

Abstract
In this work, we propose the use of regular branching of polyurethanes as a way to regulate chain dynamics and govern crystallization in highly dense hydrogen-bonded systems. As a result, robust and healable polyurethanes can be obtained. To this end, we synthesized a range of aliphatic propane diol derivatives with alkyl branches ranging from butyl (C4) to octadecanyl (C18). The series of brush polyurethanes was synthesized by polyaddition of the diols and hexamethylene diisocyanate. Polyurethanes with very short (C < 4) and very long (C = 18) brush lengths did not lead to any significant healing due to crystallization. An intermediate amorphous regime appears for polymers with middle branch lengths (C = 4 to 8) showing a fine control of material toughness. For these systems, the side chain length regulates tube dilation, and significant macroscopic healing of cut samples was observed and studied in detail using melt rheology and tensile testing. Despite the high healing degrees observed immediately after repair, it was found that samples with medium to long length brushes lost their interfacial strength at the healed site after being heated to the healing temperature for some time after the optimal time to reach full healing. Dedicated testing suggests that annealed samples, while keeping initial tackiness, are not able to completely heal the cut interface. We attribute such behavior to annealing-induced interfacial crystallization promoted by the aliphatic branches. Interestingly, no such loss of healing due to annealing was observed for samples synthesized with C4 and C7 diols, which is identified as the optimal healing regime. These results point at the positive effect of branching on healing, provided that a critical chain length is not surpassed, as well as the need to study healing behavior long after the optimal healing times.
1. Introduction
Intrinsic healable polymers, similar to biological systems, are able to sustain multiple healing events caused by physical damages even at the same location. For this reason, these systems are an attractive alternative to the damage prevention approach as a solution to obtain structures with extended lifetime while addressing the critical concerns related to raw material overconsumption and high maintenance costs.1−3
The macromolecular design of intrinsic healable polymers relies on the presence of both permanent and dynamic bonds. The permanent bonds control the mechanical properties, while the dynamic bonds enable reconstruction of crack interfaces brought into intimate contact and recovery of mechanical integrity. Cross-linking by dynamic covalent bonds4−8 or the use of hybrid dual networks9 has been reported as efficient ways to produce healable polymers with engineering-relevant mechanical properties. However, heating to relatively high temperatures (>70 °C) is required to trigger local mobility.10−12 On the other hand, many systems showing near-room-temperature healing have been created by embedding noncovalent dynamic blocks (e.g., hydrogen bonds,13,14 host–guest interactions,15,16 multiple Van der Waals interactions17,18) in low-molecular-weight polymers. However, these healable polymers and hydrogels are relatively soft with undesirably low toughness and ultimate tensile strength values, strongly limiting their applicability range in engineering applications.
One strategy to increase mechanical properties is the increase of the main-chain molecular weight, leading to entanglements that determine high material robustness. Yet, at near room temperature, chain interdiffusion that leads to randomization at crack sites is too sluggish to occur on a reasonable timescale, and a poor healing efficiency is obtained.19 Alternatively, when a polymer is designed with a high density of noncovalent dynamic units, very brittle and oriented materials are obtained.20 While this approach has been shown to be successful for the production of new-generation liquid crystals21 and microsegregated materials,22 these systems do not show any healing ability since annealing-induced crystallization and clustering hinder chain interdiffusion at mild temperature. Sufficiently fast and sufficiently complete self-healing of cracks and scratches at temperatures close to room temperature and high mechanical properties seem to be mutually exclusive, and new strategies involving modification of polymer architectures must be explored.23
To address this issue, different strategies were employed. Guan et al.24 introduced sacrificial noncovalent bonds to increase the mechanical properties of self-healing thermosets. Yanagisawa et al.25 reported polymers readily healable at room temperature and with high mechanical properties by including dense and non-directional thiourea hydrogen bonding units in the main chain. Soon after, we reported the synthesis of strong and room-temperature healing polyimides based on primary and secondary noncovalent interactions and the fundamental role of short (C8) fully aliphatic branches in regulating chain dynamics.26 Inspired by this finding, in the present study, we investigate the role of the branch length in the relaxation dynamics and crystallization kinetics of polyurethanes with a high density of hydrogen bonds. To this end, we synthesized a range of branched polyurethanes with variable dangling aliphatic chain lengths from C4 to C18 on a short repeating unit, leading to a high density of urethane units and branches. The series of brush polyurethanes was synthesized by polyaddition of branched diols with varying branch length and hexamethylene diisocyanate. Detailed calorimetry, XRD, and rheology studies clarified the effect of brush length on the aggregation state and relaxation dynamics, showing that physical properties can be finely adjusted by extending or reducing the lateral branches by a few carbon units. Tensile testing was used to assess mechanical behavior and intrinsic self-healing properties. The control of polymer architecture by regular branching in highly hydrogen-bonded systems results in a combination of decent mechanical properties (typical for polymers with a high spatial density of hydrogen bonds) and fast self-healing kinetics at near room temperature. This is attributed to the plasticization effect of side branching, and its hindrance of H-bond-induced crystallization. Moreover, we argue how critical annealing-induced crystallization upon long-term heating to ambient or near-Tg temperatures can be fatal for the healing of this polymer class when branch lengths are higher than C7, an issue that has been unaddressed in previous studies on comparable systems.25
2. Experimental Section
2.1. Materials
1-Bromobutane (99%), 1-bromooctane (99%), 1-bromononane (98%), 1-bromododecane (97%), 1-bromooctadecane (>97%), lithium aluminum hydride (LiAlH4, pellets, 95%), sodium hydride (NaH, dispersion in mineral oil, 60%), diethyl malonate (99%), dibutyltin dilaurate (DBTL, 95%), and dimethylformamide (DMF, anhydrous, 99.8%) were purchased from Sigma-Aldrich. Ethyl acetate (99%, technical), methanol (≥99.5%, technical), chloroform (≥98%, technical), deuterated chloroform (D-chloroform, 0.03% TMS), and deuterated dimethylsulfoxide (D-DMSO) were purchased from VWR Chemicals. Hexamethylene diisocyanate (HDI, ≥98%) and 1,4-butanediol (BDO, ≥99%) were purchased from TCI Europe. Hydrochloric acid (37%) was purchased from Honeywell. Magnesium sulfate (dried, contains approximately 1–2 mol water of hydration, ≥98%) was purchased from Alfa Aesar. All commercial chemicals were used as received. All reactions were carried out under a nitrogen atmosphere. The synthesis of the diols and polymers showed high reproducibility.
2.2. Synthesis of Branched Diol Monomers
Following the two-step synthesis process shown below for the case of 2,2-dibutylpropane-1,3-diol (C4DA), six branched short-length diols with different dangling chain lengths were synthesized: 2,2-dibutylpropane-1,3-diol (C4DA), 2,2-diheptylpropane-1,3-diol (C7DA), 2,2-dioctylpropane-1,3-diol (C8DA), 2,2-dinonylpropane-1,3-diol (C9DA), 2,2-didodecylpropane-1,3-diol (C12DA), and 2,2-dioctadecylpropane-1,3-diol (C18DA). As a non-branched reference, commercial 1,4-butanediol (BDO) was used (HDI_BDO). The molecular structure of the synthesized diols is represented in Scheme 1, while Table 1 shows their melting points and the nomenclature used along the text. 1H-NMR and 13C-NMR spectra of all the synthesized diols are available in Figure S1. Details about synthesis of all diols are available in the Supporting Information.
Scheme 1. Molecular Structure of the Synthesized Branched Diols.

Table 1. Diols Synthesized in This Work Including Dangling Chain Length and Melting Point As Measured by DSC.
| diol | dangling chain length (# of carbons) | melting point (°C) |
|---|---|---|
| BDO | 0 | n.a. |
| C4DA | 4 | 43 |
| C7DA | 7 | 38 |
| C8DA | 8 | 29 |
| C9DA | 9 | 36 |
| C12DA | 12 | 44 |
| C18DA | 18 | 73 |
2.2.1. Step 1: Diethyl 2,2-Dibutylmalonate (C4DE)
1-Bromobutane (59.7 g, 0.44 mol) was added to an ice-cooled and vigorously stirred suspension of sodium hydride (17.8 g, 0.45 mol) in anhydrous THF (400 mL). Subsequently, diethyl malonate (23.5 g, 0.15 mol) was added dropwise. The flask was heated to 80 °C, and the reaction mixture was refluxed overnight. The solution was then quenched using demineralized water and 10% hydrochloric acid. The salt was then dissolved in an excess of aqueous hydrochloric acid, and the reaction mixture was extracted using diethyl ether and water. The resulting organic phase was washed three times in 150 mL of demineralized water, while the aqueous fraction was extracted twice in 100 mL of diethyl ether. The remaining organic layers were combined and dried over magnesium sulfate. The product was further dried at 60 °C under vacuum overnight. The resulting crude diethyl 2,2-dibutylmalonate (C4DE) appeared as a yellowish oil (52.5 g, 0.19 mol, 66%) and was used without further purification.
2.2.2. Step 2: 2,2-Dibutylpropane-1,3-diol (C4DA)
Crude C4DE (52.5 g, 0.19 mol) was added to a stirred and ice-cooled suspension of lithium aluminum hydride (12.4 g, 0.33 mol) in anhydrous THF (400 mL). The system was subsequently heated to 80 °C and left under vigorous stirring overnight. The resulting suspension was cooled using an acetone/liquid nitrogen bath and quenched with water. The salts were then dissolved in aqueous hydrochloric acid, and the reaction mixture was extracted with diethyl ether and water. The organic phase was washed three times in 150 mL of demineralized water, and the aqueous fraction was extracted twice using 100 mL of diethyl ether. The resulting organic layers were dried over magnesium sulfate, and the solvents were evaporated in vacuo. The product was purified by column chromatography (silica gel, 450 g; hexane and ethyl acetate). Pure C4DA was obtained as a white solid (24.3 g, 88%). The synthesis completion was checked with 1H-NMR (CDCl3, 400 MHz) by the presence of the most relevant peaks:
1H-NMR (CDCl3, 400 MHz) δ: 3.56 (s, 4H); 2.32 (s, 2H); 1.23 (m, 12H); 0.90 (t, 6H). 13C-NMR (CDCl3, 400 MHz) δ: 69.50; 40.88; 30.50; 25.06; 23.60; 14.06.
2.3. Syntheses of the Brush Polyurethanes
Seven brush polyurethanes (Scheme 2) were synthesized using a single-step polymerization process by reacting hexamethylene diisocynate (HDI) with the seven diols C4DA, C7DA, C8DA, C9DA, C12DA, C18DA, and BDO discussed in section 2.2. The list of overall properties of the resulting brush polymers is shown in Table 2.
Scheme 2. Representation of Brush Polyurethane Synthesis.

Table 2. Effect of Aliphatic Dangling Chain Length on Molecular Weight Distribution, Glass Transition and Melting Temperature Measured by DSC, and Thermal Stability Measured by TGA.
| polymer | brush length (# of carbons) | Mn (kDa) | Mw (kDa) | Đ | Xn | DSC-Tg [10–20 °C min–1] (°C) | DSC-Tm [10 °C min–1] (°C) | TGA-2% wt (°C) |
|---|---|---|---|---|---|---|---|---|
| HDI_BDO | 0 | n.a.a | n.a.a | n.a.a | n.a.a | 40–43 | 165 | 281 |
| HDI_C4DA | 4 | 14.6 | 20.0 | 1.3 | 41 | 29–31 | n.a.b | 243 |
| HDI_C7DA | 7 | 21.6 | 29.0 | 1.3 | 49 | 23–26 | n.a.b | 280 |
| HDI_C8DA | 8 | 24.5 | 30.1 | 1.2 | 52 | 24–27 | n.a.b | 300 |
| HDI_C9DA | 9 | 15.6 | 21.3 | 1.3 | 32 | 19–27 | n.a.b | 298 |
| HDI_C12DA | 12 | 20.5 | 34.0 | 1.6 | 35 | 30–n.a.c | 46 | 312 |
| HDI_C18DA | 18 | n.a.a | n.a.a | n.a.a | n.a.a | n.a.c | 45 | 305 |
HDI_BDO and HDI_C18DA were not soluble in the available GPC solvents (THF, NMP).
HDI_C4DA, HDI_C7DA, HDI_C9DA, and HDI_C9DA did not present melting peaks.
HDI_C12DA and HDI_C18DA glass transitions overlapped with melting transitions.
As a mode of example, the synthesis of polymer HDI_C4DA is shown, while details of the other syntheses are given in the Supporting Information. A solution of HDI (5.382 g, 32 mmol) in anhydrous THF (9 mL, 1/3 vol.) was added to a solution of C4DA (6.019 g, 32 mmol) in anhydrous THF (18 mL, 2/3 vol.) under vigorous stirring. Subsequently, DBTL (2.02 g, 3.2 mmol) was injected into the system dropwise. The temperature was increased to 70 °C, and the reaction proceeded for 24 h. The solution was precipitated in methanol (200 mL), resulting in the separation of a viscous white solid. The solution was filtered, and the extracted precipitate was dissolved in 20 mL of chloroform and re-precipitated in methanol. The resulting viscous polymer was dried overnight at 60 °C in vacuo (7.38 g, 65%). The completion of the polyurethane synthesis was monitored by FTIR analysis through the characteristic carbonyl stretching absorption peak at ∼1170 cm–1 and the disappearance of the isocyanate absorption peak at ∼2270 cm–1. 1H-NMR confirmed the successful polyurethane synthesis through the presence of a singlet at ∼5 ppm associated with amide bond protons.
1H-NMR (CDCl3, 400 MHz) δ: 4.9 (s, 2H); 3.85 (s, 4H): 3.10 (s, 4H); 1.46 (s, 8H); 1.22 (m, 12H); 0.86 (t, 6H).
13C-NMR (CDCl3, 400 MHz) δ: 156.69; 66.58; 40.76; 39.58; 30.89; 29.81; 29.21; 26.26; 24.75; 23.42; 14.04.
The FTIR and 1H-NMR spectra of all the synthesized polymers can be found in Figure S2.
2.4. Characterization Methods
2.4.1. Infrared Spectroscopy (FTIR)
Attenuated total reflectance Fourier transform infrared spectroscopy was employed in order to follow reaction completion. Each infrared spectrum was recorded as an average of eight scans in the wavenumber range 4000–500 cm–1.
2.4.2. Proton and Carbon Nuclear Magnetic Resonance
1H-NMR and 13C-NMR spectra were recorded using a Bruker WM-400 at 25 °C using CDCl3 and DMSO-d6 as solvents.
2.4.3. Gel Permeation Chromatography
Molecular weight distributions of synthesized polymers were determined using a gel permeation chromatograph (GPC) equipped with a refractive index detector and using polystyrene standards. The solvent used was tetrahydrofuran (THF) with a polymer concentration of 1 mg/mL.
2.4.4. Thermal Analysis
Thermal properties were determined by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA was performed from room temperature to 400 °C under a nitrogen atmosphere at a 10 °C/min heating rate using a Perkin Elmer TGA 4000. DSC measurements were performed under nitrogen at 10 and 20 °C/min heating and cooling rates in the temperature range −50 to 180 °C using a Perkin Elmer Pyris Sapphire DSC. The glass transition temperature (Tg) was determined using the inflection point method.
2.4.5. Mechanical Properties by Tensile Testing
Mechanical properties were assessed using an INSTRON universal testing machine. Dog-bone specimens were tested according to the ASTM D1708 standard at a cross-head speed of 80 mm/min. The average thickness of the specimens was t = 1.3 ± 0.1 mm.
The recovery of the mechanical properties after damage (healing) was determined by healing razor-blade-cut dog-bone specimens. After cutting the dog-bone specimens into two parts at room temperature with a razor blade, these were immediately brought back into contact under gentle hand-pressure for 10 s until they were able to withstand their own weights. Subsequently, they were left to heal pressureless at their individually selected healing temperatures in a circulating air furnace. The healing temperature was selected based on the rheological study and established at 36 °C in order to maximize network mobility for all the systems in accordance to preliminary temperature sweep analyses. Healing was stopped when there was complete visual disappearance of the macroscopic damage (scar) being observed after 4 days (for HDI_C4DA) and 3 h (for HDI_C7DA and HDI_C8DA) of healing treatment. After healing, the samples were allowed to equilibrate at room temperature (near 20 °C) for 30 min prior to tensile testing. The pristine undamaged samples were tested after high-temperature shape molding (at T = 110 °C) followed by 30 min of equilibration at room temperature. At least three samples were tested for each one of the polymers studied (HDI_C4DA, HDI_C7DA, HDI_C8DA) in their pristine and healed states.
2.4.6. Melt Rheology
Oscillatory shear experiments were carried out on a strain-controlled Physica MCR 102 (Anton Paar GmbH) rheometer using a parallel plate geometry. The diameter of the plate was 8 mm, and the sample thickness was set to 1 mm. Temperature sweep analyses were carried out in the temperature range −20 to 100 °C using a heating rate of 2.8 °C/min. Isothermal frequency sweeps were performed in the range between 0.1 ≤ f ≤ 10 Hz. Both temperature sweeps and isothermal frequency sweeps were performed within the linear viscoelastic regime of the tested polymers. Frequency master curves were shifted to the reference temperature T0 = 20 °C. The shift factor aT for the construction of the master curves follows with good approximation the William–Landel–Ferry (WLF) law27 for HDI_C4DA, HDI_C7DA, and HDI_C8DA, indicating a rather simple thermo-rheological behavior. For HDI_BDO, HDI_C9DA, HDI_C12DA, and HDI_C18DA, the shift factor severely deviated from the WLF law; therefore, no frequency master curve could be constructed.
2.4.7. X-ray Diffraction Analysis
X-ray diffraction (XRD) spectra were recorded using a Rigaku MiniFlex 600 diffractometer depositing the materials on aluminum holders. The angle spans between 2θ = 1 and 60° with 0.1° increments at room temperature. The samples were rotated during the measurement in an evacuated vacuum chamber.
3. Results and Discussion
3.1. Effect of Brush Length on Polyurethane Microstructure and Dynamics
The completion of the polyurethane reaction
is confirmed by FTIR and NMR (Figure 1 and Figure S2). The incorporation
of two well-defined aliphatic brushes is confirmed by integrating
the strong multiplet peak located at δ = 1.25 ppm in the 1H-NMR spectrum. The molecular weight distribution of the polymers
is reported in Table 2. The number (Mn) molecular weight values
are in the range of 20 kDa, suggesting that these linear systems behave
as non-entangled or lightly entangled linear polymers.27 The molecular weight distribution for HDI_BDO
and HDI_C18DA could not be determined since these polymers were insoluble
in THF or NMP. Mn and Mw were found to increase with the brush length. Nevertheless,
the result is most likely an artifact of GPC analysis, which is based
on size exclusion. In this case, it is more appropriate to use as
internal standard the number-average degree of polymerization 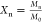 where M0 is
the molecular weight of the repeating unit.28,29 Following these criteria, it appears that the degree of polymerization
is just marginally affected by the brush length. Although the presence
of side reactions leading to cyclization and reduction of polydispersity
cannot be fully excluded, we attribute the narrow polydispersity (Đ < 2) mainly to the suspension of low-molecular-weight
oligomers in the precipitation solvent (methanol). This was confirmed
by GPC analysis of the solid residue left after centrifugation of
the methanol suspensions.
where M0 is
the molecular weight of the repeating unit.28,29 Following these criteria, it appears that the degree of polymerization
is just marginally affected by the brush length. Although the presence
of side reactions leading to cyclization and reduction of polydispersity
cannot be fully excluded, we attribute the narrow polydispersity (Đ < 2) mainly to the suspension of low-molecular-weight
oligomers in the precipitation solvent (methanol). This was confirmed
by GPC analysis of the solid residue left after centrifugation of
the methanol suspensions.
Figure 1.
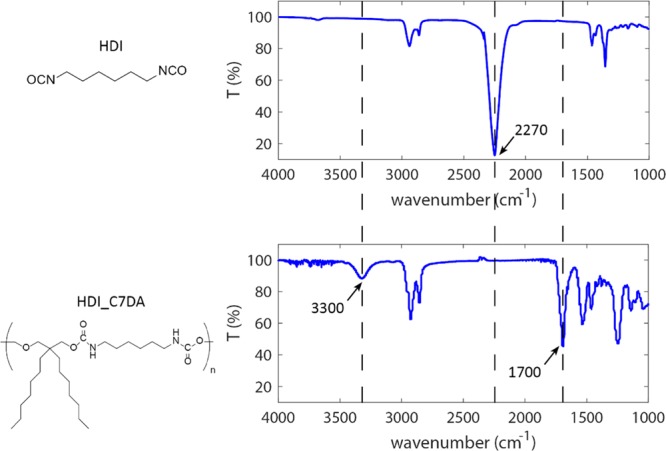
IR spectra of diisocyanate monomers (HDI) and brush polyurethane (HDI_C7DA). The figure highlights the monomer conversion through the disappearance of the characteristic isocyanate band at 2270 cm–1 and the appearance of amide and carbonyl bands at 3300 and 1700 cm–1, respectively.
Figure 2 relates the optically detectable aggregation state of the synthesized polyurethanes (before and after processing at 110 °C for 1 h) to their DSC thermograms (second heating scan) as a function of increasing brush length. All glass and melt transition temperatures are reported in Table 2. Two distinct behaviors can be observed. While the two extremes (no brushes or very long brushes) show one clear crystalline peak, all other polymers appear as amorphous thermoplastics. A total absence of brushes (HDI_BDO) yields a very fine white powder with a high degree of crystallinity explained by the presence of a high density of hydrogen bonds in the main polyurethane chain. The DSC thermogram shows a high-temperature melting peak (at 165 °C) preceded by an endotherm shoulder. As reported elsewhere for linear polyurethanes derived from α,ω-diols,30 the two melting transitions can be attributed to the melting of folded chains and the melting of the extended main-chain crystals given by directional hydrogen bonds among urethane linkages. On the other extreme of the figure (highest branch length), sample HDI_C18DA appears as a fine powder that could be thermally processed into a brittle film. Similar to HDI_BDO, DSC shows the appearance of a melting peak yet at lower temperatures (45 °C). This peak can be attributed to aliphatic side chain (brushes) crystallization as reported for aliphatic polymer chains31,32 and to alkyl side chains in comb-like polymers.33,34 Such strong interactions between the long 18-carbon aliphatic branches seem to prevent the formation of short-range H bonding between the urethane linkages as well as microphase segregation otherwise typical in segmented polyurethanes.35 The presence of well-defined crystalline phases in HDI_BDO and HDI_C18DA is further supported by XRD spectra (Figure S3).
Figure 2.

Effect of brush length on the aggregation state and melt/glass transition temperature as analyzed using DSC (second heating scan was used). In order to obtain homogeneous films, the polymers were processed at 110 °C. The camera snaps captured the polymer before and after processing. For very long brush lengths (C0DA and C18DA), semicrystalline and brittle polymers were synthesized, and no homogeneous films could be obtained. Intermediate brush lengths (C4DA, C7DA, C8DA, C9DA) led to amorphous and ductile polymers that could be processed as homogeneous films for mechanical testing. Long brushes (C12DA) also led to a semicrystalline polymer that could be processed into a homogeneous but brittle film.
The other four compositions leading to amorphous polymers (C4, C7, C8, C9) show a slight decrease of the glass transition temperature with the chain length increase (from ∼30 °C for C4 to 24 ± 1 °C for C7 and C9). In agreement with previous studies on comb polymers with variable branch length,36,37 such a decrease in Tg can be attributed to a local plasticization effect until long branches promote local crystallization (detectable already for HDI_C12DA). Interestingly, these findings are in opposition to those reported by Gerstl et al.38 who showed the absence of internal plasticization for a series of poly(alkylene oxide)s (PAOs) with different side chain lengths. Considering the similarities in terms of side chain length between our PUs and the reported PAOs, the differences in side chain-induced plasticization behavior can be attributed to profound differences in the main backbone chemical structure (e.g., absence of urethane groups and presence of ether groups in the case of PAOs) and a higher branch spacing and branches per branch point in the case of our PUs.
HDI_C12DA shows recrystallization at a cooling rate of 10 °C/min with a melt endotherm at ∼45 °C. By increasing the cooling rate to 20 °C/min, recrystallization is partially avoided, and a glass transition onset is observed just before a small endotherm peak at ∼55 °C in the second heating step. We argue that, when slow cooling is applied (10 °C/min), the flexibility of the fully aliphatic main backbone combined with the plasticization effect due to the long brushes allows macromolecular reorganization toward the most favorable thermodynamic microstructure: a semicrystalline phase of stacked aliphatic side chains. Relevantly, the measured Tm of HDI_C12DA (46 °C) coincides with the melt temperature observed for HDI_C18DA (45 °C) as an additional confirmation that, in this system, crystallization is due to side chain stacking.
Table 2 shows the onset degradation temperature at 2% weight loss. All the polymers show high-temperature stability and a degradation onset between 200 and 300 °C, in agreement with previous reports on linear polyurethanes obtained by polymerization of linear diols and linear diisocyanates.30,39 Thermal stability increases with the brush length. This trend is attributed to the higher thermal screening that long and dense side chains offer to thermally sensitive urethane linkages.40 In this sense, the higher thermal stability of HDI_BDO when compared to HDI_C4DA seems to be an exception. We argue that the improved thermal stability of HDI_BDO is due to the very high degree of hydrogen bonding and main-chain crystallinity, favored by the absence of lateral brushes. Minimal weight loss (<0.5 wt %) is observed at temperatures lower than 200 °C for all the polymers, indicating that no solvents (DMF, methanol) or unreacted monomers are entrapped at the end of the polymerization procedure.
Exemplary stress–strain curves of the amorphous and semicrystalline brush PUs are reported in Figure 3. HDI_BDO and HDI_C18DA could not be processed and tested due to their high crystallinity and brittleness. Young’s modulus (E), yield stress (σy), ultimate tensile strength (σUTS), and strain at break (εbreak) values are reported in Table S-I. When comparing HDI_C4DA, HDI_C7DA, HDI_C8DA, and HDI_C9DA, it can be seen that longer brush lengths lead to decreasing E and increasing εbreak, in analogy to what has been reported for segmented polyurethanes with decreasing hard segment content.41 The abrupt increase of εbreak and reduction of σUTS observed in HDI_C9DA are highly reproducible. HDI_C12DA does not follow the same trend and shows the highest E and σy among the analyzed polymers. We attribute this exceptional mechanical behavior to its crystallinity formed during cooling as previously discussed.
Figure 3.
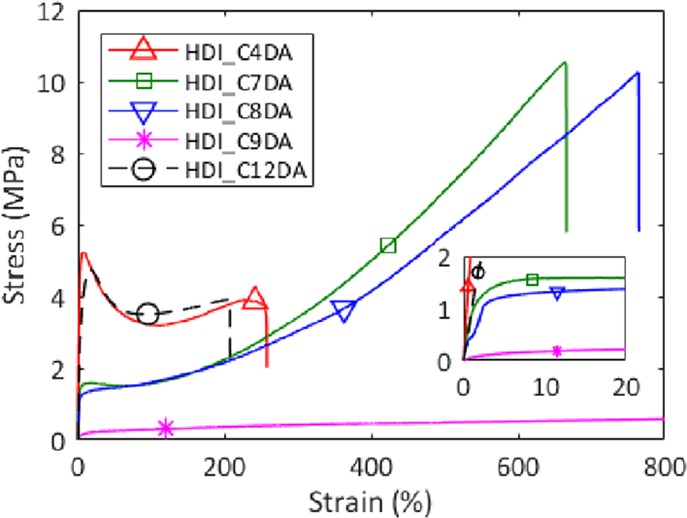
Stress–strain curves at 80 mm/min and 21 °C of HDI_C4DA, HDI_C7DA, HDI_C8DA, HDI_C9DA, and HDI_C12DA. The tests were performed after high-temperature shape molding (at T = 110 °C) followed by 30 min of equilibration at room temperature. The plot shows the effect of brush length on general tensile mechanical performances of amorphous brush polyurethanes. The inset highlights the difference in Young modulus and yield stress among the different systems.
To better understand the role of regular branching in the dynamics of the polyurethanes, we probed their macromolecular dynamics in the melt over a broad range of temperatures and frequencies using the well-known time–temperature superposition (TTS) principle. Although the applicability of the TTS principle is generally limited to fluids showing a thermorheologically simple behavior, its applicability and validity to polymers with a complex architecture and intermolecular interactions as self-healing polymers have recently been discussed in depth and applied to various polymeric systems.42−44
The linear viscoelastic master curve of HDI_C8DA is shown in Figure 4A. Frequency isotherms are shifted to the reference temperature T0 = 20 °C. The data are presented as elastic modulus (G′) and viscous modulus (G″) as a function of the angular frequency (ω). Moreover, viscoelastic data are reported in the form of the tangent of the phase angle (tanδ = G′/G″), which has been reported as being potentially more sensitive in distinguishing features of the relaxation modes associated with aliphatic branches.45−47 By following the trend of tanδ, it can be easily inferred that the spectrum resembles that of lightly entangled polymers where the partial restriction of mobility at ωcr < ω < ωβ is attributed to the existence of varying physical interactions. Frequency master curves of HDI_C4DA and HDI_C7DA were constructed shifting the data at T0 = 20 °C, showing analogous features (Figure S4). For HDI_C4DA, HDI_C7DA, and HDI_C8DA polymers, the trend of the shift factor is well fitted by the William–Landel–Ferry (WLF) law. Frequency master curves of HDI_BDO, HDI_C9DA, HDI_C12DA, and HDI_C18DA are not presented since the shift factor trend severely diverged from the WLF fit. This can be an indication of multiple dynamic phenomena occurring in the same frequency range, as will be discussed in detail further on.
Figure 4.
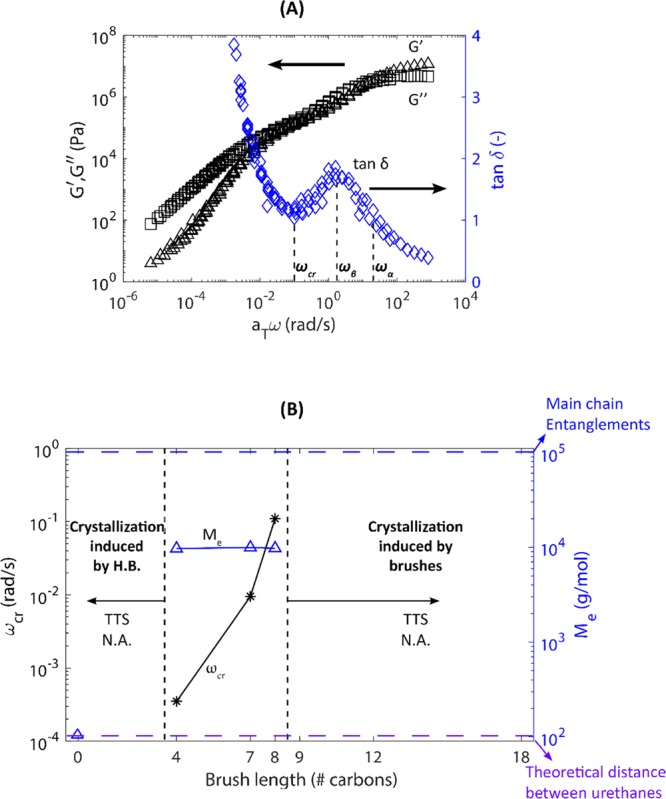
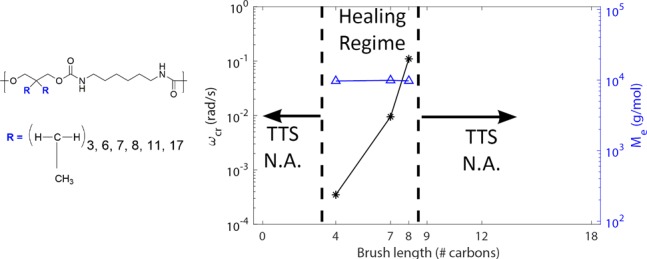
(A) Frequency sweep master curve shifted at T0 = 20 °C of HDI_C8DA. (B) Effect of brush length on the critical angular frequency (ωcr) corresponding to the minimum of tanδ in the apparent plateau region (star markers). Effect of brush length on the molecular weight between entanglements (Me) calculated by rubber elasticity theory (triangle markers). As the effect of enhanced tube dilution ωcr shifts to higher frequencies increasing the brush length, Me is unchanged since the distance among urethane linkages is unvaried for all the polymers analyzed. For brush length higher than 8, the time–temperature superposition (TTS) principle was not applicable. In the absence of brushes (HDI_BDO), TTS was not applicable. In both cases, the dependence of the shift factor on temperature severely deviated from the WLF law. Dashed lines point out the theoretical molecular weight between contiguous urethane units (dashed purple line) and the theoretical main-chain molecular weight corresponding to the occurrence of main-chain entanglements (dashed blue line).
Analysis of the values of the molecular weight
between entanglements 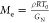 (as calculated for rubber
elasticity where
ρ is the polymer density (assumed 1 kg/L for all the systems), R is the universal gas constant, T0 is the shifting temperature, and GN is the plateau modulus as obtained from the van Gurp–Palmen
plot48,49) clarifies the origin of the physical interactions
causing the partial restriction of dynamics at ωcr < ω < ωβ. For HDI_C4DA, HDI_C7DA,
and HDI_C8DA, Me ≈ 10.000 g/mol
(Figure 4B). For all
these polymers, the length of the brushes is lower than Me (brush length ≈150 g/mol); therefore, the brushes
cannot entangle and cannot justify the high mobility restriction at
the intermediate frequency (in the region ωcr <
ω < ωβ), in a similar fashion to what
has been observed elsewhere for comb polymers with short branches.45,46 Moreover, the rather low molecular weight of these systems (Mn ≈ 20 – 34 kDa, Table 2) cannot lead to mobility restrictions
at intermediate frequencies due to main-chain entanglements as discussed
by Doi and Edwards who showed that a plateau modulus due to main-chain
entanglements sets for Mw > 4Me and becomes pronounced for Mw > 8Me26 (represented
as a dashed blue line in Figure 4B). Considering the absence of brush and main-chain
entanglements, we attribute the temporary restriction of dynamics
in the region ωcr < ω < ωβ to the presence of a physical network consisting of hydrogen bonds
among urethane linkages, in line with the design of densely branched
systems bearing effective supramolecular stickers. An additional proof
of the presence of effective supramolecular interactions comes from
the measure of G′ and G″
slopes in the terminal relaxation region (for HDI_C8DA in Figure 4A, slope G′ = 1.5, slope G″ = 0.8),
which severely deviate from slopes of ideal Rousian dynamics (2 and
1) as previously reported in comparable branched healing polymers.26
(as calculated for rubber
elasticity where
ρ is the polymer density (assumed 1 kg/L for all the systems), R is the universal gas constant, T0 is the shifting temperature, and GN is the plateau modulus as obtained from the van Gurp–Palmen
plot48,49) clarifies the origin of the physical interactions
causing the partial restriction of dynamics at ωcr < ω < ωβ. For HDI_C4DA, HDI_C7DA,
and HDI_C8DA, Me ≈ 10.000 g/mol
(Figure 4B). For all
these polymers, the length of the brushes is lower than Me (brush length ≈150 g/mol); therefore, the brushes
cannot entangle and cannot justify the high mobility restriction at
the intermediate frequency (in the region ωcr <
ω < ωβ), in a similar fashion to what
has been observed elsewhere for comb polymers with short branches.45,46 Moreover, the rather low molecular weight of these systems (Mn ≈ 20 – 34 kDa, Table 2) cannot lead to mobility restrictions
at intermediate frequencies due to main-chain entanglements as discussed
by Doi and Edwards who showed that a plateau modulus due to main-chain
entanglements sets for Mw > 4Me and becomes pronounced for Mw > 8Me26 (represented
as a dashed blue line in Figure 4B). Considering the absence of brush and main-chain
entanglements, we attribute the temporary restriction of dynamics
in the region ωcr < ω < ωβ to the presence of a physical network consisting of hydrogen bonds
among urethane linkages, in line with the design of densely branched
systems bearing effective supramolecular stickers. An additional proof
of the presence of effective supramolecular interactions comes from
the measure of G′ and G″
slopes in the terminal relaxation region (for HDI_C8DA in Figure 4A, slope G′ = 1.5, slope G″ = 0.8),
which severely deviate from slopes of ideal Rousian dynamics (2 and
1) as previously reported in comparable branched healing polymers.26
In Figure 4B, we compare the frequency shift associated with the temporary mobility restriction (ω = ωcr) of HDI_C4DA, HDI_C7DA, and HDI_C8DA following the approach proposed by Kapnistos45 for comb-like polymers bearing short aliphatic branches. ωcr shifts to higher frequencies for higher brush length. Since ωcr determines the access to terminal relaxation, we can conclude that the higher the brush length, the easier it is for the main chain to access terminal relaxation. The most likely explanation is that the aliphatic brushes act as a solvent for the main chain (tube dilation), speeding up the main-chain relaxation. The dilation effect is magnified for higher brush length, as observed elsewhere for comb polymers.45,46 Moreover, Figure 4B (triangle markers) shows that Me appears to be unaffected by the brush length. This confirms the presence of a physical network between urethane linkages. The physical cross-linking distance is unchanged by increasing the brush length since it is controlled by the length of the repeating unit (thereby, the distance among urethane linkages).
In Figure 4B, we highlight two regions in which time–temperature superposition was not applicable because the shift factor dependence on temperature severely deviated from the WLF law. For HDI_BDO (not containing any side chains), we argue that the dynamics are governed by main-chain crystallization driven by hydrogen bonds among urethane units. For branch lengths higher than 8 (HDI_C9DA, HDI_C12DA, HDI_C18DA), the inapplicability of TTS is attributed to the existence of a concurrent dynamic process due to side chain interactions and stacking.
Three idealized polymer architecture sketches are shown in Figure 5: a non-brushed highly crystalline PU (HDI_BDO), an amorphous brushed PU with effective hydrogen bonding among urethane blocks (HDI_C8DA), and a densely brushed highly crystalline PU with stacked aliphatic side chains (HDI_C18DA).
Figure 5.
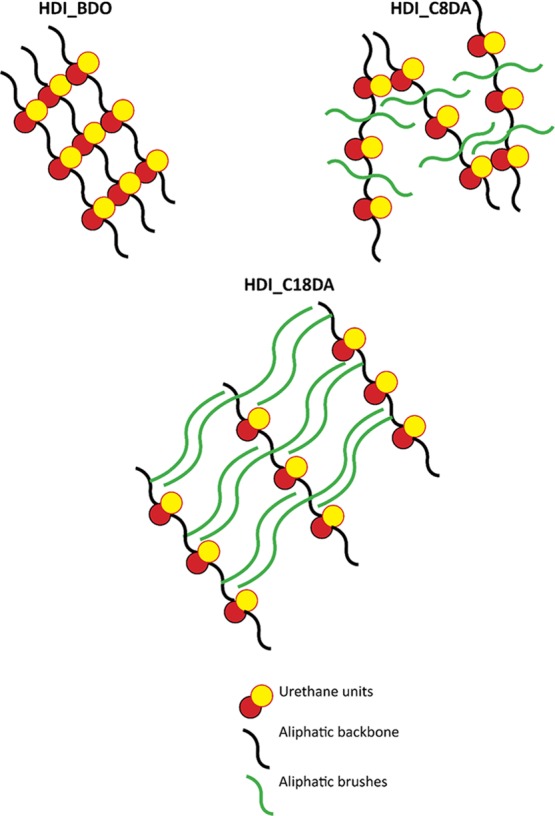
Idealized sketches of the macromolecular architecture of various brush polyurethanes with different brush lengths. In the figure, we highlight the formation of well-defined crystalline phases in the absence of aliphatic brushes (HDI_BDO) and for very long brushes (C = 18, HDI_C18DA). For medium brush length (C = 8, HDI_C8DA), a glassy phase is formed with the presence of a physical network among urethane linkages.
Melt rheology confirmed the presence of hydrogen bonding interactions among urethane units and the dilution effect operated by aliphatic side chains in amorphous brush polyurethanes. Consequently, we investigated the intrinsic self-healing property by tensile testing for all the systems for which a frequency master curve was correctly constructed.
Figure 6A shows the healing results of HDI_C4DA, HDI_C7DA, and HDI_C8DA. The healing was performed at 36 °C, corresponding to Thealing = Tmax tan δ + 5 ° C of HDI_C4DA, in order to maximize network mobility for all the systems investigated. Figure 6B effectively shows the complete recovery of the strength of the cracked interface at the end of the healing treatment for HDI_C4DA. From Figure 6A, it is evident that both E and σy are fully recovered for all optimally healed systems. Final fracture took place elsewhere in the sample rather than at the healed original fracture plane.
Figure 6.
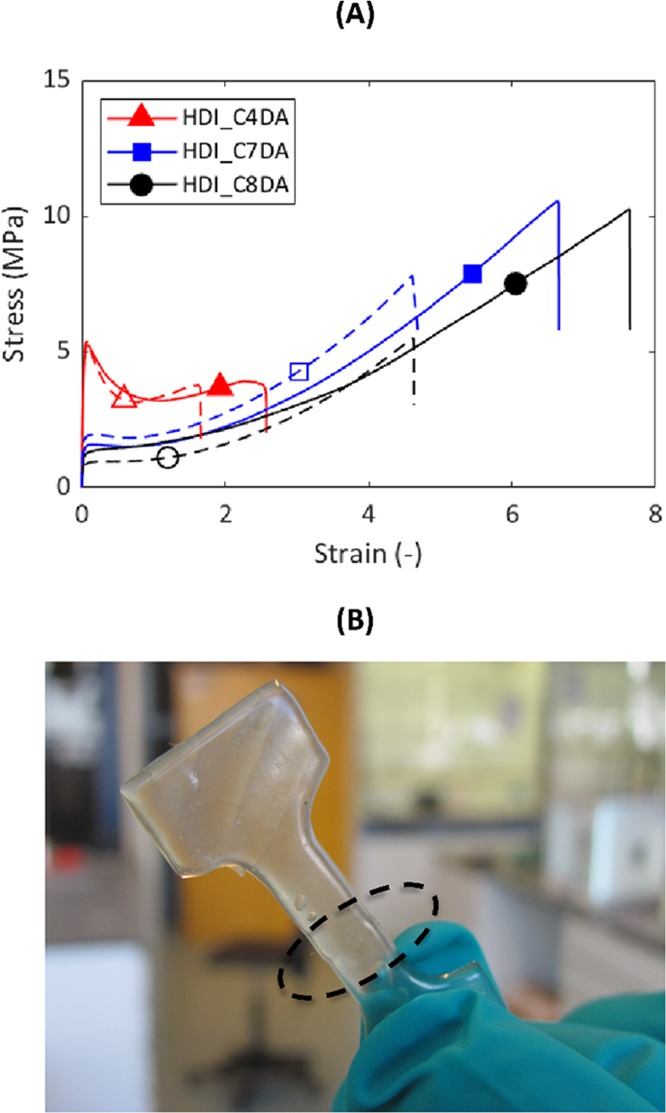
(A) Stress–strain curves at 80 mm/min and 21 °C of various healing brush polyurethanes. Pristine samples are reported as continuous lines, and healed samples are reported as dashed lines. HDI_C4DA was healed for 100 h at 36 °C. HDI_C8DA and HDI_C7DA were healed for 3 h at 36 °C. The plot shows recovery of Young’s modulus and yield stress. (B) Photo capture of HDI_C4DA after healing for 100 h at 36 °C. The black circle highlights the position of the original fracture plane.
By connecting macrorheology to tensile testing, the healing mechanism is identified as a three-stage process in a similar fashion to what Susa et al. observed for a set of self-healing polyimides.18 The first stage “self-adhesion” is governed by short-range interactions such as interfacial multiple reforming hydrogen bonding interactions among urethane units at the fractured plane. The dynamics of the reversible bond is consistent with the intermediate range of bond lifetimes previously reported for functional (and self-healing) supramolecular polymers44,50 (reversible bond lifetime τbond = 10 – 100 s) as shown in section 3.2. The second and third stages of healing (“interdiffusion” and “randomization”) are then governed by sticky Rouse reptation occurring at low frequencies, at the ultimate crossover between storage and loss moduli (see Figure 4).
The effect of the brush length on
the healing kinetics is readily
observed by comparing the mechanical characteristic of HDI_C4DA to
HDI_C7DA and HDI_C8DA in Figure 6A and is summarized in Table 3 where healing efficiency is quantified as 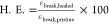 . At
equal healing temperature (Thealing =
36 °C), HDI_C8DA and HDI_C7DA
show comparable healing efficiency to HDI_C4DA (from 60 to 70% recovery
of pristine strain at break), yet in a much shorter time (3 h versus
100 h). All samples showed fracture far from the original fracture
plane. The result is in line with observations drawn in the macrorheology
study that pointed at an acceleration of the dynamics of the main
chain (ωcr shifts toward higher frequencies) with
increasing brush length, justifying rapid randomization at crack sites.
. At
equal healing temperature (Thealing =
36 °C), HDI_C8DA and HDI_C7DA
show comparable healing efficiency to HDI_C4DA (from 60 to 70% recovery
of pristine strain at break), yet in a much shorter time (3 h versus
100 h). All samples showed fracture far from the original fracture
plane. The result is in line with observations drawn in the macrorheology
study that pointed at an acceleration of the dynamics of the main
chain (ωcr shifts toward higher frequencies) with
increasing brush length, justifying rapid randomization at crack sites.
Table 3. Macroscopic Intrinsic Self-Healing Data of Amorphous Brush Polyurethane Systemsa.
| polymer | healing time at Thealing = 36 °C (h) | H.E.b (%) | broken in a different spot compared to original fracture plane |
|---|---|---|---|
| HDI_C4DA | 100 | 64 | yes |
| HDI_C7DA | 3 | 69 | yes |
| HDI_C8DA | 3 | 61 | yes |
Healing process was stopped when the healed interface was not visible.
Healing efficiency was quantified
as 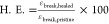 .
.
The amorphous brush polyurethanes showed high mechanical properties and rapid healing at near room temperature. However, the novel polymer architecture endowed with short yet dense aliphatic branches may highly influence the kinetics of crystallization with consequences on the intrinsic healing of these brushed polyurethanes. We investigated these effects and discussed the outcomes in the next section.
3.2. Impact of Induced Crystallization on the Healing Behavior
(Annealing-induced) crystallization is a potential threat for intrinsic healing polymers not commonly studied. Clustering and crystallization hinder chain interdiffusion and cause interfacial embrittlement, preventing randomization at crack sites, thereby affecting the healing process. The issue becomes particularly crucial for systems with a high density of noncovalent interactions since they are more prone to form ordered phases. It has been reported that polyurethanes undergo morphology reorganization and phase transition when subjected to long-term annealing.51−53 Nevertheless, no report has shown this potential threat to the healing process in self-healing polymers and polyurethanes in particular. In order to explore the possible impact of annealing-induced crystallization in our healable branched polymers, we performed a detailed annealing–crystallization analysis with special focus on the polymers that appear amorphous when cooling from the melt state.
Figure 7 shows the effect of long-term annealing at Tg on the mechanical properties of HDI_C7DA and HDI_C8DA. The analysis of traces a–d shows that, upon annealing for 170 h at Tg (= 27 °C), no substantial differences in the pristine mechanical properties and in healing efficiency are observed for HDI_C7DA. We argue that, in the observed timescale, annealing does not lead to any microstructural evolution for HDI_C7DA.
Figure 7.

HDI_C7DA mechanical tensile properties (traces a–d). No differences were observed in healing efficiency and mechanical properties upon long-term (170 h) annealing at Tg. HDI_C8DA mechanical tensile properties (traces e–k). Good healing efficiency was observed when healing fresh samples at T = 36 °C (traces e and f). No recovery was observed when healing fresh samples at Tg (trace g) as an effect of induced crystallization. An increase of strength and drastic reduction of healing efficiency were observed upon long-term (170 h) annealing at Tg and subsequent healing at T = 36 °C as an effect of induced crystallization (traces h and j). An increase of strength and good healing efficiency were observed when subjecting fresh and healed samples to long-term (170 h) annealing at Tg (trace k).
On the other hand, the evolution of the mechanical properties of HDI_C8DA reflects its dynamic character at Tg. Traces e and f show the pristine and healed mechanical properties of fresh (molded and equilibrated) samples. From these, we infer that optimal healing is obtained when healing fresh HDI_C8DA at T = 36 °C for 3 h. Trace h shows that, in contrast to what was observed for HDI_C7DA, the pristine mechanical properties of HDI_C8DA are greatly affected by the annealing procedure at Tg (170 h at 21 °C). Toughness and strength increased, while strain at break was reduced, indicating annealing induced crystallization. DSC analysis performed on a sample having received this 170 h annealing procedure confirmed the transition from a purely amorphous network (Tg = 21 °C before annealing) to a semicrystalline network (Tm = 49 °C). The newly found melting temperature nicely matches the melting temperature probed for HDI_C18DA (Tm = 45 °C). Therefore, we attributed the induced crystallinity to the stacking of the aliphatic brushes.
The semicrystalline HDI_C8DA was healed at T = 36 °C (well below the newly found Tm) to study the potential effect of crystallinity on intrinsic healing. The mechanical results upon healing are reported as trace j in Figure 7, while Figure 8 shows snapshots of the crack evolution at different healing times and corresponding DSC traces. The resulting mechanical healing efficiency dropped drastically (H.E. ≈ 30%) when compared to that of a fresh specimen, but interestingly the broken sample still showed tackiness and 2D interface recovery, even at a healing temperature that was well below the bulk melting temperature. This behavior can be attributed to local plasticization at the newly created free surface, which allows local network mobility well below the bulk melting temperature.54,55 On the other hand, the complete suppression of long-range dynamics (due to bulk crystallization) explains the persistence of the original damage scar and the low healing efficiency even when an extended healing time of 120 h was used.
Figure 8.

Photo captures showing progressive scar disappearance in semicrystalline HDI_C8DA when subjected to healing at T = Tm – 10 °C = 36 °C. The mechanical tensile property of the healed specimen corresponds to trace j of Figure 7. Time-resolved DSC analysis shows the persistence of the crystalline phase throughout the healing process.
Trace k addresses the effect of induced crystallization if the sample is annealed further once optimal healing of a fresh sample is completed. To this purpose, a fresh sample was cut, healed for 3 h at T = 36 °C, then annealed at Tg for 170 h, and tensile-tested. The mechanical properties resemble that of the semicrystalline sample (trace h) and good healing efficiency in terms of strain recovery (H.E. = 70%). In this case, both short- and long-range dynamics were accessed prior to bulk crystallization, justifying the high degree of healing in terms of strain. On the other hand, different from fresh and healed samples (traces e and f), trace k samples broke at the original damage site and showed a significant reduction of yield stress compared to the pristine sample. These effects are attributed to the loss of interfacial strength at healed sites induced by crystallization.
Since many healing systems report healing close to Tg, one question was still open: what would happen if a fresh sample of HDI_C8DA was healed for a long time at Tg? The results of this healing test are reported as trace g in Figure 7. While showing tackiness (typical of initial network mobility), at the end of the healing procedure (170 h at Tg), the healed sample suffered from embrittlement and immediately failed at the original damage site when tensile-tested. This demonstrates that no healing is possible in such a dynamic system where phase transition (from purely amorphous to semicrystalline) occurs at Tg.
Healing “reactivation” for semicrystalline HDI_C8DA was qualitatively proven by subjecting an annealed sample (170 h at Tg) to the healing temperature above the melting temperature (T = Tm + 10 °C = 60 °C). The results are reported in Figure 9A. The snapshots shows that original damage was completely restored in a very short timescale (healing time = 10 min). DSC analysis shows that the amorphous state was restored upon heating to 60 °C, indicating the reactivation of long range dynamics. The HDI_C18DA sample was healed in a similar fashion above the melting temperature (T = Tm + 10 °C = 55 °C). Even in this case, rapid scar disappearance was observed (Figure 9B), but DSC analysis at the end of the healing procedure confirmed that, in this case, bulk crystallinity is restored immediately after equilibration at room temperature.
Figure 9.
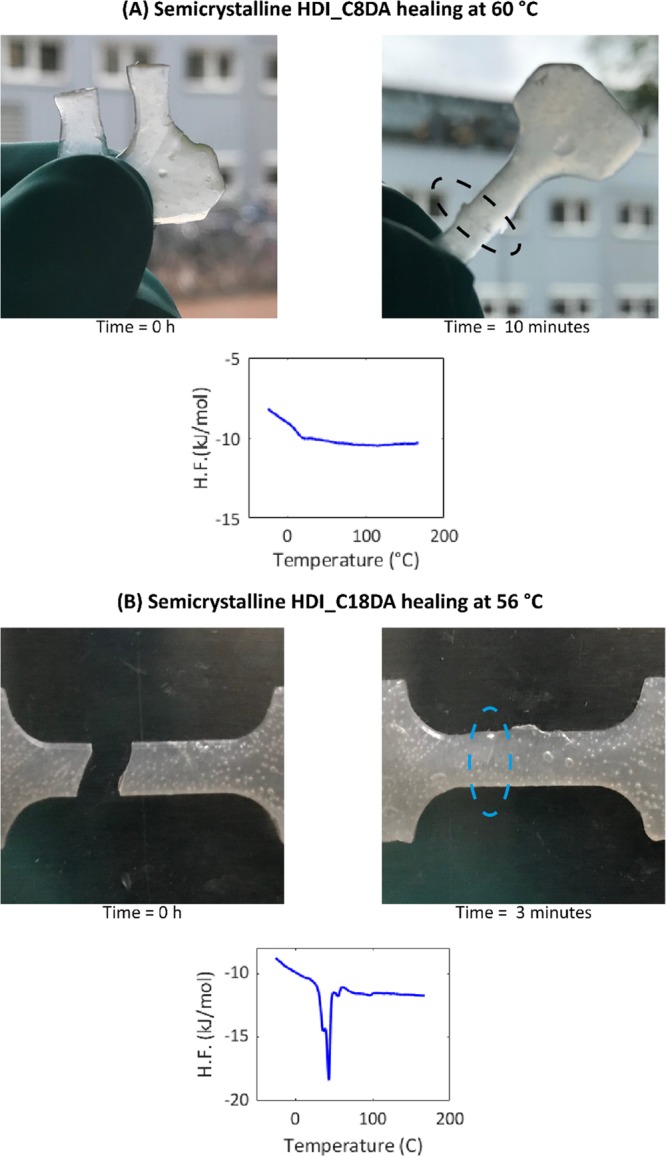
(A) Photo captures showing fast scar disappearance and optimal healing for semicrystalline HDI_C8DA upon healing at T = Tm + 10 °C = 60 °C. DSC trace shows the absence of any crystalline phase upon healing. (B) Photo captures showing fast scar disappearance and optimal healing for semicrystalline HDI_C18DA when healed at T = Tm + 10 °C = 56 °C. DSC trace shows the reformation of a crystalline phase upon healing.
The study evidenced the critical effect of annealing-induced crystallization on healing of HDI_C8DA as well as the apparent absence of crystallinity for HDI_C7DA in the observed timescale. Therefore, we performed a dedicated study on the effect of brush length on the kinetics of crystallization. To this purpose, DSC analyses were performed every 24 h while keeping the polyurethanes at their individual annealing temperatures Tann = Tmax tan δ + 5 ° C chosen as such in order to maximize network mobility. Initially, HDI_C4DA, HDI_C7DA, HDI_C8DA, and HDI_C9DA did not show the presence of any melting peak. With increasing residence time at Tann, some systems progressively underwent phase transition and crystallization, as evidenced by clear melting peaks in the DSC thermograms. Table 4 reports the used annealing temperature (Tann), the time corresponding to the end of the crystallization process (tcrys), and the melting temperature of the crystalline phase (Tm). Analyzing tcrys, we note that the decrease of brush length from 12 to 4 carbons corresponds to a progressive delay of crystallization kinetics. Crystallization seems to be absent for the short brush length (HDI_C7DA and HDI_C4DA) since no melt peak was observed on a short timescale (170 h) nor on a very long annealing timescale (1500 h). Interestingly, all brush polyurethanes showing crystallization upon annealing (HDI_C8DA, HDI_C9DA, HDI_C12DA) report melting temperatures in a common range, from 45 to 50 °C. These temperatures match well with the melting temperature of HDI_C18DA, which was attributed to side chain stacking. Therefore, we argue that crystallization with annealing in fully aliphatic brush polyurethanes occurs by side chain stacking until a limiting side chain length (C = 8) below which brushes are too short to stack and nucleate crystals while they accelerate healing kinetics. These results highlight the crucial role of polymer architecture in the design of efficient and thermally stable healing systems with high density of reversible bonds.
Table 4. Crystallization Time of Brush Polyurethanesa.
| polymer | annealing temperature (°C) | time for complete crystallization [tcrys] (h) | melting temperature [Tm] (°C) |
|---|---|---|---|
| HDI_BDO | n.a.b | n.a.b | 165 |
| HDI_C4DA | 36 | n.a.c | n.a.c |
| HDI_C7DA | 27 | n.a.c | n.a.c |
| HDI_C8DA | 21 | 170 | 49 |
| HDI_C9DA | 21 | 72 | 45 |
| HDI_C12DA | 21 | 24 | 46 |
| HDI_C18DA | n.a.b | n.a.b | 45 |
Note that the kinetics slowed down when reducing the length of the brush. The crystallization kinetics was eventually suppressed for HDI_C4DA and HDI_C7DA.
HDI_BDO and HDI_C18DA are semicrystalline when cooling from the melt state.
No crystallization was observed for HDI_C4DA and HDI_C7DA when annealing for 1500 h.
4. Conclusions
Regular branching is investigated in polyurethanes with a high density of reversible bonds as the design strategy to exploit the near-room-temperature intrinsic healing property in robust linear polymers. To this end, we synthesized a series of brush polyurethanes by facile polymerization of diols bearing branches with controlled length from C4 to C18 and commercial diisocyanate (HDI).
The length of the side aliphatic brush exerts a crucial role in regulating chain dynamics. By using melt rheology, we verified that effective hydrogen bonding interactions among urethane blocks result in a physical network. In the intermediate branch length regime, the brushes, in analogy to comb polymers with short side chains, act as a solvent, dilating the tube, therefore speeding up main-chain interdiffusion, mediating macroscopic self-healing kinetics. At a critical brush length (C = 9) and for all the higher brush lengths, the macromolecular glassy dynamics is totally suppressed by the occurrence of a crystalline phase of stacked side chains.
A detailed calorimetry study clarified the effect of brush length on annealing-induced crystallization. To this end, the polymer systems were subjected to long-term annealing at the corresponding glass transition/healing temperature. A higher brush length speeds up the crystallization kinetics. No crystallization was observed for the brush polymers synthesized with a side chain length in the range between C = 4 and 7. The result highlights that this range of brush lengths is the most promising in order to obtain intrinsic healable polymers with a high extent of reversible bond interactions and hence mechanical robustness, yet avoiding side chain crystallization, which would block the healing process. Interestingly, the polymers showing interfacial healing were the ones that met the WLF law, leading to nicely overlapping segments in the final frequency master curve.
Relevantly, we observed that, for crystallized samples, a local reactivation of mobility is still accessible at mild temperatures, below Tm. This local mobility allows partial recovery of mechanical damage in a reasonable timescale. Ultimately, total (bulk) reactivation of dynamics is observed upon healing for a very short time at temperatures above the melting temperature.
In conclusion, we demonstrated that polymer architecture control involving regular branching and a high density of reversible physical bonds can be an effective strategy to obtain near-room-temperature healable polymers with good mechanical properties, provided the side brush length is in the correct range. The approach leaves room for further optimization of both mechanical and healing properties through the control of the number of branching points per macromolecule and the use of multidentate reversible bond groups (e.g., urea groups).
Acknowledgments
The authors acknowledge the financial support of Croda Nederland BV and the Dutch National Organization for Scientific Research, Domain Applied and Engineering Sciences (NWO-TTW) under the grant number 15010. The authors acknowledge Dr. Catarina Esteves, Ingeborg Schreur-Piet, and Marco Hendrix (Eindhoven University of Technology) for their support with the melt rheology tests. Many thanks are given to Dr. Angela Smits (Croda) for helpful discussions and valuable feedback.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.9b01554.
H-NMR and C-NMR of synthesized diols and synthesized brush polyurethanes; XRD spectra of HDI_BDO and HDI_C18DA; Young’s modulus, yield stress, ultimate tensile strength, and strain at break of selected brush polyurethanes; frequency master curves of HDI_C4DA and HDI_C7DA shifted at T0 = 20 °C and low-molecular-weight HDI_C8DA; temperature sweep analyses of amorphous and semicrystalline brush polyurethanes; XRD spectra of HDI_C8DA at different annealing times; mechanical tensile test of HDI_C8DA after healing for 7 days at 21 °C (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hager M. D.; Greil P.; Leyens C.; van der Zwaag S.; Schubert U. S. Self-Healing Materials. Adv. Mater. 2010, 22, 5424–5430. 10.1002/adma.201003036. [DOI] [PubMed] [Google Scholar]
- Wool R. P. Self-Healing Materials: A Review. Soft Matter 2008, 4, 400–418. 10.1039/b711716g. [DOI] [PubMed] [Google Scholar]
- Diesendruck C. E.; Sottos N. R.; Moore J. S.; White S. R. Biomimetic Self-Healing. Angew. Chem., Int. Ed. 2015, 54, 10428–10447. 10.1002/anie.201500484. [DOI] [PubMed] [Google Scholar]
- Liu Y. L.; Chuo T. W. Self-Healing Polymers Based on Thermally Reversible Diels-Alder Chemistry. Polym. Chem. 2013, 4, 2194–2205. 10.1039/c2py20957h. [DOI] [Google Scholar]
- Nevejans S.; Ballard N.; Miranda J. I.; Reck B.; Asua J. M. The Underlying Mechanisms for Self-Healing of Poly(disulfide)s. Phys. Chem. Chem. Phys. 2016, 18, 27577–27583. 10.1039/C6CP04028D. [DOI] [PubMed] [Google Scholar]
- Ying H.; Zhang Y.; Cheng J. Dynamic Urea Bond for the Design of Reversible and Self-Healing Polymers. Nat. Commun. 2014, 5, 3218. 10.1038/ncomms4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P.; McCarthy T. J. A Surprise from 1954: Siloxane Equilibration Is a Simple, Robust, and Obvious Polymer Self-Healing Mechanism. J. Am. Chem. Soc. 2012, 134, 2024–2027. 10.1021/ja2113257. [DOI] [PubMed] [Google Scholar]
- Capelot M.; Montarnal D.; Tournilhac F.; Leibler L. Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. 10.1021/ja302894k. [DOI] [PubMed] [Google Scholar]
- Abdolahzadeh M.; C. Esteves A. C.; van der Zwaag S.; Garcia S. J. Healable Dual Organic-Inorganic Crosslinked Sol-Gel Based Polymers: Crosslinking Density and Tetrasulfide Content Effect. J. Polym. Sci. Part A: Polym. Chem. 2014, 52, 1953–1961. 10.1002/pola.27200. [DOI] [Google Scholar]
- Post W.; Cohades A.; Michaud V.; van der Zwaag S.; Garcia S. J. Healing of a Glass Fibre Reinforced Composite with a Disulphide Containing Organic-Inorganic Epoxy Matrix. Compos. Sci. Technol. 2017, 152, 85–93. 10.1016/j.compscitech.2017.09.017. [DOI] [Google Scholar]
- Chen X.; Dam M. A.; Ono K.; Mal A.; Shen H.; Nutt S. R.; Sheran K.; Wudl F. A Thermally Re-Mendable Cross-Linked Polymeric Material. Science 2002, 295, 1698–1702. 10.1126/science.1065879. [DOI] [PubMed] [Google Scholar]
- Brutman J. P.; Delgado P. A.; Hillmyer M. A. Polylactide Vitrimers. ACS Macro Lett. 2014, 3, 607–610. 10.1021/mz500269w. [DOI] [PubMed] [Google Scholar]
- Cordier P.; Tournilhac F.; Soulié-Ziakovic C.; Leibler L. Self-Healing and Thermoreversible Rubber from Supramolecular Assembly. Nature 2008, 451, 977–980. 10.1038/nature06669. [DOI] [PubMed] [Google Scholar]
- Phadke A.; Zhang C.; Arman B.; Hsu C.-C.; Mashelkar R. A.; Lele A. K.; Tauber M. J.; Arya G.; Varghese S. Rapid Self-Healing Hydrogels. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 4383–4388. 10.1073/pnas.1201122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burattini S.; Greenland B. W.; Merino D. H.; Weng W.; Seppala J.; Colquhoun H. M.; Hayes W.; MacKay M. E.; Hamley I. W.; Rowan S. J. A Healable Supramolecular Polymer Blend Based on Aromatic π-π Stacking and Hydrogen-Bonding Interactions. J. Am. Chem. Soc. 2010, 132, 12051–12058. 10.1021/ja104446r. [DOI] [PubMed] [Google Scholar]
- Nakahata M.; Takashima Y.; Yamaguchi H.; Harada A. Redox-Responsive Self-Healing Materials Formed from Host–guest Polymers. Nat. Commun. 2011, 2, 511. 10.1038/ncomms1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban M. W.; Davydovich D.; Yang Y.; Demir T.; Zhang Y.; Casabianca L. Key-and-Lock Commodity Self-Healing Copolymers. Science 2018, 362, 220–225. 10.1126/science.aat2975. [DOI] [PubMed] [Google Scholar]
- Susa A.; Bose R. K.; Grande A. M.; van der Zwaag S.; Garcia S. J. Effect of the Dianhydride/Branched Diamine Ratio on the Architecture and Room Temperature Healing Behavior of Polyetherimides. ACS Appl. Mater. Interfaces 2016, 8, 34068–34079. 10.1021/acsami.6b10433. [DOI] [PubMed] [Google Scholar]
- Adolf D.; Tirrell M.; Prager S. Molecular Weight Dependence of Healing and Brittle Fracture in Amorphous Polymers above the Entanglement Molecular Weight. J. Polym. Sci.: Polym. Phys. Ed. 1985, 23, 413–427. 10.1002/pol.1985.180230214. [DOI] [Google Scholar]
- Lebel O.; Maris T.; Perron M.-È.; Demers E.; Wuest J. D. The Dark Side of Crystal Engineering: Creating Glasses from Small Symmetric Molecules That Form Multiple Hydrogen Bonds. J. Am. Chem. Soc. 2006, 128, 10372–10373. 10.1021/ja063353s. [DOI] [PubMed] [Google Scholar]
- Stupp S. I.; LeBonheur V.; Walker K.; Li L. S.; Huggins K. E.; Keser M.; Amstutz A. Supramolecular Materials: Self-Organized Nanostructures. Science 1997, 276, 384–389. 10.1126/science.276.5311.384. [DOI] [PubMed] [Google Scholar]
- Leung L. M.; Koberstein J. T. DSC Annealing Study of Microphase Separation and Multiple Endothermic Behavior in Polyether-Based Polyurethane Block Copolymers. Macromolecules 1986, 19, 706–713. 10.1021/ma00157a038. [DOI] [Google Scholar]
- Garcia S. J. Effect of Polymer Architecture on the Intrinsic Self-Healing Character of Polymers. Eur. Polym. J. 2014, 53, 118–125. 10.1016/j.eurpolymj.2014.01.026. [DOI] [Google Scholar]
- Neal J. A.; Mozhdehi D.; Guan Z. Enhancing Mechanical Performance of a Covalent Self-Healing Material by Sacrificial Noncovalent Bonds. J. Am. Chem. Soc. 2015, 137, 4846–4850. 10.1021/jacs.5b01601. [DOI] [PubMed] [Google Scholar]
- Yanagisawa Y.; Nan Y.; Okuro K.; Aida T. Mechanically Robust, Readily Repairable Polymers via Tailored Noncovalent Cross-Linking. Science 2018, 359, 72–76. 10.1126/science.aam7588. [DOI] [PubMed] [Google Scholar]
- Susa A.; Mordvinkin A.; Saalwächter K.; van der Zwaag S.; Garcia S. J. Identifying the Role of Primary and Secondary Interactions on the Mechanical Properties and Healing of Densely Branched Polyimides. Macromolecules 2018, 51, 8333–8345. 10.1021/acs.macromol.8b01396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferry J. D.Viscoelastic Properties of Polymers; Wiley, 1980. [Google Scholar]
- Vizárová K.; Kirschnerová S.; Kačík F.; Briškárová A.; Šutý Š.; Katuščák S. Relationship between the Decrease of Degree of Polymerisation of Cellulose and the Loss of Groundwood Pulp Paper Mechanical Properties during Accelerated Ageing. Chem. Pap. 2012, 66, 1124–1129. 10.2478/s11696-012-0236-1. [DOI] [Google Scholar]
- Chiantore O.; Luda di Cortemiglia M. P.; Guaita M. Changes of Degree of Polymerisation in the Thermal Degradation of Poly(methyl Methacrylate). Polym. Degrad. Stab. 1989, 24, 113–126. 10.1016/0141-3910(89)90106-7. [DOI] [Google Scholar]
- McKiernan R. L.; Gido S. P.; Penelle J. Synthesis and Characterization of Polyethylene-like Polyurethanes Derived from Long-Chain, Aliphatic A,ω-Diols. Polymer 2002, 43, 3007–3017. 10.1016/S0032-3861(02)00096-4. [DOI] [Google Scholar]
- Kaufman H. S.; Sacher A.; Alfrey T.; Fankuchen I. Side-Chain crystallisation in Alkyl Polyacrylates. J. Am. Chem. Soc. 1948, 70, 3147–3147. 10.1021/ja01189a515. [DOI] [Google Scholar]
- Greenberg S. A.; Alfrey T. Side Chain Crystallization of N-Alkyl Polymethacrylates and Polyacrylates1. J. Am. Chem. Soc. 1954, 76, 6280–6285. 10.1021/ja01653a015. [DOI] [Google Scholar]
- Hirabayashi T.; Kikuta T.; Kasabou K.; Yokota K. Main-Chain Flexibility and Side-Chain Crystallization of Widely Spaced Comb-Like Polymers. Polym. J. 1988, 20, 693–698. 10.1295/polymj.20.693. [DOI] [Google Scholar]
- Shi H.; Zhao Y.; Dong X.; Zhou Y.; Wang D. Frustrated Crystallisation and Hierarchical Self-Assembly Behaviour of Comb-like Polymers. Chem. Soc. Rev. 2013, 42, 2075–2099. 10.1039/C2CS35350D. [DOI] [PubMed] [Google Scholar]
- He Y.; Xie D.; Zhang X. The Structure, Microphase-Separated Morphology, and Property of Polyurethanes and Polyureas. J. Mater. Sci. 2014, 49, 7339–7352. 10.1007/s10853-014-8458-y. [DOI] [Google Scholar]
- Lee Y.-L; Sung P.-H; Liu H.-T; Chou L.-C; Ku W.-H. Dangling Polymer Networks: Glass Transition of PU Elastomers. J. Appl. Polym. Sci. 1993, 49, 1013–1018. 10.1002/app.1993.070490607. [DOI] [Google Scholar]
- Yu W.; Du M.; Zhang D.; Lin Y.; Zheng Q. Influence of Dangling Chains on Molecular Dynamics of Polyurethanes. Macromolecules 2013, 46, 7341–7351. 10.1021/ma401260d. [DOI] [Google Scholar]
- Gerstl C.; Schneider G. J.; Pyckhout-Hintzen W.; Allgaier J.; Richter D.; Alegría A.; Colmenero J. Segmental and Normal Mode Relaxation of Poly(alkylene Oxide)s Studied by Dielectric Spectroscopy and Rheology. Macromolecules 2010, 43, 4968–4977. 10.1021/ma100384j. [DOI] [Google Scholar]
- Vieweg R.; Hochtler A.. Kunstoff-Handbuck. In Band VII, Polyurethane; Carl Hanser Verlag: Munchen, 1966; p 37. [Google Scholar]
- Chattopadhyay D. K.; Webster D. C. Thermal Stability and Flame Retardancy of Polyurethanes. Prog. Polym. Sci. 2009, 34, 1068–1133. 10.1016/j.progpolymsci.2009.06.002. [DOI] [Google Scholar]
- Van Bogart J. W. C.; Gibson P. E.; Cooper S. L. Structure-Property Relationships in Polycaprolactone-Polyurethanes. Jounal Polym. Sci.: Polym. Phys. Ed. 1983, 21, 65–95. 10.1002/pol.1983.180210106. [DOI] [Google Scholar]
- Müller M.; Seidel U.; Stadler R. Influence of Hydrogen Bonding on the Viscoelastic Properties of Thermoreversible Networks: Analysis of the Local Complex Dynamics. Polymer 1995, 36, 3143–3150. 10.1016/0032-3861(95)97877-I. [DOI] [Google Scholar]
- Seiffert S.; Sprakel J. Physical Chemistry of Supramolecular Polymer Networks. Chem. Soc. Rev. 2012, 41, 909–930. 10.1039/C1CS15191F. [DOI] [PubMed] [Google Scholar]
- Bose R. K.; Hohlbein N.; Garcia S. J.; Schmidt A. M.; van der Zwaag S. Connecting Supramolecular Bond Lifetime and Network Mobility for Scratch Healing in Poly(butyl Acrylate) Ionomers Containing Sodium, Zinc and Cobalt. Phys. Chem. Chem. Phys. 2015, 17, 1697–1704. 10.1039/C4CP04015E. [DOI] [PubMed] [Google Scholar]
- Kapnistos M.; Vlassopoulos D.; Roovers J.; Leal L. G. Linear Rheology of Architecturally Complex Macromolecules: Comb Polymers with Linear Backbones. Macromolecules 2005, 38, 7852–7862. 10.1021/ma050644x. [DOI] [Google Scholar]
- Kirkwood K. M.; Leal G. L.; Vlassopoulos D.; Driva P.; Hadjichristidis N. Stress Relaxation of Comb Polymers with Short Branches. Macromolecules 2009, 42, 9592–9608. 10.1021/ma900950s. [DOI] [Google Scholar]
- Kempf M.; Ahirwal D.; Cziep M.; Wilhelm M. Synthesis and Linear and Nonlinear Melt Rheology of Well-Defined Comb Architectures of PS and PpMS with a Low and Controlled Degree of Long-Chain Branching. Macromolecules 2013, 46, 4978–4994. 10.1021/ma302033g. [DOI] [Google Scholar]
- Trinkle S.; Friedrich C. Van Gurp-Palmen-Plot: A Way to Characterize Polydispersity of Linear Polymers. Rheol. Acta 2001, 40, 322–328. 10.1007/s003970000137. [DOI] [Google Scholar]
- Trinkle S.; Walter P.; Friedrich C. Van Gurp-Palmen Plot II - Classification of Long Chain Branched Polymers by Their Topology. Rheol. Acta 2002, 41, 103–113. 10.1007/s003970200010. [DOI] [Google Scholar]
- Aida T.; Meijer E. W.; Stupp S. I. Functional Supramolecular Polymers. Science 2012, 335, 813–817. 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godovsky Y. K.; Slonimsky G. L. Kinetics of Polymer Crystallization from the Melt (Calorimetric Approach). J. Polym. Sci.: Polym. Phys. Ed. 1974, 12, 1053–1080. 10.1002/pol.1974.180120603. [DOI] [Google Scholar]
- Nichetti D.; Grizzuti N. Determination of the Phase Transition Behavior of Thermoplastic Polyurethanes from Coupled rheology/DSC Measurements. Polym. Eng. Sci. 2004, 44, 1514–1521. 10.1002/pen.20148. [DOI] [Google Scholar]
- Fernández-d’Arlas B.; Baumann R. P.; Pöselt E.; Müller A. J. Influence of Composition on the Isothermal Crystallisation of Segmented Thermoplastic Polyurethanes. CrystEngComm 2017, 19, 4720–4733. 10.1039/C7CE01028A. [DOI] [Google Scholar]
- Mansfield K. F.; Theodorou D. N. Molecular Dynamics Simulation of a Glassy Polymer Surface. Macromolecules 1991, 24, 6283–6294. 10.1021/ma00023a034. [DOI] [Google Scholar]
- Baschnagel J.; Binder K. On the Influence of Hard Walls on Structural Properties in Polymer Glass Simulation. Macromolecules 1995, 28, 6808–6818. 10.1021/ma00124a016. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.