
Keywords: AMPK, diacylglycerol, IL6, insulin resistance, liver, mitophagy, Park2, Parkin, PKCε
Park2 knockout mice‐fed HFD for 1‐week were previously demonstrated to have improved hepatic insulin sensitivity. Here we demonstrate that this phenotype is associated with reduced hepatic triglyceride and diacylglycerol levels, increased very‐long chain ceramides and reductions in markers of endoplasmic reticulum stress. Hepatic AMPK activation was also increased and suggests that the underlying mechanism for improved hepatic insulin sensitivity is multi‐factorial and due to negative energy balance in Park2 knockout mice.

1. INTRODUCTION
PARK2, or Parkin, was first identified as the mutated gene responsible for autosomal recessive juvenile Parkinsonism in a small cohort of Japanese patients (Kitada et al., 1998). Parkin is now recognized as the most frequently mutated gene in familial Parkinsonism with recessive inheritance, accounting for approximately half of all cases (Bonifati, 2012). Parkin is an E3 ubiquitin ligase and member of the RING‐in‐between‐RING or RING‐HECT hybrid class of E3s (Dove & Klevit 2017) and thus catalyzes the addition of ubiquitin to specific substrate proteins. Parkin is best known for its role in mitochondrial quality control via the mitophagy pathway where it contributes to the degradation of damaged mitochondria (Youle & Narendra, 2011), but Parkin also appears to regulate a number of cellular processes unrelated to mitophagy, such as cell cycle progression, DNA binding, and transcriptional repression (da Costa et al., 2009; Gong et al., 2014). Parkin is expressed in a wide variety of cell types, including adipocytes (Lu et al., 2018), cardiomyocytes (Piquereau et al., 2013), and hepatocytes (Kim et al., 2011), suggesting functional significance outside the central nervous system. In support of this notion, a common feature of most in vivo models of Parkin deficiency is their increased susceptibility to stress in peripheral cell types; myocyte mitochondrial morphology is impaired and wing muscles are atrophied in Parkin homolog null Drosophilia (Greene et al., 2003); lipopolysaccharide‐treated Park2 knockout (KO) mice fail to recover cardiomyocyte mitochondrial respiratory capacity and cardiac contractility (Piquereau et al., 2013); and both acute and chronic exposure to alcohol induces more severe hepatocyte lipid accumulation and inflammation in Park2 KO mice (Williams, Ni, Ding, & Ding, 2015).
One of the more striking phenotypes described in the Park2 KO mouse model was their protection from diet‐induced obesity and hepatosteatosis; after six and a half weeks of high‐fat diet (HFD) feeding, Park2 KO mice weighed 30% less than controls, which was largely attributed to differences in fat mass, and liver fat was also 50% less (Kim et al., 2011). Not surprisingly, HFD‐fed Park2 KO mice displayed improved glucose and insulin tolerance when compared with obese HFD‐fed wild‐type (WT) mice, but it was unclear whether changes in liver fat and glucose homeostasis after HFD feeding were due to loss of Park2 or secondary to the protection from obesity (Kim et al., 2011). To address this question, we fed Park2 KO mice a short‐term, one‐week HFD in order to induce hepatic insulin resistance without major changes in body weight (Costa et al., 2016). Under these conditions, body fat was modestly reduced by 1.2 g or 5% in Park2 KO mice, but there was no difference in body weight. Hepatic insulin sensitivity, as assessed by hyperinsulinemic euglycemic clamp, was markedly improved in Park2 KO mice; whereas hyperinsulinemia produced only a 40% reduction in hepatic glucose production in HFD‐fed WT mice, hepatic glucose production was almost completely suppressed (~97%) by insulin in HFD‐fed Park2 KO mice (Costa et al., 2016). These data demonstrated that Park2 KO mice were protected against diet‐induced hepatic insulin resistance independent of changes in body weight, but the underlying mechanism was not addressed.
We undertook the studies described here to determine the underlying mechanism for the improved hepatic insulin sensitivity in the HFD‐fed Park2 KO mice, as well as to address outstanding questions regarding insulin sensitivity in chow‐fed animals. We evaluated key pathways commonly implicated in the pathogenesis of hepatic insulin resistance, including changes in hepatic lipid metabolites, activation of endoplasmic reticulum (ER) stress response, and alterations in inflammatory cytokine levels and signaling pathways downstream of these mechanisms. Overall, we found that hepatic triglyceride and diacylglycerol levels were reduced in Park2 KO compared with WT mice after short‐term HFD feeding, as well as markers of ER stress. Also, plasma tumor necrosis factor‐α (TNFα), interleukin‐6 (IL6) and interleukin‐10 (IL10) levels were increased in Park2 KO mice. However, the stress‐activated kinases associated with these pathways were differentially affected in that diacylglycerol‐activated protein kinase C‐ε (PKCε) was reduced in Park2 KO mouse liver, while ER stress‐associated and inflammatory‐mediated JNK and IKKβ activation were unchanged. Finally, the reduced lipid levels in Park2 KO mouse livers were associated with increased activation of the cellular energy sensor AMP kinase (AMPK), suggesting that the negative energy balance that contributed to protection from obesity during chronic HFD feeding in the Park2 KO mice manifested at the level of the hepatocyte during short‐term HFD feeding and contributed to the improved hepatic insulin sensitivity.
2. MATERIALS AND METHODS
2.1. Animal use and care
Mice were housed and studied at Yale University School of Medicine and the University of Pittsburgh according to guidelines established by the Institutional Animal Care and Use Committees at each institution. Mice were housed at 22 ± 2°C on a 12 hr light, 12 hr dark cycle with free access to food and water. Diets provided were Harlan Teklad 2018S (control rodent chow) and Research Diets D12492 (HFD, 60% kcal fat). WT and Park2 KO mice on the C57BL6/J background were sourced from the Jackson Laboratory, as previously described (Costa et al., 2016). Mice were between 12 and 14 weeks old at the time of study. Two cohorts of mice were used for the studies; cohort one underwent hyperinsulinemic euglycemic clamps after a 6‐hr morning fast for data shown in Figure 1 (more detail below); and cohort two was euthanized after a 6‐hr morning fast for dissection and collection of plasma and tissue for data reported in Figures 2, 3, 4, 5 and Table 1.
Figure 1.
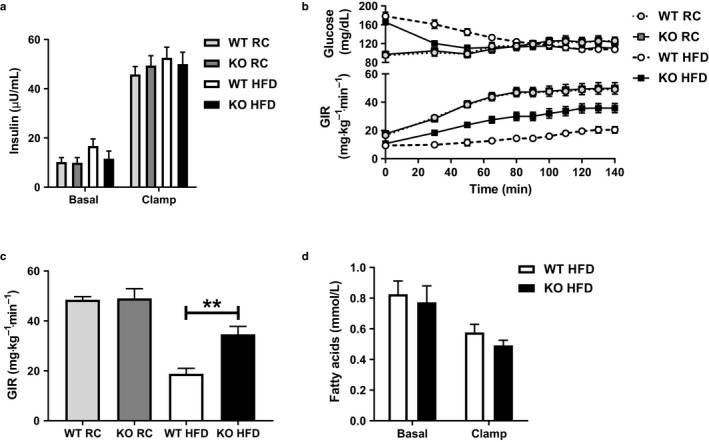
Insulin sensitivity is unchanged in chow‐fed Park2 KO mice, but improved following short‐term HFD feeding. (a) Basal (fasted) and clamped plasma insulin levels in regular chow (RC)‐ and HFD‐fed mice. (b) Plasma glucose levels and the glucose infusion rate (GIR) required to maintain euglycemia during the clamp. (c) Steady‐state GIR during the last 40 min of the clamp. (d) Basal and clamped plasma fatty acid levels in HFD‐fed mice. Data are the mean ± SEM and were analyzed by Student's t‐test. **p < .01; n = 6–8 mice/group. Data for HFD‐fed mice in panel a, b and c were previously reported (Costa et al., 2016) and are shown here for comparison to RC‐fed mice
Figure 2.
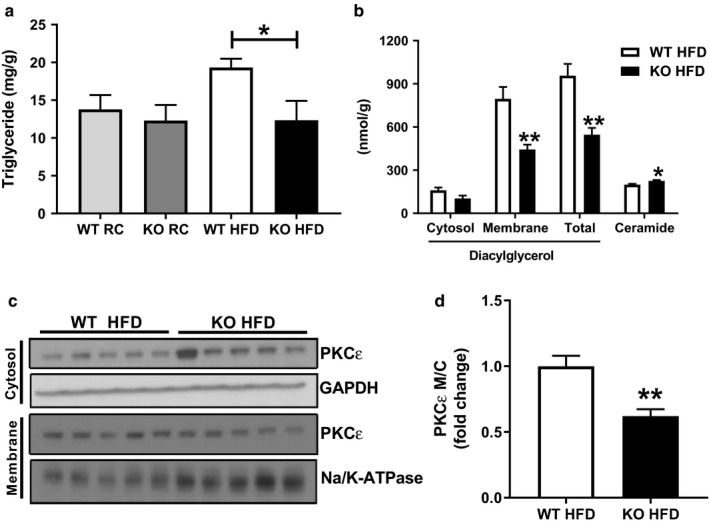
Improved hepatic insulin sensitivity in HFD‐fed Park2 KO is associated with reduced hepatosteatosis, diacylglycerol levels, and PKCε activation. (a) Liver triglyceride levels in RC‐ and HFD‐fed mice. (b) Liver diacylglycerol (cytosol, membrane‐associated and total) and ceramide levels in HFD‐fed mice. (c) Western blots for PKCε levels in cytosol and membrane fractions from liver of HFD‐fed mice, as well as GAPDH and Na/K‐ATPase loading controls. (d) PKCε activation in liver from HFD‐fed mice expressed as the quantification of the membrane to cytosol ratio of PKCε protein levels shown in C. Data are the mean ± SEM and were analyzed by Student's t‐test. *p < .05, **p < .01 ***p < .001; n = 5–6 mice/group
Figure 3.
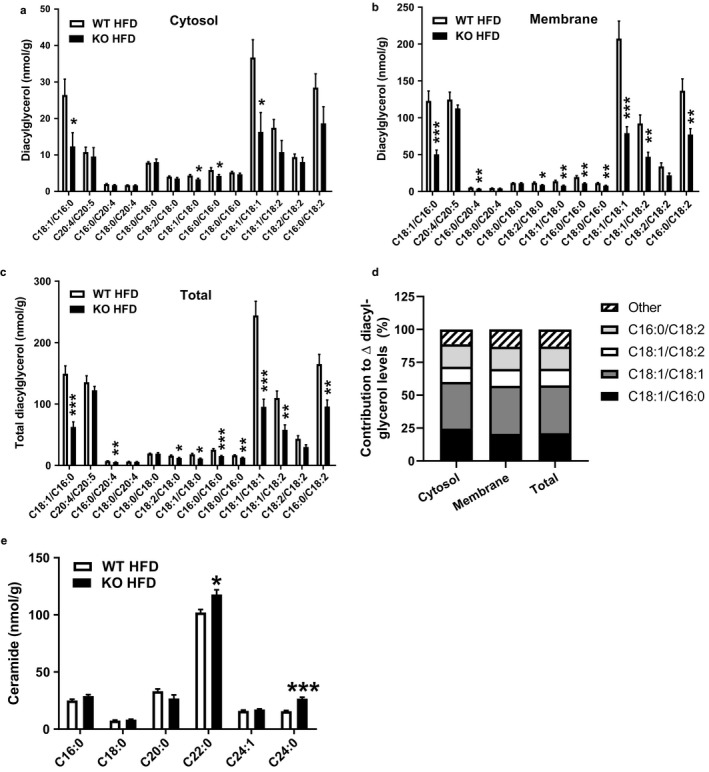
Diacylglycerol acyl chain composition in liver fractions from HFD‐fed Park2 KO and WT mice. (a) Cytosolic, (b) membrane, and (c) total diacylglycerol acyl chain species in liver of HFD‐fed mice. (d) Percent contribution of individual diacylglycerol species to changes in total levels in cytosol, membrane and total fractions. (e) Ceramide acyl chain composition. Data are the mean ± SEM and were analyzed by Student's t‐test. *p < .05, **p < .01, ***p < .001, n = 5–6 mice/group
Figure 4.
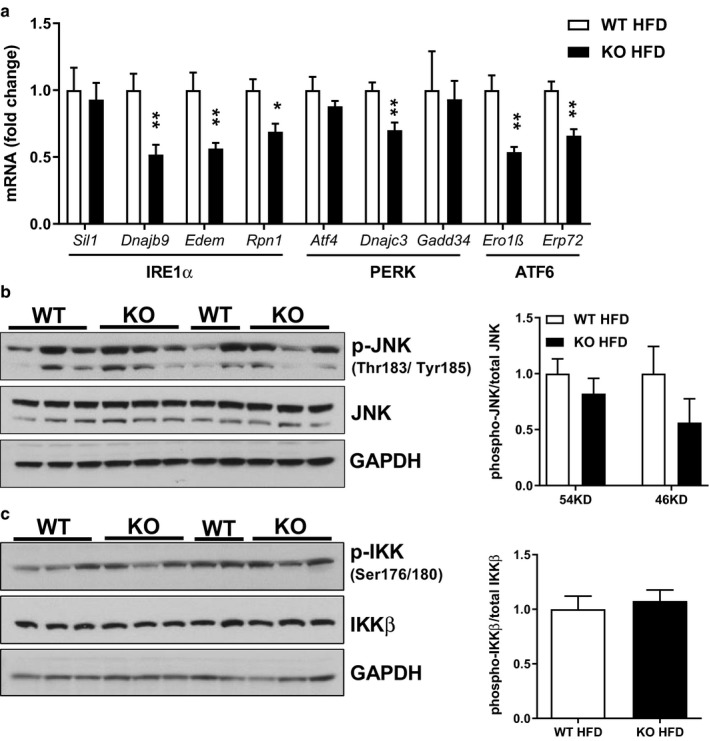
Improved hepatic insulin sensitivity in HFD‐fed Park2 KO is associated with reduced expression of transcriptional markers of ER stress, but not changes in JNK or IKKβ activation. (a) Liver mRNA levels of markers of ER stress downstream of the three branches of the UPR (IRE1α, PERK and ATF6). (b) Liver protein expression and quantification of blots for phospho‐Thr183/Tyr185 JNK relative to total JNK. (c) Liver protein expression and quantification of blots for phospho‐Ser176/180 IKKβ to total IKKβ. Data are the mean ± SEM and were analyzed by Student's t‐test. *p < .05, **p < .01; n = 5–6 mice/group
Figure 5.
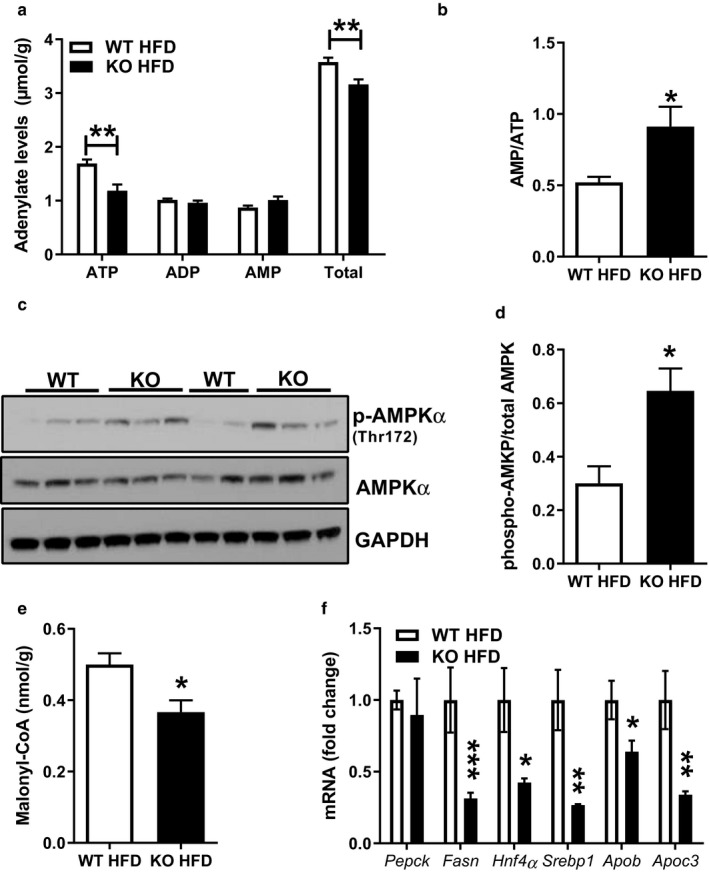
Liver AMPK activation is increased in HFD‐fed Park2 KO mice. (a) Liver adenylate levels from HFD‐fed mice. (b) Liver AMP to ATP ratio calculated using data in (a). (c) Western blots from liver of HFD‐fed mice for phospho‐AMPKα (Thr172), total AMPKα and GAPDH loading control. (d) Quantification of data in (c). (e) Liver malonyl‐CoA levels. (f) Liver mRNA levels expressed as fold change relative to WT relative expression levels using Actb for normalization. Data are the mean ± SEM and were analyzed by Student's t‐test. *p < .05, **p < .01 ***p < .001; n = 5–6 mice/group
Table 1.
Plasma cytokine and obesity/diabetes‐related hormone levels in RC‐ and HFD‐fed Park2 KO and WT mice
| Analyte (pg/ml) | WT RC (Average ± SEM) | KO RC (Average ± SEM) | p‐value WT:KO LFD | WT HFD (Average ± SEM) | KO HFD (Average ± SEM) | p‐value WT:KO HFD |
|---|---|---|---|---|---|---|
| IFNγ | 0.370 ± 0.117 | 0.262 ± 0.059 | N.S. | 0.276 ± 0.068 | 0.178 ± 0.025 | N.S. |
| IL2 | 0.462 ± 0.094 | 0.385 ± 0.054 | N.S. | 0.514 ± 0.119 | 0.403 ± 0.046 | N.S. |
| IL5 | 0.944 ± 0.184 | 1.190 ± 0.282 | N.S. | 1.826 ± 0.807 | 0.805 ± 0.055 | N.S. |
| TNFα | 5.48 ± 1.03 | 5.29 ± 0.32 | N.S. | 4.97 ± 0.43 | 8.60 ± 0.68 | p < .01 |
| IL10 | 15.13 ± 6.77 | 9.62 ± 0.99 | N.S. | 7.51 ± 0.54 | 14.75 ± 0.86 | p < .001 |
| IL6 | 2.58 ± 0.37 | 5.87 ± 1.19 | p < .05 | 3.80 ± 0.89 | 40.32 ± 13.18 | p < .05 |
| CXCL1 | 51.96 ± 6.95 | 56.83 ± 6.45 | N.S. | 59.42 ± 8.91 | 62.08 ± 8.42 | N.S. |
| GLP‐1 | 2.68 ± 1.91 | 2.06 ± 0.50 | N.S. | 20.70 ± 17.60 | 3.30 ± 1.32 | N.S. |
| Glucagon | 16.26 ± 9.81 | 12.90 ± 5.05 | N.S. | 39.60 ± 25.92 | 13.98 ± 4.03 | N.S. |
| GIP | 105.6 ± 6.0 | 115.9 ± 10.2 | N.S. | 224.1 ± 56.7 | 147.4 ± 10.9 | N.S. |
| PAI‐1 | 826.7 ± 61.8 | 994.5 ± 129.7 | N.S. | 1,088.0 ± 151.8 | 1,349.3 ± 164.0 | N.S. |
| Leptin | 1503.6 ± 217.9 | 526.2 ± 253.5 | p < .05 | 9,527.6 ± 2044.1 | 533.6 ± 159.4 | p < .001 |
| Ghrelin | 8,649 ± 1777 | 14,358 ± 2091 | p = .071 | 9,515 ± 4,438 | 12,500 ± 1988 | N.S. |
| Resistin | 151,912 ± 28,820 | 141,808 ± 9,259 | N.S. | 231,133 ± 33,850 | 161,081 ± 16,254 | p = .080 |
Upper portion: Plasma pro‐ and anti‐inflammatory cytokine levels in RC‐ and HFD‐fed mice. Lower portion: Plasma obesity/diabetes‐related hormone levels in RC‐ and HFD‐fed mice. n = 5–6 mice/group. Data are the mean ± SEM and were analyzed by Student's t‐test for RC and HFD groupings and p‐values are as shown within the table.
Bold indicates statistical significant values.
2.2. Biochemical analyses
Plasma glucose was measured by the glucose oxidase method using the Analox GM9 Glucose Analyzer. Plasma insulin was determined by radioimmunoassay kit (Linco Research) and plasma fatty acids were measured using a spectrophotometric NEFA detection reagent (Wako Diagnostics). Liver triglycerides were determined using the colormetric Infinity Triglyceride Reagent (Fisher Scientific) following tissue homogenization and lipid extraction in chloroform:methanol (2:1), followed by acidification with H2SO4 and phase separation by centrifugation. Ceramide and diacylglycerol extraction and analysis were performed as described previously (Cantley et al., 2013). Membrane and cytosolic PKCε were measured as previously described (Petersen et al., 2016). Plasma cytokines were measured by electrochemiluminescence using a Diagnostics V‐Plex Proinflammatory Panel I Mouse Kit (Meso Scale Discovery). The plasma obesity/diabetes panel was measured using a Bio‐Plex Pro Mouse Diabetes 8‐Plex Assay (BioRad). All spectrophotometric measurements were performed on a Thermo Scientific Multiskan GO. Liver malonyl‐CoA levels were measured by liquid chromatography mass spectrometry (LC/MS) using an Ultimate 3000 RSLC system connected to a Thermo Fisher Scientific Q Exactive MS (Thermo Fisher Scientific). Briefly, samples were extracted in 80% methanol and purified by silica gel cartridge (Supelco) followed by separation by ion‐pairing reversed phase chromatography by LC‐MS. The MS was equipped with an ESI source and was operated in negative mode using a scan range of 750 m/z to 1,250 m/z. Analyte identification was confirmed by high‐resolution accurate mass. Liver adenylate levels were measured by capillary electrophoresis (CE)‐MS by Human Metabolome Technologies.
2.3. Protein isolation and western blotting
Flash‐frozen liver was pulverized and homogenized in radioimmunoprecipitation assay buffer (150 mM NaCl, 1% (v/v) Triton X‐100, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS and 50 mM Tris‐HCl, pH 8.0) supplemented with HALT protease inhibitor cocktail (Thermo Fisher). Western blotting electrophoresis was carried out using the ThermoFisher Bolt System, and protein was transferred to a nitrocellulose membrane using BioRad Trans‐blot SD Semi‐dry Transfer Cell. Membranes were probed with the following antibodies and developed on a Licor Odyssey CLx with Licor IRDye goat secondary antibodies. Blots were quantified using Licor Image Studio Software (Ver. 5.2). Antibodies: α1 Na‐K‐ATPase (Abcam ab7671), PKCε (BD Laboratories 610085), GAPDH (Santa Cruz sc‐25778) and from Cell Signaling IKKα/β (pSer176/180; 2697), IKKβ (2370), AMPKα (5831), AMPKα (pThr172; 2535), SAPK/JNK (9252), SAPK/JNK (pThr183/pTyr185; 4668).
2.4. RNA isolation, cDNA synthesis and quantitative PCR
RNA was isolated from liver using a QiaShredder plus RNeasy Kit (Qiagen) and then used for cDNA synthesis using a QuantiTect Reverse Transcriptase Kit (Qiagen), both according to manufacturer's instructions. Quantitative PCR was performed using the Applied Biosystems QuantStudio3 RT‐PCR System and PowerUp SYBR Green Master Mix (Thermo Fisher). Primer sequences were designed with IDT Real Time PCR tool and were as follows: Ero1β (F CG TCCTTTAAATCCTTTGGCG; R ACACAAACCTTCTAGCCACG) Erp72 (F ATCATTGGGCTCTTTCAGGG; R AGGAACTTGGCTATTTCAGGG) ATF4 (F ATGGCGTATTAGAGGCAGC; R CTTTGTCCGTTACAGCAAC AC) Gadd34 (F GATCGCTTTTGGCAACCAG; R CAGGAGATAGAAGTTGTGGGC) Dnajc3 (F TGGAGTAAAT GCGGATGTGG; R ACGGTCGCTCTCCTATAGTATG) Dnajb9 (F AAATAAAAGCCCTGATGCTGAAG; R CCT CTTTGTCCTTTGCCATTG) Edem (F CAATGAAGGAGAAGGAGACCC; R GCATCTTCCACATCCCCTATC) Rpn1 (F TGGATGACTCTGTGGAAATGG; R TCTACAAACCGCATCTTCAGTG) Sil1 (F GATACAAAGGAGAC CAGGGAAG; R AGTCTGTAGGTTCATTCGCAC) Fasn (F CAAGCAGAATTTGTCCACCTTTAA; R TCTCTAG AGGGCTTGCACCAA) Pepck1 (F GTCACCATCACTTCCTGGAAGA; R GGTGCAGAATCGCGAGTTG) Hnf4α (F TGTTTGGTGTGAAGGTCATGATTA; R CAGACGTCCTCCTTTTCTTGTGATA) Srebp1 (F CCAGAGGGTG AGCCTGACAA; R AGCCTCTGCAATTTCCAGATCT) Apob (F ATTCGAGCACAGATGACCAG; R AGGAGTC CGATATAGCTGTGG) Apoc3 (F CTCACGACTCAATAGCTGGAG; R GGGCGTTGTCCAAACAGAAT) Actb (F‐GCAGCTCCTTCGTTGCCGGT; R‐TACAGCCCGGGGAGCATCGT). The efficiency of each primer set was determined from a four point standard curve and used to calculate relative expression using Actb as reference gene. Data are expressed as the fold‐change in relative expression relative to controls.
2.5. Hyperinsulinemic euglycemic clamps
Clamps were performed as previously described with minor modifications (Costa et al., 2016). Mice recovered for one week prior to clamp experiments following surgical implantation of an indwelling catheter in the right jugular vein. Mice were fasted 6h prior to infusion with 3‐3H‐glucose (Perkin Elmer) at a rate of 0.05 µCi min−1 for 120 min to determine basal glucose turnover. A primed infusion of insulin and 3‐3H‐glucose was administered at 7.14 mU kg−1 min−1 and 0.24 µCi/min, respectively, for 4 min, after which rates were reduced to 3 mU kg−1 min−1 insulin and 0.1 µCimin 3‐3H‐glucose. Blood was collected by tail massage for plasma measurements at set time points and a variable infusion of 20% dextrose was given to maintain euglycemia. Glucose turnover was calculated as the ratio of the 3‐3H‐glucose infusion rate to the specific activity of plasma glucose at the end of the basal infusion and during the last 40 min of the hyperinsulinemic infusion. Endogenous or hepatic glucose output represents the difference between the glucose infusion rate and the rate of glucose appearance.
2.6. Statistical analyses
Data are reported as the mean ± SEM and comparisons were made between WT and Park2 KO groups within dietary treatments using two‐tailed, unpaired Student's t‐test. A p‐value of less than 0.05 was considered significant. Analyses were performed using GraphPad Prism 8.
3. RESULTS
Earlier work demonstrated improved glucose and pyruvate tolerance in Park2 KO mice fed regular chow (RC); however, insulin sensitivity per se was not evaluated directly (Kim et al., 2011). We therefore performed hyperinsulinemic euglycemic clamps in WT and Park2 KO mice fed RC for comparison with our previously reported HFD‐fed euglycemic clamp data (Costa et al., 2016; Figure 1a–c). Fasting plasma insulin levels were similar between genotypes on either diet and clamped plasma insulin levels were matched across groups in response to the hyperinsulinemic infusion (Costa et al., 2016; Figure 1a). There was no difference in the glucose infusion rate (GIR) required to maintain euglycemia comparing RC‐fed WT and Park2 KO mice, demonstrating that whole‐body knockout of Park2 had no effect on insulin sensitivity in RC‐fed mice (Figure 1b–c). In contrast, as previously reported, Park2 KO mice were protected from HFD‐induced insulin resistance independent of changes in body weight after just one week of HFD feeding and the GIR required to maintain euglycemia was 1.8‐fold greater in Park2 KO mice (Costa et al., 2016, Figure 1b–c). The improved insulin sensitivity was primarily due to changes in hepatic glucose production, where 60% of the difference in whole‐body insulin sensitivity was attributable to changes in hepatic glucose production and 40% to peripheral glucose uptake, but the mechanism for the observed changes in hepatic insulin action were not addressed (Costa et al., 2016). Insulin regulates hepatic glucose production directly by activating the insulin receptor signaling cascade, as well as indirectly by inhibiting adipose tissue lipolysis and the supply of fatty acids, and in turn acetyl‐CoA, to liver (Perry et al., 2015). There were no differences in basal or insulin‐stimulated plasma fatty acid levels comparing HFD‐fed WT and Park2 KO mice during the clamp (Figure 1d), suggesting the changes in clamped hepatic glucose production were due to alterations in the direct, but not indirect effects of insulin on liver and thus, differences in hepatic insulin resistance.
Hepatic steatosis is associated with insulin resistance (Samuel & Shulman, 2016). We therefore measured liver triglyceride levels and found no difference between genotypes during RC feeding (Figure 2a), consistent with the lack of difference in insulin sensitivity during the euglycemic clamp (Figure 1). After one‐week HFD feeding, hepatic triglyceride levels were increased 1.4‐fold in WT mice compared with RC‐fed WT, but were unchanged in Park2 KO mice and 40% less compared with HFD‐fed WT mice (Figure 2a; p < .05). Because of the lack of differences in whole‐body insulin sensitivity and liver steatosis between WT and Park2 KO mice fed RC, we focused on HFD‐fed animals to understand the mechanistic basis for the differences in hepatic insulin sensitivity.
Accumulation of the intracellular lipid species diacylglycerol and ceramide are thought to contribute to hepatic insulin resistance by activating PKCε and protein phosphatase 2a (PP2a), respectively, resulting in inhibition of the insulin receptor signaling cascade (Petersen & Shulman, 2018). Diacylglycerol levels were 40% less in Park2 KO compared with WT mice (Figure 2b, p < .01), which was primarily due to reduction in the membrane‐associated as opposed to cytosolic pool (Figure 2b, p < .01). In contrast, ceramide levels were modestly, but significantly increased in Park2 KO compared with WT mice (Figure 2b; p < .05). Consistent with the reduction in membrane‐associated diacylglycerol levels, PKCε activity, expressed as the membrane‐to‐cytosolic ratio, was reduced by 40% in HFD‐fed Park2 KO compared with WT mice (Figure 2c and d, p < .01).
In addition to subcellular localization, the acyl chain composition of diacylglycerol species may contribute to their ability to activate PKC isoforms and inhibit insulin signaling (Bergman, Hunerdosse, Kerege, Playdon, & Perreault, 2012). While several studies explored the relationship between diacylglycerol acyl chain composition and insulin resistance in skeletal muscle (Amati et al., 2011; Bergman et al., 2012; van Hees et al., 2011; Szendroedi et al., 2014), very few reports that addressed this question in liver exist. We therefore measured both acyl chain length and saturation in diacylglycerol species from liver cytosolic and membrane fractions, as well as from the total diacylglycerol pool (Figure 3). Within the cytosolic fraction, 12 of the 13 diacylglycerol species measured were reduced in HFD‐fed Park2 KO compared with WT, but only four species (C18:1/C16:0, C18:2/C18:0, C18:1/C18:0, and C18:1/C18:1) were significantly less (Figure 3a; p < .05). Within the membrane fraction, all species of measured diacylglycerol were reduced in the Park2 KO mice, and nine of 13 of the reduced species were significantly less compared with HFD‐fed WT (Figure 3b). Results from the total diacylglycerol pool were generally similar to the membrane fraction in that all species were reduced and nine of 13 species were significantly less (Figure 3c). When looking at which acyl chain species contributed to the overall reduction in diacylglycerol content within each fraction, the combined reduction in C18:1/C16:0, C18:1/C18:1, C18:1/C18:2, and C16:0/C18:2 accounted for ~90% of the difference within each fraction (Figure 3d). We also measured acyl chain composition of ceramide species in liver and found significant increases specifically in C22:0 (p < .05) and C24:0 (p < .001) ceramide and no differences in C16:0, C18:0, C20:0, or C24:1 species (Figure 3e).
Excessive endoplasmic reticulum (ER) stress and activation of the unfolded protein response (UPR) is also associated with hepatic insulin resistance (Hotamisligil, 2010). The UPR consists of three branches referred to by the names of the trans‐ER membrane proteins that sense an increased unfolded protein load within the ER and transduce this message to the cytosol, namely the inositol‐requiring enzyme 1 (IRE1α), PRK‐like eukaryotic initiation factor 2α kinase (PERK) and activating transcription factor 6 (ATF6) branches (Zhang & Kaufman, 2004). To determine if improved hepatic insulin sensitivity in HFD‐fed Park2 KO mice was associated with changes in the ER stress response, we measured expression of a panel of transcripts downstream of each arm of the UPR. IRE1α possesses both kinase and ribonuclease activity, such that activation results in processing of the transcription factor X‐box‐binding protein‐1 (XBP1) and induction of a subset of ER stress response genes (Calfon et al., 2002; Lee, Iwakoshi, & Glimcher, 2003; Yoshida, Matsui, Yamamoto, Okada, & Mori, 2001). IRE1α/XBP1‐dependent genes Dnajb9, Edem and Rpn1 were significantly reduced in HFD‐fed Park2 KO liver, while Sil1 was not (Figure 4a). Activation of PERK results in oligomerization and autophosphorylation, and in turn increased kinase activity (Ma, Vattem, & Wek, 2002). Activated PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), resulting in translational arrest of the majority of transcripts; however, a small subset of mRNAs, including activating transcription factor 4 (Atf4) and its transcriptional targets are induced (Calfon et al., 2002). PERK‐mediated induction of Atf4 expression was not different in HFD‐fed Park2 KO compared with WT, while Dnajc3 was significantly reduced and Gadd34 was unchanged (Figure 4a). Activation of ATF6 results in its transport and cleavage in the Golgi, followed by migration of the cleaved form to the nucleus where it activates a subset of UPR genes (Haze, Yoshida, Yanagi, Yura, & Mori, 1999; Ye et al., 2000). ATF6‐dependent genes Ero1β and Erp72 were significantly reduced in HFD‐fed Park2 KO mice (Figure 4a).
In addition to lipotoxicity and ER stress, changes in local and systemic anti‐ and pro‐inflammatory cytokine levels are associated with insulin resistance (Glass & Olefsky, 2012). We therefore measured plasma levels of a panel of cytokines from WT and Park2 KO mice, as well as a panel of obesity/diabetes‐related hormones produced by endocrine cells of the intestinal tract, pancreas and adipose tissue (Table 1). IL6 levels were two‐fold greater in RC‐fed Park2 KO compared with WT mice (p < .05), and 11‐fold greater in HFD‐fed Park2 KO compared with WT mice (p < .05; Table 1). IL10 and TNFα levels were significantly greater in HFD‐fed Park2 KO compared with WT mice (two‐fold, p < .01 and 1.7‐fold, p < .001, respectively; Table 1), while there were no differences in interferon γ (IFNγ), interleukin 2 (IL2), interleukin 5 (IL5) or C‐X‐C motif chemokine ligand 1 (CXCL1) between genotypes on either diet (Table 1). Of the seven obesity/diabetes‐related hormones measured, leptin was the only factor that differed between Park2 KO and WT mice on both diets, where plasma levels were 65% and 94% less in RC‐ and HFD‐fed Park2 KO compared with WT mice, respectively (p < .05, p < .001; Table 1). There was a modest, but insignificant reduction in plasma resistin levels comparing HFD‐fed Park2 KO to WT mice (p = .08), and no detectable differences in glucagon‐like peptide 1 (GLP1), glucagon, gastric inhibitory peptide (GIP), plasminogen activator inhibitor 1 (PAI1), or ghrelin levels (Table 1).
Next, we measured phosphorylation and activation of kinases downstream of the UPR and inflammatory signaling pathways that are implicated in the pathogenesis of hepatic insulin resistance (Hotamisligil, 2010). IRE1α‐mediated activation of c‐Jun NH2‐terminal kinase (JNK) is thought to contribute to hepatic insulin resistance via phosphorylation and inhibition of insulin receptor substrate 1 (IRS1) (Ozcan et al., 2004). Despite an apparent decrease in the IRE1α arm of the UPR in HFD‐fed Park2 KO mice (Figure 4a), there was no difference in JNK phosphorylation, indicative of activation, between groups (Figure 4b). Phosphorylation and activation of the Iκβ kinase (IKKβ) via UPR and inflammatory cytokine signaling is also proposed to induce hepatic insulin resistance (Arkan et al., 2005; Cai et al., 2005); however, we found no differences in IKKβ phosphorylation between groups (Figure 4c).
As mentioned earlier, Park2 KO mice fed HFD for six and a half weeks were protected from obesity and subsequently hepatic steatosis (Kim et al., 2011). We previously demonstrated that hepatic fatty acid transport, esterification and oxidation were unchanged in Park2 KO compared with WT mice (Costa et al., 2016), suggesting that changes in energy balance that manifest at the level of the hepatocyte during short‐term HFD feeding, prior to changes in body weight, contributed to the reduced steatosis observed here. To test this idea, we measured adenylate levels, as well as activation of the cellular energy sensor AMPK in liver of HFD‐fed Park2 KO and WT mice (Figure 5). Total adenylate levels were 12% less in Park2 KO mice (p < .01), which was the result of a 30% reduction in ATP and a modest, but non‐significant increase (15%) in AMP and no change in ADP (Figure 5a). The combined increase in AMP and decrease in ATP resulted in an approximate twofold increase in the AMP to ATP ratio in HFD‐fed Park2 KO liver compared with WT mice (p < .05; Figure 5b). AMPK is subject to multiple modes of regulation, most notably allosteric activation by AMP during times of increased ATP consumption that increases the cellular AMP/ATP ratio, facilitating phosphorylation of a conserved threonine residue (T172) in the AMPKα subunit and stimulation of AMPK's kinase activity (Gowans, Hawley, Ross, & Hardie, 2013). Consistent with the observed increase in the AMP/ATP ratio, AMPKα T172 phosphorylation was 2.2‐fold greater in Park2 KO compared with WT mice (p < .05; Figure 5c–d). AMPK is a potent inhibitor of acetyl‐CoA carboxylase (ACC), the enzyme that converts acetyl‐CoA to malonyl‐CoA. Malonyl‐CoA levels were 30% less in Park2 KO mice (p < .05; Figure 5e). Lastly, we measured expression of a panel of transcripts that are typically suppressed in liver in response to AMPK activation (Viollet et al., 2006) (Figure 5f). There was no difference in Pepck expression, while Fasn, Hnf4α, Srepb1, Apob, and Apoc3 mRNA levels were all significantly reduced (p < .05 ‐ p < .001; Figure 5f).
4. DISCUSSION
Here we provide the first direct evidence demonstrating no change in whole‐body insulin sensitivity in RC‐fed Park2 KO mice, as well as mechanistic insight with regard to the improved hepatic insulin sensitivity in short‐term HFD‐fed Park2 KO mice. We find that after one‐week HFD feeding, Park2 KO mice are protected against hepatic steatosis and diacylglycerol accumulation, and that hepatic PKCε activation is subsequently reduced. Liver ceramide levels, particularly C24:0 species, which are negatively associated with hepatic insulin resistance (Montgomery et al., 2016; Park et al., 2013; Raichur et al., 2014), were increased in HFD‐fed Park2 KO mice. TNFα and IL6, which are both positively associated with insulin resistance (Hotamisligil, Shargill, & Spiegelman, 1993; Pickup, Mattock, Chusney, & Burt, 1997), were increased in plasma of HFD‐fed Park2 KO mice, inconsistent with the observed phenotype with regards to insulin sensitivity. We also observed increased plasma levels of IL10, which has been shown to inhibit the effects of IL6 with regards to insulin resistance (Kim et al., 2004), and may have contributed to the improved insulin sensitivity. Although body weight was not different in HFD‐fed WT compared with Park2 KO mice, fat mass and plasma leptin levels were reduced in Park2 KO mice, and may have contributed to the improved hepatic insulin sensitivity (Costa et al., 2016). Finally, transcriptional markers of the ER stress response were also reduced in HFD‐fed Park2 KO mice, but there was no detectable difference in downstream activation of JNK or IKKβ between groups. Together, our data suggest that reduced lipotoxicity in liver of HFD‐fed Park2 KO mice contributed to their improved hepatic insulin sensitivity, and that changes in very‐long‐chain ceramides, ER stress‐ or IL10‐mediated signaling pathways may have contributed, as well.
A large number of studies in both rodents and humans reported a positive relationship between intracellular diacylglycerol content and insulin resistance in both skeletal muscle and liver (Samuel & Shulman, 2016), similar to what we observed here. In addition to total diacylglycerol content, the localization of diacylglycerol species may influence their ability to activate novel PKC (nPKC) isoforms and inhibit insulin signaling (Petersen & Shulman, 2018). For example, hepatic knockdown of comparative gene identification‐58 (CGI‐58), a lipid droplet‐associated protein that serves as an activating co‐factor for adipose triglyceride lipase, results in diacylglyerol accumulation, yet preserved hepatic insulin sensitivity (Brown et al., 2010; Cantley et al., 2013). In contrast to typical HFD‐fed mice, where diacylglycerol accumulates in the membrane fraction, diacylglycerol in CGI‐58 knockdown mice accumulated in the lipid droplet or lipid‐associated ER fraction of the cell (Cantley et al., 2013). PCKε was similarly localized to the lipid droplet/ER compartment of CGI‐58 knockdown mice, suggesting that redistribution of PKCε away from compartments of the cell involved in insulin receptor signaling, that is the membrane associated fraction, may account for the dissociation between hepatic diacylglycerol content and insulin resistance (Cantley et al., 2013). Thus, our observation of reduced membrane‐associated diacylglycerol and PKCε in HFD‐fed Park2 KO mice is consistent with their improved hepatic insulin sensitivity (Costa et al., 2016). In addition to localization, the acyl chain composition of diacylglycerol species may contribute to their ability to activate nPKC isoforms (Madani, Hichami, Legrand, Belleville, & Khan, 2001). For example, studies in human subjects reported a positive association between saturated fatty acid diacylglycerol species and insulin resistance (Bergman et al., 2012, 2010; van Hees et al., 2011), whereas others have reported a negative association (Amati et al., 2011) or no relationship at all (Coen et al., 2010). Far fewer studies have explored the relationship between acyl chain composition of diacylglycerols and insulin resistance in liver. Within the one reported study of which we are aware, a generalized increase in most diacylglycerol species measured was observed in association with hepatic insulin resistance (Cantley et al., 2013). This is consistent with our data here, where all sub‐species of diacylglycerol measured in the membrane fraction were reduced in HFD‐fed Park2 KO mice, suggesting acyl chain composition of diacylglycerol species does not specifically contribute to differences in insulin sensitivity in HFD‐fed rodent models. Interestingly, reductions in just four acyl chain species, specifically C18:1/C16, C18:1/C18:1, C18:1/C18:2, and C16/C18:2 accounted for 90% of the reduction in total diacylglycerol within each measured cellular fraction. These same species of fatty acids account for approximately 93% of the fatty acid species found in the HFD used, suggesting hepatic diacylglycerol content is largely reflective of dietary composition.
In addition to diacylglyerol, increases in hepatic ceramide levels, particularly long‐chain ceramides composed of C16‐C22 fatty acids, are positively associated with insulin resistance (Meikle & Summers, 2017; Montgomery et al., 2016; Raichur et al., 2014). Interestingly, reductions in hepatic very‐long‐chain ceramides, such as C22:0 and C24:0, are negatively associated with insulin resistance (Montgomery et al., 2016; Park et al., 2013; Raichur et al., 2014) and may confer protection from lipid‐induced insulin resistance by modulating the ER stress response or by increasing β‐oxidation (Montgomery et al., 2016; Raichur et al., 2014). Using a liver lipidomics approach, Montgomery and colleagues measured ceramide species in five commonly studied mouse strains and found that reductions in very‐long‐chain species of C22 or greater, as well as expression of the enzyme that catalyzes their synthesis, ceramide synthase 2 (Cers2), occurred in strains susceptible to diet‐induced insulin resistance (Montgomery et al., 2016). Overexpression of Cers2 in mouse primary hepatocytes increased C22:0 and C24:0 ceramides species and conferred protection from lipid‐induced insulin resistance, which was associated with reduced markers of ER stress and no differences in JNK activation, consistent with our observations here. Similarly, Raichur and colleagues observed that reductions in very‐long‐chain ceramide levels in Cers2 haploinsufficient mice were associated with increased susceptibility to lipid‐induced insulin resistance and reduced hepatic fatty acid oxidation (Raichur et al., 2014). Increases in hepatic C22:0 and C24:0 in HFD‐fed Park2 KO mice may therefore contribute to their protection from insulin resistance by modulating the ER stress response or by promoting lipid oxidation to prevent accumulation of long‐chain ceramide species or membrane‐associated diacylglycerols.
Increased plasma levels of TNFα and IL6 are associated with insulin resistance (Shoelson, Herrero, & Naaz, 2007), inconsistent to observations made here where HFD‐fed Park2 KO mice were more insulin sensitive despite increased plasma TNFα and IL6 levels. One possibility is that the differences observed here, although significantly different statistically, may not be significant biologically given the low absolute levels of cytokines measured, although the levels we detected are similar to previous reports (Dandona et al., 1998; Kern, Ranganathan, Li, Wood, & Ranganathan, 2001; Vozarova et al., 2001). Another possibility is that the parallel increase in the anti‐inflammatory cytokine, IL10, which was increased twofold in HFD Park2 KO mice, offset the effects of the pro‐inflammatory cytokines, which was shown to occur previously with regard to IL6 (Kim et al., 2004). While the observed changes in plasma cytokines in our model may not relate to the hepatic insulin resistance phenotype, they provide additional support for recent insight into how loss of Parkin‐mediated mitophagy contributes to disease pathogenesis. Recently, a new model for how Park2 mutations and loss of mitophagy give rise to Parkinson's disease was proposed where in the absence of mitochondrial quality control, mitochondrial DNA is released from damaged mitochondria and acts as a pro‐inflammatory signal, driving local and systemic inflammation and subsequent neurodegeneration (Sliter et al., 2018). Within this model, both acute and chronic mitochondrial stressors induced a marked increase in plasma IL6 levels on the order of 1000‐fold above baseline in Park2 KO compared with WT mice (Sliter et al., 2018). Here we observed a similar, but more moderate induction of IL6 on the order of 10‐fold above baseline in HFD‐fed Park2 KO mice, as well as a twofold increase in RC‐fed animals. Our data suggest that even in the absence of a specific mitochondrial stressor, deletion of Park2 is associated with activation of an immune response and increased IL6 levels, and furthermore, that short‐term HFD feeding exacerbates this effect. Given the specific effects of a short‐term HFD on hepatic lipid accumulation and insulin resistance (Samuel et al., 2004; Turner et al., 2013), we predict that the source of the pro‐inflammatory signal in the absence of Park2 to be the liver, but this supposition requires further testing.
Given previous work demonstrating that pharmacological or genetic activation of hepatic AMPK in nutritionally stressed rodents can reduce liver triglyceride levels and improve glucose homeostasis (Cool et al., 2006; Woods et al., 2017), it is tempting to speculate that the increased AMPK activation in the HFD‐fed Park2 KO mice contributed to their improved hepatic phenotype. While we cannot rule out this possibility, the lack of differences in fatty acid uptake, oxidation or triglyceride accumulation we observed when comparing WT and Park2 KO primary hepatocytes argues against this interpretation (Costa et al., 2016). Instead, we propose that hepatic activation of AMPK reflects negative energy balance in the Park2 KO during HFD feeding that manifests over the long‐term as reduced body weight and adiposity compared with WT mice (Kim et al., 2011). Here over the short‐term, this same mechanism presents as reduced liver fat and improved hepatic insulin sensitivity. The underlying mechanism for the negative energy balance and protection from diet‐induced obesity in the Park2 KO mice remains undetermined. One unexplored explanation for this phenomenon within the current study is that changes in mitochondrial function, specifically oxidative phosphorylation, in response to loss of Park2‐mediated mitophagy contributed to the reduced hepatic energy charge. Previously, liver‐specific deletion of the electron transport chain assembly factor, apoptosis inducing factor (Aif), resulted in loss of mitochondrial complex I and ATP synthase activities. Impaired oxidative phosphorylation within this model was associated with reduced hepatic ATP levels, activation of AMPK and protection from diet‐induced obesity, similar to Park2 KO mice (Pospisilik et al., 2007). Another possibility is that increased dark cycle activity, which was reported in Park2 KO mice fed HFD for one or six and a half weeks, contributed to their energy deficit (Costa et al., 2016; Kim et al., 2011). However, no differences in energy expenditure were detected in the short‐term HFD challenge study and although energy expenditure was reportedly increased in the long‐term study, correction of the data to body weight when the models being compared differed significantly in terms of adiposity makes interpretation of the result difficult (Kaiyala & Schwartz, 2011; Kim et al., 2011). And although we demonstrated reduced intestinal lipid absorption in the Park2 KO mice, the amount of lipid detected in feces was quantitatively insufficient to account for the protection from diet‐induced obesity (Costa et al., 2016). Another possibility is that the kinetics of intestinal lipid absorption may differ in Park2 KO mice and contribute to the phenotype, as has been reported in Diacylglycerol o‐acyltransferase‐1 (Dgat1) and Acyl CoA:monoacylglycerol acyltransferase‐2 (Mgat2) KO mice (Smith et al., 2000; Yen et al., 2009). This is an intriguing hypothesis given Park2's role in Parkinson's disease where Park2 mutations give rise to loss of dopaminergic neurons in the central nervous system, begging the question as to whether similar changes in dopamine or other monoamine producing neurons in the enteric nervous system occur in Park2 KO mice during HFD feeding and thus alter intestinal motility.
In summary, we provide evidence that reduced hepatic energy balance in short‐term HFD‐fed Park2 KO mice is associated with reduced hepatosteatosis and improved hepatic insulin sensitivity. We provide further evidence supporting the hypotheses that differences in membrane‐associated diacylglycerol levels and PKCε activity, and very‐long‐chain ceramide species, contribute to alterations in hepatic insulin sensitivity. Finally, we report confirmatory evidence supporting the recent observation that loss of Park2 is associated with a pro‐inflammatory state, particularly increases in plasma IL6 levels under conditions of mitochondrial stress, suggesting that even short‐term HFD feeding presents a significant stress to hepatic mitochondria. Future studies exploring changes in hepatic mitochondrial respiratory chain function, as well as the role of Park2 in the enteric nervous system and in gastric motility may provide insight into the still unresolved mechanism for reduced energy homeostasis in Park2 KO mice during HFD‐feeding.
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
Edmunds LR, Huckestein BR, Kahn M, et al. Hepatic insulin sensitivity is improved in high‐fat diet‐fed Park2 knockout mice in association with increased hepatic AMPK activation and reduced steatosis. Physiol Rep. 2019;7:e14281 10.14814/phy2.14281
Funding information
This work was supported by funding from NIH grants K01 DK099402 and R01 DK114012 (MJJ); R01 DK116774, R01 DK119968, R01 DK114793, and P30 DK059635 (GIS), DK007052 and American Diabetes Association grant #1‐19‐PDF‐102 (LRE), and S10OD023402 (SGW).
REFERENCES
- Amati, F. , Dubé, J. J. , Alvarez‐Carnero, E. , Edreira, M. M. , Chomentowski, P. , Coen, P. M. , … Goodpaster, B. H. (2011). Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: Another paradox in endurance‐trained athletes? Diabetes, 60, 2588–2597. 10.2337/db10-1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkan, M. C. , Hevener, A. L. , Greten, F. R. , Maeda, S. , Li, Z.‐W. , Long, J. M. , … Karin, M. (2005). IKK‐beta links inflammation to obesity‐induced insulin resistance. Nature Medicine, 11, 191–198. [DOI] [PubMed] [Google Scholar]
- Bergman, B. C. , Hunerdosse, D. M. , Kerege, A. , Playdon, M. C. , & Perreault, L. (2012). Localisation and composition of skeletal muscle diacylglycerol predicts insulin resistance in humans. Diabetologia, 55, 1140–1150. 10.1007/s00125-011-2419-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman, B. C. , Perreault, L. , Hunerdosse, D. M. , Koehler, M. C. , Samek, A. M. , & Eckel, R. H. (2010). Increased intramuscular lipid synthesis and low saturation relate to insulin sensitivity in endurance‐trained athletes. Journal of Applied Physiology, 108, 1134–1141. 10.1152/japplphysiol.00684.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati, V. (2012). Autosomal recessive parkinsonism. Parkinsonism & Related Disorders, 18(Suppl 1), S4–6. 10.1016/S1353-8020(11)70004-9 [DOI] [PubMed] [Google Scholar]
- Brown, J. M. , Betters, J. L. , Lord, C. , Ma, Y. , Han, X. , Yang, K. , … Yu, L. (2010). CGI‐58 knockdown in mice causes hepatic steatosis but prevents diet‐induced obesity and glucose intolerance. Journal of Lipid Research, 51, 3306–3315. 10.1194/jlr.M010256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, D. , Yuan, M. , Frantz, D. F. , Melendez, P. A. , Hansen, L. , Lee, J. , & Shoelson, S. E. (2005). Local and systemic insulin resistance resulting from hepatic activation of IKK‐beta and NF‐kappaB. Nature Medicine, 11, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon, M. , Zeng, H. , Urano, F. , Till, J. H. , Hubbard, S. R. , Harding, H. P. , … Ron, D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature, 415, 92–96. 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- Cantley, J. L. , Yoshimura, T. , Camporez, J. P. G. , Zhang, D. , Jornayvaz, F. R. , Kumashiro, N. , … Shulman, G. I. (2013). CGI‐58 knockdown sequesters diacylglycerols in lipid droplets/ER‐preventing diacylglycerol‐mediated hepatic insulin resistance. Proceedings of the National Academy of Sciences of the United States of America, 110, 1869–1874. 10.1073/pnas.1219456110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen, P. M. , Dubé, J. J. , Amati, F. , Stefanovic‐Racic, M. , Ferrell, R. E. , Toledo, F. G. S. , & Goodpaster, B. H. (2010). Insulin resistance is associated with higher intramyocellular triglycerides in type I but not type II myocytes concomitant with higher ceramide content. Diabetes, 59, 80–88. 10.2337/db09-0988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool, B. , Zinker, B. , Chiou, W. , Kifle, L. , Cao, N. , Perham, M. , … Frevert, E. (2006). Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metabolism, 3, 403–416. 10.1016/j.cmet.2006.05.005 [DOI] [PubMed] [Google Scholar]
- Costa, D. K. , Huckestein, B. R. , Edmunds, L. R. , Petersen, M. C. , Nasiri, A. , Butrico, G. M. , … Jurczak, M. J. (2016). Reduced intestinal lipid absorption and body weight‐independent improvements in insulin sensitivity in high‐fat diet‐fed Park2 knockout mice. American Journal of Physiology. Endocrinology and Metabolism, 311, E105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Costa, C. A. , Sunyach, C. , Giaime, E. , West, A. , Corti, O. , Brice, A. , … Checler, F. (2009). Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson's disease. Nature Cell Biology, 11, 1370–1375. 10.1038/ncb1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandona, P. , Weinstock, R. , Thusu, K. , Abdel‐Rahman, E. , Aljada, A. , & Wadden, T. (1998). Tumor necrosis factor‐α in sera of obese patients: fall with weight loss. The Journal of Clinical Endocrinology & Metabolism, 83, 2907–2910. [DOI] [PubMed] [Google Scholar]
- Dove, K. K. , & Klevit, R. E. (2017). RING‐between‐RING E3 ligases: emerging themes amid the variations. Journal of Molecular Biology, 429, 3363–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, C. K. , & Olefsky, J. M. (2012). Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metabolism, 15, 635–645. 10.1016/j.cmet.2012.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, Y. , Zack, T. I. , Morris, L. G. T. , Lin, K. , Hukkelhoven, E. , Raheja, R. , … Chan, T. A. (2014). Pan‐cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nature Genetics, 46, 588–594. 10.1038/ng.2981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans, G. J. , Hawley, S. A. , Ross, F. A. , & Hardie, D. G. (2013). AMP is a true physiological regulator of AMP‐activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metabolism, 18, 556–566. 10.1016/j.cmet.2013.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, J. C. , Whitworth, A. J. , Kuo, I. , Andrews, L. A. , Feany, M. B. , & Pallanck, L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proceedings of the National Academy of Sciences of the United States of America, 100, 4078–4083. 10.1073/pnas.0737556100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haze, K. , Yoshida, H. , Yanagi, H. , Yura, T. , & Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular Biology of the Cell, 10, 3787–3799. 10.1091/mbc.10.11.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell, 140, 900–917. 10.1016/j.cell.2010.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil, G. S. , Shargill, N. S. , & Spiegelman, B. M. (1993). Adipose expression of tumor necrosis factor‐alpha: Direct role in obesity‐linked insulin resistance. Science, 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Kaiyala, K. J. , & Schwartz, M. W. (2011). Toward a more complete (and less controversial) understanding of energy expenditure and its role in obesity pathogenesis. Diabetes, 60, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern, P. A. , Ranganathan, S. , Li, C. , Wood, L. , & Ranganathan, G. (2001). Adipose tissue tumor necrosis factor and interleukin‐6 expression in human obesity and insulin resistance. American Journal of Physiology‐Endocrinology and Metabolism, 280, E745–E751. 10.1152/ajpendo.2001.280.5.E745 [DOI] [PubMed] [Google Scholar]
- Kim, H.‐J. , Higashimori, T. , Park, S.‐Y. , Choi, H. , Dong, J. , Kim, Y.‐J. , … Kim, J. K. (2004). Differential effects of interleukin‐6 and ‐10 on skeletal muscle and liver insulin action in vivo. Diabetes, 53, 1060–1067. 10.2337/diabetes.53.4.1060 [DOI] [PubMed] [Google Scholar]
- Kim, K.‐Y. , Stevens, M. V. , Akter, M. H. , Rusk, S. E. , Huang, R. J. , Cohen, A. , … Sack, M. N. (2011). Parkin is a lipid‐responsive regulator of fat uptake in mice and mutant human cells. Journal of Clinical Investigation, 121, 3701–3712. 10.1172/JCI44736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada, T. , Asakawa, S. , Hattori, N. , Matsumine, H. , Yamamura, Y. , Minoshima, S. , … Shimizu, N. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392, 605–608. 10.1038/33416 [DOI] [PubMed] [Google Scholar]
- Lee, A.‐H. , Iwakoshi, N. N. , & Glimcher, L. H. (2003). XBP‐1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and Cellular Biology, 23, 7448–7459. 10.1128/MCB.23.21.7448-7459.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, X. , Altshuler‐Keylin, S. , Wang, Q. , Chen, Y. , Henrique Sponton, C. , Ikeda, K. , … Kajimura, S. (2018). Mitophagy controls beige adipocyte maintenance through a Parkin‐dependent and UCP1‐independent mechanism. Science Signalling, 11, eaap8526 10.1126/scisignal.aap8526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, K. , Vattem, K. M. , & Wek, R. C. (2002). Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor‐2 kinase in response to endoplasmic reticulum stress. Journal of Biological Chemistry, 277, 18728–18735. 10.1074/jbc.M200903200 [DOI] [PubMed] [Google Scholar]
- Madani, S. , Hichami, A. , Legrand, A. , Belleville, J. , & Khan, N. A. (2001). Implication of acyl chain of diacylglycerols in activation of different isoforms of protein kinase C. The FASEB Journal, 15, 2595–2601. 10.1096/fj.01-0753int [DOI] [PubMed] [Google Scholar]
- Meikle, P. J. , & Summers, S. A. (2017). Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nature Reviews Endocrinology, 13, 79–91. 10.1038/nrendo.2016.169 [DOI] [PubMed] [Google Scholar]
- Montgomery, M. K. , Brown, S. H. J. , Lim, X. Y. , Fiveash, C. E. , Osborne, B. , Bentley, N. L. , … Turner, N. (2016). Regulation of glucose homeostasis and insulin action by ceramide acyl‐chain length: A beneficial role for very long‐chain sphingolipid species. Biochimica Et Biophysica Acta, 1861, 1828–1839. [DOI] [PubMed] [Google Scholar]
- Ozcan, U. , Cao, Q. , Yilmaz, E. , Lee, A.‐H. , Iwakoshi, N. N. , Ozdelen, E. , … Hotamisligil, G. S. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science, 306, 457–461. 10.1126/science.1103160 [DOI] [PubMed] [Google Scholar]
- Park, J.‐W. , Park, W.‐J. , Kuperman, Y. , Boura‐Halfon, S. , Pewzner‐Jung, Y. , & Futerman, A. H. (2013). Ablation of very long acyl chain sphingolipids causes hepatic insulin resistance in mice due to altered detergent‐resistant membranes. Hepatology, 57, 525–532. 10.1002/hep.26015 [DOI] [PubMed] [Google Scholar]
- Perry, R. J. , Camporez, J.‐P.‐ G. , Kursawe, R. , Titchenell, P. M. , Zhang, D. , Perry, C. J. , … Shulman, G. I. (2015). Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell, 160, 745–758. 10.1016/j.cell.2015.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, M. C. , Madiraju, A. K. , Gassaway, B. M. , Marcel, M. , Nasiri, A. R. , Butrico, G. , … Shulman, G. I. (2016). Insulin receptor Thr1160 phosphorylation mediates lipid‐induced hepatic insulin resistance. Journal of Clinical Investigation, 126, 4361–4371. 10.1172/JCI86013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, M. C. , & Shulman, G. I. (2018). Mechanisms of insulin action and insulin resistance. Physiological Reviews, 98, 2133–2223. 10.1152/physrev.00063.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup, J. C. , Mattock, M. B. , Chusney, G. D. , & Burt, D. (1997). NIDDM as a disease of the innate immune system: Association of acute‐phase reactants and interleukin‐6 with metabolic syndrome X. Diabetologia, 40, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Piquereau, J. , Godin, R. , Deschênes, S. , Bessi, V. L. , Mofarrahi, M. , Hussain, S. N. , & Burelle, Y. (2013). Protective role of PARK2/Parkin in sepsis‐induced cardiac contractile and mitochondrial dysfunction. Autophagy, 9, 1837–1851. 10.4161/auto.26502 [DOI] [PubMed] [Google Scholar]
- Pospisilik, J. A. , Knauf, C. , Joza, N. , Benit, P. , Orthofer, M. , Cani, P. D. , … Penninger, J. M. (2007). Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell, 131, 476–491. 10.1016/j.cell.2007.08.047 [DOI] [PubMed] [Google Scholar]
- Raichur, S. , Wang, S. T. , Chan, P. W. , Li, Y. , Ching, J. , Chaurasia, B. , … Summers, S. A. (2014). CerS2 haploinsufficiency inhibits β‐oxidation and confers susceptibility to diet‐induced steatohepatitis and insulin resistance. Cell Metabolism, 20, 919 10.1016/j.cmet.2014.10.007 [DOI] [PubMed] [Google Scholar]
- Samuel, V. T. , Liu, Z.‐X. , Qu, X. , Elder, B. D. , Bilz, S. , Befroy, D. , … Shulman, G. I. (2004). Mechanism of hepatic insulin resistance in non‐alcoholic fatty liver disease. Journal of Biological Chemistry, 279, 32345–32353. 10.1074/jbc.M313478200 [DOI] [PubMed] [Google Scholar]
- Samuel, V. T. , & Shulman, G. I. (2016). The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. Journal of Clinical Investigation, 126, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson, S. E. , Herrero, L. , & Naaz, A. (2007). Obesity, Inflammation, and Insulin Resistance. Gastroenterology, 132, 2169–2180. 10.1053/j.gastro.2007.03.059 [DOI] [PubMed] [Google Scholar]
- Sliter, D. A. , Martinez, J. , Hao, L. , Chen, X. , Sun, N. , Fischer, T. D. , … Youle, R. J. (2018). Parkin and PINK1 mitigate STING‐induced inflammation. Nature, 561, 258–262. 10.1038/s41586-018-0448-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. J. , Cases, S. , Jensen, D. R. , Chen, H. C. , Sande, E. , Tow, B. , … Farese, R. V. (2000). Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nature Genetics, 25, 87–90. 10.1038/75651 [DOI] [PubMed] [Google Scholar]
- Szendroedi, J. , Yoshimura, T. , Phielix, E. , Koliaki, C. , Marcucci, M. , Zhang, D. , … Roden, M. (2014). Role of diacylglycerol activation of PKCθ in lipid‐induced muscle insulin resistance in humans. Proceedings of the National Academy of Sciences of the United States of America, 111, 9597–9602. 10.1073/pnas.1409229111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, N. , Kowalski, G. M. , Leslie, S. J. , Risis, S. , Yang, C. , Lee‐Young, R. S. , … Bruce, C. R. (2013). Distinct patterns of tissue‐specific lipid accumulation during the induction of insulin resistance in mice by high‐fat feeding. Diabetologia, 56, 1638–1648. 10.1007/s00125-013-2913-1 [DOI] [PubMed] [Google Scholar]
- van Hees, A. M. J. , Jans, A. , Hul, G. B. , Roche, H. M. , Saris, W. H. M. , & Blaak, E. E. (2011). Skeletal muscle fatty acid handling in insulin resistant men. Obesity (Silver Spring), 19, 1350–1359. 10.1038/oby.2011.10 [DOI] [PubMed] [Google Scholar]
- Viollet, B. , Foretz, M. , Guigas, B. , Horman, S. , Dentin, R. , Bertrand, L. , … Andreelli, F. (2006). Activation of AMP‐activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. Journal of Physiology (London), 574, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozarova, B. , Weyer, C. , Hanson, K. , Tataranni, P. A. , Bogardus, C. , & Pratley, R. E. (2001). Circulating interleukin‐6 in relation to adiposity, insulin action, and insulin secretion. Obesity Research, 9, 414–417. 10.1038/oby.2001.54 [DOI] [PubMed] [Google Scholar]
- Williams, J. A. , Ni, H.‐M. , Ding, Y. , & Ding, W.‐X. (2015). Parkin regulates mitophagy and mitochondrial function to protect against alcohol‐induced liver injury and steatosis in mice. American Journal of Physiology‐Gastrointestinal and Liver Physiology, 309, G324–G340. 10.1152/ajpgi.00108.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods, A. , Williams, J. R. , Muckett, P. J. , Mayer, F. V. , Liljevald, M. , Bohlooly‐Y, M. , & Carling, D. (2017). Liver‐specific activation of AMPK prevents steatosis on a high‐fructose diet. Cell Reports, 18, 3043–3051. 10.1016/j.celrep.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J. , Rawson, R. B. , Komuro, R. , Chen, X. , Davé, U. P. , Prywes, R. , … Goldstein, J. L. (2000). ER stress induces cleavage of membrane‐bound ATF6 by the same proteases that process SREBPs. Molecular Cell, 6, 1355–1364. 10.1016/S1097-2765(00)00133-7 [DOI] [PubMed] [Google Scholar]
- Yen, C.‐L.‐ E. , Cheong, M.‐L. , Grueter, C. , Zhou, P. , Moriwaki, J. , Wong, J. S. , … Farese, R. V. (2009). Deficiency of the intestinal enzyme acyl CoA: Monoacylglycerol acyltransferase‐2 protects mice from metabolic disorders induced by high‐fat feeding. Nature Medicine, 15, 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, H. , Matsui, T. , Yamamoto, A. , Okada, T. , & Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell, 107, 881–891. 10.1016/S0092-8674(01)00611-0 [DOI] [PubMed] [Google Scholar]
- Youle, R. J. , & Narendra, D. P. (2011). Mechanisms of mitophagy. Nature Reviews Molecular Cell Biology, 12, 9–14. 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K. , & Kaufman, R. J. (2004). Signaling the unfolded protein response from the endoplasmic reticulum. Journal of Biological Chemistry, 279, 25935–25938. 10.1074/jbc.R400008200 [DOI] [PubMed] [Google Scholar]